Introduction
Metabolic aberrations are hallmarks of cancer. It
has been reported that the majority of malignant cells reprogram
their metabolism to induce anabolism via increasing glycolysis,
glutaminolysis and de novo synthesis of fatty acids through
hexokinase-II (HK2) (1), glutaminase
(GLS) (2) and fatty acid synthase
(FASN) (3) upregulation,
respectively. Furthermore, cancer progression induces a catabolic
state in patients, characterized by systemic inflammation, insulin
resistance (4), a negative energy
balance in the host (5) and
proteolysis/lipolysis to support the survival of the tumor
(6,7).
Colon cancer harbors oncogenic mutations, including
Wnt, KRAS, MYC and TP53 (8). These tumor genes reprogram the
metabolism through re-routing glucose to anabolic pathways
(9), increasing the expression of
FASN and promoting glutamine metabolism (8). These alterations may occur early in
colon cancer development to favor the tumorigenic process (10).
Our previous studies demonstrated synergy and
antitumor effects of orlistat, lonidamine and
6-diazo-5-oxo-L-norleucine (DON; termed ‘OLD’), known to inhibit
FASN, HK2 and GLS, respectively (11,12) in a
number of cancer cell lines but not in primary lung fibroblasts.
However, no studies have been reported exploring the simultaneous
effects of drug combination regimens against tumor anabolism and
host catabolism. Therefore, in the present study, the OLD scheme
supplemented with the anti-catabolic drugs growth hormone, insulin
and indomethacin (GII scheme) were used. Furthermore, the effects
of the combination of six drugs (OLD + GII schemes) in CT26.WT
cells was also investigated. The results of the present study
demonstrated that OLD and six-drug combination schemes resulted in
reduced cell viability, clonogenic capacity and cell cycle
progression, and induced apoptosis. These effects were associated
with a quiescent energetic phenotype and limited substrate
flexibility, while the three anti-catabolic drugs did not favor
malignant growth.
Materials and methods
Cell line and culture
In the present study the CT26.WT (ATCC) cell line
was employed. Cells were cultured in RPMI-1640 medium (Gibco;
Thermo Fisher Scientific, Inc.), supplemented with 10% fetal bovine
serum (Corning Inc.) and 1% streptomycin/amphotericin (Gibco;
Thermo Fisher Scientific, Inc.) at 37°C in a 5% CO2
incubator.
Drugs
Orlistat (Psicofarma, S.A., De C.V.), lonidamine
(Sigma-Aldrich; Merck KGaA), DON (Sigma-Aldrich; Merck KGaA),
growth hormone (GH; Merck KGaA), insulin (Eli Lilly & Co.) and
indomethacin (Sigma-Aldrich; Merck KGaA) were used. Orlistat and
indomethacin were dissolved in absolute ethanol, lonidamine in DMSO
(both from Sigma-Aldrich; Merck KGaA), and DON, GH and insulin in
complete medium. The compounds were used in the anti-anabolic (OLD,
orlistat + lonidamine + DON), anti-catabolic (GII, GH + insulin +
indomethacin), or six-drugs combination (OLD + GII, named 6 drugs)
schemes.
Cell viability and colony formation
ability assays
CT26.WT cells were seeded into 6-well plates
(Costar; Corning Inc.) at a density of 3×104 cells/well
in 2 ml complete medium. Following 24 h pre-incubation, cells were
treated for an additional 72 h with OLD, GII or the six-drug
combination schemes. Optimal doses of the OLD and GII schemes used
in the present study were chosen according to our previous study
(11) and pharmacokinetic data of
human studies (13–15), respectively. The OLD and GII scheme
doses are listed in Table SI.
Control cells treated with the same volume of the corresponding
drug vehicles were used to normalize each drug condition. Fresh
complete medium supplemented with drugs/vehicles was replaced every
24 h. Following 72 h, cells were detached using a 0.25%
trypsin-EDTA solution (Gibco; Thermo Fisher Scientific, Inc.) and
cell viability was evaluated via trypan blue (Life Technologies;
Thermo Fisher Scientific, Inc.) and the TC10™ Unity Automated Cell
Counter (Bio-Rad Laboratories, Inc.). The cytotoxic effect of each
treatment was expressed as the percentage of cell viability
relative to control cells. Subsequently, 1,000 cells/condition were
recovered and plated into new 6-well plates. Fresh complete medium
was replaced every 48 h for 14 days to allow colony formation.
Finally, colonies were fixed with a methanol/acetic acid solution
(3:1 v/v), dyed with a violet crystal solution (Sigma-Aldrich;
Merck KGaA) and counted with the ImageJ v2.0 software (National
Institutes of Health) (16).
Flow cytometry
A total of 3×104 cells/well were seeded
into 6-well plates and treated as mentioned above. Subsequently,
cells were recovered and dyed with propidium iodide (Sigma-Aldrich;
Merck KGaA) for 1 h. Then, 20,000 cells/sample were analyzed using
the BD FACSCanto™ II flow cytometer (BD Biosciences). Cell cycle
analysis was performed with the ModFit LT v2.0 software (Verity
Software House, Inc.). In independent assays, following treatment
for 72 h, cells were recovered and dyed with the Annexin V-FLUOS
Staining Kit (Roche). Apoptosis and necrosis rates were
simultaneously analyzed using flow cytometry (10,000 events/sample)
with the BD FACSDiva™ v6.1.3 software (BD Biosciences). Each
condition was compared to its control.
Total protein extraction, western blot
analysis and densitometry
After the cells were treated with OLD, GII and
six-drug combination and controls for 72 h, cells were washed once
with 1X PBS and then harvested with a 0.05% trypsin-0.025% EDTA
solution. Detached cells were washed once again with 1X PBS, and
proteins were extracted with radioimmunoprecipitation buffer (150
mM NaCl; 1.0% IGEPAL CA-630; 0.5% sodium deoxycholate; 0.1% SDS; 50
mM Tris, pH 8.0) in the presence of proteinase inhibitors (catalog
no. p8340; Sigma-Aldrich, Merck KGaA). Protein concentration was
determined using a bicinchoninic acid assay and the integrity was
assessed by Coomassie staining. A total of 30 µg protein was
separated by 15% SDS-PAGE and transferred onto a polyvinylidene
difluoride membrane (cat no. 162-0177; Bio-Rad Laboratories, Inc.).
