Introduction
Cutaneous T-cell lymphoma (CTCL) is a rare
malignancy of skin-homing CD4+ T-cells. The majority of
patients suffer from non-life threatening skin symptoms that,
however, significantly decrease their quality of life. In Sézary
syndrome (SS), an aggressive leukemic variant of CTCL accounting
for approximately 15% of CTCL cases, the clinical symptoms
encompass erythroderma, lymphadenopathy, hepato-and/or
splenomegaly, plantar keratoderma and alopecia (1). The prognosis is poor with overall
treatment response duration rates varying from 7.5 to 22.4 months
(2). Apart from standard therapies
e.g., extracorporeal photopheresis (ECP), photochemotherapy,
retinoids, radiation therapy, interferon (IFN)-α, low-dose
methotrexate and polychemotherapy, small-molecule inhibitors and
monoclonal antibodies (mAbs) are currently being investigated for
the treatment of this malignancy (3,4).
However, despite considerable progress being made in the management
of the disease, allogeneic stem cell transplantation is currently
the only curative option.
Recently, targeting epigenetic mechanisms has
emerged as a novel therapeutic strategy in CTCL (5,6). To
date, two inhibitors of the histone deacetylase (HDAC) family,
which comprises 18 enzymes with divergent functions, have been
registered in the treatment of CTCL. However, the response rate to
these agents remains unsatisfactory, amounting to a 30–40% overall
response, followed by the acquirement of resistance (7). Moreover, one of the issues concerning
the use of non-specific HDAC inhibitors are their gastrointestinal
adverse effects. Thus, the development of selective inhibitors of
single HDAC isoforms is considered to provide novel therapeutic
options with milder side-effects (8).
It has been demonstrated that the HDAC6 isoform is
overexpressed in primary samples from patients with CTCL (9) and that it is an attractive molecular
target in preclinical experiments using a mouse model of CTCL
(10). These data suggest that
combination schemes including HDAC6-specific inhibitors may be
successful in the treatment of CTCL. Moreover, a decrease in the
intensity of pruritus, one of the main clinical issues associated
with the management of CTCL, has been observed with the use of
non-selective HDAC inhibitors already in use (5), as well as with the selective HDAC6
inhibitor, ricolinostat, currently tested in clinical trials on
non-Hodgkin lymphoma (11).
Phosphatidylinositol 3-kinase (PI3K) inhibition is
currently being tested in clinical trials on CTCL with encouraging
results (12). PI3Ks are lipid
kinases involved in intracellular signal transduction. In humans,
four (α, β, δ and γ) catalytic subunits of PI3K exist. The δ and γ
isoforms are preferentially expressed in leukocytes, where they
control the survival and proliferation of leukocytes. Moreover,
PI3K plays a critical role in malignant transformation (13). The anti-tumor activity of PI3K
inhibition in hematological malignancies relies on the blocking of
survival signaling within tumor cells, as well as in non-malignant
cells of the microenvironment by controlling cytokine secretion and
activating the immune response (13,14).
Inhibitors of PI3K have already been successfully applied in the
management of B-cell malignancies (15,16). The
results of a clinical study on T-cell lymphoma revealed an
acceptable safety profile (12).
Moreover, preclinical evidence of both tumor cell-autonomous and
immune-mediated effects in patient-derived xenografts has been
reported (12). PI3K inhibitors have
already been demonstrated to synergize with the non-specific HDAC
inhibitor, vorinostat (17).
However, to date, at least to the best of our knowledge, no HDAC
isoform responsible for this effect has been identified. As HDAC6
is known to deacetylate and to diminish the activation of Akt, a
downstream kinase of the PI3K pathway (18), the aim of this study was to evaluate
the therapeutic potential of selective HDAC6 inhibitors in
combination with PI3K inhibitors in CTCL.
Materials and methods
Cell lines and reagents
The HH, H9 and HUT78 cell lines were a generous gift
from Dr Giandomenico Russo (Istituto Dermopatico dell'Immacolata,
IDI–IRCCS, Rome, Italy). The SeAX cell line was a generous gift
from Dr Katarzyna Izykowska (Institute of Human Genetics, Polish
Academy of Science, Poznan, Poland). 293T cells were purchased from
ATCC. The cells were maintained in RPMI-1640 medium (Invitrogen;
Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine
serum, L-glutamine (2 mM), penicillin (100 U/ml) and streptomycin
(100 µg/ml). HDAC inhibitors (ricolinostat, citarinostat and
resminostat), as well as PI3K inhibitors (duvelisib, copanlisib and
pictilisib) were purchased from Selleckchem. Inhibitors were
dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich; Merck KGaA)
or 10% trifluoric acid (Sigma-Aldrich; Merck KGaA) in the case of
copanlisib.
