Introduction
Colorectal cancer (CRC) has the second highest
cancer- associated mortality rate worldwide and is the third most
commonly diagnosed neoplasm; a total of 881,000 CRC-associated
mortalities and 1.8 million newly diagnosed cases were reported in
2018 (1). Sporadic CRC may be caused
by a hereditary mutation, such as in patients with familial
adenomatous polyposis with bi-allelic inactivation of the
adenomatous polyposis coli (APC) gene or in patients with Lynch
syndrome. The remaining cases exhibit activating mutations in
components of the Wnt signaling pathway that promote malignant cell
proliferation (2). Additionally,
sustained inflammation in the intestine, such as in patients with
inflammatory bowel disease (IBD), favors the development of
colitis-associated cancer (CAC).
During chronic inflammation in the intestine,
barrier dysfunction increases susceptibility to bacterial
infection. Furthermore, mucosal inflammation results in the
accumulation of mutations that can result in the development of
colon cancer. In CAC, inflammatory cytokines are persistently
present in the intestinal tissue, and in patients with sporadic
cancer, the administration of anti-inflammatory drugs can prevent
or delay the disease, suggesting that inflammatory processes are
involved in the initiation of tumorigenesis (3).
Cytokines serve as a means of communication among
immune, cancer and non-transformed stromal cells in the tumor
microenvironment. The signal transducer and activator of
transcription (STAT) family of proteins serve an important role in
the initiation of malignant transformation and in tumor
establishment, and have been widely studied in experimental models
and in patients with cancer (4).
Upon cytokine binding to its receptor, Janus kinases (JAKs) mediate
STAT phosphorylation. In the nucleus, STAT binds to specific DNA
sequences that result in the transcription of target genes
(5). Interleukin (IL)-6, IL-10 and
IL-23 signaling is mediated by JAK1 and recruits STAT3. Conversely,
STAT6 functions as a transcription factor in the nucleus in
response to IL-4 and IL-13 receptor binding after the activation of
JAK1 and JAK3 (5). STAT6
orchestrates numerous processes beyond immune response, including
cancer cell proliferation, apoptosis resistance, metastasis,
epithelial cell function, chromatin compaction, and DNA damage and
repair (Table I). STAT6 signaling is
frequently activated in malignant cells and regulates several genes
crucial for the immune response, inflammation and proliferation
(5). Persistent activation of STAT6
in different types of cancer results in proliferation, survival and
metastasis, as well as in decreased antitumor immunity. STAT6
polymorphisms have been identified in a subgroup of Malaysian
patients suffering from Crohn's disease (CD), as well as in a
patient cohort in Germany (6–8),
demonstrating the importance of this gene in inflammatory processes
in the colon.
 | Table I.Different roles of STAT6 in colon
cancer. |
Table I.
Different roles of STAT6 in colon
cancer.
| System/process
affected | Mechanism
implicated | First author, year
(ref.) |
|---|
| Immune system | IL-4, IL-13, IL-4R
and IL-13R are differentially expressed in the colonic mucosa of
patients with CRC according to the metastasis and survival
rates. | Formentini et
al, 2012 (28) |
|
| T cells,
macrophages and natural killer T cells exhibit increased STAT6
phosphorylation during colitis development. STAT6−/−
mice exhibit decreased secretion of IL-4, IL-5, IL-13 and
interferon-γ accompanied by disease amelioration. | Rosen et al,
2013 (29) |
|
| STAT6 signaling
contributes to the pathogenesis of colitis by inducing B
cell-dependent mast cell activation. | Hoving et
al, 2012 (30) |
|
| STAT6−/−
mice exhibit reduced tumorigenicity associated with decreased
inflammation and low mRNA levels of IL-17A and tumor necrosis
factor-α in the azoxymethane/DSS model of CAC. | Leon-Cabrera et
al, 2017 (33) |
|
| STAT6 orchestrates
epithelial cell proliferation and myeloid-derived suppressor cell
expansion by decreasing the cytotoxic activity of CD8 T cells in a
mouse model of adenomatous polyposis. | Jayakumar and
Bothwell, 2017 (34) |
|
| The STAT6-specific
inhibitor AS1517499 decreases the number of tumors and circulating
inflammatory monocytes and granulocytes in experimental CAC. | Leon-Cabrera et
al, 2017 (33) |
| Apoptosis | In DSS-induced
colitis, STAT6 deficiency alters chromatin compaction with direct
repercussions in the apoptosis of epithelial cells and mucosal
damage. | De Oliveira et
al, 2019 (35) |
|
| IL-4-dependent
STAT6 signaling pathway favors the expression of anti-apoptotic
proteins and epithelial cell growth in colon cancer in vivo
and in vitro. | Todaro et
al, 2008 (59) |
|
| Abnormal IL-4/STAT6
activation induces survivin expression that in turn increases
apoptosis resistance in colon cancer stem cells. | Di Stefano et
al, 2010 (19) |
| Epithelial
cells | IL-13 drives STAT6
phosphorylation and alters epithelial barrier function by
increasing claudin-2 activity in colitis. | Rosen et al,
2013 (29) |
|
| IL-13/STAT6
signaling pathway serves a pivotal role in reducing matriptase and
prostasin expression, two proteins involved in epithelial barrier
dysfunction. | Buzza et al,
2017 (31) |
|
| Administration of
IL-13 increases production of colonic serotonin and colitis
severity in DSS-induced colitis. | Shajib et
al, 2013 (32) |
|
| The exposure of the
colon cancer HT29 and SW480 cell lines to IL-13 enhances
EMT-promoting factor zinc finger E-box-binding homeobox 1
expression. | Cao et al,
2017 (37) |
|
| E2F transcription
factor 1 increases STAT6 expression in colon cancer cell lines that
in turn leads to expression of EMT drivers in CRC cells. | Chen et al,
2018 (40) |
|
| IL-4 downregulates
E-cadherin expression, altering cell-cell adhesion and favoring
metastasis in CRC. | Kanai et al,
2000 (43) |
|
| STAT6−/−
mice with acute murine colitis exhibit impaired wound healing and
low mucosal expression of Wnt ligands, which are important
mediators in response to epithelial injury. | Cosin-Roger et
al, 2016 (52) |
| Proliferation |
STAT6−/− mice
exhibit reduced colonic expression of cyclooxygenase-2 and nuclear
β-catenin in experimentally induced colon cancer. | Leon-Cabrera et
al, 2017 (33) |
|
| Patients with CAC
exhibit a shift from pSTAT6 to pSTAT3 in colonic epithelial and
mucosal immune cells during neoplastic transformation. | Wick et al,
2012 (36) |
| Metastasis | High STAT6
expression levels concur with lymph node metastasis and low
survival rates in patients with CCR patients. | Wang et al,
2010 (26) |
|
| IL-13Rα2 signaling
favors 11β-hydroxysteroid dehydrogenase type 2 expression, a key
enzyme involved in liver metastasis during CCR development. | Jiang et al,
2016 (38) |
|
| Activation of PI3K,
Akt and SRC in response to IL-13 is associated with increased
levels of IL-13Rα2 in highly metastatic CRC cells. | Barderas et
al, 2012 (39) |
| DNA damage | Increasing IL-4
abundance contributes to NADPH oxidase 1 expression along with
production of reactive oxygen species in human colon cancer
cells. | Liu et al,
2017 (42) |
Despite the advances in the diagnosis and treatment
of CRC, the mortality rates remain high. Therefore, there is a
requirement for the development of alternative therapeutic
strategies for this disease. Interfering with the activity of STAT6
could be a potential strategy to target the action of IL-4 and
IL-13, and the activation of other signaling pathways involved in
tumorigenesis.
