Introduction
There are two forms of genomic instability that
occur during colorectal cancer (CRC) progression: Chromosomal
instability (CIN) and microsatellite instability (MSI) (1,2). It has
been reported in 2008 that ~85% of all CRC cases worldwide exhibit
either numerical chromosomal alterations due to abnormal chromatid
cohesion, an abnormality of the spindle assembly checkpoint or a
structural abnormality; however, the definitive cause of CIN
remains unknown (3–6). By contrast, one of the mechanisms
leading to a ‘mutator phenotype’, specifically MSI, occurs due to a
loss of function in the DNA mismatch repair (MMR) system and the
abrogation of DNA fidelity (7,8).
The DNA MMR system is comprised of two recognition
complexes that detect DNA alterations (8). MutSα, a heterodimer of the MMR proteins
MutS homolog 2 (MSH2) and MSH6, recognizes base-base mismatches and
insertion/deletion (I/D) loops of <2 nucleotides, while I/D
loops of >2 nucleotides are recognized by MutSβ, an MSH2-MSH3
heterodimer (8). Germline pathogenic
variants in human MMR genes, including MSH2, MSH6, MutL
homolog 1 (MLH1) and PMS1 homolog 2 MMR system component
(PMS2), or germline deletions of epithelial cell adhesion
molecule (EPCAM), that induce the constitutional methylation
of the MSH2 promoter leading to epigenetic silencing of
MSH2, which is associated with Lynch syndrome, and the
inactivation of the DNA MMR system by somatic pathogenic variants
or the hypermethylation of human MLH1 can result in MSI-high
(MSI-H) CRC (8). Therefore,
MMR-deficient tumors are typically identified using
immunohistochemistry (IHC) to detect the loss of protein expression
of ≥1 MMR proteins (such as MLH1, MSH2, MSH6 and PMS2), and MSI
testing can be used to identify MSI-H, as the loss of functional
mutations and/or silencing via hypermethylation of DNA MMR genes
causes instability within microsatellite regions (8).
Colorectal tumors exhibiting deficient (d)MMR/MSI-H
possess certain unique characteristics; they tend to be located
within the right colon, and their histopathological features are
poorly differentiated with mucinous features and marked lymphocytic
infiltration (9–12). Several studies have reported that
patients with stage II–III CRC with proficient (p)MMR can benefit
from fluorouracil (5-FU) treatment; however, patients with tumors
that have lost their DNA MMR function do not benefit, as 5-FU
incorporated in the DNA is typically recognized by the DNA MMR
system, leading to cytotoxicity (13–17).
A previous study has revealed that patients with
stage IV CRC with dMMR/MSI-H tumors benefit more strongly from
programmed cell death-1 (PD-1) blockade than those with proficient
pMMR or non-MSI-H tumors that retain DNA MMR function (18), as hypermutable dMMR/MSI-H tumor cells
produce various types of neoantigens and induce radical T helper 1
cytotoxic immune responses under PD-1 blockade (18,19).
Therefore, understanding the frequency of patients with dMMR/MSI-H
CRC at each clinical stage is important for the selection of an
appropriate treatment.
The frequency of patients with dMMR/MSI-H CRC has
been previously estimated to be ~15% by some groups in some western
countries in the 2000s (20).
However, recent data have revealed that among 2,439 surgically
resected primary CRC cases in Japan, the frequency of patients with
MSI-H is 5.9% for stages 0-I, 8.9% for stage II, 4.0% for stage III
and 3.7% for stage IV (21), which
is similar to the frequencies of patients with dMMR/MSI-H reported
in China (5.7% for stage I, 9.9% for stage II, 4.2% for stage III
and 2.5% for stage IV) and South Korea (4.7% for stage I, 4.6% for
stage II, 5.2% for stage III and not analyzed for stage IV)
(22,23). While examining the aforementioned
data, the following observations were made: i) All the patients in
these reports were limited to surgical cases, and ii) the reports
did not consider the frequency of patients with stage 0 disease,
despite the fact that the number of patients who are being treated
using endoscopic techniques is increasing (24). Additionally, to the best of our
knowledge, the frequency of patients with dMMR/MSI-H in stage 0
lesions that can be endoscopically resected has not been previously
reported. Therefore, little is known regarding the frequency of
dMMR/MSI-H among patients with early CRC who undergo endoscopic
resection, despite knowing that DNA MMR deficiency occurs during
colorectal tumor initiation or at a very early stage of tumor
progression (8). Additionally, as
the majority of sporadic dMMR tumors possess the BRAF V600E
somatic pathogenic variant, which is an important marker of a poor
prognosis in addition to MLH1 hypermethylation (8,20,22), it
is important to characterize these parameters to allow for the
implementation of optimal treatment strategies.
In the present study, IHC was used to evaluate the
DNA MMR status of stage 0 colorectal tumors that were resected
using endoscopic submucosal dissection (ESD) or endoscopic mucosal
resection (EMR).