Primary samples
Peripheral mononuclear blood cells (PBMCs) from
patients with SS (n=4) and healthy volunteers (n=2) were isolated
from whole blood using Histopaque 1077, according to the
manufacturer's recommendations (Sigma-Aldrich; Merck KGaA). The
diagnosis of SS was established in all patients according to the
World Health Organization-European Organization for Research and
Treatment of Cancer criteria for SS. Blood was obtained from
patients with SS at the University of Zurich Biobank (EK No. 647).
This study was conducted in accordance with the principles of the
Declaration of Helsinki and was approved by the Institutional
Review Board of the University of Zurich (KEK-ZH-Nr. 2015-0209).
Patient 3 took part in NCT02953301 clinical trial [Resminostat for
Maintenance Treatment of Patients With Advanced Stage Mycosis
Fungoides (MF) or SS (RESMAIN)]. Each patient signed an informed
consent for the procedures.
Viability assays
Cell viability was assessed using flow cytometry or
ATP activity measurement. For flow cytometry, anti-human monoclonal
antibodies were used as listed: CD3 (clone BW264/56, PerCP;
Miltenyi Biotec #130-096-910), CD4 (clone VIT4; APC-Vio770;
Miltenyi Biotec #130-098-153), CD8 (clone RPA-T8, PE; BD
Biosciences). The following reagents were used to discriminate dead
cells: Annexin V (FITC; BD Biosciences), SYTOX (APC; Thermo Fisher
Scientific) and propidium iodide (Sigma-Aldrich). For ATP activity,
the Cell Titer Glo® Luminescent Cell Viability Assay
(Promega) was used. Samples were acquired on a FACSCantoII (BD
Biosciences). FCS Express 5 Flow Cytometry RUO, Origin Pro 9.1G and
GraphPad Prism 5.0 Software were used for data analysis.
Generation of sgHDAC and lentiviral
transduction
Two sgHDAC6 sequences (A and B) and sgRNA targeting
green fluorescent protein (GFP) used as a control sequence (CON)
were generated with oligonucleotide pairs (Table I) and cloned into pLenti-CRISPRv2.
293T cells seeded in 6-well plates were used for the production of
a replication-incompetent lentivirus. The cells were first
co-transfected with 0.76 µg of pLenti-CRISPRv2 and components of
2nd generation of packaging vectors, namely 0.76 µg of psPAX2 and
0.5 µg of pMD2.G, using standard calcium chloride method. At 48 h
post-transfection, the lentiviruses- containing medium was
collected and added to target cells at the volume ratio of 1:1. Two
days later, 2 µg/ml puromycin was added for an additional week.
Single clones were obtained from resistant cell pools by limiting
dilution.
 | Table I.sgRNA sequences used for the
production of lentiviral CRISPR/Cas9 knock-out. |
Table I.
sgRNA sequences used for the
production of lentiviral CRISPR/Cas9 knock-out.
| Sequence name | Sequence (5′-3′) |
|---|
| sgCON |
GGGCGAGGAGCTGTTCACCG |
| sgA |
CACCGAGGACACGCAGCGATCTAGG |
| sgB |
CACCGCCTCTAGGATAAGGATAATG |
Western blot analysis
Whole-cell protein extracts were prepared by lysing
cells with a buffer containing 1% Triton X-100, 50 mM HEPES (pH
7.4), 150 mM NaCl, 5 mM EDTA and 10% glycerol supplemented freshly
with phosphatase and protease inhibitor cocktails. Protein
concentration was measured with a Pierce BCA Protein Assay kit
(Thermo Fisher Scientific, Inc.). Samples of 5–25 µg total protein
were subjected to SDS-PAGE. Resolved proteins were transferred onto
nitrocellulose membranes, probed with specific antibodies, and
detected with Odyssey imaging system (LI-COR Biosciences). The
following primary antibodies were used: Anti-HDAC6 (D2E5; Cell
Signaling Technology, Inc.) and anti-acetylated tubulin
(polyclonal, Sigma-Aldrich; Merck KGaA). Secondary antibodies
included either HRP-conjugated antibodies from Jackson
ImmunoResearch or fluorophore-conjugated (IRDye 680RD or IRDye
800CW) antibodies from LI-COR Biosciences. For equal loading, the
control blots were re-probed with anti-β-actin-peroxidase (AC-15,
Sigma-Aldrich; Merck KGaA).
Statistical analysis
Results were plotted with GraphPad Prism.
Statistical significance was assessed by appropriate tests
specified in the figure legends, namely Mann-Whitney U test and
two-way ANOVA with multiple comparisons with Bonferroni post-hoc
test. The P-values are marked with asterisks on the charts
(*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001). The
Chou-Talalay method was used to quantify the combination index (CI)
of the tested combinations [synergistic effect (CI<1)].
