Introduction
Chronic myeloid leukaemia (CML), a malignant disease
derived from a haematopoietic stem cell clone, is characterised by
the production of the breakpoint cluster region (BCR)-ABL proto
oncogene 1 (ABL1) fusion gene and Philadelphia (Ph) chromosome,
which are present in 90% of patients and are well-confirmed
diagnostic markers for CML (1,2). The
BCR-ABL1 fusion gene encodes an oncoprotein that has an activated
tyrosine kinase domain in the ABL region, promotes cell
proliferation, causes loss of stromal adhesion and arrests
apoptosis by activating downstream pathways, including Janus
kinase-signal transducer and activator of transcription (Jak-Stat)
and Myc (2,3). Niches in the bone marrow also serve a
crucial role in the generation and development of CML (4). Currently, tyrosine kinase inhibitors
(TKIs) are applied to treat CML and strongly improve the overall
survival (OS) of patients (3,5). The
majority of patients achieve a life expectancy close to that of the
general population (4). However,
there are still some problems during treatment: i) The disease
still progresses to an advanced phase (AP) or blast crisis (BC)
following treatment with TKIs; ii) a number of patients are
resistant to TKIs; and iii) patients cannot endure the side effects
of TKIs (6). The present study
focused on the mechanisms of disease progression during treatment.
Often, resistance can lead to disease progression. Resistance to
TKI therapy occurs via BCR-ABL1-dependent and BCR-ABL1-independent
mechanisms (7,8). One of the most important factors in the
BCR-ABL1-dependent mechanism is the mutation of the BCR-ABL1 kinase
domain (KD) (7–9).
TKI resistance is defined based on the European
Leukemia Net (ELN) recommendations. Resistance can be divided into
primary and secondary categories (2). Primary resistance is characterised by
any of the following: No complete haematological response or Ph+
>95% by 3 months, BCR-ABL1 >10% or Ph+ >35% by 6 months,
or BCR-ABL1 >1%, Ph + ≥1% or complete cytogenetic response by 18
months (10). Secondary resistance
is defined by the loss of a previously documented haematological,
cytogenetic or molecular response (1).
TKI resistance has been reported to range between
25–30% among patients initially treated with imatinib (6). Most mutations in the KD lead to the
development of resistance to TKI therapy.
Most patients switched to potentially more effective
TKIs based on their mutation profile and achieved major molecular
remission, with the exception of those with the T315I mutation.
Reliable investigations are required to screen for mutations in
patients who fail TKI therapy.
Patients and methods
Patients
The present study was a retrospective analysis of
175 patients with CML treated at the Department of Hematology,
Tongji Hospital of Huazhong University of Science and Technology. A
total of 691 patients with CML visited this institution between
January 2009 and December 2016; however, only 177 patients
underwent BCR-ABL1 mutation screening. Two patients lacking
clinical information were excluded from the analysis. Thus, in the
present study, 175 patients were evaluated. The data were collected
from patients who underwent mutation screening during TKI treatment
or at diagnosis. Mutation analysis was performed as requested when
the patients exhibited resistance or were diagnosed at AP or BC.
There were nine patients diagnosed at BC and four at AP. The
definitions of the disease phases followed the ELN recommendations
(2). Patients were grouped by
BCR-ABL1 mutation status. The association between patient clinical
information and mutation status was analysed. Patient consent was
not required due to the retrospective nature of the study.