The present review describes the direct and indirect
effects of STAT6 on colon tumor transformation and discusses the
role of STAT6 in mucosal biology. Additionally, the current
treatment strategies that target the IL-4/IL-13/STAT6 axis in colon
cancer are summarized.
Overview of the IL-4/IL-13/STAT6 signaling
pathway
STAT6 serves important roles in signal transduction
throughout the cytoplasm and is a transcription factor in the
nucleus. Cytokines and growth factors bind to their cognate
receptors and activate the JAK family. Once activated, JAKs
phosphorylate docking sites in the SH2 domain of STAT molecules.
After phosphorylation, STATs form homodimers or heterodimers, which
are transported into the nucleus where they regulate the expression
of genes (5).
STAT6 activation occurs by IL-4 or IL-13 binding to
their receptors. Currently, three types of receptors have been
described. The IL-4 type I receptor is composed of the IL-4
receptor α (IL-4Rα) chain and the common γ chain (γc). The IL-4Rα
chain has a high affinity for IL-4, while the γc is a component of
receptors that bind IL-2 and IL-9. IL-4 binds to the IL-4Rα chain,
resulting in dimerization with the γc chain and receptor activation
(9). However, in human colon
carcinoma cells, it has been reported that the IL-4Rα chain does
not bind with the γc (10).
The type II receptor is composed of the IL-4Rα and
IL-13 receptor α variant 1 (IL-13Rα1) chains, and is able to bind
to both IL-4 and IL-13. IL-13 binds to the monomer IL-13Rα1 with
low affinity, and requires heterodimerization with IL-4Rα to form a
high-affinity bond. This complex recruits JAKs and leads to the
downstream activation of STAT6 (9).
Additionally, IL-13 can bind to the IL-13 receptor α
variant 2 (IL-13Rα2) chain; this receptor is a monomer that binds
IL-13 with higher affinity than IL-13Rα1 and is expressed in T
cells, B cells and endothelial cells, among others, where it has
been demonstrated to promote tumorigenesis (11). IL-13Rα2 is considered as a decoy
receptor, as it is able to bind IL-13 and prevent it from binding
to IL-13Rα1 (11).
After stimulation of the IL-4Rα receptor, JAK1-3 and
tyrosine kinase 2 (Tyk2) are phosphorylated in the cytoplasmic
tails of the receptor. Once active, JAKs phosphorylate the tyrosine
residues Y575, Y603 and Y631 on the receptor, generating docking
sites for STAT6. When monomers of STAT6 become phosphorylated on
tyrosine Y641, the C-terminal of the SH2 domain usually forms
homodimers (12) that are
translocated to the nucleus, where they can bind DNA and activate
or repress target genes. Therefore, the activity of STAT6 depends
on tyrosine phosphorylation. However, the proximity of subunits may
influence heterodimer formation. The stimulation of B cells with
interferon (IFN) type 1 promotes the generation of STAT2:STAT6
heterodimers (13).
Proper regulation of STAT6 signaling is crucial, and
specific molecules have been identified to regulate this signaling
pathway. Suppressor of cytokine signaling (SOCS) proteins,
particularly SOCS1, repress the activation of JAK1/3 and STAT6, and
affect IL-4-induced proliferation (14). STAT6 signaling induces SOCS1
expression in a negative feedback loop, inhibiting the expression
of STAT6-responsive genes (14).
Additionally, the IL-4/STAT6 signaling pathway induces SOCS3
expression in intestinal epithelial cells. Increased SOCS3 levels
in patients with ulcerative colitis (UC) are indicative of disease
exacerbation (15). STAT6 activation
is responsible for SOCS3 induction and affects the regulation of
STAT1 and STAT3 signaling (15).
Protein tyrosine phosphatase (SHP1) negatively
regulates IL-4/IL-13 signaling and STAT6 gene induction (16). Studies investigating the
overexpression of SHP1 demonstrated a decrease in IL-4/IL-13
signaling, suggesting that tyrosine phosphorylation of STAT6 is
regulated by SHP1 (16). Protein
tyrosine phosphatase 1B (PTP1B) has been demonstrated to
dephosphorylate cytoplasmic and nuclear STAT6, and attenuate its
signaling. In diffuse large B-cell lymphomas, PTP1B contributes to
STAT6 dephosphorylation, thereby enhancing tumorigenesis and
inflammatory processes (17).
Additionally, STAT6 function is regulated by methylation. When
STAT6 is methylated on arginine at the N-terminus, decreased
phosphorylation, nuclear translocation and DNA-binding activity are
observed in response to IL-4 (18).
Several target genes of STAT6 have been identified
and characterized. The STAT6 signal transduction pathway serves an
important role in mediating the biological functions of IL-4 and
IL-13 in processes associated with Th2 immune responses. During
allergic reactions, STAT6 mediates T helper 2 (Th2) cells and
eosinophil recruitment within sites of allergic inflammation, and
is involved in immunoglobulin (Ig) class switching to produce IgE
(5).
However, abnormal STAT6 activation may contribute to
the pathology of cancer by increasing the expression of proteins
involved in proliferation, migration and invasion. The IL-4/STAT6
signaling pathway increases nuclear survivin in CRC stem cells,
allowing them to evade cell death (19). Platelet-derived growth factors
(PDGFs) are secreted by human vascular endothelial cells,
epithelial cells and fibroblasts. PDGF is a survival factor that
inhibits apoptosis, promotes proliferation and stimulates
mesenchymal cell proliferation and migration (20). A previous study suggested that PDGF
and STAT6 have related functions. PDGF is able to activate STAT6 in
fibroblasts to promote their proliferation (21). The IL-13/STAT6 signaling pathway
upregulates PDGF mRNA expression levels in lung fibroblasts, and
STAT6 is required for PDGF-A and PDGF-C gene expression, with
important implications in tumor development (22). Therefore, STAT6 is a key regulatory
molecule with tumor proliferating functions that may aid the
identification of molecular targets for the treatment of colon
cancer.
STAT6 and its role in CRC
Clinically detectable IBD increases the risk of CAC;
patients with UC have an increased risk (2.4-fold) of developing
CAC (23), while patients with CD
have an increased risk (2.59-fold) of developing CAC in the next
two decades, compared with the general population (24).
Expression of the constitutively activated
IL-4/IL-13/STAT6 axis during UC, CD and in colon cancer tissues
suggests that this pathway may contribute to the underlying
pathology of these diseases. In a previous study,
immunohistochemistry analyses were performed to detect
phosphorylated STAT6 (pSTAT6) in colonic tissues of pediatric
subjects with UC or CD, and it was demonstrated that nuclear pSTAT6
was significantly upregulated in the colonic epithelium (25). Furthermore, STAT6 was significantly
upregulated in neoplastic tissues of patients with CRC, and its
expression was associated with lower survival rates and with lymph
node metastasis (26). Gene
expression levels of IL-4, IL-5 and IL-13 were significantly
upregulated in tumors compared with those in normal tissues of
patients with CRC; however, no apparent effect on clinical outcome
was observed (27). By contrast, a
study performed by Formentini et al (28) revealed that IL-4, IL-13, IL-4R and
IL-13R were expressed in CRC specimens, and that high expression
levels of IL-4, IL-4R and IL-13R were associated with a lower
frequency of lymph node metastasis, while IL-13 expression was
associated with a better overall survival rate.
IL-13-induced activation of STAT6 has been
implicated in mediating the host inflammatory cascade in UC. A
mouse model of oxazolone-induced UC demonstrated that IL-13 is
responsible for inducing the expression of the pore-forming tight
junction protein claudin-2, increasing epithelial barrier
permeability and compromising gastrointestinal barrier function
(29). In addition, epithelial
cells, T cells, macrophages and natural killer T cells exhibit
increased STAT6 phosphorylation during colitis development
(29). When colitis was induced in
STAT6-deficient mice, decreased pathology, claudin-2 expression,
and secretion of IL-4, IL-5, IL-13 and IFN-γ were observed
(29). CD4+ Th2 cells
produce IL-13, which favors IgE production by B cells, and induce
mast cell activation, suggesting that STAT6 contributes to the
pathogenesis of colitis (30).