Results
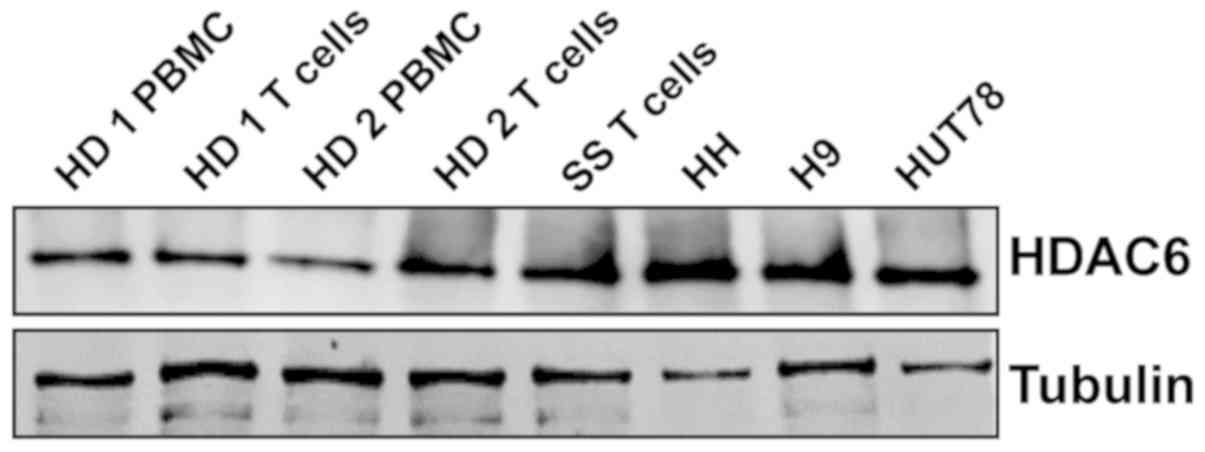
CTCL cell lines and human primary
T-cells from SS patients express HDAC6
First, the amount of HDAC6 at the mRNA and protein
level was assessed in whole cell lysates of PBMCs from healthy
donors and T-cells, as well as in CD4+ cells from one
patient with SS and established CTCL cell lines. The results
(Fig. 1) demonstrated a strong
expression of HDAC6 protein in all tested CTCL cell lines and
patient samples, despite low mRNA levels (data not shown).
HDAC6 inhibitors sensitize CTCL cells
to PI3K inhibitors
To evaluate the effect of HDAC6 inhibition on the
sensitivity to PI3K inhibitors, the viability of two CTCL cell
lines representing SS (HUT78 and HH) upon 48 h of incubation with
the tested inhibitors was examined by flow cytometry. To block
HDAC6 activity, the selective HDAC6 inhibitor, ricolinostat, was
used. HDAC6 inhibition was assessed by western blot analysis
(Fig. 2A). Three different PI3K
inhibitors were tested: the δ/γ-specific inhibitor, duvelisib, the
pan-inhibitor, copanlisib, and the pan inhibitor, pictilisib. The
results demonstrated a synergistic effect (CI<1) of increasing
concentrations of ricolinostat in combination with all tested PI3K
inhibitors in both cell lines (Fig.
2B). Moreover, in the corresponding experiments with primary
T-cells isolated from patients with CTCL, a considerable, although
not statistically significant potentiation of the anti-tumor
activity of the tested combinations was observed, as determined by
SYTOX/Annexin flow cytometry viability assay (Fig. 2C) and ATP-activity Cell Titer
Glo® proliferation assay (Fig. 2D). No such effect was observed in the
case of normal CD4+ T-cells (data not shown).