BCR-ABL1 mutation analysis
The reduction and mutation of BCR-ABL1 fusion
transcripts over time was assessed by conventional sequencing and
ultra-deep sequencing (UDS) of peripheral blood samples. UDS refers
to deep sequencing that takes advantage of a high-throughput method
and focuses on a limited genomic region; UDS can detect mutations
that cannot be identified by conventional sequencing (11). The basic principles of UDS and
conventional sequencing are similar to those of digital PCR
(1). PCR and sequencing were used to
detect mutations in the ABL1 kinase region of BCR-ABL1. Total RNA
was reverse-transcribed into cDNA and then amplified with Platinum
Taq Polymerase High Fidelity (Thermo Fisher Scientific, Inc.). The
final reaction volume (20 µl) included 10 µl Mix (Takara Bio,
Inc.), 2 µl primers, 6.5 µl RNase-free H2O and 1.5 µl
cDNA and was performed as follows: Pre-denaturation at 95°C for 10
min, denaturation at 95°C for 15 sec, annealing at 59°C for 30 sec,
extension at 72°C for 30 sec, denaturation-annealing-extension step
performed 35 cycles. The sequences of the primers (Beijing
Tianyi Huiyuan Bioscience & Technology Inc.) were as
follows: Homo BCR-ABL, forward 5′-GATGCTGACCAACTCGTGTG-3′, reverse
5′−GTTGGGGTCATTTTCACTGG-3; and GAPDH, forward
5′-CCACCATGGCAAATTCCATGGCA-3′ and reverse
5′-TCTAGACGGCAGGTCAGGTCCACC-3′. The forward primers annealed to BCR
exon b2, and the reverse primers annealed to ABL exon 10 (12). After amplification of each single
fragment read and mapping to the reference sequence, the results
were screened for mutations.
T315I is located in the ATP-binding domain (13), which is the most frequent mutation,
and is resistant to currently available TKIs. The frequency of the
T315I mutation was analysed in the present study, as well as
focusing on compound mutants and polyclonal mutants. Compound
mutations are mutations affecting two or more amino acid residues
in the same BCR-ABL molecule BCR-ABL molecule (6). Multi-TKI failure leads to the emergence
of compound mutants. To date, 60 different compound mutants in
BCR-ABL1 have been reported to be associated with TKI resistance
(14,15). Polyclonal mutations are defined as
two or more missense mutations in different BCR-ABL1 molecules
(8).
Statistical analysis
To assess the associations between two groups of
categorical variables, χ2 test was used, and age was
adjusted by a correction of continuity (in case of n>40 and
1≤T<5). OS was calculated from the identification of the
mutation until the last follow-up, death or the censor date. To
generate survival curves, the Kaplan-Meier method was applied. All
calculations were performed using SPSS 17.0 (SPSS, Inc.). P<0.05
was considered to indicate a statistically significant
difference.
Results
Patient characteristics
Table I summarises
the clinical characteristics at diagnosis and at mutation screening
of the 175 patients analysed in the present study. The
characteristics of patients are presented in Table SI. Patients were divided into groups
based on BCR-ABL1 mutation status. No differences were observed in
the clinical characteristics between the two groups, with the
exception of sex; more female patients were present in the BCR-ABL1
mutation group compared with male patients.
 | Table I.Characteristics of 175 patients with
chronic myeloid leukaemia at the time of mutation screening. |
Table I.
Characteristics of 175 patients with
chronic myeloid leukaemia at the time of mutation screening.
|
|
| BCR-ABL1
mutation |
|
|---|
|
|
|
|
|
|---|
| Variable | N | Present (n=54) | Absent (n=121) | P-value |
|---|
| Sex |
|
|
|
<0.001a |
| Male | 111 | 22 | 89 |
|
|
Female | 64 | 32 | 32 |
|
| Age, years |
|
|
| 0.320 |
| Mean, 43
(range, 3–76) | 169 | 44 (3–71) | 42 (3–76) |
|
|
Missing | 6 |
|
|
|
| Disease phase at
diagnosis |
|
|
| 0.253 |
| CP | 162 | 48 | 114 |
|
| AP | 4 | 1 | 3 |
|
| BC | 9 | 5 | 4 |
|
| Time between TKI and
mutation screen, month |
|
|
| 0.140 |
| Mean,
21.5 (range, 0–99) | 166 | 17.3 (0–92) | 23.5 (0–99) |
|
| Missing,
n | 9 | 4 | 5 |
|
| Type of
transcription |
|
|
| 0.230 |
| P210 | 167 | 50 | 117 |
|
| P230 | 8 | 4 | 4 |
|
| Choice of TKI
generation at diagnosis |
|
|
| 0.790 |
|
First | 164 | 51 | 113 |
|
|
Second | 11 | 3 | 8 |
|
| Additional
chromosome |
|
|
| 0.989 |
|
Present | 39 | 12 | 27 |
|
|
Absent | 136 | 42 | 94 |
|
Mutation analysis
Patients underwent UDS and conventional sequencing.