The membrane was blocked with 5% skim milk in 1X PBS for 1 h at
room temperature, and subsequently incubated with the antibody
against Cleaved Caspase-3 (catalog no. 9664; 1:1,000 from Cell
Signaling Technology, Inc.), and anti-actin peroxidase (A3854;
1:10,000; Sigma-Aldrich; Merck KGaA) in blocking solution (5% skim
milk in TBS + 0.1% Tween-20), overnight at 4°C. The secondary
antibody was bovine anti-rabbit, (sc-2370, Santa Cruz
Biotechnology, Inc.), which was diluted 1:1,000 and the incubation
was performed for 1 h at room temperature. Protein bands were
visualized using the chromogenic substrate Clarity Western Enhanced
Chemiluminescence Substrate (catalog no. 1705060; Bio-Rad
Laboratories, Inc.). Bands were densitometrically quantified using
the ImageJ software, version 1.50f (National Institutes of
Health).
Oxygen consumption and extracellular
acidification rates
Oxidative phosphorylation, glycolysis and fuel
flexibility were assessed by quantifying oxygen consumption (OCR)
and extracellular acidification rates (ECAR) via the Seahorse
Bioscience Extracellular Flux Analyzer XF96e (Seahorse Bioscience).
Briefly, 3×103 cells/well were seeded into XF96 culture
microplates (Seahorse Bioscience) with 100 µl complete medium using
a Viaflo Assist robot (Integra Biosciences). Following 24 h
pre-incubation, cells were treated for 14 h with the
pharmacological drug combinations or their controls. Subsequently,
1 h prior to each Seahorse assay, cells were equilibrated with
bicarbonate-free low buffered medium (Seahorse Bioscience), pH 7.4,
without any supplements, at 37°C in a non-CO2 incubator.
All required reagents for each experiment were prepared in Seahorse
assay medium and loaded into the cartridges with Viaflo Assist for
30 min into Seahorse plates as the experiment progressed. All
results were normalized to the respective controls according to
cellular confluence immediately after each assay. The cellular
confluence was measured by scanning the plate with
IncuCyte® ZOOM equipment (Essen Bioscience), and the
results were analyzed using the Wave software (Seahorse
Bioscience).
Oxidative phosphorylation and
glycolysis assays
Oxidative phosphorylation and glycolysis rates were
evaluated using the XF Cell Mito Stress Test and XF Glycolysis
Stress Test, respectively. The experimental design for both assays
was performed as described by Zaytseva et al (17). Both OCR (pmoles/min) and ECAR
(mpH/min) were measured to indicate oxidative phosphorylation,
while only ECAR was considered for glycolysis. A total of 12
measurements/assay were conducted and each drug condition was
compared to its control.
Fuel flexibility assay
Fuel flexibility assay was performed using the XF
Mito Fuel Flex Test. This test reveals the dependence of fuel
mitochondrial respiration on glucose, glutamine and fatty acids and
its ability to employ a substrate when the other two are inhibited.
The measurements were performed according to the manufacturer's
protocol and only the OCR values were considered. The injection
order of each inhibitor/pair of inhibitors required to evaluate the
capacity, dependency and energetic flexibility are presented in
Table SII. A total of 15
measurements/assay were performed and each treatment was compared
to its control.
Statistical analysis
All experiments were independently performed in
triplicate, with three internal replicates. Significant differences
were determined using multiple t-tests with Holm-Sidak correction.
The results were analyzed using the GraphPad Prism v6 software
(GraphPad Software, Inc.). Data are expressed as mean ± standard
error of the mean (SEM), and P<0.05 was considered to indicate a
statistically significant difference.
Results
The anti-anabolic scheme reduces
CT26.WT cells viability and clonogenic capacity
The present study investigated whether the treatment
of cells for 72 h with the anti-anabolic (OLD), anti-catabolic
(GII) or six-drug (OLD + GII) schemes affected cell viability. The
results demonstrated that both OLD and six-drug schemes reduced
~95% of the cell viability (Fig.
1A). Subsequently, clonogenic assays demonstrated that
following culture for 14 days the colony numbers for both OLD and
six-drug schemes were decreased to ~25% (Fig. 1C). Representative images for each
evaluated condition of cells following treatment for 72 h and
colonies from the clonogenic assays are presented in Fig. 1B and D. No statistically significant
effects were detected in cells treated with the GII scheme compared
to the control cells (Fig.
1A-D).