Therefore, IL-13 seems to drive STAT6 phosphorylation, alter
epithelial barrier function, regulate Th2 cytokine production and
mediate the inflammatory response to colitis.
The IL-13/STAT6 signaling pathway may be detrimental
for intestinal epithelial cell function. The downregulation of
matriptase and prostasin, two membrane-anchored serine proteases
important for the epithelial barrier development and for
homeostasis, was observed in a dextran sulfate sodium (DSS)-induced
model of colitis in mice and in colonic tissues from human subjects
with active UC and CD (31). When
STAT6 was inhibited by suberoylanilide hydroxamic acid (SAHA), the
expression levels of matriptase and prostasin were restored and
barrier dysfunction was decreased, implicating STAT6 signaling in
the loss of the barrier-protective protease pathway (31). Enterochromaffin cells, which are
responsible for synthesizing serotonin to increase epithelial cell
secretion, express IL-13R (32). In
a model of DSS-induced colitis, the administration of IL-13
increased colonic serotonin production and exacerbated the severity
of colitis (32). It is unclear
whether these effects are unique to colitis or increase the risk of
developing CRC. Previous studies have revealed that STAT6 serves
important roles in the early steps of colitis-associated
carcinogenesis and that it modulates inflammatory responses, as
well as controlling cell recruitment and proliferation (33,34). CAC
induction in STAT6-deficient mice (STAT6−/−) in a
azoxymethane/DSS model resulted in reduced tumorigenicity,
associated with reduced inflammation, decreased concentrations of
cyclooxygenase-2 (COX2) and nuclear β-catenin protein in the colon,
and decreased mRNA expression levels of cytokines IL-17A and tumor
necrosis factor-α (TNF-α) (33). In
addition, the number of circulating inflammatory monocytes and
granulocytes was decreased in STAT6−/− mice (33). Furthermore, STAT6 deletion in the
ApcMin/+ mouse model reduced the incidence of polyps in
the small intestine and decreased the proliferation of polyp
epithelial cells; this effect was attributed to an expansion of
myeloid-derived suppressor cells (MDSCs), implying regulation of
the antitumor T-cell response in a STAT6-dependent manner (34). Therefore, STAT6 may serve a broad
role in coordinating both polyp cell proliferation and MDSC
expansion. By contrast, Oliveira et al (35) reported that STAT6-deficient mice are
more susceptible to mucosal damage during DSS-induced colitis, and
exhibit enhanced intestinal epithelial cell apoptosis, tissue
injury and inflammatory responses. Chromatin condensation in
intestinal epithelial cells is affected by STAT6 signaling and may
protect cells from apoptosis and severe tissue damage (35). However, the role of STAT6 during
different stages of colitis and tumor cell proliferation has not
been fully elucidated.
The activation and function of STAT6 in colitis
along the continuum of inactive disease to CAC are dynamic. In a
previous study, immunohistochemistry to detect pSTAT1, pSTAT6 and
pSTAT3 in colonic epithelial and mucosal immune cells in patients
with UC and CAC was performed. A shift from predominant STAT6
activation in immune cells to STAT3 activation accompanied the
onset of dysplasia with a concomitant increase in epithelial cell
STAT3 activation in low- and high-grade tumors. STAT6 expression
was frequently detected in normal tissues, but not in CAC tissues
(36). The decrease in pSTAT6
expression in both immune cells and intestinal epithelial cells
indicates that STAT6 signaling may be detrimental in the transition
from colitis to cancer. Nevertheless, abnormal STAT6 signaling has
been implicated in pro-metastatic processes, including the
proliferation and survival of colon cancer cells (37–39).
During the epithelial-to-mesenchymal transition (EMT), epithelial
cells transform into aggressive phenotypes with enhanced migratory
capacity and invasiveness. Exposure to IL-13 enhances the
expression of the EMT-promoting factor zinc finger E-box binding
homeobox 1 (ZEB1) in the colon cancer HT29 and SW480 cell lines
(37). When STAT6 is blocked or
knocked down, the IL-13-induced EMT and ZEB1 induction in CRC cells
is reversed (37). Additionally, a
positive association between IL-13Rα1 and ZEB1 at the mRNA level
has been observed in human CRC samples, demonstrating that the
IL-13/STAT6 signaling pathway serves a critical role in promoting
EMT and the aggressiveness of CRC (37). 11β-hydroxysteroid dehydrogenase type
II (11βHSD2) is a key enzyme induced in an IL-13Rα2-dependent
manner that promotes the expression of Akt and COX2; upon
inhibiting 11βHSD2, liver metastasis is decreased, suggesting that
IL-13 may regulate malignancy via 11βHSD2 during CRC (38). In addition, highly metastatic CRC
cells express high levels of IL-13Rα2 (39). IL-13Rα2 silencing results in a
decrease in adhesion, migration, invasion and metastatic
colonization. In a previous study, the upregulation of IL13Rα2
expression in 66% of tumor samples from patients with colon cancer
was associated with late stages of progression (metastasis in lymph
nodes or liver) and a poor outcome in patients with CRC. Highly
metastatic CRC cells exhibit activation of PI3K, Akt and SRC
proto-oncogene non-receptor tyrosine kinase in response to IL-13,
supporting the role of the IL-13/IL-13R/STAT6 signaling pathway in
CRC cell invasion and metastasis (39).
Additionally, STAT6 phosphorylation is induced by
IL-4. E2F transcription factor 1 (E2F1), a critical transcription
factor for CRC development, increases STAT6 expression in colon
cancer cells and increases their susceptibility to IL-4
stimulation. E2F1 acts as an enhancer of the IL-4/STAT6 signaling
pathway and increases the expression of EMT drivers in CRC cells
(40). IL-4 is able to induce the
expression of anti-apoptotic proteins and the growth of epithelial
colon cancer cells in vivo and in vitro. IL-4
neutralization sensitizes colon carcinoma cells to chemotherapy and
to death by the TNF-related apoptosis-inducing ligand (41). Abnormal IL-4/STAT6 activation induces
the expression of survivin, a protein with an important role in
apoptosis resistance in colon cancer stem cells; the use of
leflunomide, a STAT6 inhibitor, has been shown to decrease survivin
expression and localization, thereby reducing its anti-apoptotic
effect (19). An increase in IL-4
expression contributes to an oxidant milieu, thereby increasing the
expression of NADPH oxidase 1 and reactive oxygen species,
resulting in DNA damage and the neoplastic transformation of colon
cells (42). Further evidence
indicates that IL-4 is implicated in the inhibition of colon cancer
cell-cell adhesions, acting as a negative regulator in the
expression of E-cadherin, a key component of adherent junctions,
and in maintaining epithelial cell adhesion and decreasing
invasiveness (43).
Increasing evidence demonstrates that STAT6 serves
an important role in the regulation of tumor immunity. The use of
the STAT6-specific inhibitor AS1517499 has been shown to decrease
colonic tumor load and the number of circulating inflammatory
monocytes and granulocytes in experimental models of CAC (33). The reduced number of the
aforementioned cells may be associated with decreased macrophage
infiltration and reduced tumor growth. IL-4/IL-13 cytokines, via
STAT6 activation, induce the differentiation of infiltrating
macrophages to an M2 phenotype with pro-tumoral functions. The
pharmacological inhibition of STAT6 with AS1517499 was shown to
attenuate tumor growth and liver metastasis in an orthotopic 4T1
mammary carcinoma mouse model (44).