Different results were detected by USD and conventional sequencing:
V253H/M351I/F359V/F359I vs. V253H/M351I; E255K vs. G250E/E255K;
F359V vs. none; F359V vs. none; T315I vs. none; T315I vs. none;
T315I vs. none; and K365E vs. none. Samples for mutation analysis
were collected from patients who presented with TKI resistance,
were on first-line TKIs (164 patients, 93.7%), or were on
second-line TKIs (11 patients, 6.3%). The T315I mutation was
identified to account for the largest proportion. A total of 28
patients underwent mutation screening at diagnosis, and mutations
were identified in 8 of these 28 patients (4.6%), which indicated
that the mutation exists before treatment with TKIs.
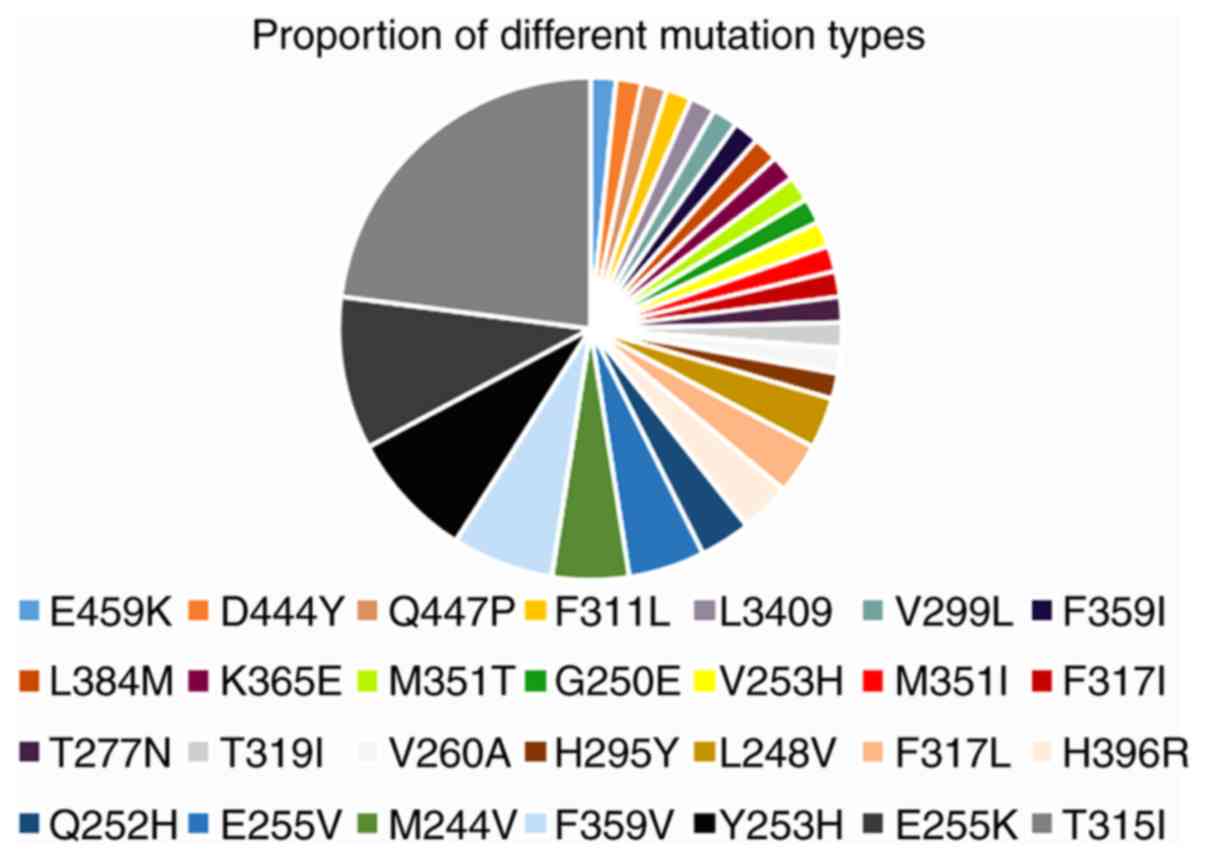
A total of 175 patients underwent BCR-ABL1 mutation
screening, and only 54 (30.9%) patients harboured mutations in the
KD, which included 28 different mutations (Fig. 1). Mutations that occurred in the
P-loop region included G250E, M244V, L248V, E255K and Y253H,
accounting for 36.1% of the mutations identified in the present
study (Table II). The proportion of
mutations in the A-loop, contact binding site and SH2 domain is
presented in Table II. The number
of patients with different types of kinase domain mutations is
presented in Table III. Of note,
eight patients harboured more than one mutation: V253H/M351I,
G250E/E255K, M351T/F359V/H396R, E255K/V299L/T315I, T315I/Y253H,