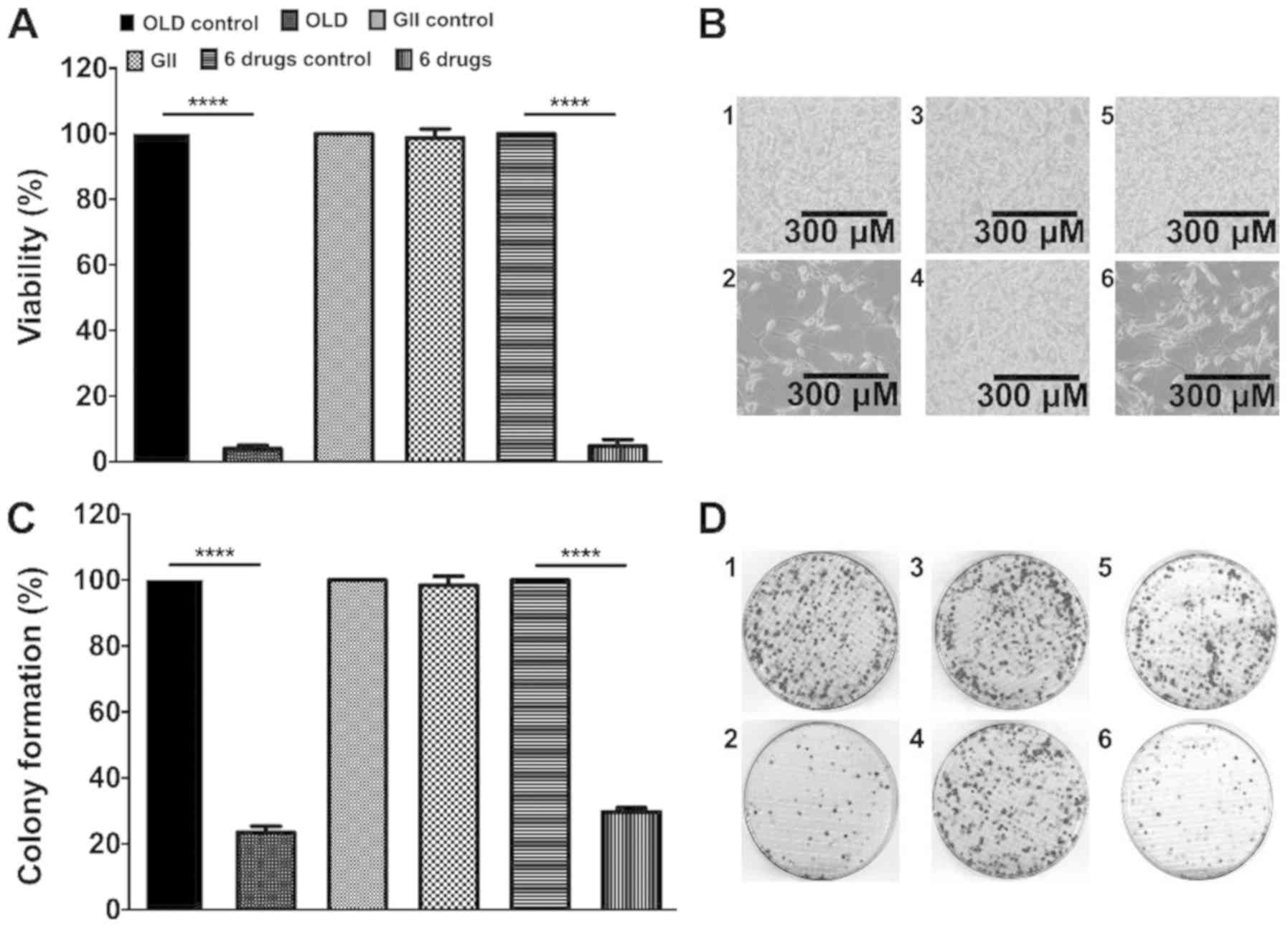 | Figure 1.The inhibition of the de novo
synthesis of fatty acids, glycolysis and glutaminolysis diminishes
cell viability and clonogenicity. (A) Percentage of cell viability
after 72 h of treatment with each scheme. (B) Treated cells with
either the OLD control (1), OLD
(2), GII control (3), GII (4),
6 drugs control (5), or 6 drugs
(6) conditions, after 72 h. (C)
Percentage of colony formation 14 days after the 72 h treatment
with each scheme. (D) CT26.WT plates from either the OLD control
(1), OLD (2), GII control (3), GII (4),
6 drugs control (5), or 6 drugs
(6) conditions, after 14 days. Data
are expressed as mean ± SEM. Scale bars, 300 µm. ****P<0.0001.
OLD, orlistat + lonidamine + DON; GII, growth hormone + insulin +
indomethacin; 6 drugs, OLD + GII. |
The anti-anabolic drug combination
inhibits cell cycle progression and induces apoptosis-related cell
death in CT26.WT-treated cells
Subsequently, the present study aimed to identify
possible treatment-induced changes on cell cycle. Both OLD and
six-drug schemes induced G0/G1 cell cycle
arrest and decreased the proportion of S phase cells. In addition,
both schemes induced sub-G0 phase in ~40% of cells,
indicating cell death (Fig. 2A-B).
Furthermore, apoptosis and necrosis rates were evaluated. OLD and
six-drug treatments promoted cell apoptosis, mainly in its early
form, without significantly increasing necrosis (Fig. 2C-D). Representative figures of cell
cycle assays (Fig. 2B) and
apoptosis/necrosis rates (Fig. 2D)
are shown for each evaluated condition. In line with cell viability
and clonogenic assays, no changes were observed in cells treated
with the GII scheme. To further corroborate that cells underwent
apoptotic death, the evaluation by western blot showed the presence
of caspase-3 following OLD and six-drug treatments but not with GII
(Fig. 2E and F).
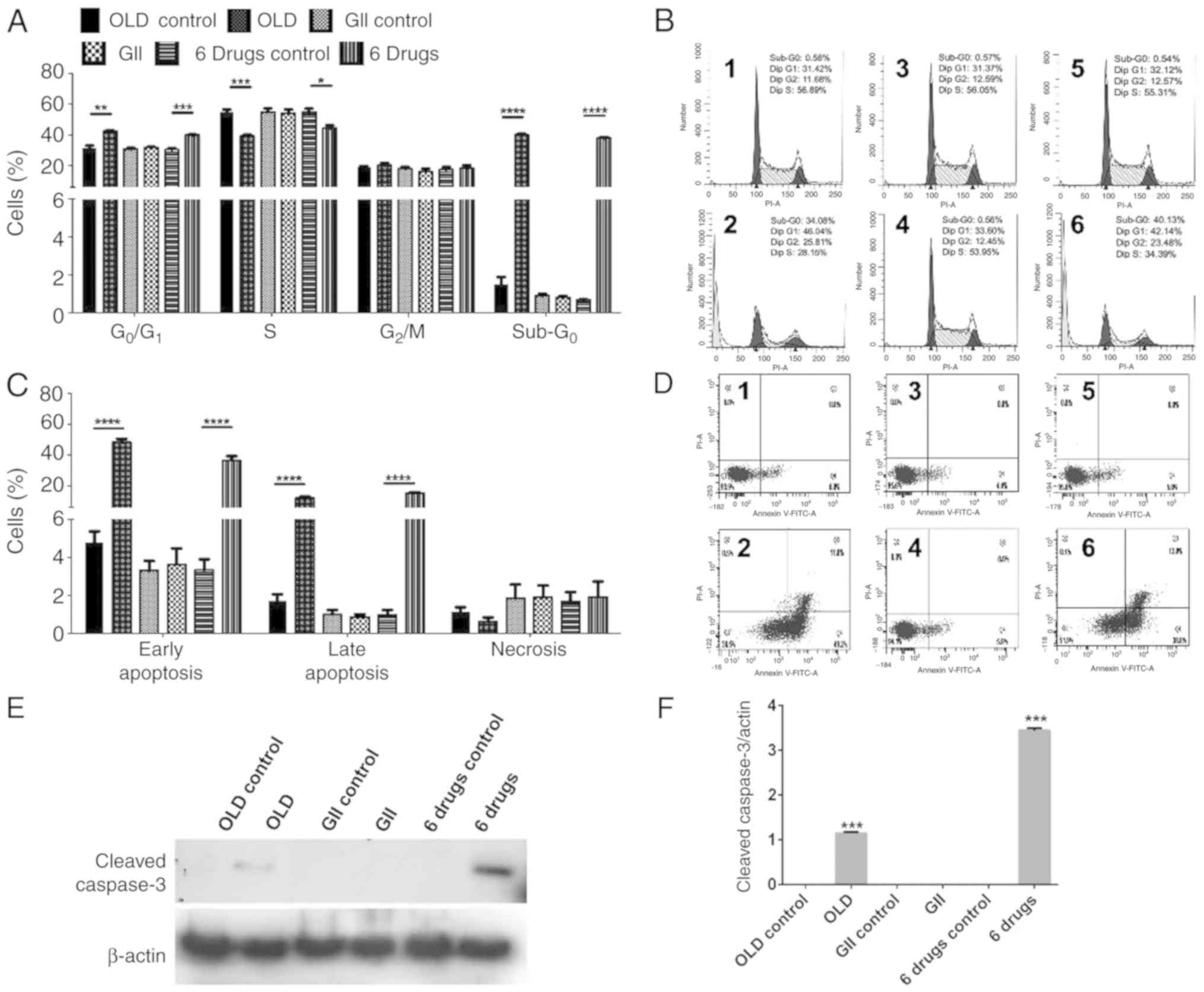 | Figure 2.The anti-anabolic drug combinations
block cell cycle progression and stimulates apoptosis. (A)
Percentage of cells in each phase of cell cycle after 72 h of
