Introduction
Tumors consist of different populations of cells,
including both transformed and untransformed cells (1). The untransformed cells include
infiltrated inflammatory cells, endothelial cells, immunocytes,
interstitial-derived smooth muscle cells, cancer-associated
fibroblasts (CAFs) and pericytes, which are herein collectively
referred to as interstitial cells (1,2).
Interstitial cells communicate among themselves and with tumor
cells, immunocytes and inflammatory cells directly through cell
contact or indirectly through exocrine/paracrine effects, the use
of proteases and regulation of the extracellular matrix (3,4). This
complicated communication network serves a key role in providing an
appropriate microenvironment to sustain tumor occurrence,
angiogenesis and metastasis (3,4). Because
of the pivotal role of the tumor stromal microenvironment in
tumorigenesis and development, identification of stromal targets
for cancer therapy is critical and may provide improved strategies
to directly target tumor cells. In cancer stromal cells, CAFs may
be required for tumor cells to proliferate and survive in
vivo (5–8). CAFs represent a heterogeneous cell
population, and their phenotype may be different from that of
normal fibroblasts, such as secreting different cytokines or
expressing different proteins (9,10).
Previous studies have indicated that fibroblasts regulate the
proliferation of cancer cells that may appear as normal cells in
the early stages of tumorigenesis (11,12).
Although the functional and phenotypical heterogeneity of CAFs
remain unclear, CAFs have been characterized as myofibroblasts, in
part according to α-smooth muscle actin (α-SMA) expression
(13).
Fibroblast activation protein-α (FAP-α), also known
as FAP or seprase, has been identified as a marker of reactive
fibroblasts in cancer (including CAFs), fibrotic lesions and
granulation tissue (14–16). FAP-α has attracted interest through
its potential role as a therapeutic target due to its regulated
expression in the stroma of cancerous lesions and the structural
evidence of its proteolytic activity (14–18).
However, its function in cancer remains largely unclear. FAP-α is
the overexpression product of CAFs and is the predominant component
of the tumor stroma (19). CAFs are
different from adult normal tissue fibroblasts and instead resemble
wound healing-associated and early human fetal fibroblasts
(19). CAFs are key regulators of
tumorigenesis; however, they are more genetically stable than
cancer cells (13). CAFs may
therefore represent more feasible therapeutic targets for tumor
immunotherapy compared with cancer cells (13).
In order to specifically target CAFs and investigate
the immunogenicity of the FAP-α protein, the present study used an
immunity method, applying H-2b positive murine bone
marrow-derived dendritic cells (DCs) transfected with a recombinant
adenovirus carrying the FAP-α gene (rAd-FAP-α), in order to induce
the antitumor immune response against subcutaneous implanted Lewis
lung carcinoma (LLC) in C57BL/6 mice.
Materials and methods
Cell line and mice
LLC cells (H-2b) were provided by The
Cell Bank of Type Culture Collection Academy of Sciences, whereas
293T cells were purchased from the American Type Culture
Collection. Cells were cultured in DMEM medium supplemented with
10% fetal bovine serum (FBS; Biological Industries) at 37°C in a
humidified incubator containing 5% CO2.
A total of 70 female C57BL/6 (H-2b) mice
(age, 7–8 weeks; weight, 18–24 g) were purchased from the
Laboratory Animal Research Institute at Tongji Medical College of
Huazhong University of Science and Technology (Wuhan, China). All
mice were kept in individual ventilated cages with food and water
ad libitum, at controlled temperature conditions (22±1°C),
with a 12 h light/dark cycle at 50% humidity. All experimental
protocols were approved by the Ethics Committee of The Second
Affiliated Hospital of Nanchang University (Nanchang, China;
approval no. NDEFYEC 175-2018) and mice were sacrificed according
to the Institutional Animal Care and Use Committee protocol.
Construction of mouse FAP-α adenovirus
vector
A cDNA-encoding mouse FAP-α of CAFs was purchased
from Open Biosystems Inc. The FAP-α fragment was amplified with
specific primers. The sense PCR primer, including a BamHI
site (underlined) was 5′-AGGTCGACTCTAGAGGATCCGCCACCATGAAGACATGGCTGAAAACTGTC-3′,
whereas the anti-sense primer, including an AgeI site
(underlined) was 5′-TCACCATGGTGGCGACCGGTCAGTCTGATAAAGAAAAGCATTGC-3′.
The amplified cDNA was digested with BamHI and AgeI,
and inserted into the GV137 plasmid (Shanghai GeneChem Co., Ltd.)
to yield GV137-FAP-α. PCR was subsequently performed to verify the
successful construction of this shuttle plasmid. FAP-α cDNA was
amplified in a 50 µl reaction containing: 5 µl 10X AmpliTaq Gold
Polymerase buffer, 5 µl dNTP (25 mM), 20 pmol (final concentration)
specific forward/reverse primers and 1 µl AmpliTaq Gold Polymerase
(Thermo Fisher Scientific, Inc.). The thermocycling conditions used
for PCR were as follows: Initial denaturation at 95°C for 10 min;
25 cycles at 95°C for 30 sec, 55°C for 30 sec, 72°C for 30 sec; and
a final extension at 72°C for 7 min. The primer sequences used for
PCR were as follows: FAP-α forward, 5′-TTCTGCCTCCTCAGTTTG-3′ and
reverse, 5′-CTGGTCGAGCTGGACGGCGACG-3′. The expected size of the
fragments was 795 bp. Following co-transfection with 4 ug the
helper plasmid [pBHGlox(δ)E1,3Cre] and 2 ug the shuttle plasmid
(GV137-FAP-α) (both from Hanheng Biotechnology Shanghai Co., Ltd;
http://www.hanbio.net) into 293T cells, the
recombinant adenovirus vector containing the mouse FAP-α cDNA
(rAd-FAP-α) was generated using the AdMax kit D (Microbix
Biosystems, Inc.). After 12 days when typical cytopathic effects
(CPE; CPE is defined as degenerative changes in cell morphology
associated with the replication of adenoviruses. These morphologic
changes, such as cell rounding and lose of cell adhesion, can be
detected via fluorescence microscopy) (20,21) were
exhibited for all cells and >50% of cells were detached from the
flask, the transfected cells were collected. Subsequently, the
transfected cells were washed once with PBS, frozen and thawed
three times at −70/37°C, respectively. The cell lysate was
centrifuged at 1,000 × g for 10 min at 4°C and the supernatant was
collected. The initial supernatant was the crude lysate that
included rAd-FAP-α, which was expanded in vitro, and
purified via density gradient centrifugation and dialysis. Briefly,
the fast sealed tube containing the prepared iodixanol gradient
(cat. no. D1556-250; Sigma-Aldrich; Merck KGaA) and the lysate was
centrifuged at 500,000 × g for 90 min at 18°C, using a rotor 70Ti
(Beckman Coulter, Inc.). In order to retrieve the virus, the tube
was fixed on a holder set at eye level and the 18 G needle was
subsequently inserted into the top of the tube to allow air to
enter. Another 18 G needle was attached to a 10 ml syringe and
inserted directly below the interface of the 60 and 40% iodixanol
layers, with the slope of the needle facing upwards. ~4 ml of
sample containing the recombinant adenovirus was extracted from the
40% iodixanol layer (20,21). The rAd-FAP-α titers were determined
using an endpoint dilution assay, as previously described (20,21). A
recombinant adenovirus containing an enhanced green fluorescent
protein (GFP) gene was generated using a similar method used to
construct the control adenovirus (rAd-c) [Hanheng Biotechnology
Shanghai Co., Ltd (20,21)].