F311L/F317L, L248V/Y253H/T315I and V260A/H295Y. However, it could
not be confirmed whether these mutations were compound or
polyclonal in this retrospective study (16). Of the eight patients with BCR-ABL1
compound or polyclonal mutations, including the T315I mutation,
three exhibited variable sensitivity to clinically available TKIs
(data not shown), suggesting that one or more TKIs may represent a
rational treatment option. These patients harboured detectable
mutations and elevated BCR-ABL1 transcription following multi-TKI
treatment. The patients with the E255K/V299L/T315I and
L248V/Y253H/T315I mutations died. At the end of the follow-up
period, the majority of the patients were in BC, treated with
combination chemotherapy and TKI with a poor response to
treatment.
| Table II.The proportion of mutations that
occur in distinct regions. |
Table II.
The proportion of mutations that
occur in distinct regions.
| Region | Mutation | Proportion, % |
|---|
| P-loop | G250E, M244V,
L248V, E255K/V, Y253H | 36.1 |
| Contact binding
site | T315I | 23 |
|
| F311L | 1.6 |
|
| F317L/I | 3.3 |
| SH2 domain | M351T/I | 3.3 |
|
| F359V | 8.2 |
| A-loop | H396R | 3.3 |
| Othera | H295Y, K356E,
V260A, Q447P, T319I, V253H, L384M, D444Y, E459K, V299L, T277N,
L3409 | 21.2 |
| Table III.Number of patients with different
types of kinase domain mutations. |
Table III.
Number of patients with different
types of kinase domain mutations.
