Introduction
Numerous cell signaling pathways are regulated by
the ubiquitin-proteasome system. Ubiquitination of the target
protein generally requires the joint action of three enzymes.
First, E1 activates the ubiquitin molecule; E2 then binds the
ubiquitin molecule; and finally, an E3 ubiquitin ligase binds to a
specific substrate and E2 covalently transfers the ubiquitin
protein to one or more lysine residues of the target protein
(1–3). Thousands of ubiquitination processes
are ongoing in a cell at any moment (4). The high specificity of this degradation
mechanism targeting specific proteins is determined mainly by the
E3 ubiquitin ligase (5). Therefore,
the E3 ligase plays a key role in degradation of the target protein
(6). E3 ubiquitin ligases with a
RING domain comprise a large group of E3 ubiquitin ligases that is
responsible for ~20% of ubiquitin-mediated protein degradation
(6); additionally, degradation
events regulated by Cullin RING ligases (CRLs), which are composed
of Cullin (Cul) proteins, account for a large proportion (7). Previous studies have shown that CRLs
not only are the primary factors in protein degradation but also
are involved in the initiation and progression of some cancers
including colorectal cancer, cholangiocarcinoma, etc (8,9).
The Cul protein family was first reported in 1996 to
form an active complex that can regulate the cell cycle (10). The human genome contains 7 Cul
proteins, including, Cul1, Cul2, Cul3, Cul4A, Cul4B, Cul5, Cul7 and
Cul9 (11). All six have conserved,
homologous Cul domains, which function to bind the subunit RING
domain proteins RING-box protein 1 (Rbx)1 or Rbx 2 in the whole
complex. In addition to Cul7 and Cul9, five other members have
three serial N-terminal Cul repeats, which are used to recognize
subunits in the E3 ubiquitin ligase complex. Since it was
discovered and defined nearly 20 years ago, subsequent studies have
revealed that the multipart E3 ligase complex consisting of this
family of proteins has an intricate structure and serves important
functions in the cell cycle, signal transduction, cell development
and other physiological processes (12,13).
Among the CRLs, CRL1 [also known as S phase kinase-
associated protein 1-Cul 1-F-box (SCF)] has been extensively
studied. The SCF complex is composed of Cul1, Rbx1, the linker
protein Skp1 and variable F-box (FBX) proteins. Within the SCF
structure, differences in the FBX proteins, determines the
specificity of substrate binding to accomplish the degradation of
different substrates. To date, 69 FBX proteins have been found and
identified in the human genome, and most can form the CRL1 complex
(14). FBX proteins can be roughly
divided into three types according to the other domains present: i)
FBXW, which contain a tryptophan-aspartic acid 40 (WD40) repeat
domain; ii) FBXL, which contain a leucine-rich repeat domain; and
iii) FBXO, which contain other domain motifs. Accumulating evidence
has indicated that dysregulation of FBX proteins is involved in the
development, angiogenesis, proliferation and metastasis of a number
of malignancies (9). F-box/WD
repeat-containing protein 7 (FBXW7) is also called AGO, hCDC4 and
SEL-10; SEL-10 was first identified from yeast, AGO was first found
in Drosophila, and CDC4 is a yeast gene (15). The SCF E3 ubiquitin ligase complex
contains FBXW7 which targets several important oncoproteins
including c-Jun, c-Myc and Notch1 etc. for ubiquitylation (Fig. 1A). The human FBXW7 gene is located on
chromosome 4 (4q31.3) and is mainly expressed in the brain, heart
and testes, but at lower levels in the liver, lungs and kidneys
(16).
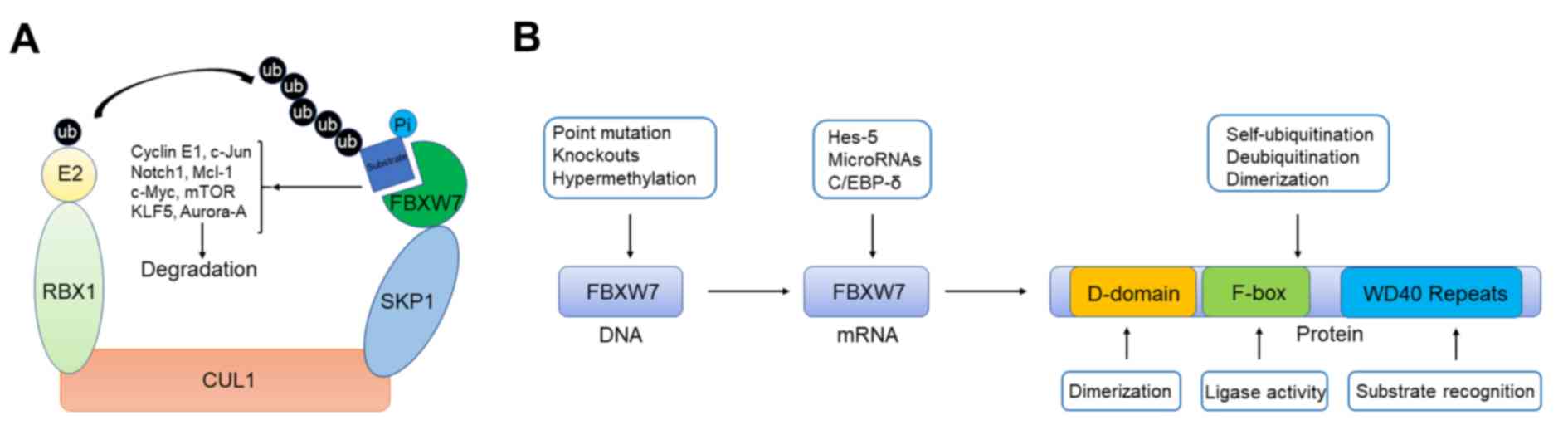 | Figure 1.Schematic illustration presenting the
functional model and regulation of FBXW7. (A) Functional model of
FBXW7. Schematic illustration showed that SCF E3 ubiquitin ligase
complex containing FBXW7 can target several important oncoproteins
including c-Jun, c-Myc, and Notch1 et al for ubiquitylation.
(B) Regulation of FBXW7 from gene to protein. FBXW7 contains a
D-domain for dimer formation and two key domains of F-box protein
family including WD repeat and F-box for substrate binding and
enzymatic activity. Regulatory mechanisms and related proteins that
induce FBXW7 dysfunction at different levels. FBXW7, F-box/WD
repeat-containing protein 7; RBX1, RING-box protein; KLF5,
Krüppel-like factor 5; E2, enzyme 2; ub, ubiquitin; Pi, phosphate;
SKP1, S phase kinase-associated protein 1; CUL1, cullin 1; Hes-5,
hairy and enhancer of split 5; C/EBP-δ, CCAAT enhancer binding
protein δ. |
The FBXW7 gene produces three mRNA transcripts
(FBXW7α, FBXW7β and FBXW7γ) through selective splicing following
transcription. The tissue expression and subcellular localization
differ among the three subtypes; FBXW7α mRNA is expressed in almost
all tissues, FBXW7β mRNA can be detected only in the brain and
testes, and FBXW7γ mRNA mainly exists in the heart and skeletal
muscle (17). FBXW7α and FBXW7β are
localized in the cytoplasm, whereas FBXW7γ is in the nucleolus
(18). FBXW7α plays the most
important role among the three subtypes and can occasionally
replace the other two (19).
However, the β and γ subtypes may also play unique roles under
specific conditions, as their abnormal localization may cause cell
functional defects (20). The human
FBXW7α contains 707 amino acid residues. The N-terminus of FBXW7α
contains an FBX motif composed of ~40 amino acids, which mainly
contributes to ubiquitin ligase activity (21). In addition, FBXW7α has a seven-tandem
repeat WD40 domain that is necessary for substrate recognition, and
a dimerization domain that is associated with the binding of FBXW7
to the substrate (Fig. 1B). FBXW7
recognizes and binds substrates with the specific phosphorylated
amino acid sequence, (L)-X-pT/pS-P-(P)-X-pS/pT/E/D (where X can be
any amino acid residue) (22). In
numerous cases, the conserved Cdc4 phosphodegron (CPD) motif of
FBXW7 substrates is phosphorylated by glycogen synthase kinase 3
(GSK3), which initiates FBXW7-mediated substrate ubiquitination
(23,24).