Generation of DCs and transduction
with rAd-FAP-α
Bone marrow (BM)-derived DCs were produced from
mouse myeloid progenitor cells as previously described (21). Briefly, two C57BL/6 mice were
sacrificed by inhaling CO2 (CO2
chamber-replacement rate of 30% for 15 min). After removing all
muscle tissues from the mice tibias and femurs, BM progenitors were
flushed out. The red blood cells (RBC) were lysed with 10 mM
ammonium chloride Tris buffer (Sigma-Aldrich; Merck KGaA) at room
temperature for 2 min. Cells were subsequently centrifuged at 300 ×
g for 5 min at room temperature and washed twice with DMEM. The
remaining BM progenitors were incubated for 2 days in 6-well
culture plates (5×106 cells/well) in DMEM medium
supplemented with 10% FBS, recombinant murine-granulocyte
macrophage colony-stimulating factor (GM-CSF; 1,000 IU/ml) and
recombinant murine-interleukin-4 (IL-4; 1,000 IU/ml; both from
PeproTech China) at 37°C in a 5% CO2 incubator. After 48
h, nonadherent granulocytes were gently removed and fresh medium
was added. A total of 50% of the culture medium was replaced with
fresh medium containing GM-CSF and IL-4 every 48 h. On day 8, the
non-adherent cells were considered as immature DCs.
The immature DCs (2×106 cells/ml) were
washed twice with 3 ml PBS and subsequently infected with either
rAd-FAP-α or rAd-c, at a multiplicity of infection (MOI) of 300.
Cells were incubated in 24-well (5×105 cells/ml) plates
in 0.5 ml of serum-free media supplemented with 1,000 IU/ml GM-CSF
and 500 IU/ml IL-4 for 2 h. Subsequently, complete media was added
and cells were incubated for an additional 2 days. The infected DCs
were harvested and washed twice with PBS, prior to subsequent
experimentation. Since the recombinant adenoviral vector included
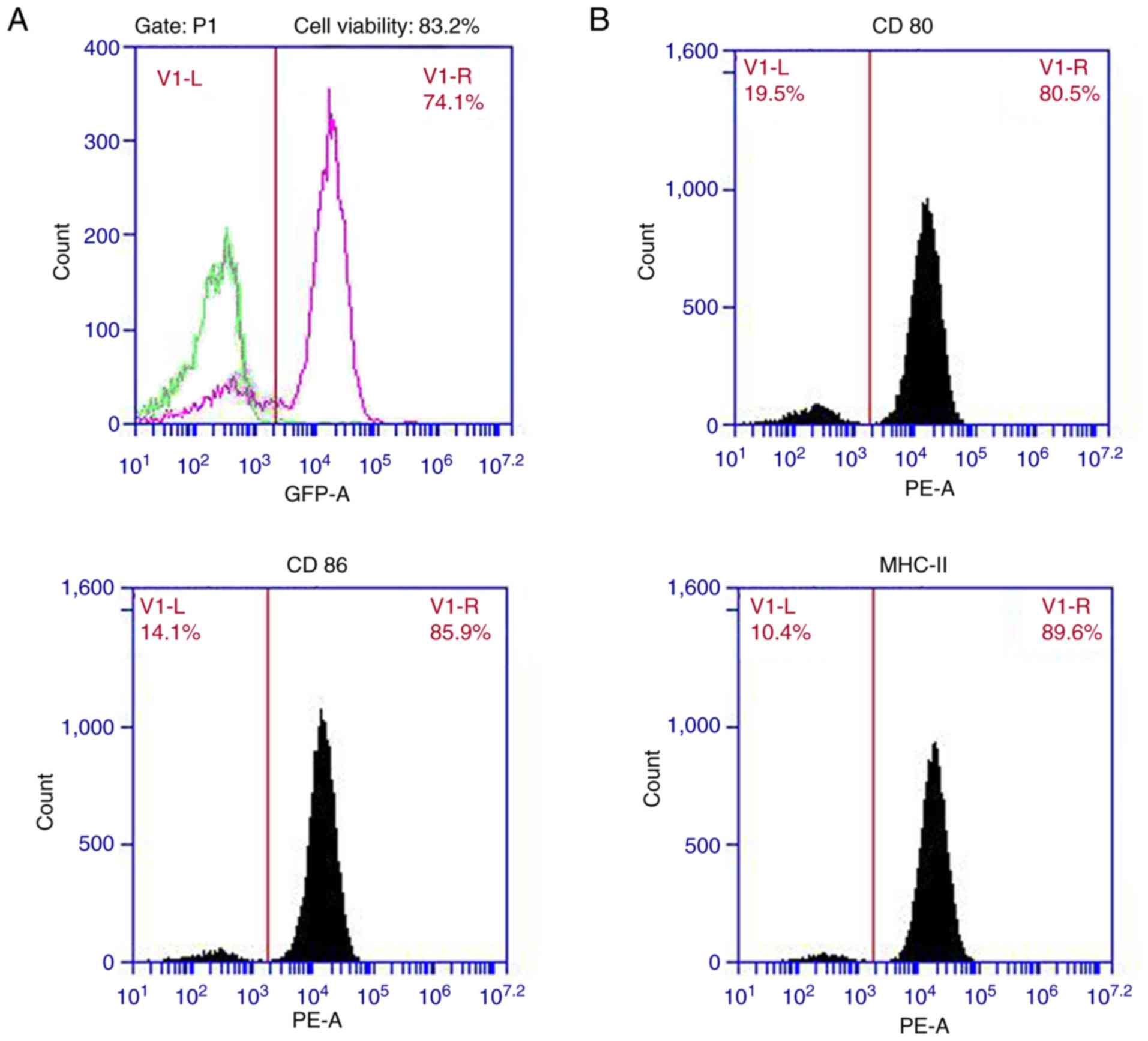
GFP (Fig. 1A; plasmid map),
transduction efficiency of DCs was determined by assessing GFP
expression via flow cytometry analysis. Cell viability was assessed
using trypan blue dye-exclusion (Sigma Aldrich; Merck KGaA).
Briefly, the cell suspension was thoroughly mixed with 0.4% trypan
blue solution at a ratio of 1:1. The number of viable and dead
cells were counted using a hemocytometer (Thermo Fisher Scientific,
Inc.), and the cell viability percentage was determined.