| Kinase domain
mutation | Number of patients
(n=61) |
|---|
| T315I | 14 |
| E255K | 6 |
| Y253H | 5 |
| F359V | 4 |
| E255V | 3 |
| M244V | 3 |
| Othera | 26 |
Survival analysis
The median follow-up time for all patients in the
present study, from the day of mutation detection to the last
follow-up date, was 22 months (range, 2–88 months). The OS rates of
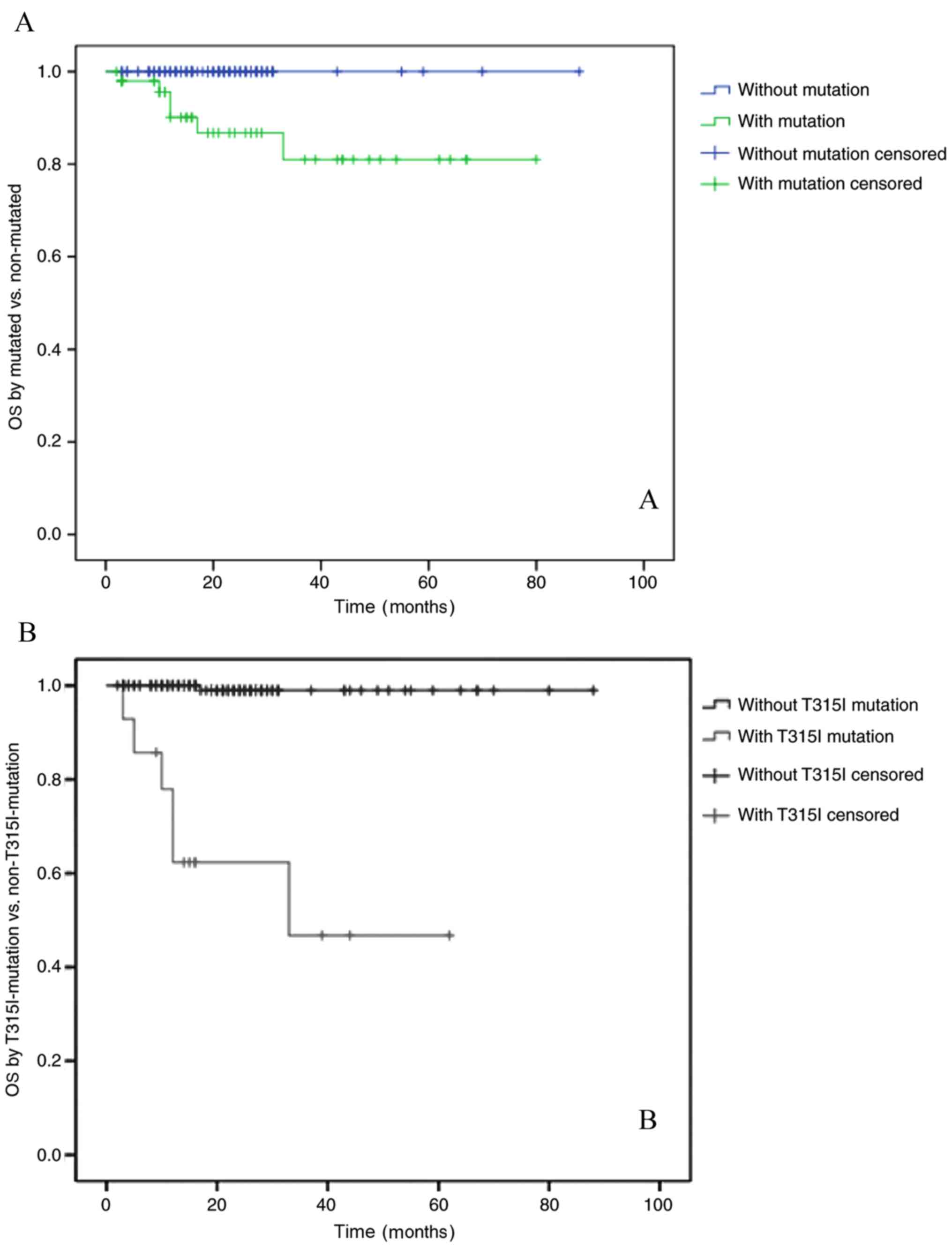
patients in the BCR-ABL1 mutation-positive and negative groups were
80 and 100%, respectively (P<0.01). The OS rates of the groups
with or without the T315I mutation were 47 and 98%, respectively
(P<0.01; Fig. 2A and B). These
results indicated that the OS rate of patients with the T315I
mutation was notably decreased.