Patients and methods
Patients
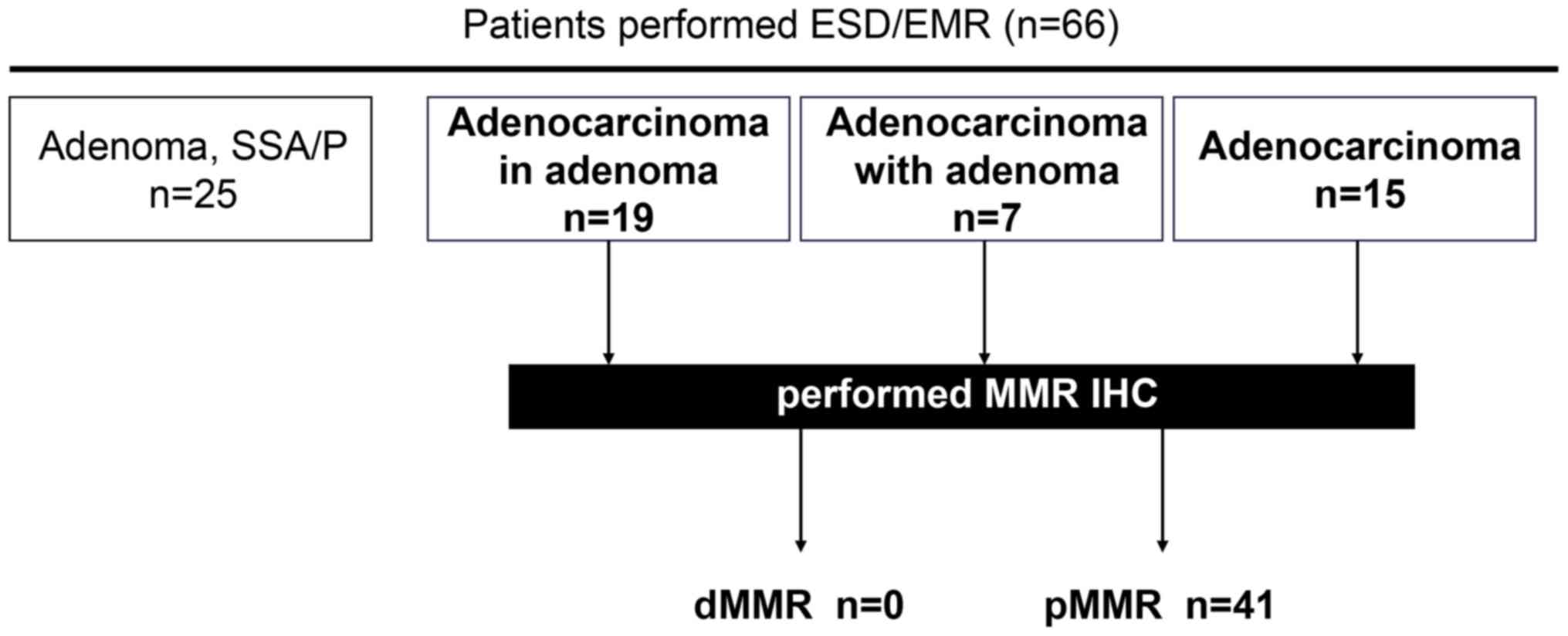
From the 66 patients with colorectal neoplasia who
underwent endoscopic resection using ESD (63 patients) or EMR (3
patients of adenoma) at the Hamamatsu University Hospital
(Hamamatsu, Japan) between April 2015 and March 2020, the DNA MMR
tumor status was evaluated using IHC in 41 patients who had been
pathologically diagnosed with stage 0 colorectal adenocarcinoma
according to pathologic tumor node metastasis staging of colorectal
carcinoma (American Joint Committee on Cancer, 8th edition)
(25) (Fig. 1). All patients provided written
informed consent, and the present study was approved by the
Institutional Review Board of the Hamamatsu University School of
Medicine (approval no. 16-084), which confirmed that the study was
in accordance with the ethical guidelines of the Helsinki
Declaration. The clinical characteristics of the patients (age,
sex, reason for undergoing colonoscopy, lifestyle, previous
personal history and family history of CRC) are shown in Table I. Tumor location, size and shape, as
well as pathological features, were recorded at the time of
colonoscopy and are shown in Table
II. Microscopic features were divided into five types: i)
Tubular adenoma; ii) adenocarcinoma with tubular adenoma occupying
≤50% of the adenocarcinoma component; iii) adenocarcinoma with
tubular adenoma occupying >50% of the adenocarcinoma component;
iv) adenocarcinoma and v) sessile serrated adenoma/polyp (SSA/P).