FBXW7 is generally recognized as a cancer suppressor
that regulates the ubiquitination and degradation of various
proliferation- and survival-related proteins, including cyclin E
(25), Notch (15), c-Myc (26) and mammalian target of rapamycin
(mTOR) (27). The mutation rate of
FBXW7 in colorectal cancer (28),
breast cancer (29) and other
malignant tumors is ~10%, whereas in some types of leukemia, the
mutation rate is as high as 60% (30). Moreover, the mutational status of
FBXW7 is directly related to the therapeutic effect of
chemotherapeutic drugs (31). In
addition, FBXW7 plays a role in embryonic development, as mice with
FBXW7 knockout mutation die on day 11, the middle stage of
embryonic development, mainly because of a developmental deformity
of the digestive organs (32). FBXW7
also serves an important role in the proliferation and maturation
of stem cell populations (33).
Transcriptional and translational regulation
of FBXW7 expression
Transcriptional regulation
On the one hand, FBXW7 affects the ubiquitination
state and protein content of various substrates; on the other hand,
this process is tightly regulated from DNA to protein expression
(Fig. 1B) (21). FBXW7 expression can be regulated at a
number of levels, including at the transcriptional level through
the regulation of multiple transcription factors. FBXW7α is
negatively regulated by CCAAT/enhancer-binding protein δ (CEBPδ), a
transcription factor involved in adipocyte differentiation as well
as inflammation reactions (34). In
mammary tumors, CEBPδ expression is induced under a hypoxic
microenvironment, which can lead to tumor metastasis by directly
binding to the promoter of FBXW7 to inhibit its expression
(35,36). Additionally, presenilin, a regulator
of Notch processing and β-catenin signaling, can also indirectly
downregulate FBXW7α mRNA expression (37). FBXW7β and FBXW7γ are upregulated in a
p53-dependent manner, and their upregulation is required for the
apoptotic response and tumor suppression induced by p53 (38).
Translational regulation
A number of previous studies have indicated that the
direct binding of multiple non-coding microRNAs (miRNAs) to the 3′
untranslated region of the mRNA can prevent protein translation of
FBXW7 (39,40). Overexpression of miRNA (miR)-223 in T
cell acute lymphoblastic leukemia (T-ALL), colorectal cancer and
gastric cancer has been reported to downregulate FBXW7 (39,40). In
T-ALL, high levels of miR-223 promote the proliferation of tumor
cells, and its inhibition increases the sensitivity to γ-secretase
inhibitor drugs (39). FBXW7 is also
repressed by miR-27a overexpression in an adenomatous polyposis
coli protein (APC) mutation-mediated murine model of colorectal
adenocarcinoma and in human high-grade colorectal adenocarcinomas
associated with preinvasive adenomas (41,42).
Inhibition of miR-27a increases FBXW7 expression and downregulates
FBXW7 substrates, which delays tumor formation in model systems and
inhibits the proliferation of colorectal cancer cells (43). miR-92a is reported to decrease the
expression levels of FBXW7 mRNA and protein, and to increase c-Myc
expression, which facilitates Myc-mediated apoptosis and
proliferation in a model of B-cell lymphoma (44). Knockdown of miR-92a suppresses cancer
cell invasion and proliferation through the upregulation of FBXW7
(45).
Additional miRNAs, including miR-548, miR-544a,
miR-367, miR-182, miR-503, miR-155-3p and miR-32, have also been
demonstrated to modulate FBXW7 activity via various mechanisms
(46–51). Although individual miRNAs are weakly
related to FBXW7 expression in patients with cancer, suggesting
that more than one miRNA is involved in the regulation of FBXW7,
miRNA-mediated suppression of FBXW7 synchronously targets the three
FBXW7 isoforms, which may influence additional FBXW7 substrates
(52).
Epigenetic regulation
The transcription and translation of FBXW7 are
regulated by epigenetics; specifically, histone modifications have
been reported to regulate FBXW7. For example, the histone
methyltransferase enhancer of zeste homolog 2 (EZH2), is associated
with epigenetic inactivation of genes, including FBXW7; EZH2
promotes trimethylation of histone H3 Lys27 residue of FBXW7, which
results in inactivation of FBXW7 gene function (53). Notably, FBXW7 has been reported to
target EZH2 for ubiquitination and degradation in pancreatic cancer
cells and to be negatively associated with the expression of EZH2
in human pancreatic cancer samples (54). In addition to histone modifications,
DNA modifications are reported to regulate FBXW7 expression. In
contrast to FBXW7α, which exhibits ubiquitous expression in cell
lines and a broad tissue distribution, FBXW7β is expressed in
specific cell lines and tissues (55). Histone and DNA modifications have
been reported to epigenetically regulate the FBXW7β promoter, which
is methylated in 51% of primary breast cancer tumors and 43% of 60
human cancer cell lines originating from brain, breast, kidney,
prostate, cervix, blood, skin, lung, thyroid and bone (56). The expression of the FBXW7β gene is
negatively associated with its methylation level (56). Patients with lymph node-positive
breast cancer with higher FBXW7 methylation levels have longer
overall survival times, although methylation of FBXW7 correlates
with high-grade tumors (56). In
addition, patients with ovarian cancer with p53 mutations have been
reported to exhibit lower FBXW7 expression compared with those with
wild-type p53, and hypermethylation of the FBXW7 promoter
associated with mutations in p53 leads to decreased FBXW7
expression (57).
Post-translational modifications of
FBXW7
Autoubiquitination
Post-translational modifications of FBXW7 are
involved in its autoubiquitination, deubiquitination, dimerization
and localization. In addition to targeting substrates for
ubiquitination and degradation, FBXW7 can also be regulated by
autoubiquitination. Peptidyl-prolyl cis-trans
isomerase NIMA-interacting 1 (Pin1) has been reported to
destabilize and downregulate FBXW7 by mediating a decrease in
dimerization and promoting FBXW7 self-ubiquitination and
degradation (58). In addition,
phosphorylation at Thr205 by extracellular signal-regulated kinase
and at Ser176 by polo-like kinase 2 leads to destabilization and
degradation (59,60). By contrast, phosphorylation mediated
by serum- and glucocorticoid-inducible kinase 1 (SGK1) and
phosphoinositide 3-kinase at Ser227 stabilizes FBXW7 but increases
ubiquitination of cyclin E, Notch and Myc (53,54).
FBXW7 stability can also be controlled by SCF-dependent mechanisms.
For instance, COP9 signalosome complex subunit 6, a member of the
COP9 signalosome complex, increases FBXW7 autoubiquitination and
proteasome-mediated degradation by regulating Cul1 neddylation
(61).
Deubiquitination
Autoubiquitination and degradation of FBXW7 can be
reversed by the deubiquitinating enzyme 28 (USP28). Overexpression
of USP28 not only allows the degradation of several FBXW7
substrates, inhibits progenitor cell proliferation and delays tumor
formation (62), but also represses
autocatalytic ubiquitination of FBXW7 (63). Genetic ablation of USP28 negatively
regulates FBXW7 and its substrates in the pancreas, liver and
lungs. In addition, abrogation of USP28 facilitates the
transformation of mouse fibroblasts due to FXBW7 destabilization
(63).
Dimerization
FBXW7 is characterized by its ability to form dimers
through a conserved D domain. Previous studies of endogenous FBXW7
with mutations preventing dimer formation have revealed that
dimerization facilitates ubiquitination of FBXW7 substrates with
low-affinity degrons, but it is unimportant to substrates with
high-affinity degrons, including cyclin E and Myc (64,65).