T315I mutation
The T315 residue is the gatekeeper, and when it is
mutated to T315I, the entrance of a TKI into the hydrophobic pocket
is blocked, while still allowing access to ATP (17). The T315I mutation, which is the most
challenging mutation, indicates a poor prognosis (18). Unfortunately, patients with the T315I
mutation are usually resistant to all available TKIs (imatinib,
bosutinib, nilotinib and dasatinib) but may benefit from ponatinib
(18–20) or allogeneic haematopoietic stem cell
transplantation (allo-HSCT). Of the 14 patients who presented with
the T315I mutation, one patient achieved complete molecular
remission after allo-HSCT (19). The
results demonstrated that patients with the T315I mutation could be
treated with allo-HSCT if a suitable donor was available. A total
of six patients were treated with a combination of a TKI and
chemotherapy and died in 1–2.5 years (18,21).
Third-generation TKIs such as ponatinib may have efficacy against
the T315I mutation (21). However,
it is difficult to obtain ponatinib in China. In the present study,
two patients were treated with ponatinib and exhibited a good
response; however, these results were temporary. Additionally,
these two patients presented with cardiovascular toxicity, which
may have been an adverse effect of ponatinib.
Karyotype analysis
Sufficient sample material was available to examine
the Karyotype. The Philadelphia chromosome (Ph) is a reciprocal
translocation between the Abelson leukemia virus (ABL) oncogene
from long arm of chromosome 9 and the breakpoint cluster region
(BCR) from long arm of chromosome 22. Additional chromosome refers
to in addition to the Philadelphia chromosome attached to other
abnormal chromosomes.
In the present study, 39 patients presented with
additional chromosomes (in addition to the Philadelphia chromosome
attached to other abnormal chromosomes), and six of these patients
had the T315I mutation. No association was identified between the
T315I mutation and additional chromosome in patients with CML
(P=0.086).
Discussion
This study analyzed data from 175 patients with CML
who underwent mutation screening. A total of 162 (92.6%) patients
were diagnosed at the chronic phase (CP), 48 of whom harboured
detectable mutations, and four were diagnosed at AP, among which
one had a mutation. A total of nine patients, who were diagnosed at
BC, were tested for mutations. However, the mutation was not
associated with disease phase.
The T315I mutation was detected in patients. Among
all patients, T315I was the most frequently detected mutation in
the present study, and this mutation was also associated with OS.
The patients who presented with the T315I mutation responded poorly
to chemotherapy. Only one patient, aged 28 years at diagnosis,
achieved molecular remission following allo-HSCT. In addition, two
patients benefited from ponatinib, although with notable toxicity.
A total of five patients died despite receiving chemotherapy.
Ponatinib has been described as an effective treatment for patients
with the T315I mutation (22). These
results were similar to those reported in the literature (6).
In the present study, the results of conventional
sequencing were different from those of UDS in eight patients. This
result confirmed that UDS exhibits higher sensitivity for mutations
compared with conventional sequencing. The K365E mutation, which
was detected by UDS, was not associated with sensitivity to any
type of TKI according to available literature. The patient with the
K365E mutation was treated with dasatinib and achieved a complete
cytogenetic response.
In the present study, eight patients presented with
more than one mutation. Of note, three patients had T315I mutations
in combination with other mutations, and two of these patients
died; however, it is unknown whether these were polyclonal or
compound mutations. The results from the present study demonstrated
that the prognosis of patients with multiple BCR-ABL1 mutations is
poor, especially for those with T315I compound or polyclonal
mutations. In the present study, the overall survival of patients
with compound mutations was not analysed due to the limited number
of cases.
The emergence of TKI resistance can be divided into
BCR-ABL-dependent and BCR-ABL-independent processes.
BCR-ABL-dependent processes includeoverexpression of the BCR-ABL
kinase and mutations in BCR-ABL domain. BCR-ABL-independent
processes include activation of alternative signalling pathways
(such as JAK/STAT) and overexpression of efflux transporters or
downregulation of influx transporters leading to low TKI levels in
the cell. Mutations in the BCR/ABL KD are the most important of
these mechanisms. Mutations in the KD leading to resistance to TKI
therapy have a much higher incidence in patients in BC compared
with patients in CP (23). Patients
with mutations in CP exhibit a higher incidence of progression to
AP or BC. Thus, mutations can provide information for patients who
do not achieve optimal responses to TKIs (10,11).