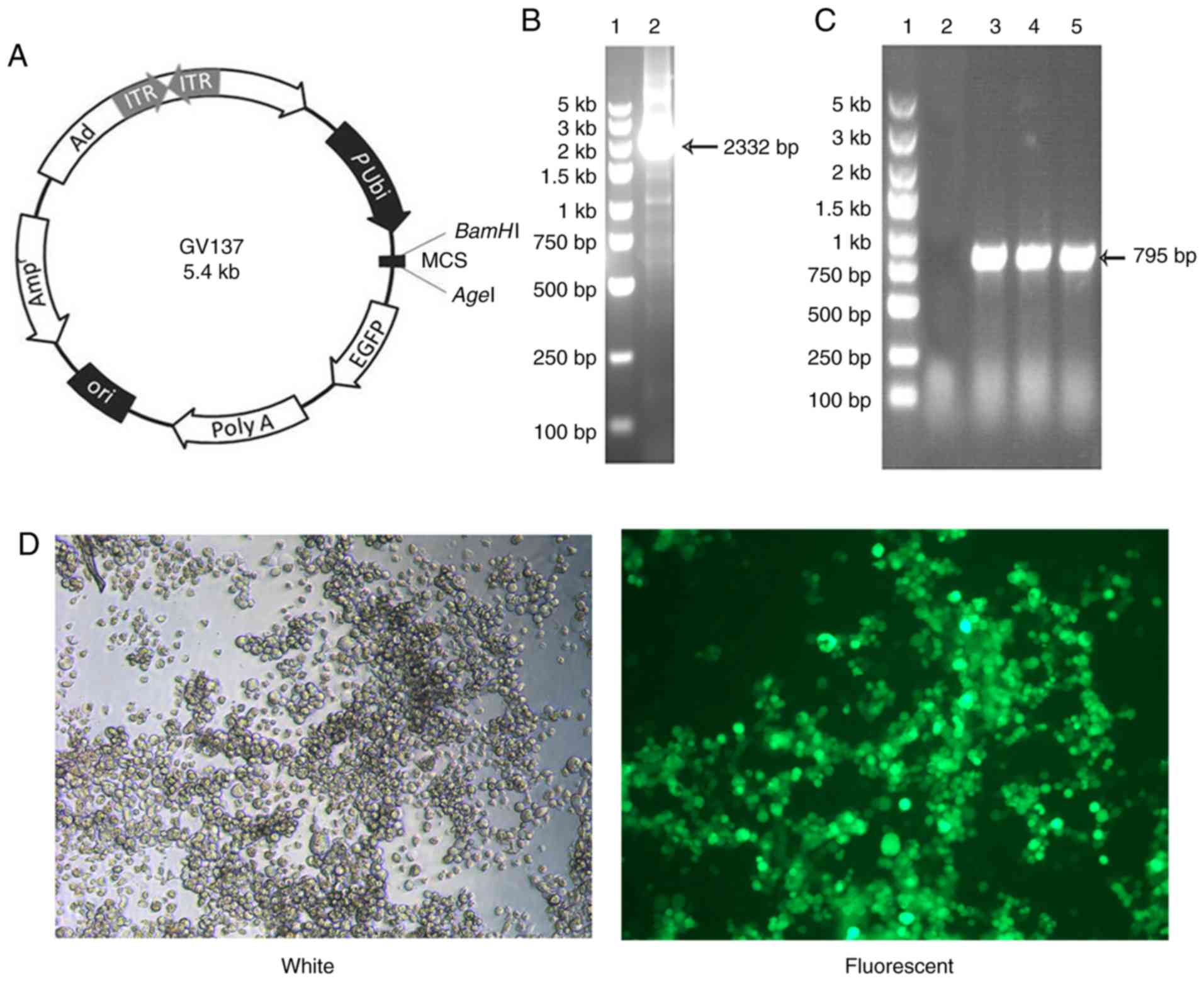 | Figure 1.Identification and construction of
adenoviral vector encoding mouse FAP-α. (A) GV137 plasmid map. (B)
Identification of the amplified FAP-α circular DNA fragment. Lane
1, DNA marker (100 bp-5 kb ladder); lane 2, amplified FAP-α PCR
products. (C) Identification of the recombinant GV137-FAP-α
plasmid. Lane 1, DNA marker (100 bp-5 kb ladder); lane 2, negative
control (empty vector control); lane 3–5, PCR products of the
recombinant GV137-FAP-α plasmid. (D) GFP expression was detected
via fluorescence microscopy (magnification ×100). FAP-α, fibroblast
activation protein-α; bp, base pair; kb, kilobase; Ad, adenovirus;
ITR, inverted terminal repeats; P Ubi, promoter from
ubiquitin gene; MCS, multiple cloning site; EGFP, enhanced green
fluorescent protein; Ori, origin of replication; Amp,
ampicillin. |
Flow cytometric analysis
On day 10, rAd-FAP-α DCs, non-infected DCs or rAd-c
DCs (106 cells/ml) were collected and resuspended in
cold FACS buffer (eBioscience; Thermo Fisher Scientific, Inc.). A
total of 100 µl DCs were immunostained with antibodies against CD80
(cat. no. 12-0801-81), CD86 (cat. no. 12-0869-42) or MHC II
(I-A/I-E; cat. no. 12-5321-81), and isotype-matched antibodies; IgG
Isotype Control (cat. no. 12-4888-81), IgG2b kappa Isotype Control
(cat. no. 12-4732-81) and IgG2b kappa Isotype Control (cat. no.
12-4031-80) in the dark for 30 min at 4°C (all 1:20 and from
eBioscience; Thermo Fisher Scientific, Inc.). The DCs were
subsequently resuspended in PBS and their phenotypes were analyzed
using a flow cytometer (BD Biosciences).
Western blotting
Total protein was extracted from rAd-FAP-α DCs,
rAd-c DCs, LLC cells or CAFs using RIPA buffer [150 mM NaCl, 100 mM
Tris (pH 8.0), 1% Triton X-100, 1% deoxycholic acid, 0.1% SDS, 5 mM
EDTA, 10 mM NaF, 1 mM sodium vanadate, 2 mM leupeptin, 2 mM
aprotinin, 1 mM phenylmethylsulfonyl fluoride and 1 mM
dithiothreitol] (eBioscience; Thermo Fisher Scientific, Inc.) on
ice for 30 min. Protein concentration was determined using the BCA
Protein Assay kit (Thermo Fisher Scientific, Inc.). Proteins (20
µg) were separated by 6% SDS-PAGE and transferred onto
Hybond-polyvinylidene difluoride membranes. Membranes were blocked
with 5% non-fat milk in PBS at room temperature for 1 h and
subsequently incubated with primary antibodies against FAP-α
(1:1,000; cat. no. NB110-85534; Novus Biologicals, Ltd.) and GAPDH
(1:1,000; cat. no. NB100-56875; Novus Biologicals, Ltd.) for 2 h at
room temperature. Membranes were washed three times in TBST (5
min/wash) and further incubated with an alkaline
phosphatase-conjugated goat anti-rabbit IgG secondary antibody
(1:2,500; cat. no. ADI-SAB-301-J; Enzo Life Sciences Inc.) for 1 h
at room temperature. Protein bands were visualized using the ECL
western blot analysis system (GE Healthcare).