Tumors with microscopic features of types ii), iii) or iv)
including a cancer component, were further examined using IHC to
identify the status of DNA MMR expression.
 | Table I.Clinical characteristics of patients
(n=41). |
Table I.
Clinical characteristics of patients
(n=41).
|
Characteristics | Values |
|---|
| Median age (range),
years | 71.2 (44–84) |
| Sex, n (%) |
|
Male | 27 (65.85) |
|
Female | 14 (34.15) |
| Purpose of
colonoscopy, n (%) |
|
Screening | 3 (7.32) |
|
Anemia | 1 (2.44) |
|
FOB | 21 (51.22) |
| CT/PET
abnormality | 2 (4.88) |
|
Post-CRC operation | 6 (14.63) |
| Bloody
stool | 6 (14.63) |
|
Abnormal bowel movement | 2 (4.88) |
| Cigarette smoking,
n (%) |
|
Never | 17 (41.46) |
|
Former | 19 (46.34) |
|
Current | 5 (12.20) |
| Alcohol intake,
g/day, n (%) |
| 0 | 26 (63.41) |
|
0-20 | 9 (21.95) |
|
21-40 | 0 (0.00) |
|
≥41 | 4 (9.76) |
|
Unknown | 2 (4.88) |
| History of cancer,
n (%) |
| + | 10 (24.39) |
| – | 31 (75.61) |
| Family history, n
(%) |
| No
family history | 12 (29.27) |
|
FDR | 16 (39.02) |
|
SDR | 5 (12.20) |
|
Unknown | 8 (19.51) |
| Table II.Pathological features of patients
(n=41). |
Table II.
Pathological features of patients
(n=41).
| Features | n (%) |
|---|
| Location |
|
Cecum | 4 (9.76) |
|
Ascending colon | 7 (17.07) |
|
Transverse colon | 2 (4.88) |
|
Descending colon | 2 (4.88) |
| Sigmoid
colon | 10 (24.39) |
|
Rectum | 16 (39.02) |
| Side location |
|
Right | 13 (31.71) |
|
Left | 28 (68.29) |
| Morphology |
| LST-G
(Homo) | 6 (14.63) |
| LST-G
(Mix) | 8 (19.51) |
| LST-NG
(Flat) | 7 (17.07) |
| LST-NG
(PD) | 1 (2.44) |
|
Protruded type | 18 (43.90) |
|
Superficial elevated type | 1 (2.44) |
| Tumor size |
| <10
mm | 2 (4.88) |
| 10–20
mm | 13 (31.71) |
| 20–30
mm | 13 (31.71) |
| >30
mm | 12 (29.27) |
|
Piecemeal resection | 1 (2.44) |
| Pathology |
|
Adenocarcinoma in adenoma | 19 (46.34) |
|
Adenocarcinoma with
adenoma | 7 (17.07) |
|
Adenocarcinoma | 15 (36.59) |
Endoscopic surgery
Colorectal ESD was indicated for the following
lesions requiring an endoscopic en bloc resection: i) Lesions for
which an en bloc resection using a snare EMR would be difficult to
apply, such as non-granular laterally spreading tumors (LST-NG),
particularly LST-NG pseudo-depressed type tumors, lesions with a
VI-type pit pattern, carcinoma with shallow T1 submucosal (SM)
invasion, large depressed-type tumors and large protruded-type
lesions suspected of being carcinomas; ii) mucosal tumors with
submucosal fibrosis; iii) sporadic localized tumors in patients
with chronic inflammation, such as ulcerative colitis; and iv)
local residual or recurrent early carcinomas after endoscopic
resection (24). EMR was performed
in cases where ESD was initially scheduled but EMR was subsequently
found to be preferable based on endoscopic observations made on the
day of treatment. ESD was performed using an S-O clip®
(Zeon Medical Inc.), DualKnife™ (Olympus Corporation) or the Clutch
Cutter® (Fujifilm Holdings Corporation). EMR was
performed using the Captivator II (Boston Scientific Corporation).
EMR and ESD were performed by board-certified fellows of the Japan
Gastroenterological Endoscopy Society working at the Hamamatsu
University Hospital.
IHC staining
Tissues were collected under the supervision of an
experienced pathologist. Staining for the expression of 4 MMR
proteins (MLH1, MSH2, PMS2 and MSH6) was performed using 10%
formalin-fixed for 6–48 h at room temperature, paraffin-embedded
blocks cut into 4-µm thick serial sections. The slides were stained
using an automated procedure. Briefly, the slides were dewaxed by
heating at 55°C for 30 min, followed by three 5-min washes using
xylene. Subsequently, the tissues were rehydrated using a series of
5-min washes in 100, 95 and 80% ethanol, and distilled water.