Currently, a number of options of therapeutic TKI are available,
depending on the mutations associated with TKI resistance (24,25).
Acquired mutations prevent TKIs from binding or lead to a decrease
in TKI sensitivity (7). Subsets of
these mutations, which have been reported in certain TKI-resistant
cases, remain sensitive to one or more first-generation (imatinib)
or second-generation (nilotinib, dasatinib, bosutinib and
ponatinib) TKIs (2). Distinct
mutations exhibit different levels of sensitivity to TKIs. In the
present study, TKIs were selected based on the nature of the
mutation; for example, dasatinib has activity against T315I and
F317L/C, whereas T315I and F359V/C have limited sensitivity to
nilotinib (19,26–28).
However, the unique mutation T315I affects the topology of the ATP
binding region and is not responsive to any available TKI, except
ponatinib (17). Thus, the detection
of mutations can aid with selecting the TKI and influence the
prognosis of patients with CML, who can be switched to different
TKIs based on the detected mutation.
CML can be regarded as a chronic illness due to the
contribution of TKIs; however, a small number of patients still
experience disease progression. Thus, more clinical case analyses
are required to better understand CML.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JL collected and analyzed data about patients and
methods, and completed the draft. HY, XX and SY collected data
about the patients and helped analyzing data. LM made substantial
contributions to conception and design of the study and revised and
wrote the final version of the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Machova Polakova K, Kulvait V, Benesova A,
Linhartova J, Klamova H, Jaruskova M, de Benedittis C, Haferlach T,
Baccarani M, Martinelli G, et al: Next-generation deep sequencing
improves detection of BCR-ABL1 kinase domain mutations emerging
under tyrosine kinase inhibitor treatment of chronic myeloid
leukemia patients in chronic phase. J Cancer Res Clin Oncol.
141:887–899. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kaleem B, Shahab S, Ahmed N and Shamsi TS:
Chronic myeloid leukemia-prognostic value of mutations. Asian Pac J
Cancer Prev. 17:7415–7423. 2015. View Article : Google Scholar
|
|
3
|
Apperley JF: Chronic myeloid leukaemia.
Lancet. 385:1447–1459. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Loscocco F, Visani G, Galimberti S, Curti
A and Isidori A: BCR-ABL independent mechanisms of resistance in
chronic myeloid leukemia. Front Oncol. 9:9392019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pasic I and Lipton JH: Current approach to
the treatment of chronic myeloid leukaemia. Leuk Res. 55:65–67.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Eide CA and O'Hare T: Chronic myeloid
leukemia: Advance in understanding disease biology and mechanisms
of resistance to tyrosine kinase inhibitors. Curr Hematol Malig
Rep. 10:158–166. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Soverini S, de Benedittis C, Mancini M and
Martinelli G: Mutations in the BCR-ABL1 kinase domain and elsewhere
in chronic myeloid leukemia. Clin Lymphoma Myeloma Leuk S1.
(Suppl):S120–S128. 2015. View Article : Google Scholar
|
|
8
|
Yang K and Fu LW: Mechanisms of resistance
to BCR-ABL TKIs and the therapeutic strategies: A review. Crit Rev
Oncol Hematol. 93:277–292. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Balabanov S, Braig M and Brümmendorf TH:
Current aspects in resistance against tyrosine kinase inhibitors in
chronic myelogenous leukemia. Drug Discov Today Technol. 11:89–99.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Baccarani M, Castagnetti F, Gugliotta G
and Rosti G: A review of the European LeukemiaNet recommendations
for the management of CML. Ann Hematol. 94 (Suppl 2):S141–S147.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Soverini S, De Benedittis C, Mancini M and
Martinelli G: Present and future of molecular monitoring in chronic
myeloid leukaemia. Br J Haematol. 173:337–349. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pagnano KB, Bendit I, Boquimpani C, De
Souza CA, Miranda EC, Zalcberg I, Larripa I, Nardinelli L, Silveira
RA, Fogliatto L, et al: BCR-ABL mutations in chronic myeloid
leukemia treated with tyrosine kinase inhibitors and impact on
survival. Cancer Invest. 33:451–458. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cagnetta A, Garuti A, Marani C, Cea M,
Miglino M, Rocco I, Palermo C, Fugazza G, Cirmena G, Colombo N, et
al: Evaluating treatment response of chronic myeloid leukemia:
Emerging science and technology. Curr Cancer Drug Targets.