Dimer-deficient mutants of CDC4 in yeast variant show increased
autocatalytic ubiquitination and instability in vivo
(66). In addition, isomerization
mediated by Pin1 and phosphorylation of human FBXW7 at Ser205
repress its dimerization and enhance its autoubiquitination
(58).
Location
Abnormal cellular distribution of FBXW7 impairs its
interaction with substrates. For example, nucleophosmin is
necessary for the nucleolar localization of FBX7γ and is often
mutated in acute myelogenous leukemia, leading to FBX7γ instability
and an increase in c-Myc expression (67). Additionally, phosphorylation of
FBXW7α at Ser10 inhibits one of its nuclear localization signals
(68).
Genetic alterations of FBXW7 in cancers
The important role of FBXW7 in tumor suppression has
been confirmed by different genetic alterations of FBXW7 in various
human cancers. Functional silencing of FBXW7 by deletion, mutation
and hypermethylation ultimately leads to tumorigenesis and cancer
progression (69). Although rare,
FBXW7 mutations occur often in breast, pancreatic, gastric and
cervical cancers but rarer promoter hypermethylation and genetic
deletion of FBXW7 occurs in bladder, cervical and breast cancers
(65). However, missense point
mutations are the most common type of genetic alterations in FBXW7
and are observed on three arginine residues (R465, R479 and R505)
at the β propeller (65). FBXW7 is
usually expressed as a functional wild-type protein because the
second wild-type allele is retained. Consistent with this
observation, mouse models with monoallelic deletion of FBXW7 show a
milder tumor phenotype compared with biallelic gene deletions
(70). Therefore, biallelic FBXW7
mutations are assumed to silence the function of the wild-type
protein as dominant negative alleles (65).
In the intestine and the hematopoietic system,
knock-in mice with a heterozygous FBXW7 mutation show accelerated
tumorigenesis compared to mice with heterozygous wild-type FBXW7
(FBXW7+/−) (70,71). The hematopoietic stem cells in
FBXW7Mut/+ mice show a significant increase in Myc but
do not exhibit the characteristic hyperproliferative phenotype of
those in FBXW7−/− animals (70). Mice with T-cell leukemias induced by
an activated Notch allele show accumulation of sterol regulatory
element-binding protein 1 and Myc (70). Moreover, additional depletion of p53
does not promote the onset of disease, indicating the functional
difference between complete loss and mutation of FBXW7 (70). By contrast, the observation that only
Krüppel-like factor 5 (KLF5) and homeobox protein TGIF1 (TGIF1),
instead of most tested FBXW7 substrates, are substantially affected
by mutated FBXW7, reveals that the influence of FBXW7 mutations on
substrate regulation is strongly context dependent (71). Although FBXW7 mutations enhance
intestinal tumorigenesis driven by the multiple intestinal
neoplasia, a mutant allele of APC, the levels of Notch, Jun and Myc
remain normal in these mice with developing adenomas (71). Therefore, heterozygous mutations in
FBXW7 may promote tumorigenesis by the regulation of non-canonical
substrates such as TGIF1 and KLF5.
Substrates and mechanisms of FBXW7 involved
in tumor suppression
Ubiquitin ligases play a biological role by
affecting the expression levels of specific target proteins. The
most common mechanism by which ubiquitin ligases participate in
cancer processes is through regulating the content of cell
cycle-related factors. FBXW7 can effectively recognize a variety of
substrates, such as cyclin E, KLF5, mTOR, Aurora A, c-Myc, c-Jun
and induced myeloid leukemia cell differentiation protein Mcl-1
(MCL-1). Most of these protein substrates are involved in the
regulation of cell cycle processes or homeostasis. Additionally,
they are the expressed products of oncogenes or potential oncogenes
(Fig. 2; Table I).
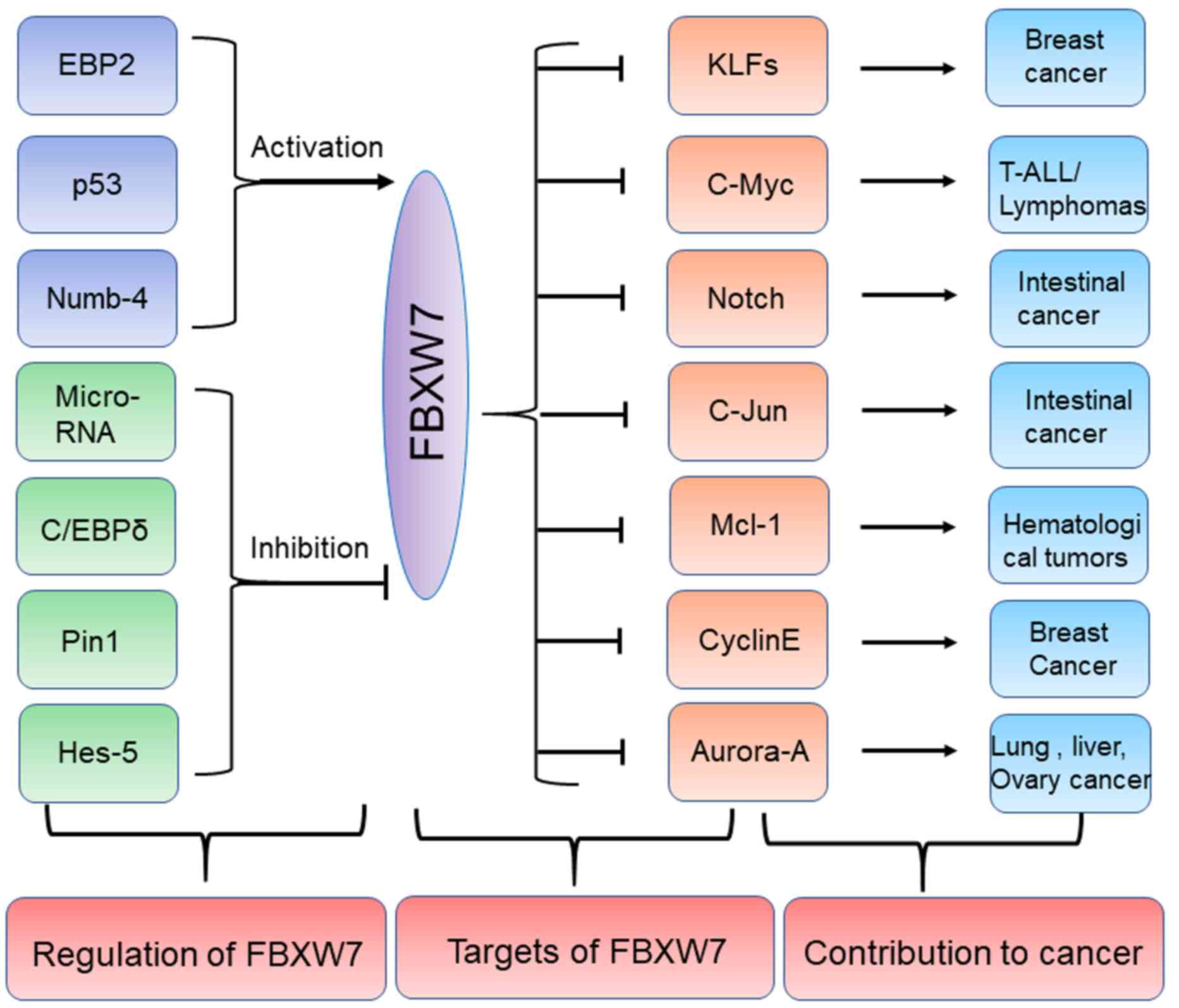 | Figure 2.Regulators and substrates of FBXW7 in
human cancers. Several upstream proteins including EBP2, p53 and
Numb-4 can positively regulate FBXW7 while other upstream
regulators including Pin1, Hes-5 and C/EBPδ etc. can negatively
regulate FBXW7. The specific substrates of FBXW7 including c-Myc,
c-Jun and Mcl-1 can promote development of some tumors including
lymphomas, intestinal cancer and hematological tumors. FBXW7,
F-box/WD repeat-containing protein 7; EBP2, eIF4E-binding protein
2; C/EBP-δ, CCAAT enhancer binding protein δ; Pin1, peptidyl-prolyl
cis-trans isomerase NIMA-interacting 1; Hes-5, hairy and
enhancer of split 5; KLFs, Krüppel-like factor; T-ALL, T cell acute
lymphoblastic leukemia; Mcl-1, induced myeloid leukemia cell
differentiation protein Mcl-1. Numb-4, NUMB endocytic adaptor
protein. |
 | Table I.Substrates targeted by F-box/WD
repeat-containing protein 7. |
Table I.