Endogenous peroxidase activity was blocked by treatment with 3%
hydrogen peroxide for 10 min at room temperature. Following
incubation with Protein Block reagent [StartingBlock™ (TBS)
Blocking Buffer; cat. no. 37542; Thermo Fisher Scientific Inc.] for
5 min at room temperature and washing with TBS twice, the slides
were incubated with the following mouse monoclonal antibodies:
Anti-MLH1 (clone G168-728; 1:50; cat. no. 554073 BD Biosciences),
anti-MSH2 (clone FE11; 1:10; cat. no. NA27; Merck KGaA), anti-PMS2
(clone A16-4; 1:50; cat. no. 556415; BD Biosciences) and anti-MSH6
(clone 44/MSH6;1:20; cat. no. 610918; BD Biosciences) for 30 min at
room temperature, and then incubated with dextran polymer
conjugated with goat anti-mouse immunoglobulin G and horseradish
peroxidase (ChemMate Envision kit; Dako; Agilent Technologies,
Inc.) for 30 min at room temperature. The antigen-antibody complex
was visualized using 3,3-diaminobenzidine tetrahydrochloride and
was counterstained with hematoxylin for 1 min at room temperature
using an autostainer (Histostainer; Nichirei Biosciences Inc.). The
slides were examined by a light microscope (magnifications, ×100
and ×400).
Evaluation of MMR status
The MMR status was evaluated as previously reported
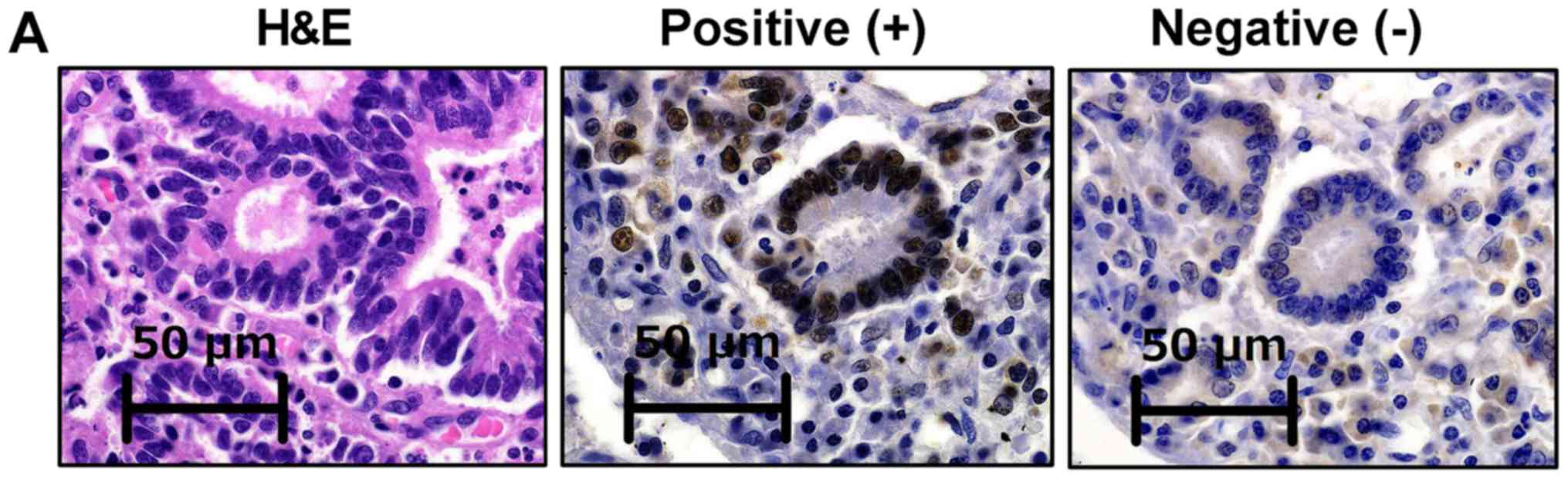
(26). Briefly, tumors were
considered to be negative for MLH1, MSH2, PMS2 or MSH6 expression
when there was a complete absence of nuclear staining in the tumor
cells, while nuclear staining of normal colonic crypt epithelium
adjacent to the tumor, lymphoid cells and stromal cells served as
internal positive controls (Fig.
2A). Tumors lacking MLH1, MSH2, PMS2 or MSH6 expression were
considered to possess a dMMR status. As MLH1 is required to
stabilize PMS2, but PMS2 is not required to stabilize MLH1, tumors
lacking both MLH1 and PMS2 expression exhibit a loss of MLH1
functionally, followed by PMS2 destabilization. When the loss of
both MSH2 and MSH6 expression is detected, this indicates that MSH2
functional loss is followed by MSH6 degradation, as MSH2 is
required to stabilize MSH6 (26).
