Introduction
Esophageal cancer (EC) is a complex and
heterogeneous gastrointestinal malignancy, which is ranked 7th with
respect to incidence rate and 6th for overall mortality rate in
2018 for 36 cancer types, leading to >400,000 deaths each year,
globally (1). Esophageal
adenocarcinoma (EADC) and esophageal squamous cell carcinoma (ESCC)
are the most common subtypes of EC (2). Cases of ESCC are primarily reported in
lower income regions, such as South-Eastern and Central Asia, and
the global incidence rate for ESCC is higher compared with that in
EADC (3–5). There are various different approaches
to treating EC, such as chemotherapy, radiotherapy, surgery and a
combination treatment; however, the prognosis of patients with EC
is still a challenge. Developing effective chemoprevention and
chemotherapeutics to treat EC is difficult, as its etiology varies
from person to person, such as tobacco and alcohol in North America
and betel quid in India (6).
Inefficient treatments result in poor 5-year survival rate and,
even following advanced radical esophagectomy, the 5-year survival
rate of patients with EC in 2018 is still <20% in China
(7).
The primary cause of the low survival rate and poor
prognosis for EC is early invasion and metastasis, which is a
biological characteristic of cancer cells (8). The occurrence of invasion and
metastasis involves complex multiple-step processes, numerous
signaling pathways and regulation by transcription and growth
factors, such as TGF-β, Snail, ZEB and bHLH (9–12), and
the tumor microenvironment (13,14).
Epithelial-mesenchymal transition (EMT) is an important process
during tumor cell development and metastasis, which involves the
transformation of polar epithelial cells into active mesenchymal
cells, which can migrate freely between the cell stroma (15). EMT is marked by the loss of
epithelioid cell polarity and the acquisition of mesenchymal cell
characteristics, with reduced expression of typical epithelial cell
markers (such as E-cadherin) and increased mesenchymal cell markers
(such as N-cadherin) (15). This
phenotypic transformation releases tumor cells from intercellular
connections, enabling non-invasive tumor cells to acquire invasive
capacity, thereby promoting local invasion and distant tumor
metastasis (16).
Numerous studies have indicated that EMT is
regulated by autophagy, a critical cell survival process through
which aging organelles and misfolded proteins are degraded
(17,18). Although previous studies (19,20)
showed that dihydroartemisinin (DHA), the major active metabolite
of artemisinin, might have an antitumor activity in various cancer
models, whether DHA could regulate the EMT and migration of EC
cells remains unknown. In the present study, a series of
experiments were conducted to investigate the migration inhibition
and underlying molecular mechanisms of DHA. Exploring the molecular
mechanism could provide new perspectives and therapeutic targets
for clinical therapy in patients with EC.
Materials and methods
Cell culture and reagents
The TE-1 and Eca109 EC cell lines were purchased
from the Library of Typical Culture of the Chinese Academy of
Sciences. The cells were cultured in Dulbecco's modified Eagle's
medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.) containing 10%
fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) in a
37°C incubator (95% air and 5% CO2). DHA was dissolved
in DMSO (both Sigma-Aldrich; Merck KGaA) to a final concentration
of 20 µg/ml and stored at −20°C, in the dark until further use.
Immunofluorescence assay
EC cells were harvested using 0.25% trypsin-EDTA
(HyClone; Cytiva) and seeded into a 24-well plate (1×105
cells/well) to adhere overnight. The cells were then transfected
with 800 ng pcDNA3.1-LC3-GFP, a plasmid expressing green
fluorescent protein (GFP) and microtubule-associated protein
1A/1B-light chain 3 (LC3) (coding sequence of GFP and human LC3
were sub-cloned into pcDNA3.1(V790-20, Invitrogen; Thermo Fisher
Scientific, Inc.), hereafter referred to as GFP-LC3) using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). The culture medium was replaced following 6 h of
incubation at 37°C and 2 µg/ml DHA was added prior to incubation
for a further 24 h. The samples were fixed with 4% paraformaldehyde
at room temperature for 15 min and permeabilized with 0.1% Triton-X
100 buffer for 10 min, and the slides were subsequently stained
with DAPI (Beyotime Institute of Biotechnology) for 5 min at room
temperature.
Wound-healing assay
A total of 1×106 cells/well were seeded
into a 6-well plate and cultured overnight to adhere. The cell
monolayers were scratched with a 1-ml pipette tip, and the culture
medium was replaced with fresh medium containing 3% FBS to maintain
cell survival. DHA was added at concentrations of 1, 2 and 5 µg/ml,
and the cells were incubated for a further 24 h in a 37°C incubator
(95% air and 5% CO2). Images were captured at 0- and
24-h time points by fluorescence microscopy (magnification, ×100),
and the wound-closure area was calculated using ImageJ software
v1.51d (National Institutes of Health).
Migration assay
The migration assay was performed using 24-well
plates with Transwell inserts (filter membrane pore-size, 8-µm;
Corning, Inc.). Briefly, 2×104 cells in FBS-free medium
were added to the upper chambers of the Transwell inserts, while
conditioned medium with 20% FBS was added to the lower chambers.