Substrates targeted by F-box/WD
repeat-containing protein 7.
| Author, year | Substrate | Genomic
location | Function/signalling
pathways | Molecular size
(amino acids) | Phospho-degron
(phosphorylation sites) | Putative modifying
kinase | (Refs.) |
|---|
| Finkin et
al, 2008 | Aurora-A | 20q13 | Protein kinase;
cell cycle | 403 | LGTVYREL | GSK3 | (78) |
| Klotz et al,
2009 | Cyclin E1 | 19q12 | Cyclin, cell
cycle | 410 | LLTPPQSG | GSK3 | (74) |
| Tsunematsu et
al, 2004 | Notch1 | 9q34 | Transcription
factor; Notch signalling pathway | 2,555 | FLTPSPES | Cdk8 | (84) |
| Yang-Yen et
al, 2006 | Mcl-1 | 1q23 | BCL2-family
protein; cell survival | 350 | IMSPEEEL,
DGSLPSTP | GSK3 | (86) |
| Welcker et
al, 2004 | c-Myc | 8q24 | Transcription
factor; cell proliferation | 439 | LPTPPLSP | GSK3 | (87) |
| Mao et al,
2008 | mTOR | 1p36 | Protein kinase;
PI3K signalling pathway | 2,549 | LLTPSIHL | – | (27) |
| Liu et al,
2010 | KLF5 | 13q22 | Transcription
factor; adipocyte differentiation and lipid metabolism | 457 | PPSPPSSE, LNTPDLDM,
NLTPPPSY | GSK3 | (92) |
| Wei et al,
2005 | c-Jun | 1p32 | Transcription
factor; cell proliferation | 331 | GETPPLSP | GSK3 | (23) |
Cyclin E
Cyclin E binds and activates the cell
cycle-dependent protein kinase cyclin-dependent kinase 2 (CDK2) to
promote transition of the cell cycle from G1 to S phase
(72). Increased expression of CDK2
can cause chromosomal instability and accelerate the occurrence of
cancer (73). Cyclin E is expressed
mainly in tissues exhibiting vigorous cell division, in direct
contrast with FBXW7, which is expressed mainly in non-proliferative
tissues (74). Cyclin E has a
typical CPD, in which phosphorylation of Thr380 and Ser384 is
necessary for degradation (32).
FBXW7 can catalyze the degradation of cyclin E2 in addition to E1,
the most common cyclin E subtype. Cyclin E2 contains two typical
CPDs, that are specifically recognized by FBXW7, and the
double-site phosphorylation of Thr392 and Ser396 initiates
ubiquitination and proteasomal degradation (74), explaining why the decrease in FBXW7
expression and the increase in cyclin E in primary invasive breast
cancer lead to the appearance of a large number of cells with
chromosomal polyploidy (75).
Aurora A
Aurora A (also known as serine/threonine protein
kinase 15) serves a role in mitosis and meiosis by regulating the
phosphorylation of specific substrates, and its activity is the
highest during G2/S phase transition (76). Aurora A is overexpressed in a variety
of tumors, resulting in abnormal centrosome expansion, increased
chromosomal instability and eventual carcinogenic transformation
(76,77). FBXW7-deficient HCT116 and HeLa cells
exposed to vincristine, paclitaxel and spindle toxin exhibit
extensive mitotic delay and nuclear duplication, which are
important causes of polyploidy. Loss of FBXW7 can increase the
content of cyclin E and Aurora A, but the increase of single cyclin
E or Aurora A could not cause drug-induced polyploidy. This
observation shows that the increase in cyclin E and Aurora A is a
cause of polyploidy (78).
Notch
Notch is a highly conserved signaling system in
multicellular organisms, playing an important role in cell
proliferation, differentiation and apoptosis (79). In mammals there are four isoforms of
Notch receptors, all of which are single-pass transmembrane
receptors in which the N-terminus is located outside the cell and
accounts for most of the structure and only a small part
(C-terminus) is inside the cell (80). When a ligand binds to the
extracellular domain of the Notch receptor, it can induce a
proteolysis reaction and release the intracellular domain (81). The intracellular domain of the Notch
receptor acts as a transcription factor to regulate the expression
of specific genes (81).
Overactivation of the Notch signaling pathway can cause abnormal
cell proliferation and cancer (82).
Ubiquitination of the intracellular domain of Notch 1 and Notch 4
by FBXW7 significantly weakens Notch signal transduction, whereas
inhibition of FBXW7 enhances Notch-mediated activation of
downstream signaling (83). FBXW7
gene knockout results in the abolishment of Notch 4 signal
transmission, which can lead to abnormal development of blood
vessels in mouse embryos and may result in death at ~11 days
(84). Similarly, mice with
brain-specific FBXW7 knockout succumb after birth owing to the
accumulation of Notch 1 and Notch 3 proteins in the brain, which
results in increased expression of target genes and abnormal
differentiation of neural stem cells, causing abnormal brain
development and morphology in these mice (85).
MCL-1
MCL1 is an antiapoptotic protein of the BCL2 family
that can promote cancer development by reducing apoptosis (86). A number of hematological tumors, such
as B-cell lymphoma and chronic myeloid leukemia, exhibit an
abnormal increase in MCL1 expression, which is considered an
important cause of chemotherapeutic resistance. In normal cells,
the half-life of the MCL1 protein is short, and it is easily
degraded by ubiquitination modification. However, in tumor cells,
the MCL1 protein content is increased, although a detailed
understanding of this mechanism is lacking. MCL1 can be
phosphorylated by GSK3, which initiates ubiquitination and
degradation of MCL1 (86). Loss of
FBXW7 in a human T-ALL cell line is accompanied by an increase in
MCL1 content. This cell line is sensitive to a variety of kinase
inhibitors, such as sorafenib but is resistant to ABT-737, an
inhibitor of BCL2 (22). When FBXW7
function is restored or MCL1 is lost, sensitivity to ABT-737 can be
restored. Therefore, these findings confirm that the increase in
the MCL1 protein content caused by FBXW7 deficiency is a mechanism
underlying tumor chemoresistance (22). In addition, paclitaxel treatment
induces phosphorylation modification of MCL1, which can be
recognized by FBXW7, leading to MCL1 ubiquitination and
degradation, consequently increasing apoptosis (31). By contrast, when FBXW7 is inactivated
or expression of FBXW7 is decreased, the protein stability of MCL1
is increased. Correspondingly, resistance to microtubule-targeted
drugs, such as paclitaxel, is increased in tumor patients, and the
its chemotherapeutic effect is significantly reduced (31).
c-Myc
c-Myc is an important oncogenic protein that can
regulate cell growth and division, serving a number of roles in
human cancer. In lymphoid tumor cell lines, mutation of the Thr58
site in c-Myc is the most frequent mutation and results in the
failure of FBXW7 to regulate c-Myc protein content, accumulation of
c-Myc protein and eventual tumor development (87). In addition to GSK3, NEMO-like kinase
(NLK) also regulates the c-Myc protein content. NLK can directly
bind to c-Myc and catalyze the phosphorylation of multiple
C-terminal sites. This modification promotes ubiquitination and
proteasomal degradation of FBXW7. Mutation of these sites can
abrogate the c-Myc-FBXW7 interaction and protein ubiquitination
(88).
