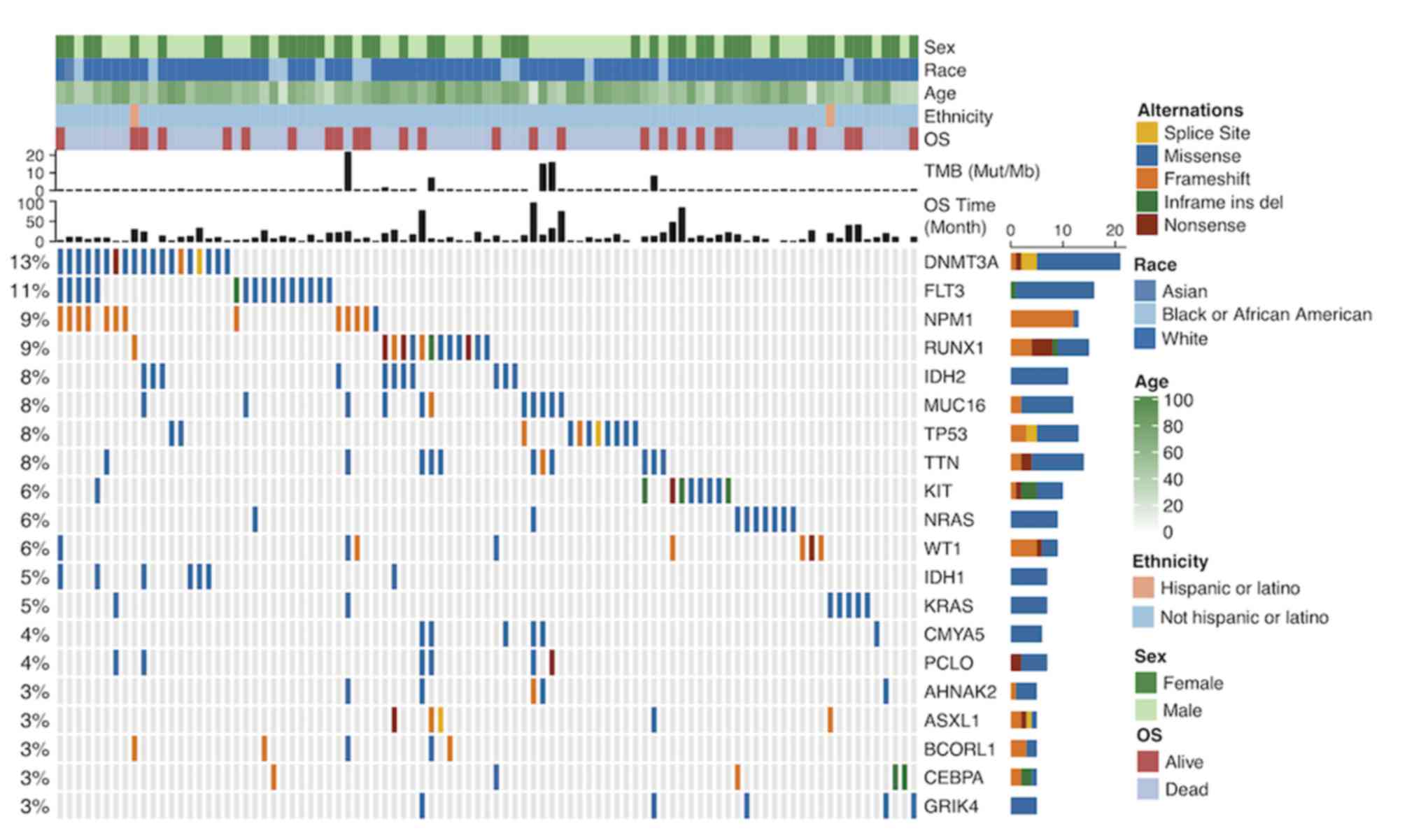
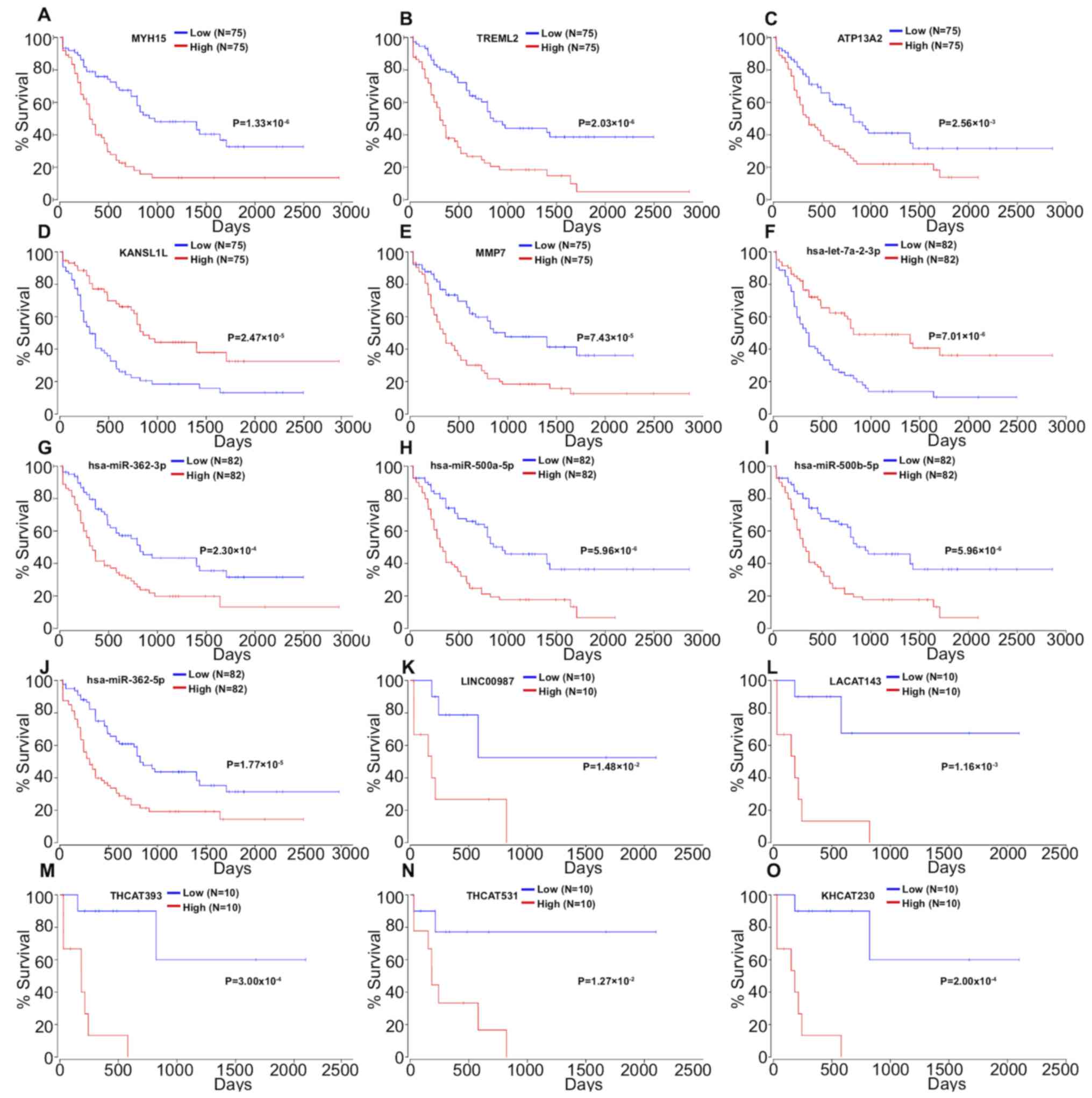
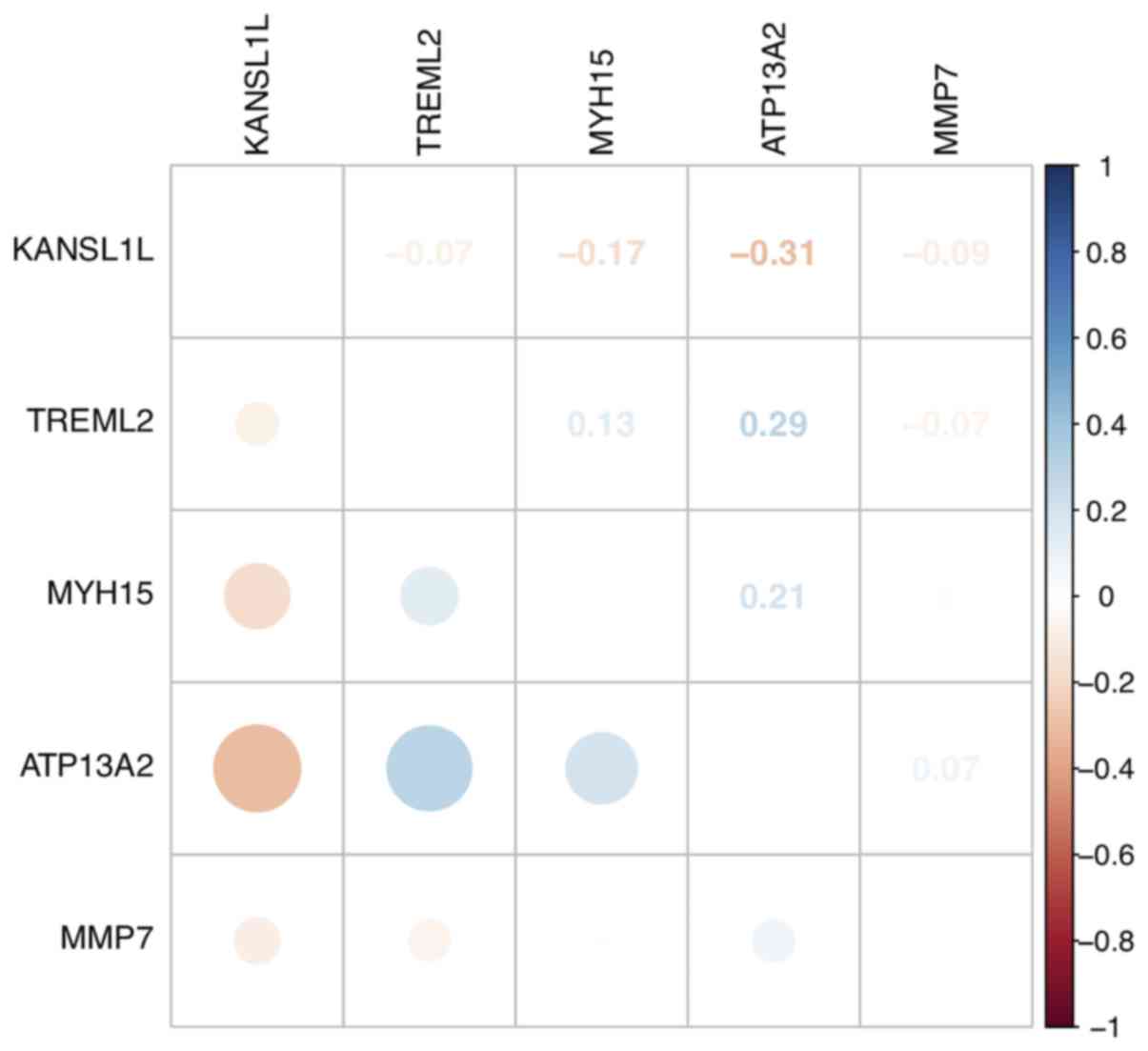
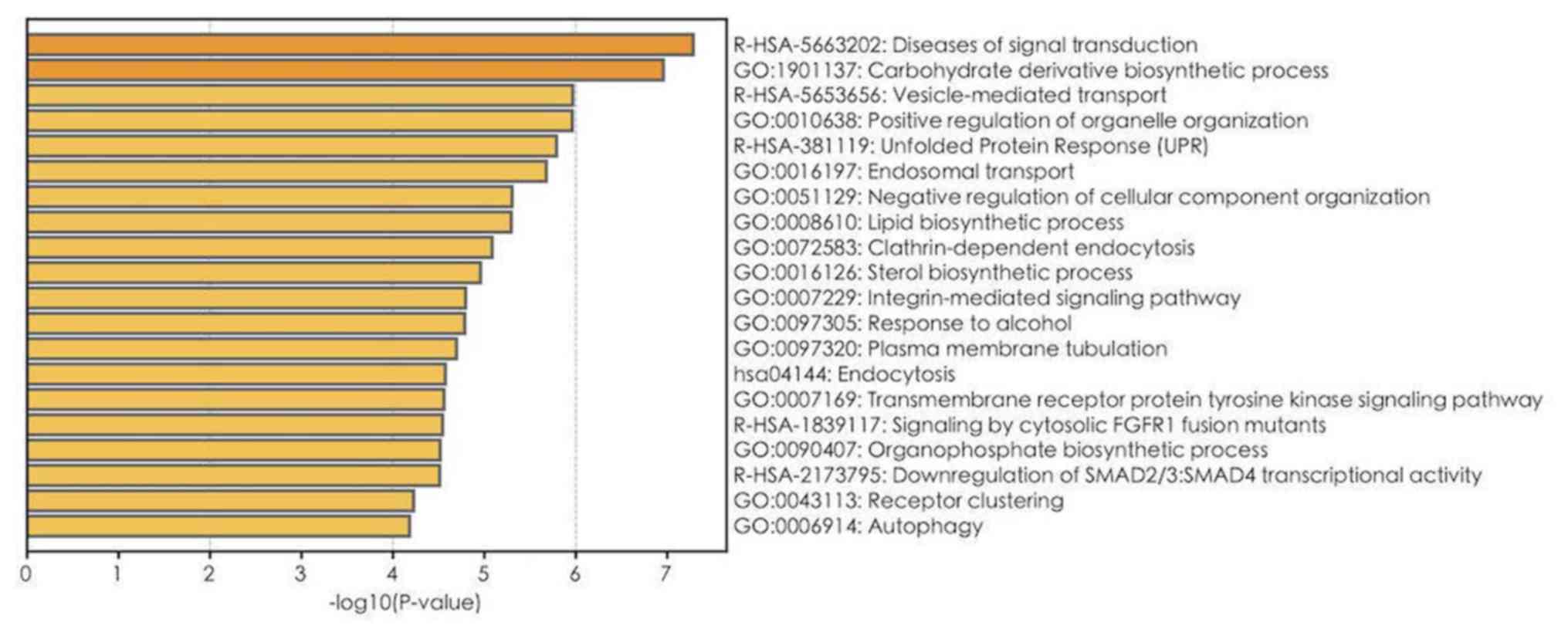
|
1
|
Tallman MS, Wang ES, Altman JK, Appelbaum
FR, Bhatt VR, Bixby D, Coutre SE, De Lima M, Fathi AT, Fiorella M,
et al: Acute myeloid leukemia, version 3.2019, NCCN clinical
practice guidelines in oncology. J Natl Compr Canc Netw.
17:721–749. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
National Cancer Institute, . SEER cancer
statistics review, 1975–2015: Overview, median age at diagnosis.
September
10–2018
|
|
3
|
Juliusson G: Older patients with acute
myeloid leukemia benefit from intensive chemotherapy: An update