treatment with each scheme. (B) ModFit diagrams showing cell cycle
distribution from either the OLD control (1), OLD (2),
GII control (3), GII (4), 6 drugs control (5), or 6 drugs (6) conditions. (C) Percentage of cells
either alive, on early apoptosis, on late apoptosis, or on
necrosis, after 72 h of treatment with each scheme. (D) Diva
diagrams showing alive (Q1), necrotic (Q2), late apoptotic (Q3), or
early apoptotic (Q4) cells, from either the OLD control (1), OLD (2),
GII control (3), GII (4), 6 drugs control (5), or 6 drugs (6) conditions. (E and F) Western blot
evaluation (E) and densitometric analysis (F) of cleaved caspase-3
among all the schemes. Data are expressed as mean ± SEM.
*P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. OLD,
orlistat + lonidamine + DON; GII, growth hormone + insulin +
indomethacin; 6 drugs, OLD + GII. |
The anti-anabolic drug combination
affects the energetic metabolism of CT26.WT cells
As the employed compounds modulated metabolism, the
cells energy production was subsequently investigated. Therefore,
both OCR and ECAR rates were evaluated. First, the treatment
timepoint that caused prolonged metabolic alterations was
determined. The results indicated that cells treated with the
compounds for 14 h exhibited the same effects on oxidative
phosphorylation and glycolysis compared with those noted following
treatment for 72 h (data not shown). Therefore, 14 h treatment was
selected for metabolic analysis.
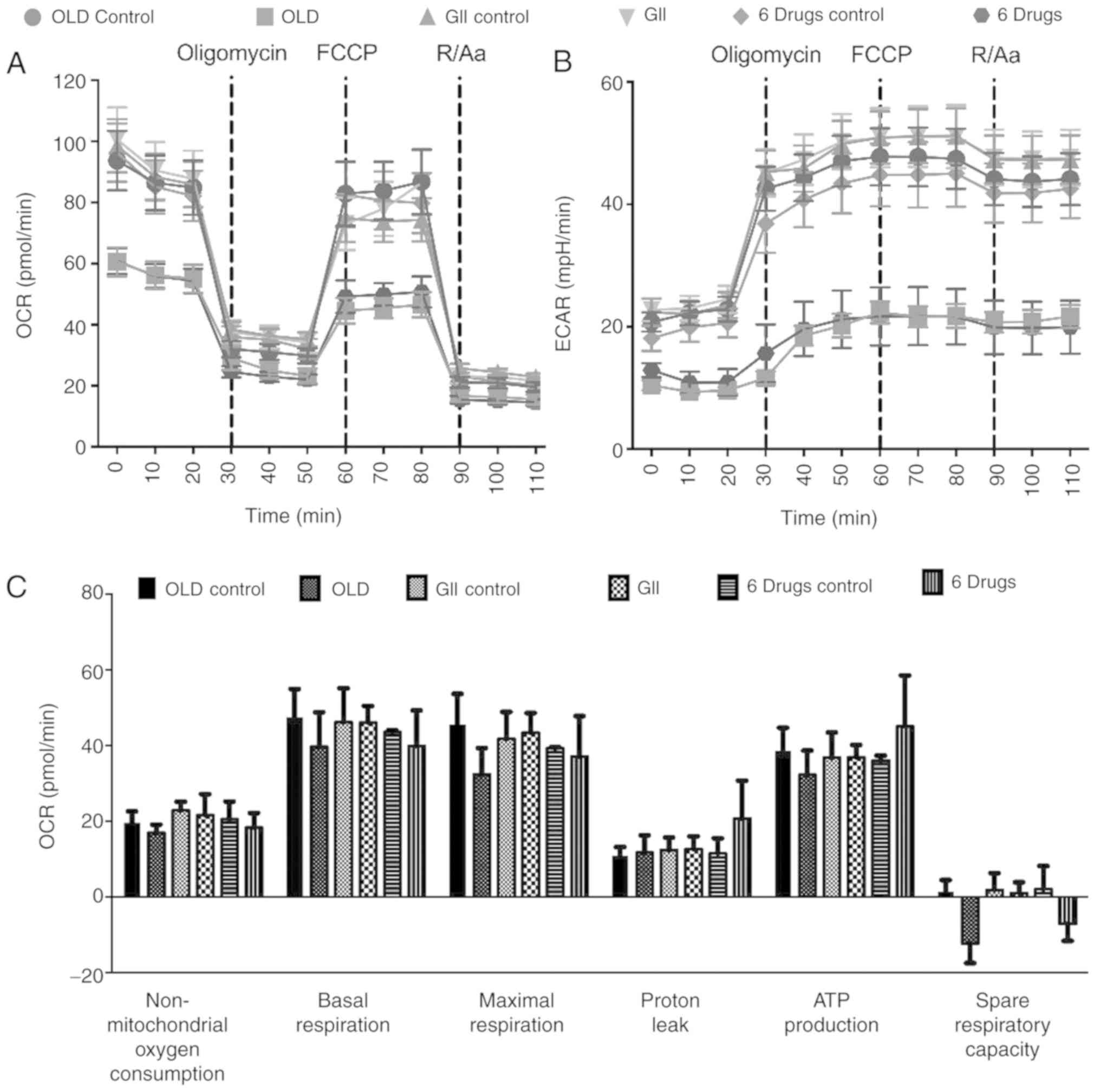
The evaluation of oxidative phosphorylation
demonstrated that both OLD and six-drug schemes showed reduced OCR
and ECAR rates. These effects were preserved following sequential
injections of oligomycin, carbonyl
cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) and
rotenone/antimycin A (Fig. 3A and
B). Although no significant changes were observed in any of the
individual oxidative phosphorylation parameters, treated cells
failed to increase OCR beyond basal levels after FCCP injection,
resulting in decreased OCR values in the spare respiratory capacity
of OLD- and six-drug-treated cells (Fig.