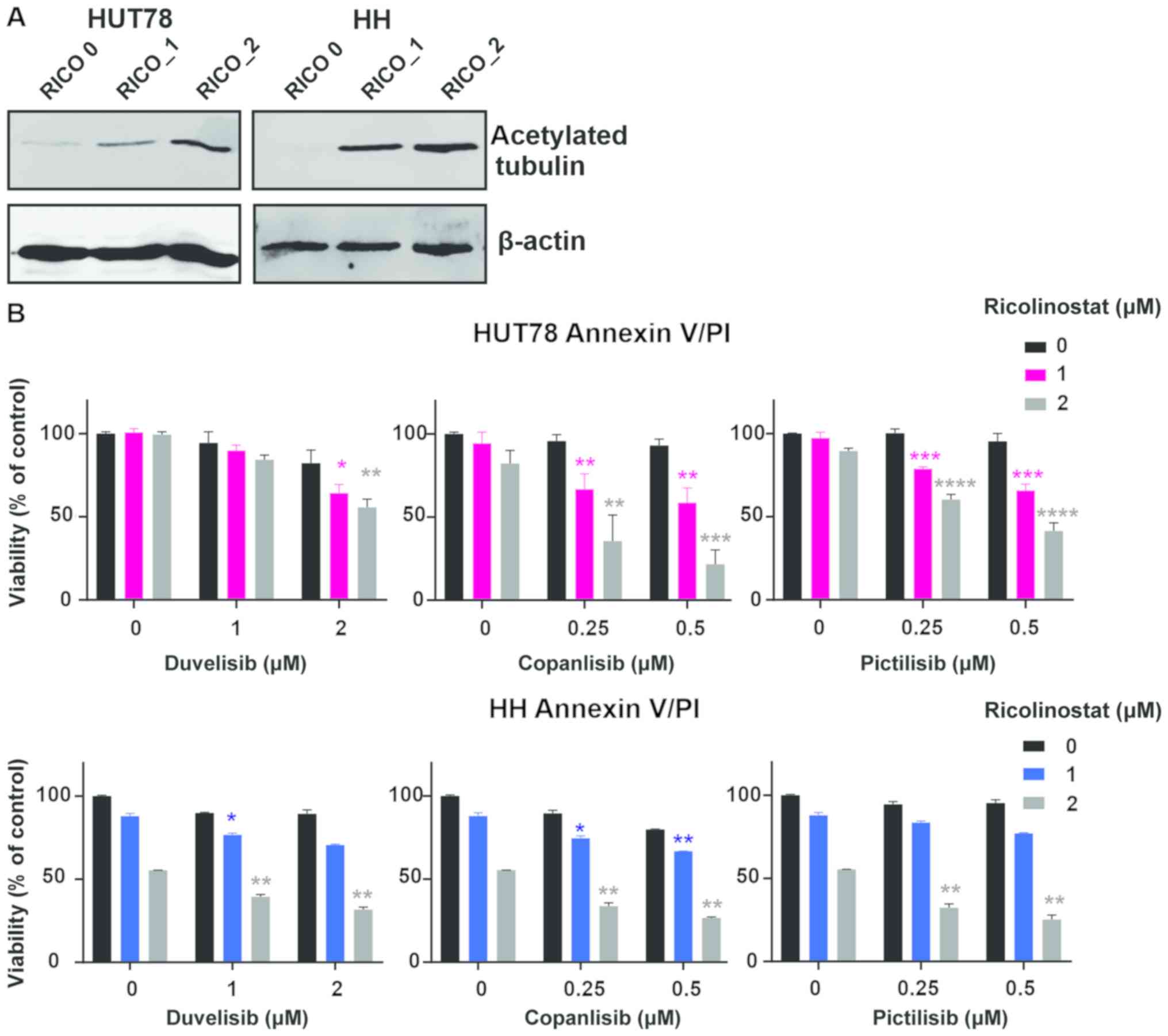 | Figure 2.HDAC6 inhibitors sensitize CTCL cells
to PI3K inhibitors. (A and B) Established CTCL cell lines (HUT78
and HH) were co-incubated for 48 h with increasing concentrations
of the HDAC6 specific inhibitor, ricolinostat, and the following
PI3K inhibitors: The non-selective inhibitor, copanlisib, the
γ/δ-selective inhibitor, duvelisib, and the pan-inhibitor
pictilisib. (A) Levels of acetylated tubulin (a hallmark of HDAC6
inhibition) were assessed by western blot analysis in whole-cell
lysates of HUT78 and HH cell lines incubated with 0, 1 and 2 µM
ricolinostat (RICO_0,1,2). β-actin was used as a loading control.
(B) Cell death was assessed by flow cytometry using propidium
iodide or SYTOX combined with Annexin V and proliferation was
assessed by ATP quantification with Cell Titer Glo®.
Statistical significance was assessed using the Mann-Whitney U
test; *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
HDAC6 inhibitors sensitize CTCL cells to PI3K inhibitors. (C and D)
HDAC6 inhibitors sensitize CTCL primary cells to PI3K inhibitors.