Abnormal localization of c-Myc proteins can cause
its accumulation and may lead to tumorigenesis (63). USP28 can bind to FBXW7 and inhibit
ubiquitination modification of c-Myc by the latter, thus increasing
the protein stability of c-Myc (89). DNA damage caused by ultraviolet
radiation can reduce c-Myc protein content due to dissociation of
USP28 and FBXW7, thus increasing FBXW7-mediated ubiquitination and
degradation of c-Myc protein. Because both cyclin E and c-Myc are
positive regulators of the cell cycle, decreases in their levels
can cause cell cycle exit. However, FBXW7 deficiency increases the
protein levels of these two factors and promotes cell cycle
re-entry and G1/S phase transition, which is conducive
to cell division (90). This
mechanism is an important reason for cancer driven by FBXW7
mutations.
mTOR
mTOR is a protein kinase that promotes cell growth
and division by regulating protein synthesis and cell autotropism.
The increase in mTOR content and activity is a common feature of
tumorigenesis, and mTOR is widely used as a drug target in tumor
therapy (27). In human breast
cancer cell lines and patients with primary breast cancer, FBXW7
can contain a gene deletion or a functional inactivation mutation,
which leads to an increase in the mTOR protein content and
activation of its downstream signaling pathway (27). The sensitivity of breast cancer cells
harboring wild-type FBXW7 to rapamycin is significantly increased,
which suggests that loss of FBXW7 function is a mechanism
underlying the resistance of tumor patients to mTOR pathway
inhibitors (27).
KLF5
KLF5 is a development-related transcription factor
that can promote the expression of multiple development-related
genes and may serve a role in cell proliferation, cell cycle,
apoptosis and cell migration and differentiation (73). KLF5 is overexpressed in various
cancers and can promote proliferation and deterioration of breast
cells (91). KLF5 is a short-lived
protein that can be rapidly degraded by ubiquitination. KLF5
contains three CPDs recognized by FBXW7; simultaneous mutation of
these sites can inhibit the interaction and ubiquitination of FBXW7
and KLF5, whereas the point mutations in FBXW7 can significantly
delay degradation of the KLF5 protein, leading to accumulation of
KLF5 in cells (92). The binding and
ubiquitination of KLF5 and FBXW7 are dependent on phosphorylation
of KLF5 at Ser303 by GSK3β (93). In
tumor cells, mutation or abnormal activation of FBXW7 results in a
decrease in the ubiquitination and the protein degradation of KLF5.
In turn, excessive accumulation of KLF5 promotes the occurrence of
cancer by increasing the expression of target genes.
Therapeutic exploitation of FBXW7
signaling
Given the substantial tumor control exhibited by
FBXW7 in mouse models, therapeutic exploitation of FBXW7 and its
related pathways for several types of cancer has attracted intense
interest. Various oncogenic transcription factors and oncoproteins
are degraded through FBXW7-mediated ubiquitination. Most substrates
with increased levels due to FBXW7 silencing mutations are
oncoproteins. Such substrates are likely to alter regulation of the
cell cycle, perturb stress response and rewire metabolic pathways
(28). Previous studies have
reported that the accumulation of c-Myc and MCL1 induce synthetic
lethal interactions between non-functionalFBXW7 and tumor necrosis
factor-like death ligands or mitotic inhibitors in tumor cells
(31,94). Similarly, FBXW7 mutant cells with
increased c-Myc are more sensitive to suppression of additional
enzymes, such as CDK1, and energy-sensing enzymes, such as
AMP-activated protein kinase (95,96).
Alternatively, post-translational modifications are important for
FBXW7 E3 ligase activity, suggesting that aberrant alteration of
upstream signaling might impair the antitumor effect of FBXW7. One
possibility is to target deubiquitinases, considering that USP36
and USP28 can antagonize FBXW7 activity. Growth of established
tumors can be inhibited by acute decrease of USP28, showing that
the development of tumors depends on USP28 (56), and that certain types of tumor could
be effectively treated using inhibition of USP28 with small
molecules. USP7 inhibitors successfully stabilize p53 and promote
apoptosis in myeloma cells that are not sensitive to available
therapies (97). Additionally,
several studies have demonstrated that loss of p53, or mutation or
deletion of FBXW7 promotes tumorigenesis (10,98).
Since frequent mutant FBXW7 alleles result in impaired substrate
recognition, recovering substrate binding by FBXW7 may effectively
inhibit tumor development. This may be potentially achieved in a
manner analogous to p53, in which mutant forms of the protein can
be reactivated by small molecules (99). Moreover, since mutant FBXW7 has the
wild-type allele in most human tumors, several enzymes, such as
Pin1 and SGK1, which modulate the activity of FBXW7, may provide a
reasonable approach for tumor suppression by increasing FBXW7
activity (53,100). Therefore, restoring the antitumor
function of FBXW7 by blocking oncogenic upstream mediators may be
an effective therapeutic strategy.
Conclusions
Ubiquitination of specific proteins plays an
important role in tumor initiation, development, metastasis and
chemoresistance. Currently, the drugs designed to target
ubiquitin-modified molecules exhibit good potential in cancer
treatment. FBXW7 has been suggested to have tumor-suppressive
effects in various tumors. On the one hand, mutation of FBXW7 or a
reduction in its activity promotes the occurrence and progression
of tumors; on the other hand, it also increases the chemoresistance
of tumors. These developments are important to improving our
understanding of tumor occurrence mechanisms, the development of
diagnostic reagents, and the optimization and design of therapeutic
drugs. In the study of FBXW7, a series of important problems remain
to be solved. For example, are there still unidentified FBXW7
substrates? Is there a network mechanism underlying the regulation
of multiple substrates by FBXW7? What are the precise mechanisms of
FBXW7 regulation? In conclusion, the present review examined the
crucial role and molecular mechanism of FBXW7 in tumor inhibition,
and may offer a putative therapeutic approach for multiple
cancers.
Acknowledgements
Not applicable.
Funding
This work was jointly supported by The National
Science Foundation of China (grant nos. 81871950, 81702871 and
81602085), The Shanghai Municipal Commission of Health and Family
Planning (grant nos. 20154Y0090 and 2018YQ06) and The Shanghai
Sailing Program (grant no. 16YF1401800).
Availability of data and materials
Not applicable.
Authors' contributions
XY, ZZ, XX, SJ and WX were involved in the
conception of the review. ZZ, QH, WL and ML were involved in the
writing of the article. QS, GF, ZY, and YQ were involved in
critically revising and proofreading the manuscript. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hershko A and Ciechanover A: The ubiquitin
system. Annu Rev Biochem. 67:425–479. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schulman BA and Harper JW: Ubiquitin-like
protein activation by E1 enzymes: The apex for downstream
signalling pathways. Nat Rev Mol Cell Biol. 10:319–331. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wenzel DM, Stoll KE and Klevit RE: E2s:
Structurally economical and functionally replete. Biochem J.
433:31–42. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Deng L, Meng T, Chen L, Wei W and Wang P:
The role of ubiquitination in tumorigenesis and targeted drug
discovery. Signal Transduct Target Ther. 5:112020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zheng N and Shabek N: Ubiquitin ligases:
Structure, function, and regulation. Annu Rev Biochem. 86:129–157.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Deshaies RJ and Joazeiro CA: RING domain
E3 ubiquitin ligases. Annu Rev Biochem. 78:399–434. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zimmerman ES, Schulman BA and Zheng N:
Structural assembly of cullin-RING ubiquitin ligase complexes. Curr
Opin Struct Biol. 20:714–721. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Diaz VM and de Herreros AG: F-box
proteins: Keeping the epithelial-to-mesenchymal transition (EMT) in
check. Semin Cancer Biol. 36:71–79. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zheng N, Zhou Q, Wang Z and Wei W: Recent
advances in SCF ubiquitin ligase complex: Clinical implications.