3C).
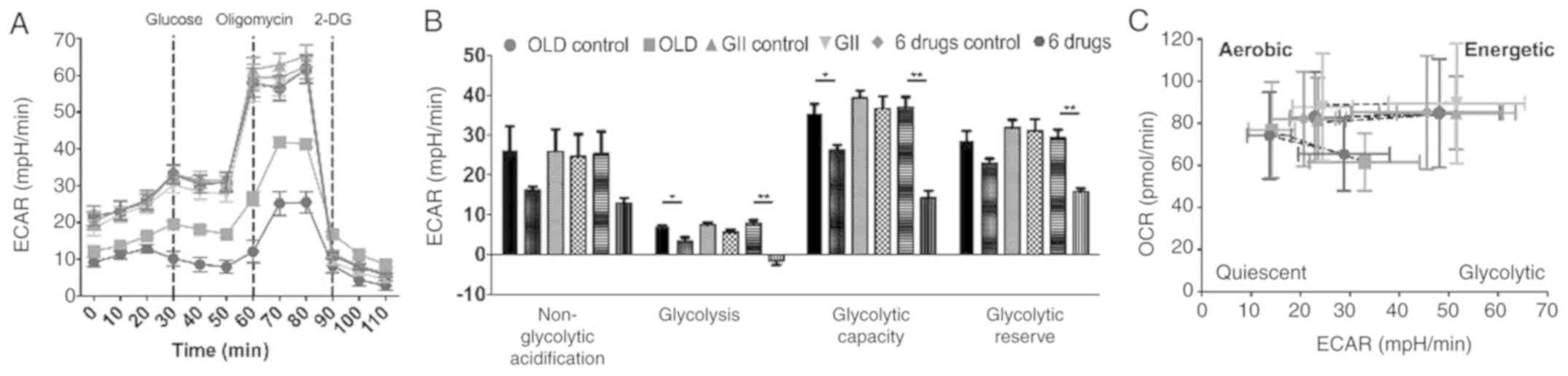
However, glycolysis analysis showed reduced ECAR
following treatment with OLD or six-drug schemes. This effect was
more pronounced in cells treated with six-drug combination
(Fig. 4A) and this scheme exhibited
significantly higher effects on individual parameters of
glycolysis. Therefore, glycolysis, glycolytic capacity and
glycolytic reserve were reduced (Fig.
4B). Besides, energetic phenotype charts were constructed using
data emerged from the oxidative phosphorylation assays, including
basal and FCCP-induced maximal mitochondrial stress measurements.
The results demonstrated that cells treated with either OLD or
six-drug schemes exhibited decreased basal OCR and ECAR rates and a
quiescent metabolic phenotype following treatment with FCCP
(Fig. 4C). However, the GII
treatment did not increase OCR and ECAR values as compared with
those noted in the control groups.
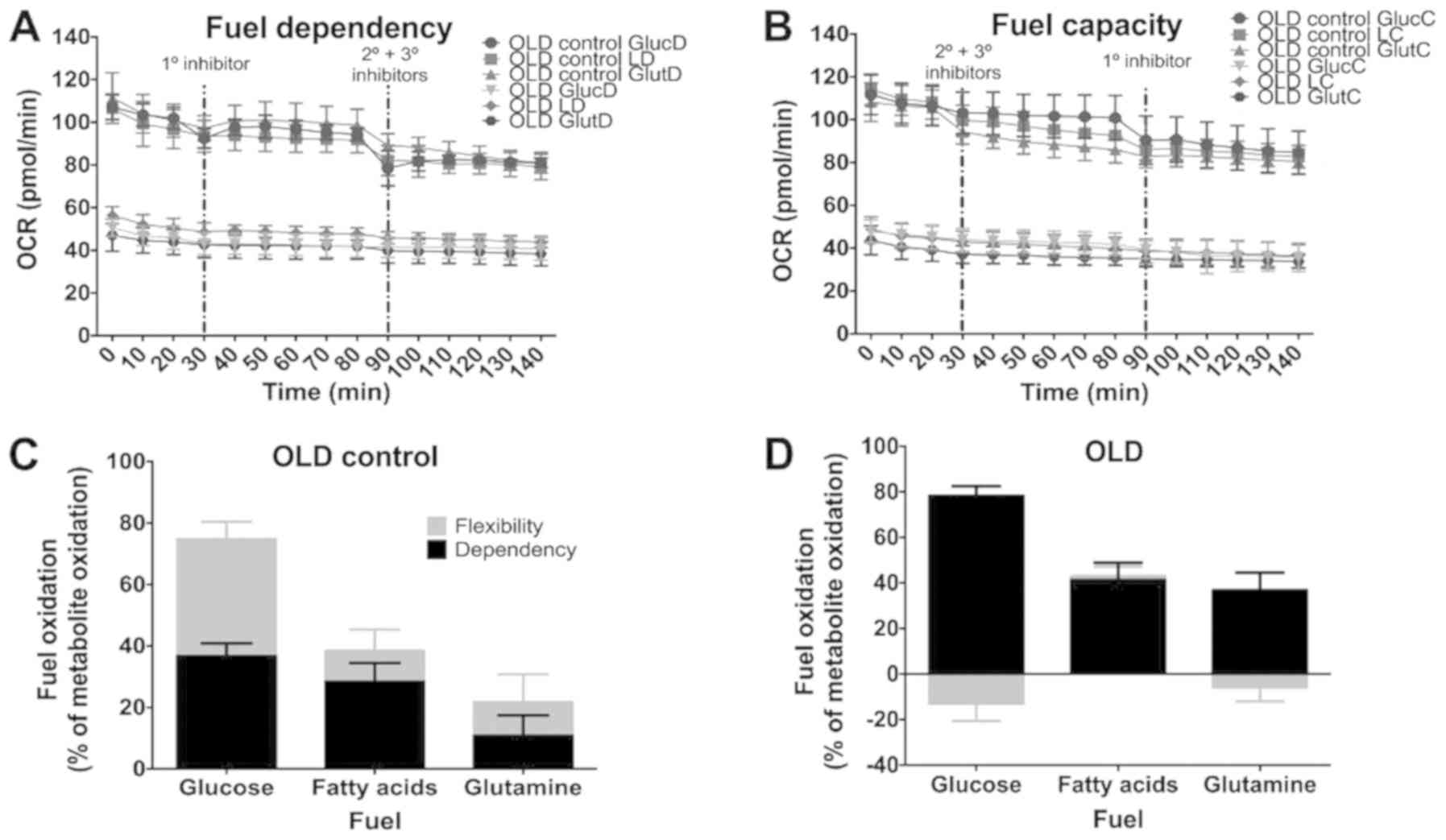
OLD limits the fuel flexibility in
CT26.WT-treated cells
Since the OLD scheme inhibited glycolysis,
glutaminolysis and de novo synthesis of fatty acids, the
dependence and capacity of treated cells to alter their preferred
energetic substrate when the three metabolic pathways were blocked,
were further investigated (Fig. 5A and
B). The results obtained revealed that control cells exhibited
increased glucose and decreased glutamine dependency and
flexibility (Fig. 5C). Therefore,
the dependency rate on basal conditions was 36.72, 28.5 and 10.83%
for glucose, fatty acids and glutamine, respectively. In addition,
the flexibility rates were 38.21, 11.02 and 10.04%, respectively.
However, following treatment with OLD scheme for 14 h, cells
dramatically changed their metabolic flexibility from glucose and
glutamine and increased their dependency on glucose (Fig. 5D). The metabolic dependency from both
glutamine and fatty acids increased to comparable levels, at 41.37
and 36.9%, respectively. Finally, the flexibility from glucose,
glutamine and fatty acids was reduced to approximately negative
values.
Discussion
The present study demonstrated that the simultaneous
use of OLD inhibitors and the six-drug scheme exhibited antitumor
effects in vitro, associated with reduced cellular energy
production.
Currently, targeting the abnormal metabolism in
cancer as a novel form of therapy is gaining momentum (18,19).
Although cancer metabolism inhibitors have not yet been applied in
routine clinical practice, pre-clinical and early-phase clinical
studies with lonidamine and DON demonstrate they were
well-tolerated in cancer patients (20). Intravenous orlistat has not being
tested in humans, but it was well-tolerated in mice at
therapeutical plasma concentration (21,22).
The present study aimed to investigate the antitumor