Tumors with maintained expression levels of MLH1, MSH2, PMS2 and
MSH6 were considered to possess a pMMR status. For all cases, the
diagnoses were confirmed under the supervision of an experienced
pathologist.
 | Figure 2.IHC staining of DNA MMR proteins in
tissues from patients with colorectal cancer. (A) Tumors were
considered negative for MLH1, MSH2, PMS2 or MSH6 expression when
there was a complete absence of nuclear staining within the tumor
cells, while positive staining was confirmed in normal epithelial
cells and lymphocytes used as controls. Tumors lacking MLH1, MSH2,
PMS2 or MSH6 expression were considered to possess a dMMR status,
while tumors that maintained the expression of MLH1, MSH2, PMS2 and
MSH6 were considered to exhibit a pMMR status. (B) A patient with a
germline MLH1 pathogenic variant possessed a tumor lacking
both MLH1 and PMS2 expression. (C) A patient with a tumor lacking
MSH2 and MSH6 expression and a suspected disruption of MSH2
possessed a germline MSH2 pathogenic variant. A patient with
(D) sporadic advanced and (E) endoscopically resected early stage
CRC without a germline pathogenic variant in the DNA MMR genes
presented a tumor that expressed all four analyzed MMR proteins.
Scale bar, 50 µm. IHC, immunohistochemistry; H&E, hematoxylin
and eosin; MMR, mismatch repair; MLH1, MutL homolog 1; MSH2/6, MutS
homolog 2/6; PMS2, PMS1 homolog 2 MMR system component. |
Results
Confirmation of appropriate IHC
staining for DNA MMR proteins in CRC
As shown in Fig. 2, a
patient with a germline MLH1 pathogenic variant determined
by direct sequencing possessed a tumor lacking both MLH1 and PMS2
expression (Fig. 2B), and another
patient with a tumor lacking MSH2 and MSH6 expression, and a
suspected disruption of MSH2, possessed a germline MSH2
pathogenic variant (Fig. 2C). A
third patient with sporadic advanced (Fig. 2D) and stage 0 CRC (Fig. 2E) without germline pathogenic
variants of the DNA MMR genes possessed a tumor exhibiting the
expression of all four analyzed MMR. The present results indicated
that the current IHC method functions appropriately, and IHC was
subsequently performed on early stage CRC specimens resected using
ESD or EMR (Fig. 2E).
DNA MMR system is not disrupted in
very early stage CRC specimens resected using ESD
Prior to endoscopic resection, consent was obtained
from 66 patients, including 3 who underwent EMR and 63 who
underwent ESD. In one patient, the tumor was large and the
operability was poor, ultimately requiring a piecemeal resection.
Among the 66 patients, 25 cases were histologically diagnosed as
non-adenocarcinoma (3 cases of SSA/P and 22 cases of adenoma) and
41 cases as adenocarcinoma (Fig. 1).
Among the 41 patients with adenocarcinoma, the number of patients
with tumors in the left-sided colorectum (descending colon, sigmoid
colon and rectum) is higher compared with those with tumors in the
right-sided colon (cecum, ascending colon and transverse colon)
(Table II), which is similar to
previous reports (27–30).
As tumor MSI testing and/or IHC staining of MMR
proteins for screening of Lynch syndrome is currently performed for
patients with CRC (31), IHC
staining of MMR proteins was performed in the 41 cases diagnosed
with colorectal adenocarcinoma. Notably, none of the 41
endoscopically resected specimens of early stage CRC exhibited a
dMMR status (Fig. 1).
Discussion
Once dMMR occurs, affected tumors develop more
rapidly compared with those tumors that progress via the CIN
pathway; tumors possessing dMMR progress within 1–3 years, while
CIN tumors progress over decades (32). Therefore, the detection and resection
of CRC at a very early stage is important for a complete treatment.
Notably, the present study has revealed that DNA MMR function was
not disrupted in stage 0 CRC. To the best of our knowledge,
although the frequencies of tumor dMMR among patients with
advanced-stage CRC have been previously reported (21–23), the
MMR status of endoscopically resected, very early stage CRC has not
been previously reported.
The first step for MSI/dMMR in sporadic CRC is
epigenetic silencing of the MLH1 gene through the
hypermethylation of both its promoters (33,34).
Menigatti et al (35)
reported that 70% of the pre-cancerous aberrant crypt foci (ACF)
lesions are methylated at the promoter region of MLH1,
suggesting that most ACF may have lost their ability to express the
MLH1 protein, and this occurs in early stage CRC. However, when the
aforementioned data (35) were more
carefully examined, the median methylation levels for the
MLH1 gene were considered to be so low that the
downregulation of MLH1 protein expression was likely limited to
single cells or crypts, as noted by another study (36), in which it was further speculated
that these alterations are very difficult to identify using IHC.
Although there may be a time lag between MLH1 promoter
methylation and the loss of MLH1 protein expression, the present
results suggested that the resection of tumors prior to the loss of
MLH1 expression may reduce the likelihood of recurrence, which was
not shown in the present study; however, if the resected tumor is
diagnosed as dMMR, careful surveillance will be required.