Biochim Biophys Acta. 1866:12–22. 2016.PubMed/NCBI
|
|
10
|
Bai C, Sen P, Hofmann K, Ma L, Goebl M,
Harper JW and Elledge SJ: SKP1 connects cell cycle regulators to
the ubiquitin proteolysis machinery through a novel motif, the
F-box. Cell. 86:263–274. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pei XH, Bai F, Li Z, Smith MD, Whitewolf
G, Jin R and Xiong Y: Cytoplasmic CUL9/PARC ubiquitin ligase is a
tumor suppressor and promotes p53-dependent apoptosis. Cancer Res.
71:2969–2977. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nguyen HC, Wang W and Xiong Y: Cullin-RING
E3 ubiquitin ligases: Bridges to destruction. Subcell Biochem.
83:323–347. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Harper JW and Tan MK: Understanding
cullin-RING E3 biology through proteomics-based substrate
identification. Mol Cell Proteomics. 11:1541–1550. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nakagawa T, Nakayama K and Nakayama KI:
Knockout mouse models provide insight into the biological functions
of CRL1 components. Adv Exp Med Biol. 1217:147–171. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gupta-Rossi N, Le Bail O, Gonen H, Brou C,
Logeat F, Six E, Ciechanover A and Israel A: Functional interaction
between SEL-10, an F-box protein, and the nuclear form of activated
Notch1 receptor. J Biol Chem. 276:34371–34378. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jin J, Cardozo T, Lovering RC, Elledge SJ,
Pagano M and Harper JW: Systematic analysis and nomenclature of
mammalian F-box proteins. Genes Dev. 18:2573–2580. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Matsumoto A, Onoyama I and Nakayama KI:
Expression of mouse Fbxw7 isoforms is regulated in a cell cycle- or
p53-dependent manner. Biochem Biophys Res Commun. 350:114–119.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Welcker M, Orian A, Grim JE, Eisenman RN
and Clurman BE: A nucleolar isoform of the Fbw7 ubiquitin ligase
regulates c-Myc and cell size. Curr Biol. 14:1852–1857. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Grim JE, Gustafson MP, Hirata RK, Hagar
AC, Swanger J, Welcker M, Hwang HC, Ericsson J, Russell DW and
Clurman BE: Isoform- and cell cycle-dependent substrate degradation
by the Fbw7 ubiquitin ligase. J Cell Biol. 181:913–920. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Welcker M, Larimore EA, Frappier L and
Clurman BE: Nucleolar targeting of the fbw7 ubiquitin ligase by a
pseudosubstrate and glycogen synthase kinase 3. Mol Cell Biol.
31:1214–1224. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yumimoto K and Nakayama KI: Recent insight
into the role of FBXW7 as a tumor suppressor. Semin Cancer Biol.
Feb 27–2020.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Inuzuka H, Shaik S, Onoyama I, Gao D,
Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, et al:
SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for
ubiquitylation and destruction. Nature. 471:104–109. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wei W, Jin J, Schlisio S, Harper JW and
Kaelin WG Jr: The v-Jun point mutation allows c-Jun to escape
GSK3-dependent recognition and destruction by the Fbw7 ubiquitin
ligase. Cancer Cell. 8:25–33. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Welcker M, Singer J, Loeb KR, Grim J,
Bloecher A, Gurien-West M, Clurman BE and Roberts JM: Multisite
phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol
Cell. 12:381–392. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Moberg KH, Bell DW, Wahrer DC, Haber DA
and Hariharan IK: Archipelago regulates Cyclin E levels in
Drosophila and is mutated in human cancer cell lines.
Nature. 413:311–316. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yada M, Hatakeyama S, Kamura T, Nishiyama
M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K and
Nakayama KI: Phosphorylation-dependent degradation of c-Myc is
mediated by the F-box protein Fbw7. EMBO J. 23:2116–2125. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mao JH, Kim IJ, Wu D, Climent J, Kang HC,
DelRosario R and Balmain A: FBXW7 targets mTOR for degradation and
cooperates with PTEN in tumor suppression. Science. 321:1499–1502.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rajagopalan H and Lengauer C: hCDC4 and
genetic instability in cancer. Cell Cycle. 3:693–694. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rajagopalan H, Jallepalli PV, Rago C,
Velculescu VE, Kinzler KW, Vogelstein B and Lengauer C:
Inactivation of hCDC4 can cause chromosomal instability. Nature.
428:77–81. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Maser RS, Choudhury B, Campbell PJ, Feng
B, Wong KK, Protopopov A, O'Neil J, Gutierrez A, Ivanova E, Perna
I, et al: Chromosomally unstable mouse tumours have genomic
alterations similar to diverse human cancers. Nature. 447:966–971.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wertz IE, Kusam S, Lam C, Okamoto T,
Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, et al:
Sensitivity to antitubulin chemotherapeutics is regulated by MCL1
and FBW7. Nature. 471:110–114. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tetzlaff MT, Yu W, Li M, Zhang P, Finegold
M, Mahon K, Harper JW, Schwartz RJ and Elledge SJ: Defective
cardiovascular development and elevated cyclin E and Notch proteins
in mice lacking the Fbw7 F-box protein. Proc Natl Acad Sci USA.
101:3338–3345. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang Z, Inuzuka H, Fukushima H, Wan L, Gao
D, Shaik S, Sarkar FH and Wei W: Emerging roles of the FBW7 tumour
suppressor in stem cell differentiation. EMBO Rep. 13:36–43. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Balamurugan K, Sharan S, Klarmann KD,
Zhang Y, Coppola V, Summers GH, Roger T, Morrison DK, Keller JR and
Sterneck E: FBXW7α attenuates inflammatory signalling by
downregulating C/EBP δ and its target gene Tlr4. Nat Commun.
4:16622013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kourtis N, Moubarak RS, Aranda-Orgilles B,
Lui K, Aydin IT, Trimarchi T, Darvishian F, Salvaggio C, Zhong J,
Bhatt K, et al: FBXW7 modulates cellular stress response and
metastatic potential through HSF1 post-translational modification.
Nat Cell Biol. 17:322–332. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yumimoto K, Akiyoshi S, Ueo H, Sagara Y,
Onoyama I, Ueo H, Ohno S, Mori M, Mimori K and Nakayama KI: F-box
protein FBXW7 inhibits cancer metastasis in a non-cell-autonomous
manner. J Clin Invest. 125:621–635. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rocher-Ros V, Marco S, Mao JH, Gines S,
Metzger D, Chambon P, Balmain A and Saura CA: Presenilin modulates
EGFR signaling and cell transformation by regulating the ubiquitin
ligase Fbw7. Oncogene. 29:2950–2961. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Grinkevich VV, Nikulenkov F, Shi Y, Enge
M, Bao W, Maljukova A, Gluch A, Kel A, Sangfelt O and Selivanova G:
Ablation of key oncogenic pathways by RITA-reactivated p53 is
required for efficient apoptosis. Cancer Cell. 31:724–726. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mansour MR, Sanda T, Lawton LN, Li X,
Kreslavsky T, Novina CD, Brand M, Gutierrez A, Kelliher MA,
Jamieson CH, et al: The TAL1 complex targets the FBXW7 tumor
suppressor by activating miR-223 in human T cell acute
lymphoblastic leukemia. J Exp Med. 210:1545–1557. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kumar V, Palermo R, Talora C, Campese AF,
Checquolo S, Bellavia D, Tottone L, Testa G, Miele E, Indraccolo S,
et al: Notch and NF-kB signaling pathways regulate miR-223/FBXW7
axis in T-cell acute lymphoblastic leukemia. Leukemia.