effects of the combination of lonidamine, an inhibitor of
glycolysis (23); DON, an inhibitor
of glutaminolysis (24); and
orlistat, which inhibits the de novo synthesis of fatty
acids (25). It has been reported
that these anabolic pathways are hyper-functioning in malignancies,
thus these drug schemes were used to inhibit cancer anabolism
(18,19). In addition, tumor progression induces
a pro-inflammatory and catabolic state in the host, resulting in
cancer-associated cachexia (26,27).
Therefore, the OLD scheme in combination with GH, insulin and
indomethacin (GII scheme) was employed to reduce systemic
inflammation and lipolysis, and to promote protein biosynthesis and
glucose internalization (28–35).
OLD and six-drug schemes decreased cancer growth by
inhibiting cell cycle progression at G0/G1
and S phases, and promoting cell apoptosis. To the best of our
knowledge, the application of a triple drug scheme that inhibits
tumor anabolism has not yet been reported. A study demonstrated
that the concomitant treatment with lonidamine and DON showed
higher anti-leukemia effects compared to those noted when each
inhibitor was administered separately (36). Additionally, in non-small cell lung
cancer combined lonidamine and glutaminase inhibitor-968 treatment
induced potent cytotoxicity and growth inhibition (37). Regarding the GII combination
treatment, although the GH-cancer relationship has been a subject
of discussion (38), previous in
vitro and in vivo studies have demonstrated that it does
not exhibit pro-tumor effects (39–41).
Furthermore, although insulin promotes malignant cell proliferation
and invasiveness in vitro (42,43),
however, contradictory effects have been reported regarding cancer
survival and risk in patients (44–46).
According to these studies, in the present model, GII treatment did
not show pro-tumor effects, although GII-treated cells expressed GH
and insulin receptors, as well as cyclooxygenase (data not shown).
All these molecules are targeted by the GII treatment scheme.
Therefore, the absence of tumor-promoting effects cannot be
attributed to the lack of GH and insulin receptors, and
cyclooxygenase expression.
Interestingly, the OLD and six-drug schemes
significantly reduced glycolysis, as demonstrated by diminished
ECAR values. These findings suggested that OLD and six-drug, but
not GII treatment, shifted the energy response to a more quiescent
metabolic state. The extent of glycolysis inhibition was equivalent
to that noted in a previous study with lonidamine treatment in
non-small cell lung cancer, employing the Seahorse methodology as
well (35). On the other hand, OLD
treatment increased cellular dependence on the oxidation of glucose
and glutamine, and reduced fuel flexibility on both substrates.