This result was associated with a decrease in tumor-associated
macrophages displaying the M2 phenotype, suggesting that IL-4
blockade inhibits tumor angiogenesis and growth by reprograming
macrophages to a less aggressive phenotype (45). In addition, the deletion of the STAT6
gene was demonstrated to improve antitumor immunity in the same
model via a type 1 CD4+ response (46).
Autocrine production of IL-4 by colon cancer cells
protects against apoptosis and chemotherapy-induced cell death
(47–50). It was found that IL-4 was highly
expressed in cancer-initiating cells obtained from patients with
CRC and was associated with low immunogenic profiles; blocking IL-4
increased Th1-type CD8+ T cells responses in
vitro (51). Previous studies
also revealed that STAT6 maintains mucosal homeostasis by
sustaining myeloid cells. STAT6−/− mice with acute
murine colitis exhibited delayed wound healing and decreased
mucosal expression levels of Wnt ligands, which are important
responses to epithelial injury (52). The administration of M2 macrophages
to STAT6−/− mice promoted mucosal repair through the
activation of the Wnt signaling pathway; however, mutations that
cause aberrant Wnt signaling are responsible for polyp development
in the small intestine and colon (52). In the APCmin/+ mouse
model, which recapitulates the disease observed in patients with
familial APC, STAT6 promoted the expansion of MDSCs and decreased
the cytotoxicity of CD8 T cells, contributing to intestinal
tumorigenesis (34). The
identification of the direct functions of STAT6 on
immunosurveillance and epithelial cell homeostasis may serve as the
basis for the development of novel immunotherapy strategies.
The aforementioned results demonstrate that the
IL-4/IL-13/STAT6 axis may serve a role in inflammation, tumor cell
proliferation, cancer cell survival and metastasis, and that the
interference of its effects in colitis or in tumor cells may
present a novel strategy for the treatment of CRC (Fig. 1; Table
I).
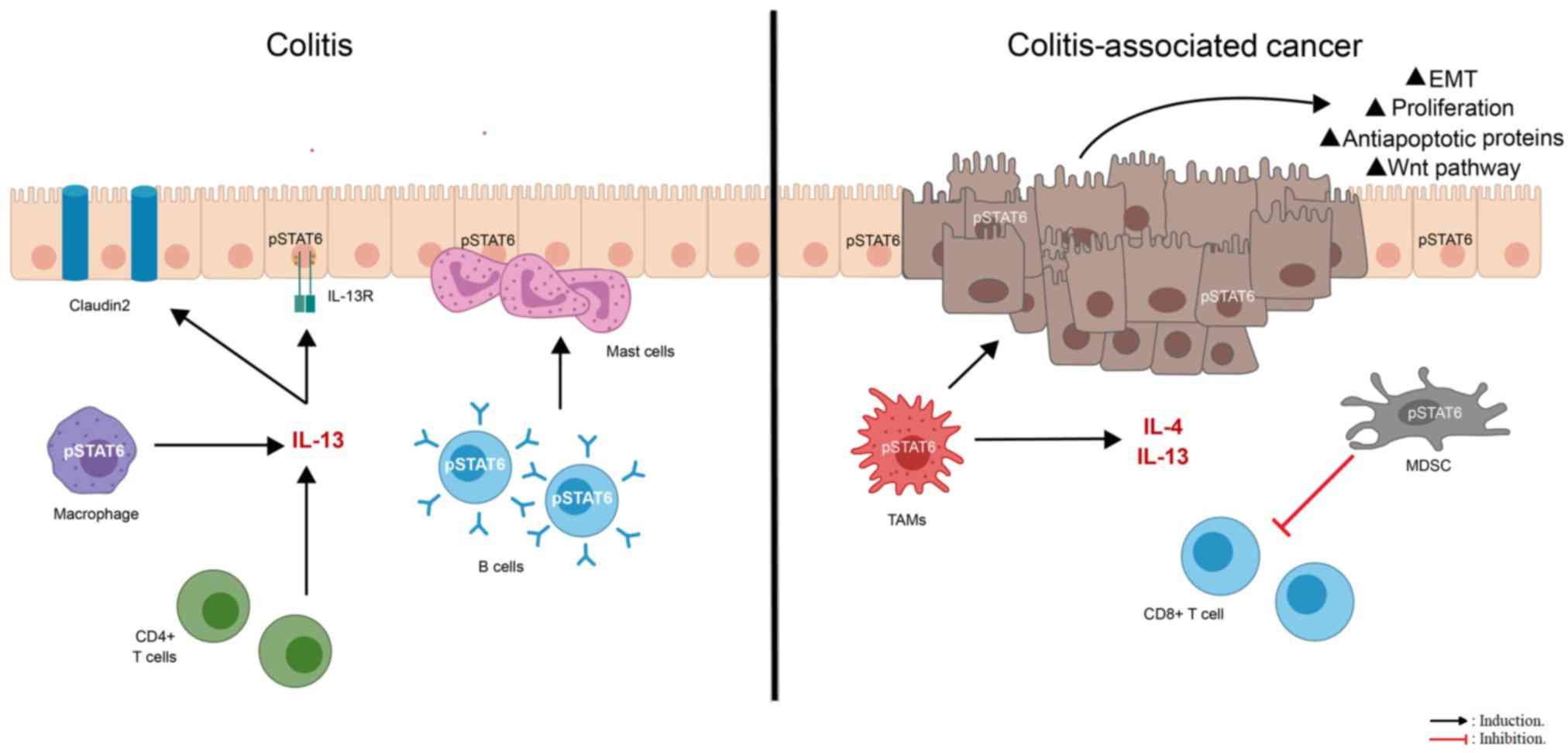 | Figure 1.STAT6 modulates pro-colitis and
pro-cancer responses. The IL-13/STAT6 signaling pathway appears to
be detrimental to the function of intestinal epithelial cells in
subjects with ulcerative colitis or Crohn's disease, in which
nuclear pSTAT6 is significantly increased in the colonic
epithelium. IL-13 signaling alters the expression of proteins,
including the pore-forming tight junction protein claudin-2,
matriptase, prostasin and serotonin, involved in epithelial barrier
permeability and gastrointestinal barrier function via STAT6. In
addition, IL-13 produced by CD4+ T helper 2 cells favors
STAT6 phosphorylation in mast cells, B cells and macrophages during
the development of colitis, suggesting that STAT6 signaling in
immune cells favors the progression of the disease. In the tumor
microenvironment, STAT6 coordinates both polyp cell proliferation
and myeloid-derived suppressor cell expansion. Furthermore, the
IL-4/STAT6 signaling pathway induces the expression of
anti-apoptotic proteins and the growth of epithelial colon cancer
cells. STAT6 activation prompts the differentiation of infiltrating
macrophages into a tumor-associated phenotype with pro-tumoral
functions; these M2 macrophages promote mucosal repair through the
activation of the Wnt signaling pathway. However, mutations that
cause aberrant Wnt signaling are responsible for polyp development
in the small intestine and colon. Additionally, the exposure to
IL-13 enhances the expression of epithelial-to-mesenchymal
transition-promoting factors, resulting in a decrease in adhesion,
migration, invasion and metastatic colonization. pSTAT6,
phosphorylated signal transducer and activator of transcription 6;
IL-R, interleukin receptor; TAMs, tumor-associated macrophages;
MDSC, myeloid-derived suppressor cell. |
Targeting STAT6 for colon cancer
therapy
To the best of our knowledge, the use of specific
STAT6 inhibitors in preclinical and clinical studies for colon
cancer has not been reported. However, in experimental models and
cancer cell lines, STAT6 inhibition decreases tumor cell
proliferation, survival, adhesion, invasion and metastasis,
suggesting that STAT6 inhibition may serve as a therapeutic target
in colon cancer.