28:2324–2335. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lerner M, Lundgren J, Akhoondi S, Jahn A,
Ng HF, Akbari Moqadam F, Oude Vrielink JA, Agami R, Den Boer ML,
Grander D and Sangfelt O: MiRNA-27a controls FBW7/hCDC4-dependent
cyclin E degradation and cell cycle progression. Cell Cycle.
10:2172–2183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang Q, Li DC, Li ZF, Liu CX, Xiao YM,
Zhang B, Li XD, Zhao J, Chen LP, Xing XM, et al: Upregulation of
miR-27a contributes to the malignant transformation of human
bronchial epithelial cells induced by SV40 small T antigen.
Oncogene. 30:3875–3886. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Spruck C: MiR-27a regulation of SCF(Fbw7)
in cell division control and cancer. Cell Cycle. 10:3232–3233.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Olive V, Sabio E, Bennett MJ, De Jong CS,
Biton A, McGann JC, Greaney SK, Sodir NM, Zhou AY, Balakrishnan A,
et al: A component of the mir-17-92 polycistronic oncomir promotes
oncogene-dependent apoptosis. Elife. 2:e008222013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhou C, Shen L, Mao L, Wang B, Li Y and Yu
H: MiR-92a is upregulated in cervical cancer and promotes cell
proliferation and invasion by targeting FBXW7. Biochem Biophys Res
Commun. 458:63–69. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang P, Cao L, Fan P, Mei Y and Wu M:
LncRNA-MIF, a c-Myc-activated long non-coding RNA, suppresses
glycolysis by promoting Fbxw7-mediated c-Myc degradation. EMBO Rep.
17:1204–1220. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu X, Ma J, Xu F and Li L: TINCR
suppresses proliferation and invasion through regulating
miR-544a/FBXW7 axis in lung cancer. Biomed Pharmacother. 99:9–17.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang Y, Liu Z, Yao B, Li Q, Wang L, Wang
C, Dou C, Xu M, Liu Q and Tu K: Long non-coding RNA CASC2
suppresses epithelial-mesenchymal transition of hepatocellular
carcinoma cells through CASC2/miR-367/FBXW7 axis. Mol Cancer.
16:1232017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Li L, Sarver AL, Khatri R, Hajeri PB,
Kamenev I, French AJ, Thibodeau SN, Steer CJ and Subramanian S:
Sequential expression of miR-182 and miR-503 cooperatively targets
FBXW7, contributing to the malignant transformation of colon
adenoma to adenocarcinoma. J Pathol. 234:488–501. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tang B, Lei B, Qi G, Liang X, Tang F, Yuan
S, Wang Z, Yu S and He S: MicroRNA-155-3p promotes hepatocellular
carcinoma formation by suppressing FBXW7 expression. J Exp Clin
Canc Res. 35:932016. View Article : Google Scholar
|
|
51
|
Xia W, Zhou J, Luo H, Liu Y, Peng C, Zheng
W and Ma W: MicroRNA-32 promotes cell proliferation, migration and
suppresses apoptosis in breast cancer cells by targeting FBXW7.
Cancer Cell Int. 17:142017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xu W, Taranets L and Popov N: Regulating
Fbw7 on the road to cancer. Semin Cancer Biol. 36:62–70. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mo JS, Ann EJ, Yoon JH, Jung J, Choi YH,
Kim HY, Ahn JS, Kim SM, Kim MY, Hong JA, et al: Serum- and
glucocorticoid-inducible kinase 1 (SGK1) controls Notch1 signaling
by downregulation of protein stability through Fbw7 ubiquitin
ligase. J Cell Sci. 124:(Pt 1). 100–112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Schulein C, Eilers M and Popov N:
PI3K-dependent phosphorylation of Fbw7 modulates substrate
degradation and activity. FEBS Lett. 585:2151–2157. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Cao J, Ge MH and Ling ZQ: Fbxw7 tumor
suppressor: A vital regulator contributes to human tumorigenesis.
Medicine (Baltimore). 95:e24962016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Akhoondi S, Lindstrom L, Widschwendter M,
Corcoran M, Bergh J, Spruck C, Grander D and Sangfelt O:
Inactivation of FBXW7/hCDC4-β expression by promoter
hypermethylation is associated with favorable prognosis in primary
breast cancer. Breast Cancer Res. 12(R105)2010.PubMed/NCBI
|
|
57
|
Kitade S, Onoyama I, Kobayashi H, Yagi H,
Yoshida S, Kato M, Tsunematsu R, Asanoma K, Sonoda K, Wake N, et
al: FBXW7 is involved in the acquisition of the malignant phenotype
in epithelial ovarian tumors. Cancer Sci. 107:1399–1405. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Min SH, Lau AW, Lee TH, Inuzuka H, Wei S,
Huang P, Shaik S, Lee DY, Finn G, Balastik M, et al: Negative
regulation of the stability and tumor suppressor function of Fbw7
by the Pin1 prolyl isomerase. Mol Cell. 46:771–783. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ji S, Qin Y, Shi S, Liu X, Hu H, Zhou H,
Gao J, Zhang B, Xu W, Liu J, et al: ERK kinase phosphorylates and
destabilizes the tumor suppressor FBW7 in pancreatic cancer. Cell
Res. 25:561–573. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cizmecioglu O, Krause A, Bahtz R, Ehret L,
Malek N and Hoffmann I: Plk2 regulates centriole duplication
through phosphorylation-mediated degradation of Fbxw7 (human Cdc4).
J Cell Sci. 125:(Pt 4). 981–992. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Chen J, Shin JH, Zhao R, Phan L, Wang H,
Xue Y, Post SM, Ho Choi H, Chen JS, Wang E, et al: CSN6 drives
carcinogenesis by positively regulating Myc stability. Nat Commun.
5:53842014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Diefenbacher ME, Popov N, Blake SM,
Schulein-Volk C, Nye E, Spencer-Dene B, Jaenicke LA, Eilers M and
Behrens A: The deubiquitinase USP28 controls intestinal homeostasis
and promotes colorectal cancer. J Clin Invest. 124:3407–3418. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Schulein-Volk C, Wolf E, Zhu J, Xu W,
Taranets L, Hellmann A, Janicke LA, Diefenbacher ME, Behrens A,
Eilers M and Popov N: Dual regulation of Fbw7 function and
oncogenic transformation by Usp28. Cell Rep. 9:1099–1109. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Welcker M, Larimore EA, Swanger J,
Bengoechea-Alonso MT, Grim JE, Ericsson J, Zheng N and Clurman BE:
Fbw7 dimerization determines the specificity and robustness of
substrate degradation. Genes Dev. 27:2531–2536. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Davis RJ, Welcker M and Clurman BE: Tumor
suppression by the Fbw7 ubiquitin ligase: Mechanisms and
opportunities. Cancer Cell. 26:455–464. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Tang X, Orlicky S, Lin Z, Willems A,
Neculai D, Ceccarelli D, Mercurio F, Shilton BH, Sicheri F and
Tyers M: Suprafacial orientation of the SCFCdc4 dimer accommodates
multiple geometries for substrate ubiquitination. Cell.
129:1165–1176. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bonetti P, Davoli T, Sironi C, Amati B,
Pelicci PG and Colombo E: Nucleophosmin and its AML-associated
mutant regulate c-Myc turnover through Fbw7 gamma. J Cell Biol.
182:19–26. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Durgan J and Parker PJ: Regulation of the
tumour suppressor Fbw7α by PKC-dependent phosphorylation and
cancer-associated mutations. Biochem J. 432:77–87. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Akhoondi S, Sun D, von der Lehr N,
Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D,
Marth C, et al: FBXW7/hCDC4 is a general tumor suppressor in human
cancer. Cancer Res. 67:9006–9012. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
King B, Trimarchi T, Reavie L, Xu L,
Mullenders J, Ntziachristos P, Aranda-Orgilles B, Perez-Garcia A,
Shi J, Vakoc C, et al: The ubiquitin ligase FBXW7 modulates
leukemia-initiating cell activity by regulating MYC stability.