These findings are well-suited to a more quiescent energetic
phenotype.
In summary, the present study demonstrated that the
concomitant blockade with OLD scheme exhibited antitumor effects
and dramatically affected the energetic machinery of CT26.WT colon
cancer cells. Currently, several novel inhibitors of glycolysis,
glutaminolysis and de novo synthesis of fatty acids, as well
as anti-cachectic agents, are under pre-clinical and clinical
investigation. This study revealed the antitumor effects of OLD
treatment, in order to inhibit tumor anabolism, and the lack of
pro-tumor effects of the GII scheme.
Supplementary Material
Supporting Data
Acknowledgements
Dr Alejandro Schcolnik-Cabrera is a student
belonging to the Plan de Estudios Combinados en Medicina (PECEM),
UNAM. The authors would like to thank Ms. Rocío Morales-Bárcenas
from the National Cancer Institute (Mexico City, Mexico) for her
technical support with the flow cytometer.
Funding
The current work was funded by CONACyT (grant no.
140654) and supported by the CONACyT scholarship (no. 439704),
provided to Alejandro Schcolnik-Cabrera.
Availability of data and materials
All data generated or analyzed during this study are
included in the present article.
Author's contributions
ASC performed all the experiments, analyzed the
data, and wrote the manuscript. MJ helped to perform clonogenic
assays. ASC, ACB, GDG, DL, SH, ART, JDC and ADG designed the
experiments. ADG conceived the project, wrote the manuscript and
together with DL, SH and ART, provided infrastructure and research
facilities to execute the experiments. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
This work was approved by the Ethic and Scientific
committees of the National Cancer Institute of Mexico (protocol
nos. 017/009/IBI and CEI/1055/17), in Mexico City, Mexico.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mathupala SP, Ko YH and Pedersen PL:
Hexokinase II: Cancer's double-edged sword acting as both
facilitator and gatekeeper of malignancy when bound to
mitochondria. Oncogene. 25:4777–4786. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Erickson JW and Cerione RA: Glutaminase: A
hot spot for regulation of cancer cell metabolism? Oncotarget.
1:734–740. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Menendez JA and Lupu R: Fatty acid
synthase and the lipogenic phenotype in cancer pathogenesis. Nat
Rev Cancer. 7:763–777. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guttridge DC, Mayo MW, Madrid LV, Wang CY
and Baldwin AS Jr: NF-kappaB-induced loss of MyoD messenger RNA:
Possible role in muscle decay and cachexia. Science. 289:2363–2366.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tayek JA and Brasel JA: Failure of
anabolism in malnourished cancer patients receiving growth hormone:
A clinical research center study. J Clin Endocrinol Metab.
80:2082–2087. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
DeBerardinis RJ and Cheng T: Q's next: The
diverse functions of glutamine in metabolism, cell biology and
cancer. Oncogene. 29:313–324. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tijerina AJ: The biochemical basis of
metabolism in cancer cachexia. Dimens Crit Care Nurs. 23:237–243.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
La Vecchia S and Sebastián C: Metabolic
pathways regulating colorectal cancer initiation and progression.
Semin Cell Dev Biol. 98:63–70. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lewis NE and Abdel-Haleem AM: The
evolution of genome-scale models of cancer metabolism. Front
Physiol. 4:2372013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Satoh K, Yachida S, Sugimoto M, Oshima M,
Nakagawa T, Akamoto S, Tabata S, Saitoh K, Kato K, Sato S, et al:
Global metabolic reprogramming of colorectal cancer occurs at
adenoma stage and is induced by MYC. Proc Natl Acad Sci USA.
114:E7697–E7706. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schcolnik-Cabrera A, Dominguez-Gómez G,
Chávez-Blanco A, Ramírez-Yautentzi M, Morales-Bárcenas R,
Chávez-Díaz J, Taja-Chayeb L and Dueáas-González A: A combination
of inhibitors of glycolysis, glutaminolysis and de novo fatty acid
synthesis decrease the expression of chemokines in human colon
cancer cells. Oncol Lett. 18:6909–6916. 2019.PubMed/NCBI
|
|
12
|
Cervantes-Madrid D and Dueñas-González A:
Antitumor effects of a drug combination targeting glycolysis,
glutaminolysis and de novo synthesis of fatty acids. Oncol Rep.
34:1533–1542. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maimaiti M, Tanahashi Y, Mohri Z and
Fujieda K: Development of a bioassay system for human growth
hormone determination with close correlation to immunoassay. J Clin
Lab Anal. 26:328–335. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
de la Peña A, Riddle M, Morrow LA, Jiang
HH, Linnebjerg H, Scott A, Win KM, Hompesch M, Mace KF, Jacobson
JG, et al: Pharmacokinetics and pharmacodynamics of high-dose human
regular U-500 insulin versus human regular U-100 insulin in healthy
obese subjects. Diabetes Care. 34:2496–2501. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Manvelian G, Daniels S and Altman R: A
phase I study evaluating the pharmacokinetic profile of a novel,
proprietary, nano-formulated, lower-dose oral indomethacin.
Postgrad Med. 124:197–205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schneider CA, Rasband WS and Eliceiri KW:
NIH Image to ImageJ: 25 years of image analysis. Nat Methods.
9:671–675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zaytseva YY, Harris JW, Mitov MI, Kim JT,
Butterfield DA, Lee EY, Weiss HL, Gao T and Evers BM: Increased
expression of fatty acid synthase provides a survival advantage to
colorectal cancer cells via upregulation of cellular respiration.