From the point of view of the occurrence of dMMR,
patients with pre-cancerous lesions, including patients with SSA/P
with cytological dysplasia (SSAD), require careful surveillance
after resection. Among the three main types of serrated lesions,
namely hyperplastic polyps, traditional serrated adenoma (TSA) and
SSA/P, TSA is reportedly associated with KRAS mutations and
DNA methylation (37). During
progression to SSA/P, a BRAF gene mutation is thought to
occur first, followed by the expression of a CpG island methylator
phenotype (38). It must be noted
that previous clinical reports have demonstrated that MSI-H/dMMR
has not been identified in hyperplastic polyps, TSA or SSA/P, but
has been reported in SSAD only (39,40).
Additionally, SSAD is the only pre-cancerous colorectal lesion in
which MLH1 is methylated (41). Furthermore, the frequency of MSI is
reportedly low in Japan (9,42,43), and
it is possible that no cases were observed in the present study as
this was not measured.
The present study presents some limitations: i) IHC
for DNA MMR was performed only for cancerous lesions and not
pre-cancerous lesions; ii) DNA was not extracted for a mutation
search; and iii) the methylation profile was not evaluated. These
points require further investigation in future studies. However, to
the best of our knowledge, the present study is the first to focus
on the MMR status in endoscopic resection cases, and it may
represent the first step towards the use of dMMR status evaluation
to help select the appropriate treatment.
In conclusion, dMMR was evaluated in endoscopically
resected early stage CRC specimens, and dMMR was not detected in
any of the cases. Regarding the development and progression of CRC,
the results of the present study suggested that changes in MMR may
not be involved in early tumor development, and the current
findings are important for future medical care and understanding
the pathophysiology of CRC. Future molecular biology and molecular
genetic studies should further clarify the present findings.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Japan Society
for the Promotion of Science (KAKENHI grant no. 19K08392), the
Takeda Science Foundation and Hamamatsu University School of
Medicine (HUSM) Grant-in-Aid.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
MI conceived the study. SB performed and evaluated
the immunohistochemistry data. MI, TS, MK, ST, MY, YH, TF, HM, SO,
MM and KS analyzed and interpreted the data. TS, MI, HM, MM and KS
drafted the manuscript and revised it critically for important
intellectual content. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Hamamatsu University School of Medicine (approval no.
16-084), and all the patients provided written informed
consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MSI-H
|
microsatellite instability-high
|
|
CRC
|
colorectal cancer
|
|
d/pMMR
|
deficient/proficient mismatch
repair
|
|
CIN
|
chromosomal instability
|
|
IHC
|
immunohistochemistry
|
|
EMR
|
endoscopic mucosal resection
|
|
ESD
|
endoscopic submucosal dissection
|
References
|
1
|
Lengauer C, Kinzler KW and Vogelstein B:
Genetic instabilities in human cancers. Nature. 396:643–649. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kinzler KW and Vogelstein B: Lessons from
hereditary colorectal cancer. Cell. 87:159–170. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Iwaizumi M, Shinmura K, Mori H, Yamada H,
Suzuki M, Kitayama Y, Igarashi H, Nakamura T, Suzuki H, Watanabe Y,
et al: Human Sgo1 downregulation leads to chromosomal instability
in colorectal cancer. Gut. 58:249–260. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kahyo T, Iwaizumi M, Shinmura K, Matsuura
S, Nakamura T, Watanabe Y, Yamada H and Sugimura H: A novel
tumor-derived SGOL1 variant causes abnormal mitosis and unstable
chromatid cohesion. Oncogene. 30:4453–4463. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Matsuura S, Kahyo T, Shinmura K, Iwaizumi
M, Yamada H, Funai K, Kobayashi J, Tanahashi M, Niwa H, Ogawa H, et
al: SGOL1 variant B induces abnormal mitosis and resistance to
taxane in non-small cell lung cancers. Sci Rep. 3:30122013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Grady WM: Genomic instability and colon
cancer. Cancer Metastasis Rev. 23:11–27. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Grilley M, Holmes J, Yashar B and Modrich
P: Mechanisms of DNA-mismatch correction. Mutat Res. 236:253–267.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Grady WM and Carethers JM: Genomic and
epigenetic instability in colorectal cancer pathogenesis.
Gastroenterology. 135:1079–1099. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ishikubo T, Nishimura Y, Yamaguchi K,
Khansuwan U, Arai Y, Kobayashi T, Ohkura Y, Hashiguchi Y, Tanaka Y
and Akagi K: The clinical features of rectal cancers with
high-frequency microsatellite instability (MSI-H) in Japanese
males. Cancer Lett. 216:55–62. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kadowaki S, Kakuta M, Takahashi S,
Takahashi A, Arai Y, Nishimura Y, Yatsuoka T, Ooki A, Yamaguchi K,
Matsuo K, et al: Prognostic value of KRAS and BRAF mutations in
curatively resected colorectal cancer. World J Gastroenterol.