Cell. 153:1552–1566. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Davis H, Lewis A, Behrens A and Tomlinson
I: Investigation of the atypical FBXW7 mutation spectrum in human
tumours by conditional expression of a heterozygous propellor tip
missense allele in the mouse intestines. Gut. 63:792–799. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ju Y, Yu A, Sun X, Wu D and Zhang H:
Glucosamine, a naturally occurring amino monosaccharide, inhibits
A549 and H446 cell proliferation by blocking G1/S transition. Mol
Med Rep. 8:794–798. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Dong JT and Chen C: Essential role of KLF5
transcription factor in cell proliferation and differentiation and
its implications for human diseases. Cell Mol Life Sci.
66:2691–2706. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Klotz K, Cepeda D, Tan Y, Sun D, Sangfelt
O and Spruck C: SCF(Fbxw7/hCdc4) targets cyclin E2 for
ubiquitin-dependent proteolysis. Exp Cell Res. 315:1832–1839. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Byrd KN, Huey B, Roydasgupta R, Fridlyand
J, Snijders AM and Albertson DG: FBXW7 and DNA copy number
instability. Breast Cancer Res Treat. 109:47–54. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Liu X, Zhang Y, Wu S, Xu M, Shen Y, Yu M,
Fan J, Wei S, Xu C, Huang L, et al: Palmatine induces G2/M phase
arrest and mitochondrial-associated pathway apoptosis in colon
cancer cells by targeting AURKA. Biochem Pharmacol. 175:1139332020.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zhang H, Bao J, Zhao S, Huo Z and Li B:
MicroRNA-490-3p suppresses hepatocellular carcinoma cell
proliferation and migration by targeting the aurora kinase A gene
(AURKA). Arch Med Sci. 16:395–406. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Finkin S, Aylon Y, Anzi S, Oren M and
Shaulian E: Fbw7 regulates the activity of endoreduplication
mediators and the p53 pathway to prevent drug-induced polyploidy.
Oncogene. 27:4411–4421. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
McIntyre B, Asahara T and Alev C: Overview
of basic mechanisms of notch signaling in development and disease.
Adv Exp Med Biol. 1227:9–27. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Nowell CS and Radtke F: Notch as a tumour
suppressor. Nat Rev Cancer. 17:145–159. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Bray SJ: Notch signalling in context. Nat
Rev Mol Cell Biol. 17:722–735. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Pancewicz J, Taylor JM, Datta A, Baydoun
HH, Waldmann TA, Hermine O and Nicot C: Notch signaling contributes
to proliferation and tumor formation of human T-cell leukemia virus
type 1-associated adult T-cell leukemia. Proc Natl Acad Sci USA.
107:16619–16624. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Wu G, Lyapina S, Das I, Li J, Gurney M,
Pauley A, Chui I, Deshaies RJ and Kitajewski J: SEL-10 is an
inhibitor of notch signaling that targets notch for
ubiquitin-mediated protein degradation. Mol Cell Biol.
21:7403–7415. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Tsunematsu R, Nakayama K, Oike Y,
Nishiyama M, Ishida N, Hatakeyama S, Bessho Y, Kageyama R, Suda T
and Nakayama KI: Mouse Fbw7/Sel-10/Cdc4 is required for notch
degradation during vascular development. J Biol Chem.
279:9417–9423. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Matsumoto A, Onoyama I, Sunabori T,
Kageyama R, Okano H and Nakayama KI: Fbxw7-dependent degradation of
Notch is required for control of ‘stemness’ and neuronal-glial
differentiation in neural stem cells. J Biol Chem. 286:13754–13764.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Yang-Yen HF: Mcl-1: A highly regulated
cell death and survival controller. J Biomed Sci. 13:201–204. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Welcker M, Orian A, Jin J, Grim JE, Harper
JW, Eisenman RN and Clurman BE: The Fbw7 tumor suppressor regulates
glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein
degradation. Proc Natl Acad Sci USA. 101:9085–9090. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Kanei-Ishii C, Nomura T, Takagi T,
Watanabe N, Nakayama KI and Ishii S: Fbxw7 acts as an E3 ubiquitin
ligase that targets c-Myb for nemo-like kinase (NLK)-induced
degradation. J Biol Chem. 283:30540–30548. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Popov N, Wanzel M, Madiredjo M, Zhang D,
Beijersbergen R, Bernards R, Moll R, Elledge SJ and Eilers M: The
ubiquitin-specific protease USP28 is required for MYC stability.
Nat Cell Biol. 9:765–774. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Li M, Ouyang L, Zheng Z, Xiang D, Ti A, Li
L, Dan Y, Yu C and Li W: E3 ubiquitin ligase FBW7α inhibits
cholangiocarcinoma cell proliferation by downregulating c-Myc and
cyclin E. Oncol Rep. 37:1627–1636. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Qin J, Zhou Z, Chen W, Wang C, Zhang H, Ge
G, Shao M, You D, Fan Z, Xia H, et al: BAP1 promotes breast cancer
cell proliferation and metastasis by deubiquitinating KLF5. Nat
Commun. 6:84712015. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Liu N, Li H, Li S, Shen M, Xiao N, Chen Y,
Wang Y, Wang W, Wang R, Wang Q, et al: The Fbw7/human CDC4 tumor
suppressor targets proproliferative factor KLF5 for ubiquitination
and degradation through multiple phosphodegron motifs. J Biol Chem.
285:18858–18867. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zhao D, Zheng HQ, Zhou Z and Chen C: The
Fbw7 tumor suppressor targets KLF5 for ubiquitin-mediated
degradation and suppresses breast cell proliferation. Cancer Res.
70:4728–4738. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Rottmann S, Wang Y, Nasoff M, Deveraux QL
and Quon KC: A TRAIL receptor-dependent synthetic lethal
relationship between MYC activation and GSK3beta/FBW7 loss of
function. Proc Natl Acad Sci USA. 102:15195–15200. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Goga A, Yang D, Tward AD, Morgan DO and
Bishop JM: Inhibition of CDK1 as a potential therapy for tumors
over-expressing MYC. Nat Med. 13:820–827. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Liu L, Ulbrich J, Muller J, Wustefeld T,
Aeberhard L, Kress TR, Muthalagu N, Rycak L, Rudalska R, Moll R, et
al: Deregulated MYC expression induces dependence upon AMPK-related
kinase 5. Nature. 483:608–612. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Chauhan D, Tian Z, Nicholson B, Kumar KG,
Zhou B, Carrasco R, McDermott JL, Leach CA, Fulcinniti M, Kodrasov
MP, et al: A small molecule inhibitor of ubiquitin-specific
protease-7 induces apoptosis in multiple myeloma cells and
overcomes bortezomib resistance. Cancer Cell. 22:345–358. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Grim JE, Knoblaugh SE, Guthrie KA, Hagar
A, Swanger J, Hespelt J, Delrow JJ, Small T, Grady WM, Nakayama KI
and Clurman BE: Fbw7 and p53 cooperatively suppress advanced and
chromosomally unstable intestinal cancer. Mol Cell Biol.
32:2160–2167. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Selivanova G and Wiman KG: Reactivation of
mutant p53: Molecular mechanisms and therapeutic potential.
Oncogene. 26:2243–2254. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Csizmok V, Montecchio M, Lin H, Tyers M,
Sunnerhagen M and Forman-Kay JD: Multivalent interactions with Fbw7
and Pin1 facilitate recognition of c-Jun by the SCFFbw7
Ubiquitin Ligase. Structure. 26:28–39.e2. 2018. View Article : Google Scholar : PubMed/NCBI
|