In vivo immunization of mice and tumor
challenge study
Female C57BL/6 mice (H-2b) were used in
the in vivo experiments. In the prophylactic study, mice
were initially vaccinated and then injected with tumor cells. Mice
were divided into three groups (n=10 mice/group) and were immunized
subcutaneously (s.c.) with rAd-FAP-α DCs, rAd-c DCs or DCs alone
(5×105 cells/mouse), three times into the left flanks at
6 day intervals. Following the last immunization, after 1 week,
5×105 LLC cells were injected s.c. into the right flanks
of the mice and the tumor of mice were observed for 8 weeks.
In the therapeutic efficacy study, mice were
initially injected with tumor cells and then vaccinated. A total of
5×105 LLC cells were s.c. injected into the right flank
of each mouse. Mice began treatment when the tumor diameter reached
4–6 mm (at day 7). The mice were divided into three groups (n=10
mice/group) and injected s.c. with rAd-FAP-α DCs, rAd-c DCs or DCs
alone (5×105 cells/mouse), three times into the left
flanks at 4 day intervals. The shortest diameter (width) and the
longest diameter (length) of each tumor were measured using a
digital caliper every 2 days to determine the antitumor effect.
Tumor volume was calculated as follows: V = length ×
width2 ×0.52. All mice were sacrificed according to the
Institutional Animal Care and Use Committee protocol when they
became moribund or when the tumor diameter reached 20 mm, which
were recorded as the date of death in the survival studies. The
overall survival rates of the mice were assessed using SPSS 20.0
software (IBM Corp.).
CAFs cultures and immunofluorescence
staining
LLC CAFs were obtained from the s.c. implanted LLC
tumor by collagenase treatment, following separation with meshing
by a nylon filter and adhesion of plastics, as previously described
(1,2). CAFs were incubated in DMEM media
supplemented with 20% FBS at 37°C and passaged every 3–4 days.
After 5 passages, cells were fixed with 4% paraformaldehyde
(BioSharp Life Sciences) for 15 min, and subsequently permeabilized
with 0.2% Triton-X-100 (Beijing Dingguo Changsheng Biotechnology
Co., Ltd.) for 15 min at room temperature. Cell were blocked with
10% goat serum (Biological Industries) at room temperature for 1 h
and incubated with the following primary antibodies: Rabbit
anti-α-smooth muscle actin (1:500; cat. no. NBP1-30894; Novus
Biological, Ltd.), anti-FAP-α (1:500; cat. no. MAB9727; Novus
Biological, Ltd.), anti-vimentin (1:500; cat. no. NBP1-97670; Novus
Biological) or anti-pan cytokeratin (1:200; cat. no. NB600-579;
Novus Biological) overnight at 4°C. Cells were washed three times
with PBS and subsequently incubated with a goat anti-rabbit IgG
secondary antibody (1:500; cat no. SA00001-2; ProteinTech Group,
Inc.) for 1 h at 37°C. The nuclei of the CAFs were stained with
DAPI (ProteinTech Group, Inc.) and cell images were captured using
an OLYMPUS IX73 fluorescence microscope (magnification × 400;
Olympus Corporation).
Cytotoxic T lymphocyte (CTL)
assay
A CytoTox 96® non-radioactive
cytotoxicity assay (Promega Corporation) was performed, according
to the manufacturer's protocol to assess cytotoxic activity.
Briefly, C57BL/6 mice (n=9; 3/group) were immunized s.c. with
rAd-FAP-α DCs, rAd-c DCs or DCs alone (5×105
cells/mouse), two times at 6 day intervals to evaluate whether
rAd-FAP-α DCs could induce antitumor specific CTLs. After the last
immunization on day 6 mice were sacrificed. Splenocytes were
obtained by gentle disruption of the spleen of the mice and
filtration using a Falcon®40 µm Nylon cell strainer
(Corning, Inc.). Subsequently, the splenocytes were treated with
RBC lysis solution and further cultured at 37°C in complete medium
for 90 min. Nonadherent splenocytes (3×106 cells/ml)
were harvested and re-stimulated in 24-well culture plates using
mitomycin C (Sigma-Aldrich; Merck KGaA)-treated CAFs
(106 cells/ml), in the presence of recombinant murine
IL-2 (10 U/ml; PeproTech). These splenocytes [effector cells (E)]
were incubated at 37°C for 5 days. After 5 days, E were cultured
with CAFs [target cells (T)] in round-bottom 96-well culture plates
according to the E:T ratios 10:1, 30:1 and 50:1. After 4 h, the
plate was centrifuged at 250 × g for 5 min at 4°C and the
supernatants were collected. Lactate dehydrogenase maximal release
was determined according to completely lysed T. T cultured without
E were considered for spontaneous release and used as the negative
controls. The percentage of cytotoxicity (specific lysis) was
measured using the following formula: [(experimental
release-spontaneous release)/(maximum release-spontaneous release)]
×100.
Statistical analysis
Statistical analysis was performed using SPSS 20.0
software (IBM Corp.) and data are presented as the mean ± standard
deviation. ANOVA followed by Bonferroni's post-hoc test were used
to compare differences between multiple groups and two groups.
Survival analysis of mice was performed using the log-rank test,
followed by Bonferroni's post-hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Construction of recombinant adenovirus
carrying FAP-α
The cDNA encoding mouse FAP-α was amplified using
primers. The amplified DNA fragment corresponded to the expected
size segment (2,332 bp; Fig. 1B).
FAP-α cDNA fragments were inserted into the GV137 shuttle plasmid
(Fig. 1A) to yield GV137-FAP-α,
which was verified via PCR analysis. PCR amplification produced the
expected size of the band (795 bp), confirming therefore that the
recombinant GV137-FAP-α plasmid contained the target segment
(Fig. 1C). The 293T cells were
co-transfected with the GV137-FAP-α shuttle plasmid and
pBHGlox(δ)E1,3Cre helper plasmid, including the adenovirus
backbone. The CPE was assessed via fluorescence microscopy, which
verified production of the recombinant adenovirus (rAd-FAP-α) 10
days post-cotransfection. rAd-FAP-α was subsequently infected with
293T cells, and GFP expression was detected 2–3 days post-infection
via fluorescence microscopy (Fig.
1D). The purified rAd-FAP-α particle titer was
2.2×1010 TCID50/ml.
In vitro recombinant adenoviral
infection of DCs
On day 7 of DC culture, DCs were infected with
rAd-FAP-α, and the efficiency of infection was assessed. The
positive rate of GFP detected via flow cytometry analysis was
~74.1% (Fig. 2A). MOI 300 was used
for gene transduction in the present study as cell viability was
>80%.