PBMCs from a leukemic patient with CTCL were isolated using
standard Ficoll centrifugation. (C) Cells were then co-incubated
for 48 h in the presence of IL-2 and IL-15 stimulation with the
HDAC 1/3/6-specific inhibitor, resminostat, or the HDAC6-specific
molecules, ricolinostat and citarinostat (all at a 4 µM
concentration), in combination with the PI3K inhibitors,
copanlisib, pictilisib and duvelisib (1 µM). Cell viability was
assessed by flow cytometry following Annexin V/SYTOX staining of
CD3+ CD4+ cells. The figure shows the
percentages of necrotic (upper panel) and apoptotic (lower panel)
cells. (D) In addition, ATP activation was assessed using Cell
Titer Glo®. HDAC6 inhibitors sensitize CTCL cells to
PI3K inhibitors. (E and F) HDACi-based therapy sensitizes primary
CTCL cells to PI3K inhibitors. PBMCs from leukemic patients with
CTCL ‘on and off’ therapy with (E) the HDAC pan-inhibitor,
vorinostat, and (F) the HDAC1/3/6 specific inhibitor, ricolinostat,
were then co-incubated for 48 h in the presence of IL-2 and IL-15
with 1 and 2 µM PI3K inhibitors, copanlisib, pictilisib and
duvelisib. Cell viability was assessed by ATP quantification using
Cell Titer Glo®. Statistical significance was assessed
by two-way ANOVA with multiple comparisons; *P<0.05. RICO,
ricolinostat; CITA, citarinostat; RESMI, resminostat; CTCL,
cutaneous T-cell lymphoma; HDAC6, histone deacetylase 6. |
Treatment with HDAC inhibitors
sensitizes primary patient cells to PI3K
To assess the clinical relevance of these
observations, the effects of the clinically available pan-HDAC
inhibitor, vorinostat (Fig. 2E), and
the clinically tested selective HDAC1/3/6 inhibitor, ricolinostat
(Fig. 2F), on the efficacy of PI3K
inhibition were examined. In these experiments, the in vitro
sensitivity to PI3K inhibitors of primary cells isolated from
patients treated in vivo with HDAC inhibitors was assessed.
In the vorinostat-treated patient (Patient 2), a significant
sensitization to all tested PI3K inhibitors was observed, while
treatment with ricolinostat did not exert a significant effect in
Patient 3. However, it should be noted that Patient 3 received
ricolinostat only twice, while Patient 2 was continuing vorinostat
on a regular basis.
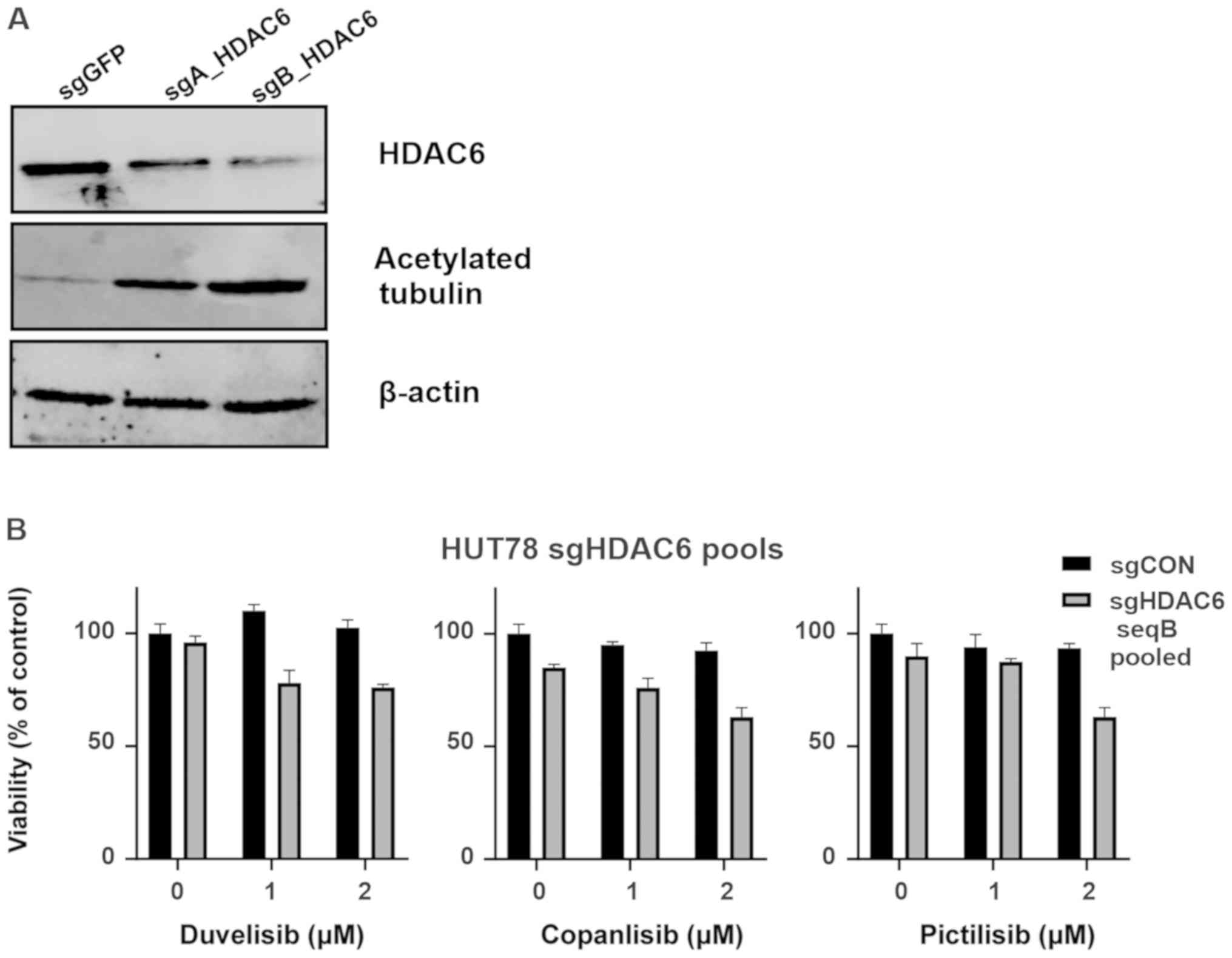
HDAC6 knockout sensitizes the HUT78
CTCL cell line to PI3K inhibitors
In order to determine whether the observed
sensitization to PI3K relies on the specific inhibition of HDAC6,
HUT78 cells were stably transduced using CRISPR/Cas9 sgRNA
constructs targeting HDAC6. Following puromycin selection, 2 cell
lines (HUT78-sgA_HDAC6 and HUT78-sgB_HDAC6) were obtained that were
both characterized by increased levels of acetylated tubulin (a
surrogate marker for HDAC6 inhibition). The HUT78-sgB_HDAC6 cells
exhibited decreased HDAC6 levels and profoundly increased tubulin
acetylation (Fig. 3A); thus, these
cells were used to examine the efficacy of PI3K inhibitors. Some
sensitization to all tested PI3K inhibitors was observed; however,