21:1275–1283. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lochhead P, Kuchiba A, Imamura Y, Liao X,
Yamauchi M, Nishihara R, Qian ZR, Morikawa T, Shen J, Meyerhardt
JA, et al: Microsatellite instability and BRAF mutation testing in
colorectal cancer prognostication. J Natl Cancer Inst.
105:1151–1156. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nitsche U, Friess H, Agha A, Angele M,
Eckel R, Heitland W, Jauch KW, Krenz D, Nüssler NC, Rau HG, et al:
Prognosis of mucinous and signet-ring cell colorectal cancer in a
population-based cohort. J Cancer Res Clin Oncol. 142:2357–2366.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ribic CM, Sargent DJ, Moore MJ, Thibodeau
SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R,
Shepherd LE, et al: Tumor microsatellite-instability status as a
predictor of benefit from fluorouracil-based adjuvant chemotherapy
for colon cancer. N Engl J Med. 349:247–257. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Carethers JM, Smith EJ, Behling CA, Nguyen
L, Tajima A, Doctolero RT, Cabrera BL, Goel A, Arnold CA, Miyai K,
et al: Use of 5-fluorouracil and survival in patients with
microsatellite-unstable colorectal cancer. Gastroenterology.
126:394–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tajima A, Hess MT, Cabrera BL, Kolodner RD
and Carethers JM: The mismatch repair complex hMutS alpha
recognizes 5-fluorouracil-modified DNA: Implications for
chemosensitivity and resistance. Gastroenterology. 127:1678–1684.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Iwaizumi M, Tseng-Rogenski S and Carethers
JM: DNA mismatch repair proficiency executing 5-fluorouracil
cytotoxicity in colorectal cancer cells. Cancer Biol Ther.
12:756–764. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tajima A, Iwaizumi M, Tseng-Rogenski S,
Cabrera BL and Carethers JM: Both hMutSα and hMutSβ DNA mismatch
repair complexes participate in 5-fluorouracil cytotoxicity. Plos
One. 6:e281172011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Le DT, Durham JN, Smith KN, Wang H,
Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et
al: Mismatch repair deficiency predicts response of solid tumors to
PD-1 blockade. Science. 357:409–413. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Le DT, Uram JN, Wang H, Bartlett BR,
Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et
al: PD-1 blockade in tumors with mismatch-repair deficiency. N Engl
J Med. 372:2509–2520. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Boland CR and Goel A: Microsatellite
instability in colorectal cancer. Gastroenterology.
138:2073–2087.e3. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fujiyoshi K, Yamamoto G, Takenoya T,
Takahashi A, Arai Y, Yamada M, Kakuta M, Yamaguchi K, Akagi Y,
Nishimura Y, et al: Metastatic pattern of stage IV colorectal
cancer with high-frequency microsatellite instability as a
prognostic factor. Anticancer Res. 37:239–247. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guo TA, Wu YC, Tan C, Jin YT, Sheng WQ,
Cai SJ, Liu FQ and Xu Y: Clinicopathologic features and prognostic
value of KRAS, NRAS and BRAF mutations and DNA mismatch repair
status: A single-center retrospective study of 1,834 Chinese
patients with Stage I–IV colorectal cancer. Int J Cancer.
145:1625–1634. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim CG, Ahn JB, Jung M, Beom SH, Kim C,
Kim JH, Heo SJ, Park HS, Kim JH, Kim NK, et al: Effects of
microsatellite instability on recurrence patterns and outcomes in
colorectal cancers. Br J Cancer. 115:25–33. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tanaka S, Kashida H, Saito Y, Yahagi N,
Yamano H, Saito S, Hisabe T, Yao T, Watanabe M, Yoshida M, et al:
JGES guidelines for colorectal endoscopic submucosal
dissection/endoscopic mucosal resection. Dig Endosc. 27:417–434.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu Q, Luo D, Cai S, Li Q and Li X: P-TNM
staging system for colon cancer: Combination of P-stage and AJCC
TNM staging system for improving prognostic prediction and clinical
management. Cancer Manag Res. 10:2303–2314. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kawazoe A, Shitara K, Kuboki Y, Bando H,
Kojima T, Yoshino T, Ohtsu A, Ochiai A, Togashi Y, Nishikawa H, et
al: Clinicopathological features of 22C3 PD-L1 expression with
mismatch repair, Epstein-Barr virus status, and cancer genome
alterations in metastatic gastric cancer. Gastric Cancer. 22:69–76.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shiga H, Ohba R, Matsuhashi T, Jin M,
Kuroha M, Endo K, Moroi R, Kayaba S and Iijima K: Feasibility of
colorectal endoscopic submucosal dissection (ESD) carried out by
endoscopists with no or little experience in gastric ESD. Dig
Endosc. 29 (Suppl 2):58–65. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Iacopini F, Saito Y, Bella A, Gotoda T,
Rigato P, Elisei W, Montagnese F, Iacopini G and Costamagna G:
Colorectal endoscopic submucosal dissection: Predictors and
neoplasm-related gradients of difficulty. Endosc Int Open.