Effect of rAd-FAP-α infection on
phenotype of DCs
The present study assessed the DC surface markers,
MHC class II, CD86 and CD80, using flow cytometric analysis. The
results demonstrated that the expression levels of CD80, CD86 and
MHC class II in rAd-FAP-α-infected DCs were 80.5, 85.9 and 89.6%,
respectively (Fig. 2B). No
significant differences in the expression of the surface markers
were observed between rAd-FAP-α-infected DCs and rAd-c-infected DCs
(data not shown).
CAFs cultures
CAFs were obtained from s.c. implanted LLC tumors by
collagenase digestion and passaged every 4 days (Fig. 3A). After 4 passages, CAFs nuclei was
stained using DAPI, >95% of CAFs positively expressed vimentin
(Fig. 3B), α-SMA (Fig. 3C) and FAP-α (Fig. 3D) as demonstrated via immunostaining
analysis.
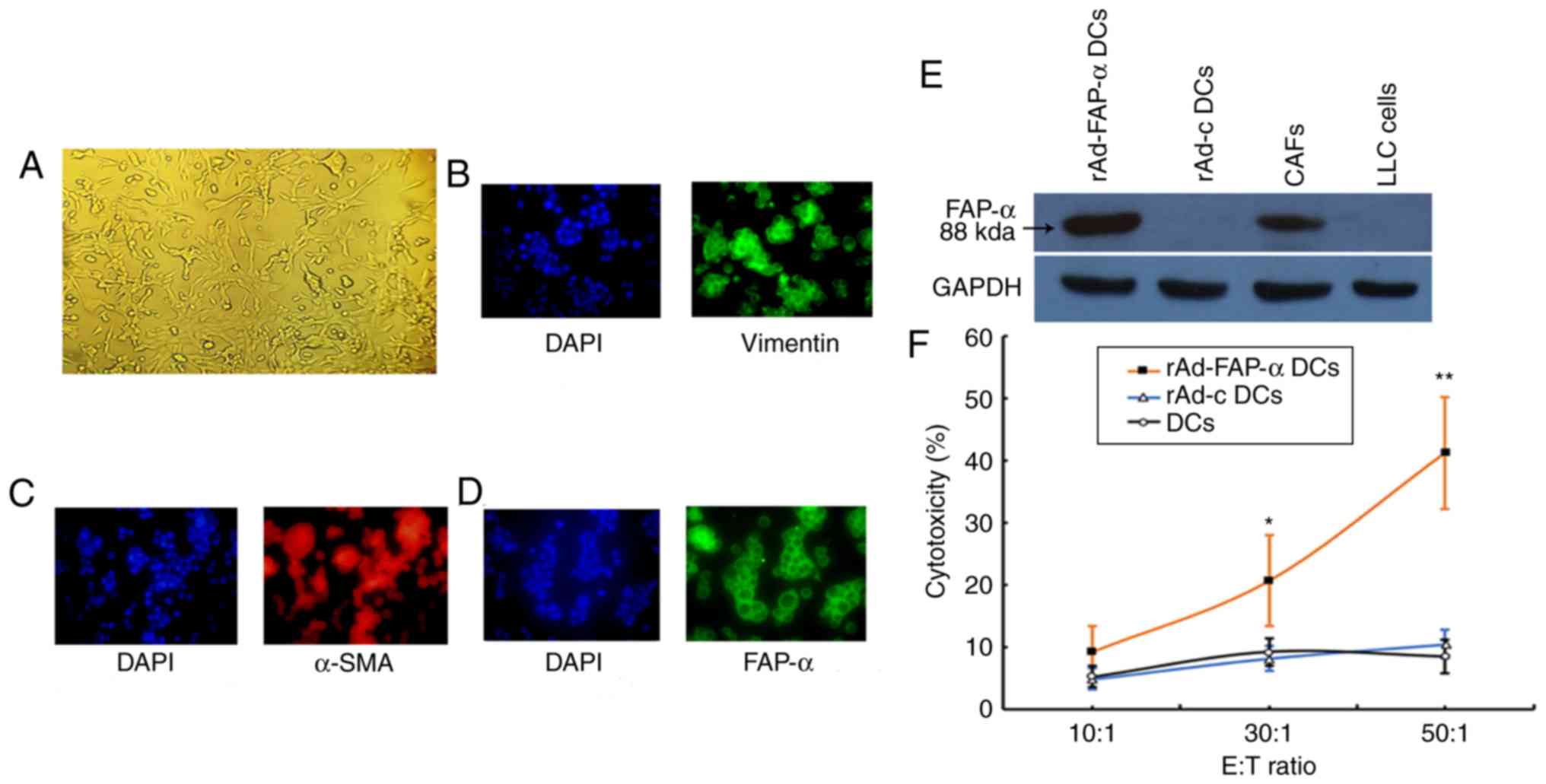 | Figure 3.Identification of CAFs, western blot
analysis of FAP-α expression and induction of CTL by immunizing
mice with rAd-FAP-α DCs. (A) CAFs (magnification ×200), DAPI
staining of CAFs nuclei and (B) vimentin, (C) α-SMA and (D) FAP-α
immunofluorescence staining of CAFs (magnification ×400). (E)
Western blot analysis of FAP-α expression. LLC cells and rAd-c DCs
were used as the negative controls, while GAPDH was used as the
standard internal control. (F) Induction of tumor-specific
cytotoxic T lymphocyte activity, following vaccination of mice with
rAd-FAP-α DCs. *P<0.01, **P<0.001 vs. rAd-c DCs group and DCs
group. CAFs, cancer-associated associated fibroblasts; FAP-α,
fibroblast activation protein-α; DCs, dendritic cells; α-SMA,
α-smooth muscle actin. |
FAP-α expression in CAFs and rAd-FA-α
DCs
Previous studies have reported that FAP-α is
expressed in CAFs but not in cancer cells (22,23). In
order to determine whether FAP-α displays a similar expression
pattern in the experimental systems of the present study, FAP-α
expression was assessed in a series of CAFs, LLC cells, rAd-c DCs
and rAd-FAP-α DCs. The results demonstrated that mouse FAP-α
expression was overexpressed in rAd-FAP-α DCs and CAFs; however, no
expression was detected in rAd-c DCs and LLC cells (Fig. 3E). This suggests that rAd-FAP-α DCs
vaccine had been successfully constructed, the CAFs used in present
study were appropriate to be targeted for immunotherapy and LLC
cells cannot be targeted by CTL induced by rAd-FAP-α DCs vaccine in
subsequent experiments due to lack of FAP-α expression.
Tumor-specific CTL activity
The effector cells from C57BL/6 mice immunized with
rAd-FAP-α DCs exhibited significantly cytotoxic effects in CTLs
against the targets cells (CAFs) at E:T ratios of 30:1 and 50:1;
however, the control effector cells from C57BL/6 mice immunized
with non-transduced DCs or rAd-c DCs exhibited minimal cytotoxicity
(Fig. 3F). Taken together, these
results indicated that rAd-FAP-α DCs may have the ability to induce
the production of specific CTLs against FAP-α-positive CAFs.