the effect did not reach statistical significance (Fig. 3B). Therefore, single clones were
obtained from the HUT78-sgB_HDAC6 cells by limiting dilution. Two
obtained clones (sg9 and sg10) were found to exhibit HDAC6 knockout
(Fig. 3C). Again, clones were tested
for their sensitivity to PI3K inhibition. Both the sg9 and sg10
clones were characterized with a significantly increased
sensitivity to PI3K inhibition (Fig.
3D).
Discussion
SS represents an aggressive leukemic variant of CTCL
with a poor prognosis. Despite their established position in the
treatment of CTCL, HDAC inhibitors do not lead to durable
remissions. However, preclinical data suggest the potential role of
HDAC inhibitors in combination treatment. As the pan-HDACi
inhibitor, vorinostat, has already been demonstrated to sensitize
CTCL to PI3K inhibition, this study examined whether this mode of
action may result from the inhibition of a single HDAC isoform,
HDAC6. HDAC6 is an isoform shown to be overexpressed in CTCL and a
druggable target with demonstrated preclinical efficacy in B-cell
malignancies. HDAC6 inhibition using its small-molecule inhibitor,
ricolinostat, has been shown to exert a direct tumor-killing
effect, as well as to sensitize malignant cells to a variety of
drugs with different mechanisms of action (11,19,20).
Importantly, data from clinical trials suggest its improved safety
profile when compared to non-specific HDAC inhibitors (21). PI3K inhibition has recently been
proposed as a novel treatment option for CTCL. In this study, it
was demonstrated that the anti-tumor effects of PI3K inhibitors can
be further potentiated by HDAC6 inhibition. In this study setting,
a synergistic effect of HDAC6 inhibition was observed, combined
with three different both isoform-specific, as well as pan-PI3K
inhibitors on CTCL established cell lines. Moreover, an additive
effect was observed on primary CD4+ cells obtained from
patients with SS, that are regularly observed to be
apoptosis-resistant. In this study, using both pharmacological
(HDAC6-selective inhibitor), as well as genetic tools (CRISRP/Cas9
system) it was demonstrated that the observed sensitization to PI3K
inhibition relies on HDAC6 blockage. Although the mechanistic basis
of this phenomenon requires additional functional experiments, the
combination of PI3K with HDAC6 inhibitors seems to be a rational
approach with which to enhance the cytotoxic effects in CTCL. The
results of this study contribute to the opening of new avenues for
the use of specific HDAC-inhibitors in CTCL (22). To date, the expression of HDAC2 has
been linked to a more aggressive phenotype of CTCL (9). In addition, the selective targeting of
HDAC3 may be an option for CTCL, as it has been shown to disrupt
the DNA replication of rapidly cycling CTCL cells (23). Moreover, quisinostat, an inhibitor
with the highest potency to inhibit HDAC1, has demonstrated
encouraging results in modifying the phenotype of the HUT78 cell
line (24). Therefore, inhibitors of
class I HDACs seem to be promising in this type of malignancy.