DHA (2 µg/ml), 3-MA (2 mmol/l; cat. no. HY-19312; MedChemExpress)
or a combination of both were then added to the chambers, and the
cells were incubated at 37°C for 24 h. The non-migrated cells were
removed with cotton swabs, and the cells on the underside of the
filter membrane were washed with PBS and fixed with 4%
paraformaldehyde at room temperature for 15 min. The chambers were
subsequently stained with 0.1% crystal violet buffer for 30 min at
room temperature, following washing with PBS. Migration was
determined using the mean number of migratory cells from 20 visual
fields with light microscope at ×10 magnification.
Western blot analysis
A total of 1×106 cells/well were
inoculated into a 6-well plate and left to adhere at 37°C
overnight. The cells were treated with various concentrations of
DHA (1.25, 2.5 and 5.00 µg/ml) or 2 mmol/l 3-MA or a combined
treatment for 24 h, following removal of the culture medium. The
cells were then lysed on ice with radioimmunoprecipitation assay
lysis buffer (Sangon Biotech Co., Ltd.) and the supernatant was
collected following centrifugation (12,000 × g for 10 min) at 4°C.
Protein concentration was determined using a bicinchoninic acid kit
(Sangon Biotech Co., Ltd.). Equal amounts of protein (15 µg per
well) were electrophoresed using 10% sodium dodecyl
sulfate-polyacrylamide gels and transferred to PVDF membranes. The
membranes were blocked with 1× EZ-Block A in PBS solution at 23°C
for 1 h (Sangon Biotech Co., Ltd.). Subsequently, the membranes
were incubated with LC3 (cat. no. L8918), sequestosome 1 (SQSTM1;
cat. no. P0067), and β-actin antibodies (cat. no. SAB5600204) (all
Sigma-Aldrich; Merck KGaA; dilution, 1:1,000), E-cadherin (cat. no.
3195S), N-cadherin (cat. no. 13116S), vimentin (cat. no. 5741S),
p-mTOR (cat. no. 5536S), mTOR (cat. no. 2983S), AKT (cat. no.
4685S), phosphorylated (p)-Akt (cat. no. 13038S) (all Cell
Signaling Technology, Inc.; dilution, 1:1,000) for 12 h at 4°C.
Next, the membranes were incubated with HRP-labeled goat
anti-rabbit IgG secondary antibodies (ZSGB-BIO; cat. no. ZB2301;
dilution, 1:10,000) for 2 h at room temperature. The protein
visualization was mediated by Western Blot Chemiluminescence HRP
Substrate kit (Takara Bio, Inc.; cat. no. T7101A). The grey value
was detected using ImageJ software v1.51d (National Institutes of
Health).
Constitutively active (CA)-Akt
overexpression assay
A total of 9×105 cells were seeded in
6-well plates overnight, and 5 µg CA-Akt plasmid (cat. no. 14751;
Addgene, Inc.) and pcDNA3.1 empty vector (cat. no. V790-20,
Invitrogen; Thermo Fisher Scientific, Inc.) was transfected into
the cells using Lipofectamine® 3000 (Invitrogen; Thermo
Fisher Scientific, Inc.) when the cell fusion rate reached 85%. The
cell culture was replaced and the cells was treated with DHA (2
µg/ml) or DMEM medium, 12 h later. Following 24 h of DHA treatment
western blot analysis and wound-healing assays were performed as
aforementioned.
Statistical analysis
The data are presented as the mean ± standard
deviation. SPSS v19.0 (IBM Corp.) was used to conduct all
statistical analyses. An unpaired student's t test was used to
compare two groups or one-way ANOVA followed by Tukey's post hoc
test for multiple comparisons. P<0.05 was considered to indicate
a statistically significant difference. GraphPad Prism v8.01
(GraphPad Software, Inc.) was used to produce the figures.
Results
DHA inhibits the migration of EC
cells
To understand the effect of DHA on tumor invasion
and metastasis, a cell migration assay was performed. The migratory
capacity of Eca-109 and TE-1 EC cells was markedly inhibited by
DHA. DHA-treated cells possessed a markedly larger wound width
compared with those of the control group, and the width of the
wound was concentration-dependent (Fig.
1A and C). For the Transwell assays, the number of migratory
cells in the DHA group was significantly decreased in both Eca109
(P=0.006) and TE-1 (P=0.003) cells (Fig.
1B and D), which was consistent with the results of the wound
healing assay. These findings demonstrate that DHA can inhibit the
migration of EC cells in a concentration-dependent manner.