from the Swedish acute leukemia registry. Clin Lymphoma Myeloma
Leuk. 11 (Suppl 1):S54–S59. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
DiNardo C and Lachowiez C: Acute myeloid
leukemia: From mutation profiling to treatment decisions. Curr
Hematol Malig Rep. 14:386–394. 2019.PubMed/NCBI
|
|
5
|
Stuani L, Sabatier M and Sarry JE:
Exploiting metabolic vulnerabilities for personalized therapy in
acute myeloid leukemia. BMC Biol. 17:572019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pagana L, Pulsoni A, Tosti ME, Avvisati G,
Mele L, Mele M, Martino B, Visani G, Cerri R, Di Bona E, et al:
Clinical and biological features of acute myeloid leukaemia
occurring as second malignancy: GIMEMA archive of adult acute
leukaemia. Br J Haematol. 112:109–117. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pulsoni A, Pagano L, Lo Coco F, Avvisati
G, Mele L, Di Bona E, Invernizzi R, Leoni F, Marmont F, Mele A, et
al: Clinicobiological features and outcome of acute promyelocytic
leukemia occurring as a second tumor: The GIMEMA experience. Blood.
100:1972–1976. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tomczak K, Czerwińska P and Wiznerowicz M:
The cancer genome atlas (TCGA): An immeasurable source of
knowledge. Contemp Oncol (Pozn). 19:A68–A77. 2015.PubMed/NCBI
|
|
9
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Sci Signal. 6:pl12013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Alboukadel K, Marcin K and Przemyslaw B:
Drawing Survival Curves using ‘ggplot2’. R package version 0.4.6.
September
9–2019
|
|
11
|
Chambers J: Programming with R. Software
for data analysis. Springer-Verlag. (New York, NY). 2008.
View Article : Google Scholar
|
|
12
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhou Y, Zhou B, Pache L, Chang M,
Khodabakhshi AH, Tanaseichuk O, Benner C and Chanda SK: Metascape
provides a biologist-oriented resource for the analysis of
systems-level datasets. Nat Commun. 10:15232019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wickham H: ggplot2. Wiley
Interdisciplinary Reviews: Computational Statistics. 3:180–185.