13:779–790. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shah NP, Skaggs BJ, Branford S, Hughes TP,
Nicoll JM, Paquette RL and Sawyers CL: Sequential ABL kinase
inhibitor therapy selects for compound drug-resistant BCR-ABL
mutations with altered oncogenic potency. J Clin Invest.
117:2562–2569. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Khorashad JS, Kelley TW, Szankasi P, Mason
CC, Soverini S, Adrian LT, Eide CA, Zabriskie MS, Lange T, Estrada
JC, et al: BCR-ABL1 compound mutations in tyrosine kinase
inhibitor-resistant CML: Frequency and clonal relationships. Blood.
121:489–498. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gibbons DL, Pricl S, Posocco P, Laurini E,
Fermeglia M, Sun H, Talpaz M, Donato N and Quintás-Cardama A:
Molecular dynamics reveal BCR-ABL1 polymutants as a unique
mechanism of resistance to PAN-BCR-ABL1 kinase inhibitor therapy.
Proc Natl Acad Sci USA. 111:3550–3555. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Miller GD, Bruno BJ and Lim CS: Resistant
mutations in CML and Ph+ALL-role of ponatinib. Biologics.
8:243–254. 2014.PubMed/NCBI
|
|
18
|
Poch Martell M, Sibai H, Deotare U and
Lipton JH: Ponatinib in the therapy of chronic myeloid leukemia.
Expert Rev Hematol. 9:923–932. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kujak C and Kolesar JM: Treatment of
chronic myelogenous leukemia. Am J Health Syst Pharm. 73:113–120.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Modugno M: New resistance mechanisms for
small molecule kinase inhibitors of Abl kinase. Drug Discov Today
Technol. 11:5–10. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Haznedaroglu IC: Drug therapy in the
progressed CML patient with multi-TKI failure. Mediterr J Hematol
Infect Dis. 7:e20150142015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Patel AB, O'Hare T and Deininger MW:
Mechanisms of resistance to ABL kinase inhibition in chronic
myeloid leukemia and the development of next generation ABL kinase
inhibitors. Hematol Oncol Clin North Am. 31:589–612. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Apperley JF: Part I: Mechanisms of
resistance to imatinib in chronic myeloid leukemia. Lancet Oncol.
8:1018–1029. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Corbin AS, La Rosée P, Stoffregen EP,
Druker BJ and Deininger MW: Several BCR-ABL kinase domain mutants
associated with imatinib mesylate resistance remain sensitive to
imatinib. Blood. 101:4611–4614. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hochhaus A, Schenk T, Erben P, Ernst T, La
Rosée P and Müller MC: Cause and management of therapy resistance.
Best Pract Res Clin Haematol. 22:367–379. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hehlmann R, Saußele S, Voskanyan A and
Silver RT: Management of CML-blast crisis. Best Pract Res Clin
Haematol. 29:295–307. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Soverini S, De Benedittis C, Mancini M and
Martinelli G: Best practices in chronic myeloid leukemia monitoring
and management. Oncologist. 21:626–633. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Morozova EV, Vlasova YY, Pryanishnikova
MV, Lepik KV and Afanasyev BV: Efficacy of dasatinib in a CML
patient in blast crisis with F317L mutation: A case report and
literature review. Biomarker Insights. 10 (Suppl 3):S43–S47.
2015.
|