Oncotarget. 6:18891–18904. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pavlova NN and Thompson CB: The Emerging
Hallmarks of Cancer Metabolism. Cell Metab. 23:27–47. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Clem BF, O'Neal J, Klarer AC, Telang S and
Chesney J: Clinical development of cancer therapeutics that target
metabolism. QJM. 109:367–372. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cervantes-Madrid D, Romero Y and
Dueñas-González A: Reviving lonidamine and
6-diazo-5-oxo-l-norleucine to be used in combination for metabolic
cancer therapy. BioMed Res Int. 2015:6904922015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kridel SJ, Axelrod F, Rozenkrantz N and
Smith JW: Orlistat is a novel inhibitor of fatty acid synthase with
antitumor activity. Cancer Res. 64:2070–2075. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schcolnik-Cabrera A, Chávez-Blanco A,
Domínguez-Gómez G, Taja-Chayeb L, Morales-Barcenas R,
Trejo-Becerril C, Perez-Cardenas E, Gonzalez-Fierro A and
Dueñas-González A: Orlistat as a FASN inhibitor and multitargeted
agent for cancer therapy. Expert Opin Investig Drugs. 27:475–489.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bhutia YD, Babu E and Ganapathy V:
Re-programming tumour cell metabolism to treat cancer: No lone
target for lonidamine. Biochem J. 473:1503–1506. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Thangavelu K, Chong QY, Low BC and
Sivaraman J: Structural basis for the active site inhibition
mechanism of human kidney-type glutaminase (KGA). Sci Rep.
4:38272014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pemble CW IV, Johnson LC, Kridel SJ and
Lowther WT: Crystal structure of the thioesterase domain of human
fatty acid synthase inhibited by Orlistat. Nat Struct Mol Biol.
14:704–709. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schcolnik-Cabrera A, Chávez-Blanco A,
Domínguez-Gómez G and Dueñas-González A: Understanding tumor
anabolism and patient catabolism in cancer-associated cachexia. Am
J Cancer Res. 7:1107–1135. 2017.PubMed/NCBI
|
|
27
|
Mondello P, Mian M, Aloisi C, Famà F,
Mondello S and Pitini V: Cancer cachexia syndrome: Pathogenesis,
diagnosis, and new therapeutic options. Nutr Cancer. 67:12–26.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Manson JM and Wilmore DW: Positive
nitrogen balance with human growth hormone and hypocaloric
intravenous feeding. Surgery. 100:188–197. 1986.PubMed/NCBI
|
|
29
|
Ward HC, Halliday D and Sim AJ: Protein
and energy metabolism with biosynthetic human growth hormone after
gastrointestinal surgery. Ann Surg. 206:56–61. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cersosimo E, Pisters PW, Pesola G, Rogatko
A, Vydelingum NA, Bajorunas D and Brennan MF: The effect of graded
doses of insulin on peripheral glucose uptake and lactate release
in cancer cachexia. Surgery. 109:459–467. 1991.PubMed/NCBI
|
|
31
|
Pearlstone DB, Wolf RF, Berman RS, Burt M
and Brennan MF: Effect of systemic insulin on protein kinetics in
postoperative cancer patients. Ann Surg Oncol. 1:321–332. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Moley JF, Morrison SD and Norton JA:
Insulin reversal of cancer cachexia in rats. Cancer Res.
45:4925–4931. 1985.PubMed/NCBI
|
|
33
|
Noguchi Y, Nomura K, Yoshikawa T, Fukuzawa
K, Makino T, Tsuburaya A and Matsumoto A: Role of insulin
resistance in decreasing lipoprotein lipase activity in
tumor-bearing rats. Surg Today. 26:271–275. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hitt A, Graves E and McCarthy DO:
Indomethacin preserves muscle mass and reduces levels of E3 ligases
and TNF receptor type 1 in the gastrocnemius muscle of
tumor-bearing mice. Res Nurs Health. 28:56–66. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lundholm K, Daneryd P, Körner U, Hyltander
A and Bosaeus I: Evidence that long-term COX-treatment improves
energy homeostasis and body composition in cancer patients with
progressive cachexia. Int J Oncol. 24:505–512. 2004.PubMed/NCBI
|
|
36
|
Griffiths M, Keast D, Patrick G, Crawford
M and Palmer TN: The role of glutamine and glucose analogues in
metabolic inhibition of human myeloid leukaemia in vitro. Int J
Biochem. 25:1749–1755. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Meijer TWH, Peeters WJM, Dubois LJ, van
Gisbergen MW, Biemans R, Venhuizen JH, Span PN and Bussink J:
Targeting glucose and glutamine metabolism combined with radiation
therapy in non-small cell lung cancer. Lung Cancer. 126:32–40.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cohen P, Clemmons DR and Rosenfeld RG:
Does the GH-IGF axis play a role in cancer pathogenesis? Growth
Horm IGF Res. 10:297–305. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen JY, Liang DM, Gan P, Zhang Y and Lin
J: In vitro effects of recombinant human growth hormone on growth
of human gastric cancer cell line BGC823 cells. World J
Gastroenterol. 10:1132–1136. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liang DM, Chen JY, Zhang Y, Gan P, Lin J
and Chen AB: Effects of recombinant human growth hormone on growth
of human gastric carcinoma xenograft model in nude mice. World J
Gastroenterol. 12:3810–3813. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Harrison LE, Blumberg D, Berman R, Ng B,
Hochwald S, Brennan MF and Burt M: Effect of human growth hormone
on human pancreatic carcinoma growth, protein, and cell cycle
kinetics. J Surg Res. 61:317–322. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lu CC, Chu PY, Hsia SM, Wu CH, Tung YT and
Yen GC: Insulin induction instigates cell proliferation and
metastasis in human colorectal cancer cells. Int J Oncol.
50:736–744. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen X, Liang H, Song Q, Xu X and Cao D:
Insulin promotes progression of colon cancer by upregulation of
ACAT1. Lipids Health Dis. 17:1222018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dulskas A, Patasius A,
Linkeviciute-Ulinskiene D, Zabuliene L and Smailyte G: A cohort
study of antihyperglycemic medication exposure and survival in
patients with gastric cancer. Aging (Albany NY). 11:7197–7205.
2019.PubMed/NCBI
|
|
45
|
Baglia ML, Cui Y, Zheng T, Yang G, Li H,
You M, Xu L, Murff H, Gao YT, Zheng W, et al: Diabetes medication
use in association with survival among patients of breast,
colorectal, lung, or gastric cancer. Cancer Res Treat. 51:538–546.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Karlstad O, Starup-Linde J, Vestergaard P,
Hjellvik V, Bazelier MT, Schmidt MK, Andersen M, Auvinen A, Haukka
J, Furu K, et al: Use of insulin and insulin analogs and risk of
cancer - systematic review and meta-analysis of observational
studies. Curr Drug Saf. 8:333–348. 2013. View Article : Google Scholar : PubMed/NCBI
|