The use of the STAT6-specific inhibitor AS1517499
reduced tumor growth and signs of the disease in mice with CRC
(33,53). In vivo, STAT6 inhibition
reduced colonic tumor load and colon tissue damage, corresponding
with a decrease in STAT6 phosphorylation in the intestine (33). In an orthotopic 4T1 mammary carcinoma
mouse model, the use of AS1517499 attenuated tumor growth and early
liver metastasis (44). Similarly,
in primary epithelial cells from patients with prostate cancer,
exposure to AS1517499 decreased IL-4-induced colony formation
(54). Upregulation of microRNA
(miRNA/miR)-361 and miR-135b is associated with a decrease in STAT6
expression (55). The common
intravenous anesthetic agent propofol has been associated with a
reduction in tumor-associated inflammation and with the ability to
induce miRNAs to suppress STAT6 expression (55). The treatment of the CRC SW480 and RKO
cell lines with propofol was shown to increase the expression
levels of miR-135b and miR-361, which are STAT6-targeting miRNAs,
and decrease cell proliferation and migration, suggesting that
propofol interferes with the IL-13/STAT6 signaling pathway
(55). Nevertheless, the in
vivo effect of propofol requires further investigation. A
preclinical model utilizing small interfering RNA (siRNA) to
specifically suppress STAT6 expression in lung epithelial cells was
reported (56); the study stated
that the intranasal application of STAT6 siRNAs attenuated allergic
airway inflammation, demonstrating that STAT6 may be targeted in
specific tissues. Therefore, further studies evaluating the
possibility of suppressing STAT6 signaling in the colonic
epithelium are required. The use of STAT6 inhibitors results in the
inhibition of type I and type II IL-4Rs, and therefore, the
negative implications for normal immune functions should be
determined.
Targeting IL-4, IL-13, IL-4R and IL-13Rα2 in
colon cancer therapy
IL-4 and IL-13 bind to their receptors and activate
the JAK/STAT6 signaling pathway. Therefore, the use of molecules
that can block binding or impair the signaling of these cytokines
to prevent STAT6 activation may be clinically relevant. Previous
studies have revealed that blocking IL-4 and IL-13, as well as
their receptors, has different consequences on tumor development
(Fig. 2).
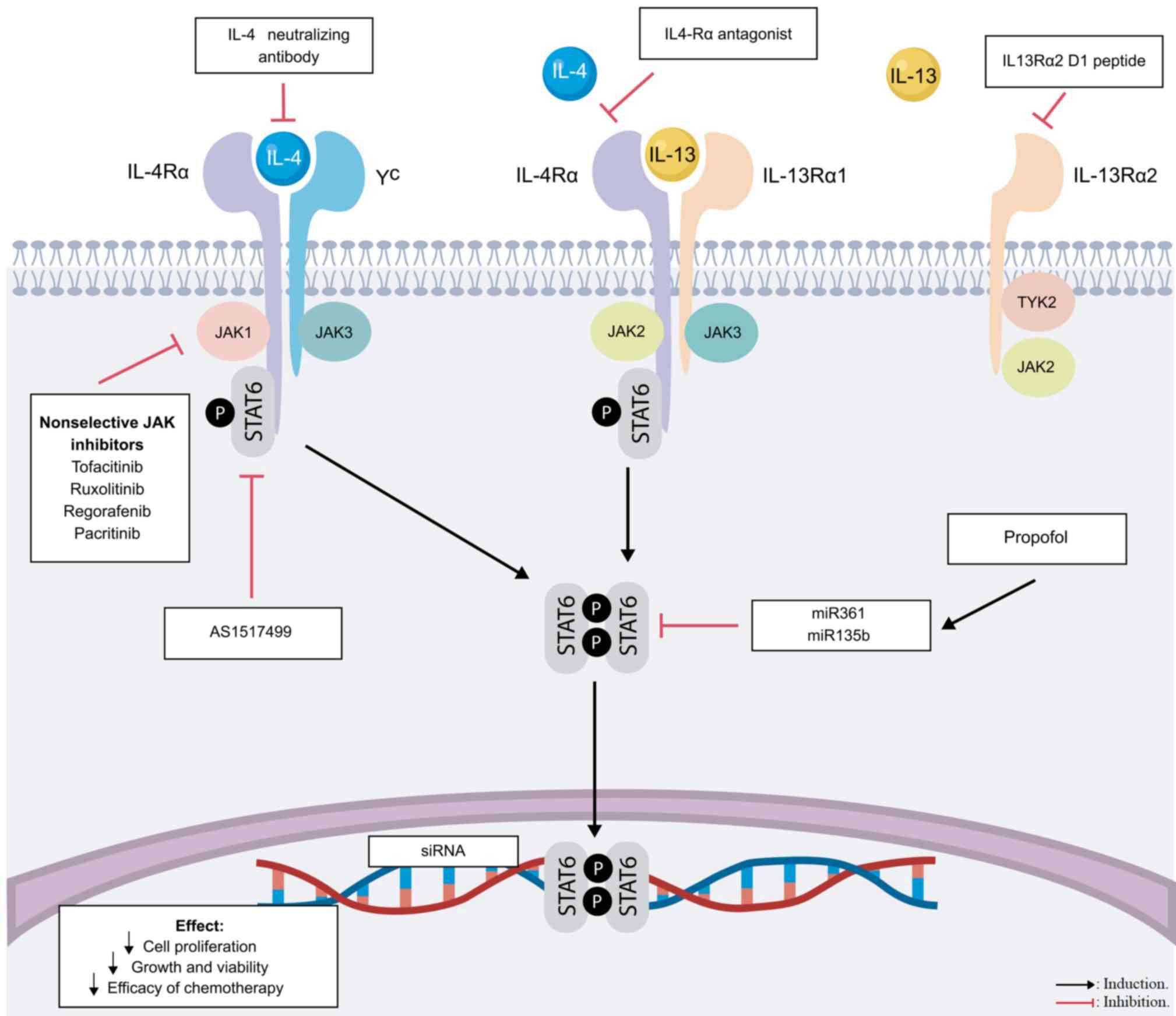 | Figure 2.Targeting the IL-4/IL-13/STAT6
signaling pathway for cancer therapy. Upon cytokine-receptor
binding, JAK1, JAK2 or tyrosine kinase 2 are recruited and
activated, inducing the phosphorylation of STAT6. STAT6 homodimers
translocate to the nucleus and bind DNA sequences at genes involved
in apoptosis, proliferation and the immune response. Various drug
targets have been evaluated to block IL-4, IL-4R, IL-13Rα1 or
STAT6. Points of potential therapeutic inhibition of the pathway
are indicated. STAT6, signal transducer and activator of
transcription 6; IL-R, interleukin receptor; JAK, Janus kinase;
miR, microRNA; siRNA, small interfering RNA; γc, γ chain. |
The synthetic IL-13Rα2 D1 peptide inhibits
IL-13-mediated STAT6 activation through IL-13Rα1, and blocks IL-13
binding to IL-13Rα2, decreasing IL-13 signaling; the administration
of IL-13Rα2 D1 peptide was previously shown to repress tumor
growth, invasion and proliferation in a metastatic CRC mouse model
(57).
The direct administration of anti-IL-4 antibody in
combination with chemotherapy suppresses the growth and viability
of the Caco cell line; this effect is associated with a decrease in
the expression levels of the CRC stem cell marker CD133, suggesting
that anti-IL-4 therapy may enhance the efficacy of chemotherapy
regimens (58). When conventional
drugs, such as 5-fluorouracil or oxaliplatin, are combined with an
IL-4 neutralizing antibody or an inhibitory form of IL-4, the
efficacy of chemotherapy is enhanced in both mature cancer cells
and cancer-stem like cells (59).