Antitumor efficacy of rAd-FAP-α
DCs
In order to determine whether rAd-FAP-α DCs exhibit
an antitumor function in vivo, the present study used LLC
bearing mouse models [LLC(H-2b) in C57BL/6 mice
(H-2b)]. The results demonstrated that vaccination with
rAd-FAP-α DCs induced significant antitumor effects and led to 100%
mice survival compared with the rAd-c DCs and DCs groups (Fig. 4A).
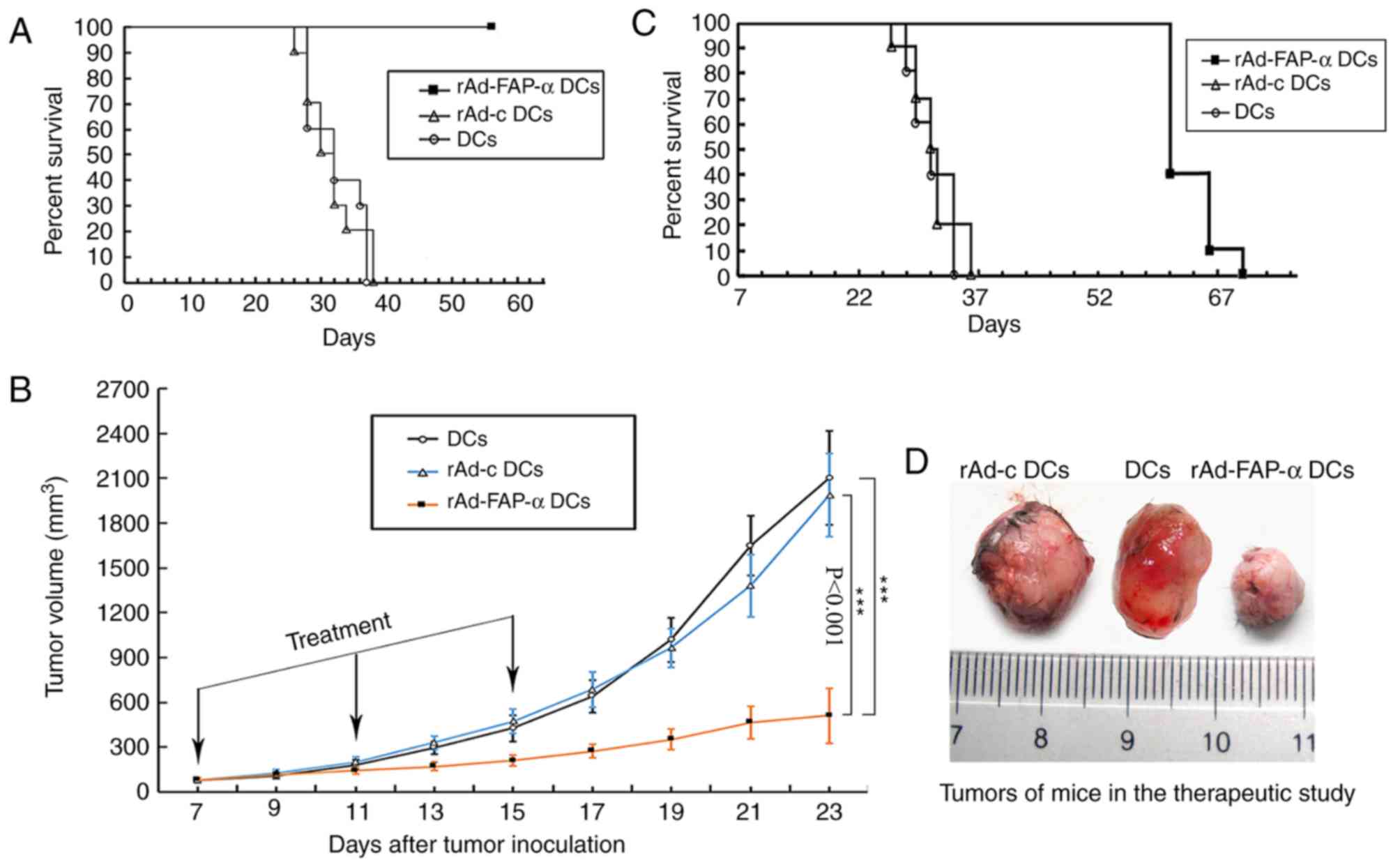 | Figure 4.Efficacy of rAd-FAP-α DCs in
antitumor progression and prolongation of mouse survival time.
C57BL/6 mice were vaccinated s.c. with rAd-FAP-α DCs, rAd-c DCs or
DCs. (A) In the prophylactic study, mice were injected s.c. with
5×105 LLC cells 1 week after the last vaccination. Mice
were subsequently divided into three groups and overall survival
time was assessed for 8 weeks. (B) In the therapeutic efficacy
study, LLC cells (5×105) were initially injected s.c.
into the right flank of mice on the first day. After 7 days, mice
were subsequently divided into three groups, according to tumor
size. Tumor volumes were measured every 2 days, while (C) mice
survival rates were observed for 70 days. (D) Tumors of mice in the
therapeutic study, following s.c. injection with LLC cells at day
23. ***P<0.001 vs. rAd-c DCs group and DCs group. rAd-FAP-α DCs,
recombinant adenovirus-fibroblast activation protein-α dendritic
cells; s.c., subcutaneously; LLC, Lewis lung cancer. |
Considering the results from the prophylactic study,
an in vivo therapeutic experiment was performed to determine
the therapeutic effects of vaccination with rAd-FAP-α DCs. The
results demonstrated that vaccination with rAd-FAP-α DCs
significantly inhibited tumor growth compared with the rAd-c DCs or
DCs groups, during the observation period of 23 days, following
s.c. injection with LLC cells (days 13–23, all P<0.001; Fig. 4B). Furthermore, the tumor volumes in
the rAd-FAP-α DCs group were significantly decreased compared with
the rAd-c DCs and DCs groups at days 13–23 (Fig. 4C). The representative tumor volume
image is from day 23 (Fig. 4D).
However, no significant difference was observed in the tumor volume
between the rAd-c DCs group and the DCs group. A total of 4/10 mice
immunized with rAd-FAP-α DCs survived until day 61 of the
observation period, while all mice in the other two groups either
died of natural causes or were euthanized (P<0.001; Fig. 4C). Taken together, the results of the
present study suggested that immunization with rAd-FAP-α DCs may
inhibit tumor growth and increase the survival rates of LLC-bearing
mice. However, this effect failed to completely eradicate the tumor
in the present study.
Discussion
The discovery that human melanomas could express a
type of non-mutated tumor-associated antigen, which can
spontaneously induce CD8+ T cell responses was a major
advancement in the study of tumor immunology (24). However, therapeutic vaccines using
such antigens lack efficiency in controlling tumor growth (25–27).