Indeed, domatinostat (4SC-202), a novel inhibitor of class I HDACs,
has been demonstrated to effectively inhibit the growth of CTCL
cells (25). It is anticipated that
in the future, novel isoform-specific compounds will emerge.
Acknowledgements
This abstract was presented at the EORTC Cutaneous
Lymphoma Task Force Meeting 2018 (September 27, 2018-September 29,
2018; St. Gallen, Switzerland), and was published as Abstract no.
025.
Funding
The present study was supported by the European
Commission Horizon 2020 Program 692180- STREAMH2020-TWINN-2015 (to
MB, JS, MD and MW), the Polish National Science Centre
2015/18/E/NZ6/00702 (to MB, JS and MW), EMBO (short-term fellowship
no. 7637 to MB), the Ministry of Science and Higher Education
(2019/94/DIR/NN3 to MB) the Promedica Stiftung (1406/M and 1412/M,
both to EG), the Swiss Cancer Research Foundation (KFS-4243-08-2017
to EG) and the Clinical Research Priority Program (CRPP) of the
University of Zurich (to EG). The funders had no role in the study
design, data collection and analysis, decision to publish, or
preparation of the manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JD, MD, DI, YTC, CI, AS and KM performed the
experiments and analyzed the data. MB and NMZ performed the
experiments and analyzed the data, and were involved in the
manuscript preparation. EG and MW contributed to the conception and
design of the study, were involved in drafting the manuscript and
data interpretation, and provided expert advice and guidance
throughout the study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was conducted in accordance with
the principles of the Declaration of Helsinki and approved by the
Institutional Review Board of the University of Zurich (KEK-ZH-Nr.
2015-0209). Each patient provided written informed consent for the
procedures.
Patient consent for publication
The patients provided consent for the publication of
the results.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CTCL
|
cutaneous T-cell lymphoma
|
|
HDAC6
|
histone deacetylase 6
|
|
mAbs
|
monoclonal antibodies
|
|
PBMC
|
peripheral mononuclear blood cells
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
SS
|
Sézary syndrome
|
References
|
1
|
Mangold AR, Thompson AK, Davis MD, Saulite
I, Cozzio A, Guenova E, Hodak E, Amitay-Laish I, Pujol RM,
Pittelkow MR and Gniadecki R: Early clinical manifestations of
Sezary syndrome: A multicenter retrospective cohort study. J Am
Acad Dermatol. 77:719–727. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Janiga J, Kentley J, Nabhan C and Abdulla
F: Current systemic therapeutic options for advanced mycosis
fungoides and Sezary syndrome. Leuk Lymphoma. 59:562–577. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guenova E, Hoetzenecker W, Rozati S,
Levesque MP, Dummer R and Cozzio A: Novel therapies for cutaneous
T-cell lymphoma: What does the future hold? Expert Opin Investig
Drugs. 23:457–467. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Saulite I, Hoetzenecker W, Weidinger S,
Cozzio A, Guenova E and Wehkamp U: Sezary syndrome and atopic
dermatitis: Comparison of immunological aspects and targets. Biomed
Res Int. 2016:97175302016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Moskowitz AJ and Horwitz SM: Targeting
histone deacetylases in T-cell lymphoma. Leuk Lymphoma.
58:1306–1319. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Quaglino P, Maule M, Prince HM, Porcu P,
Horwitz S, Duvic M, Talpur R, Vermeer M, Bagot M, Guitart J, et al:
Global patterns of care in advanced stage mycosis fungoides/Sezary
syndrome: A multicenter retrospective follow-up study from the
cutaneous lymphoma international consortium. Ann Oncol. 30:4942019.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wilcox RA: Cutaneous T-cell lymphoma: 2017
update on diagnosis, risk-stratification, and management. Am J
Hematol. 92:1085–1102. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
West AC and Johnstone RW: New and emerging
HDAC inhibitors for cancer treatment. J Clin Invest. 124:30–39.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marquard L, Gjerdrum LM, Christensen IJ,
Jensen PB, Sehested M and Ralfkiaer E: Prognostic significance of
the therapeutic targets histone deacetylase 1, 2, 6 and acetylated
histone H4 in cutaneous T-cell lymphoma. Histopathology.