Similarly, it has been shown that when IL-4 and IL-13 responses are
inhibited by an IL-4Rα antagonist in nude mice injected with colon
cancer spheroids, the tumor response to chemotherapeutic drugs is
enhanced (60). The increased
production of IL-4 in cancer cells may favor a death-resistant
phenotype, and limiting the production and/or signaling of IL-4 may
be an alternative approach for treatment-resistant cells.
The antitumor effect of doxorubicin has been widely
reported (61). In a previous study,
the liposomal form of doxorubicin was conjugated with a ligand of
the atherosclerotic plaque-specific peptide-1 (AP1), a peptide
characterized by its ability to bind IL-4R; Yang et al
(62) proposed that AP1-conjugated
liposomal doxorubicin exhibits an increased and selective cytotoxic
effect on CRC cells, and has potential as a targeted anticancer
therapy. Collectively, the aforementioned studies demonstrated that
the inhibition of IL-4, IL-4R, IL-13Rα1 and IL-13Rα2 may be
beneficial for colon cancer.
Clinical trials targeting JAKs in colon
cancer therapy
It is well known that IL-4R, γc and IL-13Rα1
activate JAK1/2 and Tyk2. STAT6 is phosphorylated following JAK
activation and forms homodimers that translocate to the nucleus.
JAK molecules have been targeted therapeutically to treat
rheumatoid arthritis, psoriasis and IBD (63,64). The
inhibition of the JAK/STAT inflammatory pathway may be an
alternative for the treatment of CRC. However, to the best of our
knowledge, JAK inhibitors have not been approved by the Food and
Drug Administration (FDA) for the treatment of colon cancer.
In addition to activating STAT6, JAKs mediate a
number of cytokine receptor responses (3). While several cytokines are involved in
inflammatory responses that may promote cancer development, other
cytokines regulate processes involved in mucosal healing, barrier
function and immunosurveillance (3–5). For
example, the inhibition of JAK1 may alter the signaling of IL-2,
IL-7, IL-9, IL-6 and IL-10 (4).
Changes in IL-10, an important anti-inflammatory cytokine, may
affect intestinal homeostasis. In addition, the function of T cells
and natural killer cells may be altered as a result of changes in
cytokine signaling (3,4). The inhibition of JAKs in response to
IL-4 mediates alterations in Akt, ERK and mTOR, which are involved
in several processes (12).
Therefore, it is difficult to discern and control the effect of JAK
inhibition, particularly in cancer development, where the tumor and
stromal cells are actively interacting through cytokines.
Consequently, the use of JAK inhibitors for the treatment of colon
cancer is not widespread. Ruxolitinib, an oral selective inhibitor
of JAK1/2, is approved by the FDA for use in myelofibrosis
(65). Ruxolitinib was tested in a
phase 2 study in combination with regorafenib, an oral
multi-targeted kinase inhibitor, in patients with advanced and
metastatic adenocarcinoma of the colon or rectum (66). However, there was no significant
difference in the overall survival rate or progression-free
survival rate between the regorafenib + placebo vs. regorafenib +
ruxolitinib groups (66).
Nevertheless, the treatment was administered in the advanced stages
of tumor development, and the trial was terminated early per
sponsor decision; therefore, further investigation is required,
particularly during early stages of CRC.
Pacritinib, an oral inhibitor of JAKs and other
kinases, was administrated to patients with metastatic CRC;
however, the study did not produce conclusive results as the trial
was discontinued prior to completion (64). Additionally, the use of tofacitinib,
a JAK1/3 inhibitor widely used for the treatment of rheumatoid
arthritis, was found to be associated with an increase in lung
metastasis accompanied with a decrease in natural killer cell
number in a mouse model of colon cancer (67). Therefore, the development of
selective tissue-specific inhibitors may overcome these types of
complications.
Conclusions
Several studies have revealed that the
IL-4/IL-13/STAT6 axis contributes to the pathology of IBD and colon
cancer. Nuclear pSTAT6 is significantly increased in colonic
epithelium and in neoplastic tissues obtained from patients with
CRC, and its expression is associated with lower survival rates.
Additionally, STAT6 seems to regulate mechanisms that promote the
proliferation, survival, invasion and metastasis of tumor cells, as
well as the suppression of antitumor immunity. Inhibiting STAT6
signaling in experimental models and cancer cell lines has resulted
in a decrease in cancer-associated processes. Therefore, STAT6 may
serve as a potential target in the treatment of colon cancer.
However, to the best of our knowledge, STAT6 inhibitors in
preclinical and clinical studies for colon cancer have not been
reported. The indirect inhibition of the STAT6 signaling pathway
through IL-4, IL-13 or JAKs may inhibit carcinogenesis, but it may
also result in side effects as a consequence of alterations in the
immune response. Therefore, preclinical and clinical studies
investigating specific STAT6 inhibitors with a targeted organ
distribution are required to evaluate the potential of STAT6 as a
target for colon cancer therapy.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from
Support Program for Research Projects and Technological Innovation
(PAPIIT)(grant no. IA204218) and National Council for Science and
Technology (CONACYT)(grant no. A1-S23944).
Availability of data and materials
Not applicable.
Authors' contributions
YDR and SLC contributed to the conception of the
study, writing the manuscript and performing the literature search.
VC and GVG collected and analyzed data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Authors' information
YDR is a doctoral student from the Biomedical
Sciences Doctorate Program, at the National University of Mexico
and received a National Council for Science and Technology
fellowship (606590).
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kuipers EJ, Grady WM, Lieberman D,
Seufferlein T, Sung JJ, Boelens PG, van de Velde CJ and Watanabe T:
Colorectal cancer. Nat Rev Dis Primers. 1:150652015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
West NR, McCuaig S, Franchini F and Powrie
F: Emerging cytokine networks in colorectal cancer. Nat Rev
Immunol. 15:615–629. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: A leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hebenstreit D, Wirnsberger G, Horejs-Hoeck
J and Duschl A: Signaling mechanisms, interaction partners, and
target genes of STAT6. Cytokine Growth Factor Rev. 17:173–188.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chua KH, Ng JG, Ng CC, Hilmi I, Goh KL and
Kee BP: Association of NOD1, CXCL16, STAT6 and TLR4 gene
polymorphisms with Malaysian patients with Crohn's disease. Peerj.
4:e18432016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Klein W, Tromm A, Folwaczny C, Hagedorn M,
Duerig N, Epplen J, Schmiegel W and Griga T: The G2964A
polymorphism of the STAT6 gene in inflammatory bowel disease. Dig
Liver Dis. 37:159–161. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xia B, Crusius JB, Wu J, Zwiers A, van
Bodegraven AA and Pena AS: Signal transducer and activator of
transcription 6 gene G2964A polymorphism and inflammatory bowel
disease. Clin Exp Immunol. 131:446–450. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mueller TD, Zhang JL, Sebald W and Duschl
A: Structure, binding, and antagonists in the IL-4/IL-13 receptor
system. Biochim Biophys Acta. 1592:237–250. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Murata T, Noguchi PD and Puri RK:
Receptors for interleukin (IL)-4 do not associate with the common
gamma chain, and IL-4 induces the phosphorylation of JAK2 tyrosine
kinase in human colon carcinoma cells. J Biol Chem.
270:30829–30836. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sengupta S, Thaci B, Crawford AC and
Sampath P: Interleukin-13 receptor alpha 2-targeted glioblastoma
immunotherapy. Biomed Res Int. 2014:9521282014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mikita T, Campbell D, Wu P, Williamson K
and Schindler U: Requirements for interleukin-4-induced gene
expression and functional characterization of Stat6. Mol Cell Biol.