2011. View
Article : Google Scholar
|
|
15
|
Gu Z, Eils R and Schlesner M: Complex
heatmaps reveal patterns and correlations in multidimensional
genomic data. Bioinformatics. 32:2847–2849. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ilyas AM, Ahmad S, Faheem M, Naseer MI,
Kumosani TA, Al-Qahtani MH, Gari M and Ahmed F: Next generation
sequencing of acute myeloid leukemia: Influencing prognosis. BMC
Genomics. 16 (Suppl 1):S52015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ding L, Ley TJ, Larson DE, Miller CA,
Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan
MD, et al: Clonal evolution in relapsed acute myeloid leukaemia
revealed by whole-genome sequencing. Nature. 481:506–510. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Martelli MP, Sportoletti P, Tiacci E,
Martelli MF and Falini B: Mutational landscape of AML with normal
cytogenetics: Biological and clinical implications. Blood Rev.
27:13–22. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tomasson MH, Xiang Z, Walgren R, Zhao Y,
Kasai Y, Miner T, Ries RE, Lubman O, Fremont DH, McLellan MD, et
al: Somatic mutations and germline sequence variants in the
expressed tyrosine kinase genes of patients with de novo acute
myeloid leukemia. Blood. 111:4797–4808. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lindsley RC, Mar BG, Mazzola E, Grauman
PV, Shareef S, Allen SL, Pigneux A, Wetzler M, Stuart RK, Erba HP,
et al: Acute myeloid leukemia ontogeny is defined by distinct
somatic mutations. Blood. 125:1367–1376. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cancer Genome Atlas Research Network, .
Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A,
Hoadley K, Triche TJ Jr, Laird PW, et al: Genomic and epigenomic
landscapes of adult de novo acute myeloid leukemia. N Engl J Med.
368:2059–2074. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Marcucci G, Haferlach T and Döhner H:
Molecular genetics of adult acute myeloid leukemia: Prognostic and
therapeutic implications. J Clin Oncol. 29:475–486. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu H, Li P, Shao N, Ma J, Ji M, Sun X, Ma
D and Ji C: Aberrant expression of Treg-associated cytokine IL-35
along with IL-10 and TGF-β in acute myeloid leukemia. Oncol Lett.
3:1119–1123. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen J, Mu Q, Li X, Yin X, Yu M, Jin J, Li
C, Zhou Y, Zhou J, Suo S, et al: Homoharringtonine targets Smad3
and TGF-β pathway to inhibit the proliferation of acute myeloid
leukemia cells. Oncotarget. 8:40318–40326. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
De Visser KE and Kast WM: Effects of
TGF-beta on the immune system: Implications for cancer
immunotherapy. Leukemia. 13:1188–1199. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lin HK, Bergmann S and Pandolfi PP:
Deregulated TGF-beta signaling in leukemogenesis. Oncogene.
24:5693–5700. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Massagué J: TGFβ signalling in context.
Nat Rev Mol Cell Biol. 13:616–630. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Massagué J: TGFbeta in cancer. Cell.
134:215–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ruscetti FW, Akel S and Bartelmez SH:
Autocrine transforming growth factor-beta regulation of
hematopoiesis: Many outcomes that depend on the context. Oncogene.
24:5751–5763. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Matsunawa M, Ishii Y, Kasukabe T, Tomoyasu
S, Ota H and Honma Y: Cotylenin A-induced differentiation is
independent of the transforming growth factor-beta signaling system
in human myeloid leukemia HL-60 cells. Leuk Lymphoma. 47:733–740.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Macdonald D, Reiter A and Cross NC: The
8p11 myeloproliferative syndrome: A distinct clinical entity caused
by constitutive activation of FGFR1. Acta Haematol. 107:101–107.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ren M, Li X and Cowell JK: Genetic
fingerprinting of the development and progression of T-cell
lymphoma in a murine model of atypical myeloproliferative disorder
initiated by the ZNF198-fibroblast growth factor receptor-1
chimeric tyrosine kinase. Blood. 114:1576–8154. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jackson CC, Medeiros LJ and Miranda RN:
8p11 myeloproliferative syndrome: A review. Hum Pathol. 41:461–476.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yu J, Chen L, Cui B, Widhopf GF II, Shen
Z, Wu R, Zhang L, Zhang S, Briggs SP and Kipps TJ: Wnt5a induces
ROR1/ROR2 heterooligomerization to enhance leukemia chemotaxis and
proliferation. J Clin Invest. 126:585–598. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Agarwal A, Das K, Lerner N, Sathe S, Cicek
M, Casey G and Sizemore N: The AKT/IκB kinase pathway promotes
angiogenic/metastatic gene expression in colorectal cancer by
activating nuclear factor-κB and β-catenin. Oncogene. 24:1021–1031.
2005. View Article : Google Scholar : PubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBIPubMed/NCBI
|