53:267–277. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mishra A, La Perle K, Kwiatkowski S,
Sullivan LA, Sams GH, Johns J, Curphey DP, Wen J, McConnell K, Qi
J, et al: Mechanism, consequences, and therapeutic targeting of
abnormal IL15 signaling in cutaneous T-cell lymphoma. Cancer
Discov. 6:986–1005. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Amengual JE, Prabhu SA, Lombardo M, Zullo
K, Johannet PM, Gonzalez Y, Scotto L, Serrano XJ, Wei Y, Duong J,
et al: Mechanisms of acquired drug resistance to the HDAC6
selective inhibitor ricolinostat reveals rational drug-drug
combination with ibrutinib. Clin Cancer Res. 23:3084–3096. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Horwitz SM, Koch R, Porcu P, Oki Y,
Moskowitz A, Perez M, Myskowski P, Officer A, Jaffe JD, Morrow SN,
et al: Activity of the PI3K-δ, γ inhibitor duvelisib in a phase 1
trial and preclinical models of T-cell lymphoma. Blood.
131:888–898. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vanhaesebroeck B, Guillermet-Guibert J,
Graupera M and Bilanges B: The emerging mechanisms of
isoform-specific PI3K signalling. Nat Rev Mol Cell Biol.
11:329–341. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kaneda MM, Messer KS, Ralainirina N, Li H,
Leem CJ, Gorjestani S, Woo G, Nguyen AV, Figueiredo CC, Foubert P,
et al: PI3Kγ is a molecular switch that controls immune
suppression. Nature. 539:437–442. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Balakrishnan K, Peluso M, Fu M, Rosin NY,
Burger JA, Wierda WG, Keating MJ, Faia K, O'Brien S, Kutok JL and
Gandhi V: The phosphoinositide-3-kinase (PI3K)-delta and gamma
inhibitor, IPI-145 (Duvelisib), overcomes signals from the
PI3K/AKT/S6 pathway and promotes apoptosis in CLL. Leukemia.
29:1811–1822. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Flinn IW, Hillmen P, Montillo M, Nagy Z,
Illes A, Etienne G, Delgado J, Kuss BJ, Tam CS, Gasztonyi Z, et al:
The phase 3 DUO trial: Duvelisib vs ofatumumab in relapsed and
refractory CLL/SLL. Blood. 132:2446–2455. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wozniak MB, Villuendas R, Bischoff JR,
Aparicio CB, Martinez Leal JF, de La Cueva P, Rodriguez ME,
Herreros B, Martin-Perez D, Longo MI, et al: Vorinostat interferes
with the signaling transduction pathway of T-cell receptor and
synergizes with phosphoinositide-3 kinase inhibitors in cutaneous
T-cell lymphoma. Haematologica. 95:613–621. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Iaconelli J, Lalonde J, Watmuff B, Liu B,
Mazitschek R, Haggarty SJ and Karmacharya R: Lysine deacetylation
by HDAC6 regulates the kinase activity of AKT in human neural
progenitor cells. ACS Chemical Biology. 12:2139–2148. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cosenza M, Civallero M, Marcheselli L,
Sacchi S and Pozzi S: Ricolinostat, a selective HDAC6 inhibitor,
shows anti-lymphoma cell activity alone and in combination with
bendamustine. Apoptosis. 22:827–840. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bobrowicz M, Dwojak M, Pyrzynska B,
Stachura J, Muchowicz A, Berthel E, Dalla-Venezia N, Kozikowski M,
Siernicka M, Miazek N, et al: HDAC6 inhibition upregulates CD20
levels and increases the efficacy of anti-CD20 monoclonal
antibodies. Blood. 130:1628–1638. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vogl DT, Raje N, Jagannath S, Richardson
P, Hari P, Orlowski R, Supko JG, Tamang D, Yang M, Jones SS, et al:
Ricolinostat, the first selective histone deacetylase 6 inhibitor,
in combination with bortezomib and dexamethasone for relapsed or
refractory multiple myeloma. Clin Cancer Res. 23:3307–3315. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lopez AT, Bates S and Geskin L: Current
status of HDAC inhibitors in cutaneous T-cell lymphoma. Am J Clin
Dermatol. 19:805–819. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wells CE, Bhaskara S, Stengel KR, Zhao Y,
Sirbu B, Chagot B, Cortez D, Khabele D, Chazin WJ, Cooper A, et al:
Inhibition of histone deacetylase 3 causes replication stress in
cutaneous T cell lymphoma. PLoS One. 8:e689152013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mehul B, Perrin A, Grisendi K, Galindo AN,
Dayon L, Menigot C, Rival Y and Voegel JJ: Mass spectrometry and
DigiWest technology emphasize protein acetylation profile from
quisinostat-treated HuT78 CTCL cell line. J Proteomics.
187:126–143. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wobser M, Weber A, Glunz A, Tauch S, Seitz
K, Butelmann T, Hesbacher S, Goebeler M, Bartz R, Kohlhof H, et al:
Elucidating the mechanism of action of domatinostat (4SC-202) in
cutaneous T cell lymphoma cells. J Hematol Oncol. 12:302019.
View Article : Google Scholar : PubMed/NCBI
|