16:5811–5820. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gupta S, Jiang M and Pernis AB: IFN-alpha
activates Stat6 and leads to the formation of Stat2:Stat6 complexes
in B cells. J Immunol. 163:3834–3841. 1999.PubMed/NCBI
|
|
14
|
Dickensheets H, Vazquez N, Sheikh F,
Gingras S, Murray PJ, Ryan JJ and Donnelly RP: Suppressor of
cytokine signaling-1 is an IL-4-inducible gene in macrophages and
feedback inhibits IL-4 signaling. Genes Immun. 8:21–27. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li Y, Deuring J, Peppelenbosch MP, Kuipers
EJ, de Haar C and van der Woude CJ: STAT1, STAT6 and adenosine
3′,5′-cyclic monophosphate (cAMP) signaling drive SOCS3 expression
in inactive ulcerative colitis. Mol Med. 18:1412–1419. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hanson EM, Dickensheets H, Qu CK, Donnelly
RP and Keegan AD: Regulation of the dephosphorylation of Stat6.
Participation of Tyr-713 in the interleukin-4 receptor alpha, the
tyrosine phosphatase SHP-1, and the proteasome. J Biol Chem.
278:3903–3911. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lu X, Malumbres R, Shields B, Jiang X,
Sarosiek KA, Natkunam Y, Tiganis T and Lossos IS: PTP1B is a
negative regulator of interleukin 4-induced STAT6 signaling. Blood.
112:4098–4108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen W, Daines MO and Hershey GK:
Methylation of STAT6 modulates STAT6 phosphorylation, nuclear
translocation, and DNA-binding activity. J Immunol. 172:6744–6750.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Di Stefano AB, Iovino F, Lombardo Y,
Eterno V, Höger T, Dieli F, Stassi G and Todaro M: Survivin is
regulated by interleukin-4 in colon cancer stem cells. J Cell
Physiol. 225:555–561. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang F, Wang D, Yao YL and Wang M: PDGF
signaling in cancer progression. Int J Clin Exp Med. 10:9918–9929.
2017.
|
|
21
|
Patel BK, Wang LM, Lee CC, Taylor WG,
Pierce JH and LaRochelle WJ: Stat6 and Jak1 are common elements in
platelet-derived growth factor and interleukin-4 signal
transduction pathways in NIH 3T3 fibroblasts. J Biol Chem.
271:22175–22182. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ingram JL, Antao-Menezes A, Mangum JB,
Lyght O, Lee PJ, Elias JA and Bonner JC: Opposing actions of Stat1
and Stat6 on IL-13-induced up-regulation of early growth response-1
and platelet-derived growth factor ligands in pulmonary
fibroblasts. J Immunol. 177:4141–4148. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jess T, Rungoe C and Peyrin-Biroulet L:
Risk of colorectal cancer in patients with ulcerative colitis: A
meta-analysis of population-based cohort studies. Clin
Gastroenterol Hepatol. 10:639–645. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
von Roon AC, Reese G, Teare J,
Constantinides V, Darzi AW and Tekkis PP: The risk of cancer in
patients with Crohn's disease. Dis Colon Rectum. 50:839–855. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rosen MJ, Frey MR, Washington MK,
Chaturvedi R, Kuhnhein LA, Matta P, Revetta FL, Wilson KT and Polk
DB: STAT6 activation in ulcerative colitis: A new target for
prevention of IL-13-induced colon epithelial cell dysfunction.
Inflamm Bowel Dis. 17:2224–2234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang CG, Ye YJ, Yuan J, Liu FF, Zhang H
and Wang S: EZH2 and STAT6 expression profiles are correlated with
colorectal cancer stage and prognosis. World J Gastroenterol.
16:2421–2427. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tosolini M, Kirilovsky A, Mlecnik B,
Fredriksen T, Mauger S, Bindea G, Berger A, Bruneval P, Fridman WH,
Pagès F and Galon J: Clinical impact of different classes of
infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in
patients with colorectal cancer. Cancer Res. 71:1263–1271. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Formentini A, Braun P, Fricke H, Link KH,
Henne-Bruns D and Kornmann M: Expression of interleukin-4 and
interleukin-13 and their receptors in colorectal cancer. Int J
Colorectal Dis. 27:1369–1376. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rosen MJ, Chaturvedi R, Washington MK,
Kuhnhein LA, Moore PD, Coggeshall SS, McDonough EM, Weitkamp JH,
Singh AB, Coburn LA, et al: STAT6 deficiency ameliorates severity
of oxazolone colitis by decreasing expression of claudin-2 and
Th2-inducing cytokines. J Immunol. 190:1849–1858. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hoving JC, Kirstein F, Nieuwenhuizen NE,
Fick LC, Hobeika E, Reth M and Brombacher F: B Cells that produce
immunoglobulin E mediate Colitis in BALB/c mice. Gastroenterology.
142:96–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Buzza MS, Johnson TA, Conway GD, Martin
EW, Mukhopadhyay S, Shea-Donohue T and Antalis TM: Inflammatory
cytokines down-regulate the barrier-protective prostasin-matriptase
proteolytic cascade early in experimental colitis. J Biol Chem.
292:10801–10812. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shajib MS, Wang HQ, Kim JJ, Sunjic I, Ghia
JE, Denou E, Collins M, Denburg JA and Khan WI: Interleukin 13 and
Serotonin: Linking the immune and endocrine systems in murine
models of intestinal inflammation. PLoS One. 8:e727742013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Leon-Cabrera SA, Molina-Guzman E,
Delgado-Ramirez YG, Vázquez-Sandoval A, Ledesma-Soto Y,
Pérez-Plasencia CG, Chirino YI, Delgado-Buenrostro NL,
Rodríguez-Sosa M, Vaca-Paniagua F, et al: Lack of STAT6 attenuates
inflammation and drives protection against early steps of
colitis-associated colon cancer. Cancer Immunol Res. 5:385–396.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jayakumar A and Bothwell ALM: Stat6
promotes intestinal tumorigenesis in a mouse model of adenomatous
polyposis by expansion of MDSCs and inhibition of cytotoxic CD8
response. Neoplasia. 19:595–605. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
De Oliveira T, Ramakrishnan M, Diamanti
MA, Ziegler PK, Brombacher F and Greten FR: Loss of Stat6 affects
chromatin condensation in intestinal epithelial cells causing
diverse outcome in murine models of inflammation-associated and
sporadic colon carcinogenesis. Oncogene. 38:1787–1801. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wick EC, LeBlanc RE, Ortega G, Robinson C,
Platz E, Pardoll DM, Iacobuzio-Donahue C and Sears CL: Shift from
pStat6 to pStat3 predominance is associated with inflammatory bowel
disease-associated dysplasia. Inflamm Bowel Dis. 18:1267–1274.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cao H, Zhang J, Liu H, Wan L, Zhang H,
Huang Q, Xu E and Lai M: IL-13/STAT6 signaling plays a critical
role in the epithelial-mesenchymal transition of colorectal cancer
cells. Oncotarget. 7:61183–61198. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jiang L, Cheng Q, Zhang B and Zhang M:
IL-13 induces the expression of 11βHSD2 in IL-13Rα2 dependent
manner and promotes the malignancy of colorectal cancer. Am J
Transl Res. 8:1064–1072. 2016.PubMed/NCBI
|
|
39
|
Barderas R, Bartolome RA,
Fernandez-Acenero MJ, Torres S and Casal JI: High expression of
IL-13 receptor α2 in colorectal cancer is associated with invasion,
liver metastasis, and poor prognosis. Cancer Res. 72:2780–2790.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen J, Gong C, Mao H, Li Z, Fang Z, Chen
Q, Lin M, Jiang X, Hu Y, Wang W, et al: E2F1/SP3/STAT6 axis is
required for IL-4-induced epithelial-mesenchymal transition of
colorectal cancer cells. Int J Oncol. 53:567–578. 2018.PubMed/NCBI
|
|
41
|
Todaro M, Lombardo Y, Francipane MG, Alea
MP, Cammareri P, Iovino F, Di Stefano AB, Di Bernardo C, Agrusa A,
Condorelli G, et al: Apoptosis resistance in epithelial tumors is
mediated by tumor-cell-derived interleukin-4. Cell Death Differ.