5:E839–E846. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee EJ, Lee JB, Lee SH and Youk EG:
Endoscopic treatment of large colorectal tumors: Comparison of
endoscopic mucosal resection, endoscopic mucosal
resection-precutting, and endoscopic submucosal dissection. Surg
Endosc. 26:2220–2230. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Saito Y, Fukuzawa M, Matsuda T, Fukunaga
S, Sakamoto T, Uraoka T, Nakajima T, Ikehara H, Fu KI, Itoi T, et
al: Clinical outcome of endoscopic submucosal dissection versus
endoscopic mucosal resection of large colorectal tumors as
determined by curative resection. Surg Endosc. 24:343–352. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Benson AB III, Venook AP, Al-Hawary MM,
Cederquist L, Chen YJ, Ciombor KK, Cohen S, Cooper HS, Deming D,
Engstrom PF, et al: NCCN Guidelines Insights: Colon Cancer, Version
2.2018. J Natl Compr Canc Netw. 16:359–369. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nguyen LH, Goel A and Chung DC: Pathways
of colorectal carcinogenesis. Gastroenterology. 158:291–302. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Toyota M, Ahuja N, Ohe-Toyota M, Herman
JG, Baylin SB and Issa JP: CpG island methylator phenotype in
colorectal cancer. Proc Natl Acad Sci USA. 96:8681–8686. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kane MF, Loda M, Gaida GM, Lipman J,
Mishra R, Goldman H, Jessup JM and Kolodner R: Methylation of the
hMLH1 promoter correlates with lack of expression of hMLH1 in
sporadic colon tumors and mismatch repair-defective human tumor
cell lines. Cancer Res. 57:808–811. 1997.PubMed/NCBI
|
|
35
|
Menigatti M, Truninger K, Gebbers JO,
Marbet U, Marra G and Schär P: Normal colorectal mucosa exhibits
sex- and segment-specific susceptibility to DNA methylation at the
hMLH1 and MGMT promoters. Oncogene. 28:899–909. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shen L, Kondo Y, Rosner GL, Xiao L,
Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR,
Einspahr JG, et al: MGMT promoter methylation and field defect in
sporadic colorectal cancer. J Natl Cancer Inst. 97:1330–1338. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
McCarthy AJ, Serra S and Chetty R:
Traditional serrated adenoma: An overview of pathology and emphasis
on molecular pathogenesis. BMJ Open Gastroenterol. 6:e0003172019.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nazemalhosseini Mojarad E, Kuppen PJ,
Aghdaei HA and Zali MR: The CpG island methylator phenotype (CIMP)
in colorectal cancer. Gastroenterol Hepatol Bed Bench. 6:120–128.
2013.PubMed/NCBI
|
|
39
|
O'Brien MJ, Yang S, Mack C, Xu H, Huang
CS, Mulcahy E, Amorosino M and Farraye FA: Comparison of
microsatellite instability, CpG island methylation phenotype, BRAF
and KRAS status in serrated polyps and traditional adenomas
indicates separate pathways to distinct colorectal carcinoma end
points. Am J Surg Pathol. 30:1491–1501. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Murakami T, Akazawa Y, Yatagai N, Hiromoto
T, Sasahara N, Saito T, Sakamoto N, Nagahara A and Yao T: Molecular
characterization of sessile serrated adenoma/polyps with
dysplasia/carcinoma based on immunohistochemistry, next-generation
sequencing, and microsatellite instability testing: a case series
study. Diagn Pathol. 13:882018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Oono Y, Fu K, Nakamura H, Iriguchi Y,
Yamamura A, Tomino Y, Oda J, Mizutani M, Takayanagi S, Kishi D, et
al: Progression of a sessile serrated adenoma to an early invasive
cancer within 8 months. Dig Dis Sci. 54:906–909. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Asaka S, Arai Y, Nishimura Y, Yamaguchi K,
Ishikubo T, Yatsuoka T, Tanaka Y and Akagi K: Microsatellite
instability-low colorectal cancer acquires a KRAS mutation during
the progression from Dukes' A to Dukes' B. Carcinogenesis.
30:494–499. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ikeda M, Yamanaka T, Yamazaki K, Yamaguchi
K, Muro K, Kusumoto T, Uetake H, Sato T, Kato T, Nishina T, et al:
Validation study of the 12-gene Recurrence Score (RS) in patients
(pts) with stage II and III colon cancer (CC) without adjuvant
chemotherapy; Sunrise Study. Ann Oncol. 26 (Suppl 4):iv1042015.
View Article : Google Scholar
|