Previous studies reported that tumors may induce immunological
tolerance (25) or lose expression
of the tumor cell surface antigen upon tumor progression (26,27);
however, these hypotheses fail to explain the occurrence of
systemic tumor immune responses in patients with tumors that have
been immunized using such antigens, alongside the fact that these
immune responses cannot maintain or induce tumor regression,
although the tumor continues to express tumor antigens and MHC
class I (28,29). These findings indicate that
immunosuppression in the tumor microenvironment could be a major
reason for the poor efficacy of therapeutic vaccination. The tumor
microenvironment includes numerous types of stromal cells. Tumor
cells are embedded in the tumor stroma, which is the connective
tissue framework of several types of solid tumors. Thus, tumoral
stroma cells may be a major determinant in the immune suppression
of the tumor microenvironment (6,30).
Certain mesenchymal-derived stromal cells, including peritumoral
and intratumoral CAFs, may be identified according to the
expression of the type II membrane glycoprotein with intrinsic
dipeptidyl-peptidase FAP-α (30),
which is associated with immune suppression and tumor growth
(31–33). FAP-α is a type II transmembrane
protein that belongs to the serine integral membrane peptidases
(SIMPs) family. SIMPs also contain the cell surface serine protease
dipeptidyl peptidase IV (DPPIV, also known as CD26) and dipeptidyl
peptidase IIX (34,35). These peptidases are active on the
cell surface and are specific and inducible for proline-containing
peptides (34,35). According to previous clinical trials
where the enzymatic activity of FAP-α was targeted (36,37), and
previous studies demonstrating FAP-α specific upregulation in
>90% of pancreatic, colon, breast and lung cancers (23,38),
treatment through targeting FAP-α may be considered as an effective
therapeutic method for patients with various types of tumor.
In order to assess the antitumor immune effect of
targeting FAP-α, the present study designed a recombinant
adenovirus containing the mouse FAP-α with the aim of transducing
DCs and immunizing C57BL/6 mice. The results of the present study
demonstrated that CTLs produced in mice using the vaccine targeting
FAP-α may recognize the mouse FAP-α-derived epitope and induce a
significant cytotoxic effect. In order to determine the antitumor
immunotherapy efficacy of rAd-FAP-α DCs vaccine in vivo, the
present study used LLC bearing mouse models. The results
demonstrated that vaccination with rAd-FAP-α DCs markedly increased
the antitumor protection effects (in the prophylactic immunotherapy
experiment, 100% for LLC tumor) and markedly delayed the growth of
the established LLC tumor. However, rAd-c DCs had no significant
effects on the antitumor protection efficacy of mice. Previous
studies have also demonstrated the use of anti-stromal
immunotherapy by targeting FAP-α (39–41) and
reported results similar to those from the present study.
In summary, the present study demonstrated that
targeting a surface type II membrane glycoprotein of CAFs named
FAP-α, which is overexpressed specifically and selectively on CAFs,
may inhibit tumor growth in a mouse LLC model. FAP-α may be
considered as a potential target for killing or destroying CAFs
within the tumor stromal microenvironment, and may be used to
develop immunogenic tumor vaccines. However, successful application
of FAP-α-targeted immune therapy in patients with lung cancer will
require the development of more efficient vaccination protocols,
and assessment in other lung cancer models. Further investigation
is also required to determine the underlying mechanism by which
targeting the FAP-α could inhibit tumor growth in vivo,
either by direct inhibition of the CAFs or by appendant damages,
such as secretion of cytokines (leading to a local inflammatory
response) and a decrease in infiltrative immunosuppressive cells in
the tumor microenvironment.
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant nos. 81160294 and 81960425) and
the key projects of Science and Technology Department of Jiangxi
Province (grant no. 20181BBH80005).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
JX designed the present study, analyzed and
interpreted the data, and drafted the initial manuscript. SY, LP,
HL, LN performed the experiments. HX and XG performed flow
cytometric analysis and collated the relevant literature. MY and FD
analyzed the data and revised the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Ethics Committee of The Second Affiliated Hospital of Nanchang
University (Nanchang, China; approval no. NDEFYEC 175-2018), and
mice were sacrificed according to the Institutional Animal Care and
Use Committee protocol.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lai D, Ma L and Wang F: Fibroblast
activation protein regulates tumor-associated fibroblasts and
epithelial ovarian cancer cells. Int J Oncol. 41:541–550. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Giannoni E, Bianchini F, Calorini L and
Chiarugi P: Cancer associated fibroblasts exploit reactive oxygen
species through a proinflammatory signature leading to epithelial
mesenchymal transition and stemness. Antioxid Redox Signal.
14:2361–2371. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Puré E and Lo A: Can targeting stroma pave
the way to enhanced antitumor immunity and immunotherapy of solid
tumors? Cancer Immunol Res. 4:269–278. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu T, Zhou L, Li D, Andl T and Zhang Y:
Cancer-associated fibroblasts build and secure the tumor
microenvironment. Front Cell Dev Biol. 7:602019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Erez N, Truitt M, Olson P, Arron ST and
Hanahan D: Cancer-associated fibroblasts are activated in incipient
neoplasia to orchestrate tumor-promoting inflammation in an
NF-kappaB-dependent manner. Cancer Cell. 17:135–147. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ohshio Y, Teramoto K, Hanaoka J, Tezuka N,
Itoh Y, Asai T, Daigo Y and Ogasawara K: Cancer-associated
fibroblast-targeted strategy enhances antitumor immune responses in
dendritic cell-based vaccine. Cancer Sci. 106:134–142. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Barron DA and Rowley DR: The reactive
stroma microenvironment and prostate cancer progression. Endocr
Relat Cancer. 19:R187–R204. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gascard P and Tlsty TD:
Carcinoma-associated fibroblasts: Orchestrating the composition of
malignancy. Genes Dev. 30:1002–1019. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Valkenburg KC, de Groot AE and Pienta KJ:
Targeting the tumour stroma to improve cancer therapy. Nat Rev Clin
Oncol. 15:366–381. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Elenbaas B and Weinberg RA: Heterotypic
signaling between epithelial tumor cells and fibroblasts in
carcinoma formation. Exp Cell Res. 264:169–184. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xing F, Saidou J and Watabe K: Cancer
associated fibroblasts (CAFs) in tumor microenvironment. Front
Biosci (Landmark Ed). 15:166–179. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bhowmick NA, Neilson EG and Moses HL:
Stromal fibroblasts in cancer initiation and progression. Nature.
432:332–337. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Santos AM, Jung J, Aziz N, Kissil JL and
Puré E: Targeting fibroblast activation protein inhibits tumor
stromagenesis and growth in mice. J Clin Invest. 119:3613–3625.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Edosada CY, Quan C, Tran T, Pham V,
Wiesmann C, Fairbrother W and Wolf BB: Peptide substrate profiling
defines fibroblast activation protein as an endopeptidase of strict
Gly(2)-Pro(1)-cleaving specificity. FEBS Lett. 580:1581–1586. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Edosada CY, Quan C, Wiesmann C, Tran T,
Sutherlin D, Reynolds M, Elliott JM, Raab H, Fairbrother W and Wolf
BB: Selective inhibition of fibroblast activation protein protease
based on dipeptide substrate specificity. J Biol Chem.