15:762–772. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu H, Antony S, Roy K, Juhasz A, Wu Y, Lu
J, Meitzler JL, Jiang G, Polley E and Doroshow JH: Interleukin-4
and interleukin-13 increase NADPH oxidase 1-related proliferation
of human colon cancer cells. Oncotarget. 8:38113–38135. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kanai T, Watanabe M, Hayashi A, Nakazawa
A, Yajima T, Okazawa A, Yamazaki M, Ishii H and Hibi T: Regulatory
effect of interleukin-4 and interleukin-13 on colon cancer cell
adhesion. Br J Cancer. 82:1717–1723. 2000.PubMed/NCBI
|
|
44
|
Binnemars-Postma K, Bansal R, Storm G and
Prakash J: Targeting the Stat6 pathway in tumor-associated
macrophages reduces tumor growth and metastatic niche formation in
breast cancer. FASEB J. 32:969–978. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Linde N, Lederle W, Depner S, van Rooijen
N, Gutschalk CM and Mueller MM: Vascular endothelial growth
factor-induced skin carcinogenesis depends on recruitment and
alternative activation of macrophages. J Pathol. 227:17–28. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ostrand-Rosenberg S, Sinha P, Clements V,
Dissanayake SI, Miller S, Davis C and Danna E: Signal transducer
and activator of transcription 6 (Stat6) and CD1: Inhibitors of
immunosurveillance against primary tumors and metastatic disease.
Cancer Immunol Immunother. 53:86–91. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Conticello C, Pedini F, Zeuner A, Patti M,
Zerilli M, Stassi G, Messina A, Peschle C and De Maria R: IL-4
protects tumor cells from anti-CD95 and chemotherapeutic agents via
up-regulation of antiapoptotic proteins. J Immunol. 172:5467–5477.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Francipane MG, Alea MP, Lombardo Y, Todaro
M, Medema JP and Stassi G: Crucial role of interleukin-4 in the
survival of colon cancer stem cells. Cancer Res. 68:4022–4025.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Li BH, Yang XZ, Li PD, Yuan Q, Liu XH,
Yuan J and Zhang WJ: IL-4/Stat6 activities correlate with apoptosis
and metastasis in colon cancer cells. Biochem Biophys Res Commun.
369:554–560. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li BH, Xu SB, Li F, Zou XG, Saimaiti A,
Simayi D, Wang YH, Zhang Y, Yuan J and Zhang WJ: Stat6
activity-related Th2 cytokine profile and tumor growth advantage of
human colorectal cancer cells in vitro and in vivo. Cell Signal.
24:718–725. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Volonte A, Di Tomaso T, Spinelli M, Todaro
M, Sanvito F, Albarello L, Bissolati M, Ghirardelli L, Orsenigo E,
Ferrone S, et al: Cancer-initiating cells from colorectal cancer
patients escape from T cell-mediated immunosurveillance in vitro
through membrane-bound IL-4. J Immunol. 192:523–532. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Cosin-Roger J, Ortiz-Masia D, Calatayud S,
Hernandez C, Esplugues JV and Barrachina MD: The activation of Wnt
signaling by a STAT6-dependent macrophage phenotype promotes
mucosal repair in murine IBD. Mucosal Immunol. 9:986–998. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mendoza-Rodriguez MG, Sanchez-Barrera CA,
Callejas BE, García-Castillo V, Beristain-Terrazas DL,
Delgado-Buenrostro NL, Chirino YI, León-Cabrera SA, Rodríguez-Sosa
M, Gutierrez-Cirlos EB, et al: Use of STAT6 Phosphorylation
inhibitor and trimethylglycine as new adjuvant therapies for
5-fluorouracil in colitis-associated tumorigenesis. Int J Mol Sci.
21(pii): E21302020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Nappo G, Handle F, Santer FR, McNeill RV,
Seed RI, Collins AT, Morrone G, Culig Z, Maitland NJ and Erb HHH:
The immunosuppressive cytokine interleukin-4 increases the
clonogenic potential of prostate stem-like cells by activation of
STAT6 signalling. Oncogenesis. 6:e3422017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Xu K, Tao W and Su Z: Propofol prevents
IL-13-induced epithelial-mesenchymal transition in human colorectal
cancer cells. Cell Biol Int. 42:985–993. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Healey GD, Lockridge JA, Zinnen S, Hopkin
JM, Richards I and Walker W: Development of pre-clinical models for
evaluating the therapeutic potential of candidate siRNA targeting
STAT6. PLoS One. 9:e903382014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bartolome RA, Jaen M and Casal JI: An
IL13Rα2 peptide exhibits therapeutic activity against metastatic
colorectal cancer. Br J Cancer. 119:940–949. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gharib AF, Shalaby SM, Raafat N, Fawzy WMS
and Abdel Hakim NH: Assessment of neutralizing interleukin-4 effect
on CD133 gene expression in colon cancer cell line. Cytokine.
97:66–72. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Todaro M, Perez Alea M, Scopelliti A,
Medema JP and Stassi G: IL-4-mediated drug resistance in colon
cancer stem cells. Cell Cycle. 7:309–313. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Todaro M, Alea MP, Di Stefano AB,
Cammareri P, Vermeulen L, Iovino F, Tripodo C, Russo A, Gulotta G,
Medema JP and Stassi G: Colon cancer stem cells dictate tumor
growth and resist cell death by production of interleukin-4. Cell
Stem Cell. 1:389–402. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Rivankar S: An overview of doxorubicin
formulations in cancer therapy. J Cancer Res Ther. 10:853–858.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yang CY, Liu HW, Tsai YC, Tseng JY, Liang
SC, Chen CY, Lian WN, Wei MC, Lu M, Lu RH, et al: Interleukin-4
receptor-targeted liposomal doxorubicin as a model for enhancing
cellular uptake and antitumor efficacy in murine colorectal cancer.
Cancer Biol Ther. 16:1641–1650. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
O'Shea JJ, Kontzias A, Yamaoka K, Tanaka Y
and Laurence A: Janus kinase inhibitors in autoimmune diseases. Ann
Rheum Dis. 72 (Suppl 2):ii111–ii115. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Regenbogen T, Chen L, Trinkaus K,
Wang-Gillam A, Tan BR, Amin M, Pedersen KS, Park H, Suresh R, Lim
KH, et al: Pacritinib to inhibit JAK/STAT signaling in refractory
metastatic colon and rectal cancer. J Gastrointest Oncol.
8:985–989. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Pardanani A and Tefferi A: How I treat
myelofibrosis after failure of JAK inhibitors. Blood. 132:492–500.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Fogelman D, Cubillo A, Garcia-Alfonso P,
Mirón MLL, Nemunaitis J, Flora D, Borg C, Mineur L, Vieitez JM,
Cohn A, et al: Randomized, double-blind, phase two study of
ruxolitinib plus regorafenib in patients with relapsed/refractory
metastatic colorectal cancer. Cancer Med. 7:5382–5393. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Shimaoka H, Takeno S, Maki K, Sasaki T,
Hasegawa S and Yamashita Y: A cytokine signal inhibitor for
rheumatoid arthritis enhances cancer metastasis via depletion of NK
cells in an experimental lung metastasis mouse model of colon
cancer. Oncol Lett. 14:3019–3027. 2017. View Article : Google Scholar : PubMed/NCBI
|