281:7437–7444. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jansen K, Heirbaut L, Cheng JD, Joossens
J, Ryabtsova O, Cos P, Maes L, Lambeir AM, De Meester I, Augustyns
K, et al: Selective inhibitors of fibroblast activation protein
(FAP) with a (4-Quinolinoyl)-glycyl-2-cyanopyrrolidine scaffold.
ACS Med Chem Lett. 4:491–496. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Meadows SA, Edosada CY, Mayeda M, Tran T,
Quan C, Raab H, Wiesmann C and Wolf BB: Ala657 and conserved active
site residues promote fibroblast activation protein endopeptidase
activity via distinct mechanisms of transition state stabilization.
Biochemistry. 46:4598–4605. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ostermann E, Garin-Chesa P, Heider KH,
Kalat M, Lamche H, Puri C, Kerjaschki D, Rettig WJ and Adolf GR:
Effective immunoconjugate therapy in cancer models targeting a
serine protease of tumor fibroblasts. Clin Cancer Res.
14:4584–4592. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jia J, Martin TA, Ye L and Jiang WG: FAP-α
(Fibroblast activation protein-α) is involved in the control of
human breast cancer cell line growth and motility via the FAK
pathway. BMC Cell Biol. 15:162014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nyberg-Hoffman C, Shabram P, Li W, Giroux
D and Aguilar-Cordova E: Sensitivity and reproducibility in
adenoviral infectious titer determination. Nat Med. 3:808–811.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xie J, Xiong L, Tao X, Li X, Su Y, Hou X
and Shi H: Antitumor effects of murine bone marrow-derived
dendritic cells infected with xenogeneic livin alpha recombinant
adenoviral vectors against lewis lung carcinoma. Lung Cancer.
68:338–345. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee J, Fassnacht M, Nair S, Boczkowski D
and Gilboa E: Tumor immunotherapy targeting fibroblast activation
protein, a product expressed in tumor-associated fibroblasts.
Cancer Res. 65:11156–11163. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Puré E and Blomberg R: Pro-tumorigenic
roles of fibroblast activation protein in cancer: Back to the
basics. Oncogene. 37:4343–4357. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
van der Bruggen P, Traversari C, Chomez P,
Lurquin C, De Plaen E, Van den Eynde B, Knuth A and Boon T: A gene
encoding an antigen recognized by cytolytic T lymphocytes on a
human melanoma. Science. 254:1643–1647. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Willimsky G, Czéh M, Loddenkemper C,
Gellermann J, Schmidt K, Wust P, Stein H and Blankenstein T:
Immunogenicity of premalignant lesions is the primary cause of
general cytotoxic T lymphocyte unresponsiveness. J Exp Med.
205:1687–1700. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dunn GP, Koebel CM and Schreiber RD:
Interferons, immunity and cancer immunoediting. Nat Rev Immunol.
6:836–848. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Speiser DE, Baumgaertner P, Barbey C,
Rubio-Godoy V, Moulin A, Corthesy P, Devevre E, Dietrich PY,
Rimoldi D, Liénard D, et al: A novel approach to characterize
clonality and differentiation of human melanoma-specific T cell
responses: Spontaneous priming and efficient boosting by
vaccination. J Immunol. 177:1338–1348. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rosenberg SA, Sherry RM, Morton KE,
Scharfman WJ, Yang JC, Topalian SL, Royal RE, Kammula U, Restifo
NP, Hughes MS, et al: Tumor progression can occur despite the
induction of very high levels of self/tumor antigen-specific CD8 +
T cells in patients with melanoma. J Immunol. 175:6169–6176. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Valmori D, Souleimanian NE, Tosello V,
Bhardwaj N, Adams S, O'Neill D, Pavlick A, Escalon JB, Cruz CM,
Angiulli A, et al: Vaccination with NY-ESO-1 protein and CpG in
montanide induces integrated antibody/Th1 responses and CD8 T cells
through cross-priming. Proc Natl Acad Sci USA. 104:8947–8952. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Garin-Chesa P, Old LJ and Rettig WJ: Cell
surface glycoprotein of reactive stromal fibroblasts as a potential
antibody target in human epithelial cancers. Proc Natl Acad Sci
USA. 87:7235–7239. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dolznig H, Schweifer N, Puri C, Kraut N,
Rettig WJ, Kerjaschki D and Garin-Chesa P: Characterization of
cancer stroma markers: In silico analysis of an mRNA expression
database for fibroblast activation protein and endosialin. Cancer
Immun. 5:102005.PubMed/NCBI
|
|
32
|
Scarfò I and Maus MV: Current approaches
to increase CAR T cell potency in solid tumors: Targeting the tumor
microenvironment. J Immunother Cancer. 5:282017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Orimo A, Gupta PB, Sgroi DC,
Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL
and Weinberg RA: Stromal fibroblasts present in invasive human
breast carcinomas promote tumor growth and angiogenesis through
elevated SDF-1/CXCL12 secretion. Cell. 121:335–348. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen WT and Kelly T: Seprase complexes in
cellular invasiveness. Cancer Metastasis Rev. 22:259–269. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rosenblum JS and Kozarich JW: Prolyl
peptidases: A serine protease subfamily with high potential for
drug discovery. Curr Opin Chem Biol. 7:496–504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hofheinz RD, al-Batran SE, Hartmann F,
Hartung G, Jäger D, Renner C, Tanswell P, Kunz U, Amelsberg A,
Kuthan H and Stehle G: Stromal antigen targeting by a humanised
monoclonal antibody: An early phase II trial of sibrotuzumab in
patients with metastatic colorectal cancer. Onkologie. 26:44–48.
2003.PubMed/NCBI
|
|
37
|
Scott AM, Wiseman G, Welt S, Adjei A, Lee
FT, Hopkins W, Divgi CR, Hanson LH, Mitchell P, Gansen DN, et al: A
phase I dose-escalation study of sibrotuzumab in patients with
advanced or metastatic fibroblast activation protein-positive
cancer. Clin Cancer Res. 9:1639–1647. 2003.PubMed/NCBI
|
|
38
|
Kawase T, Yasui Y, Nishina S, Hara Y,
Yanatori I, Tomiyama Y, Nakashima Y, Yoshida K, Kishi F, Nakamura M
and Hino K: Fibroblast activation protein-α-expressing fibroblasts
promote the progression of pancreatic ductal adenocarcinoma. BMC
Gastroenterol. 15:1092015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Brennen WN, Rosen DM, Wang H, Isaacs JT
and Denmeade SR: Targeting carcinoma-associated fibroblasts within
the tumor stroma with a fibroblast activation protein-activated
prodrug. J Natl Cancer Inst. 104:1320–1334. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Schreiber H and Rowley DA: Cancer.
Awakening immunity. Science. 330:761–762. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kraman M, Bambrough PJ, Arnold JN, Roberts
EW, Magiera L, Jones JO, Gopinathan A, Tuveson DA and Fearon DT:
Suppression of antitumor immunity by stromal cells expressing
fibroblast activation protein-alpha. Science. 330:827–830. 2010.
View Article : Google Scholar : PubMed/NCBI
|