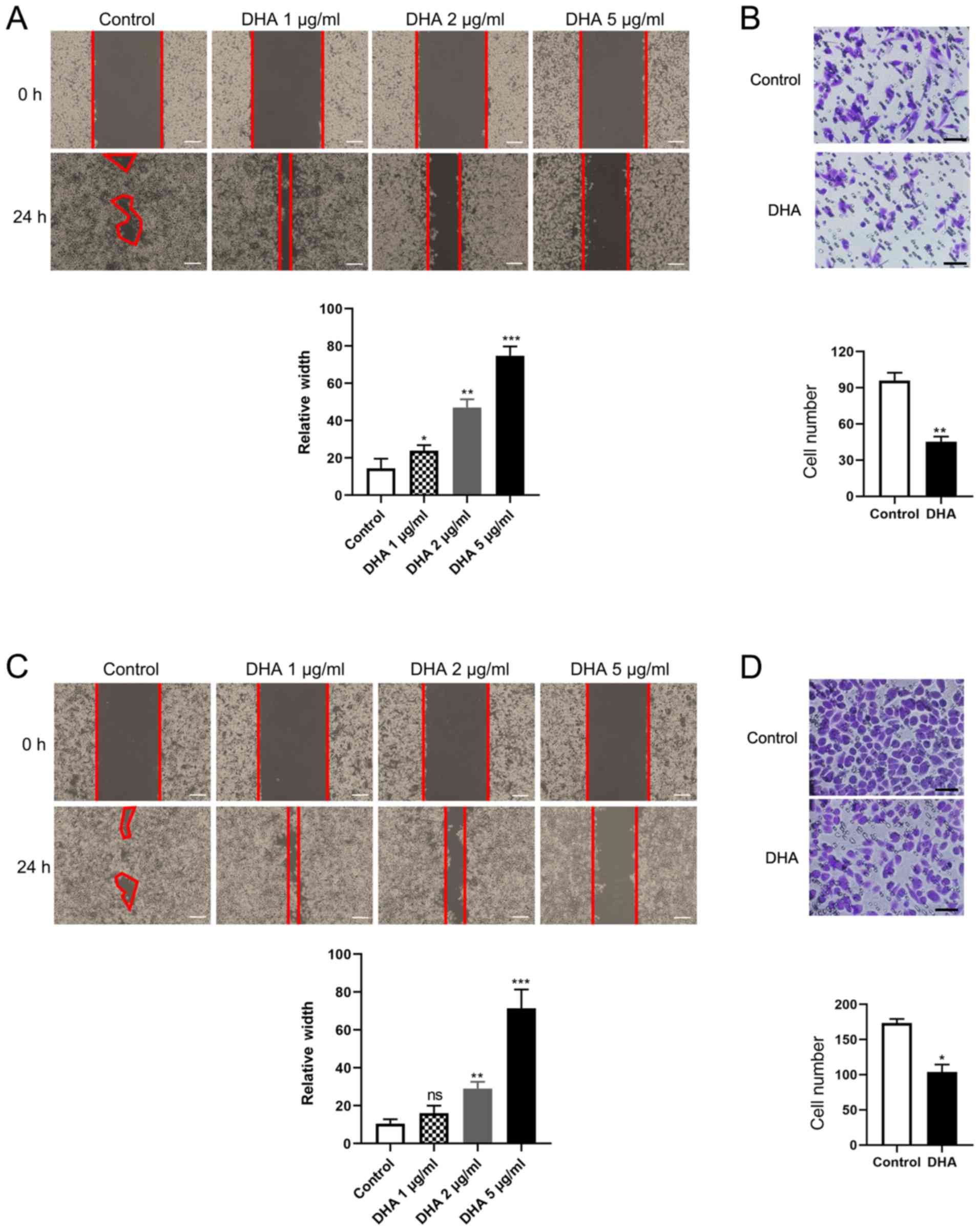 | Figure 1.DHA inhibits esophageal cancer cell
migration in a concentration-dependent manner. (A) A total of
1×106Eca-109 cells were seeded into 6-well plates and
incubated overnight, following which the monolayers were scratched
with 1-ml tips and the cells were cultured with media containing 3%
FBS, and treated with 1, 2 or 5 µg/ml DHA for 24 h. Images of wound
closure distances were captured 0 and 24 h post-treatment. Scale
bar, 100 µm. (B) 2×104 Eca-109 cells were inoculated
into Transwell chambers with serum-free medium, treated with 2
µg/ml DHA for 24 h, and then fixed and stained. Migration ability
was measured using the mean number of cells in 20 visual fields.
Scale bar, 50 µm. (C) A total of 1×106 TE-1 cells were
seeded into 6-well plates and incubated overnight, the description
of wound healing assay was similar as above. Scale bar, 100 µm. (D)
2×104 TE-1 cells were inoculated into Transwell chambers
with serum-free medium and performed as above. Migration ability
was measured using the mean number of cells in 20 visual fields.
Scale bar, 50 µm. Data are represented as the mean ± standard
deviation from 3 independent experiments. *P<0.05, **P<0.01
and ***P<0.001 vs. control. DHA, dihydroartemisinin. |
DHA activates autophagy in TE-1 and
Eca109 cells
To investigate the underlying mechanism by which DHA
inhibits the migration of EC cells, its effects on the levels of
proteins associated with autophagy, a crucial process for tumor
cell survival under starvation and stress (13), were investigated. EC cells were
transfected with a GFP-LC3 plasmid, and the puncta of LC3 were
markedly increased following DHA treatment in both TE-1 (P=0.039)
and Eca109 (P=0.012) cells (Fig.
2A). Western blot analysis also revealed a significant increase
in LC3 protein expression, and a significant decrease in the
protein levels of SQSTM, with increasing concentrations of DHA in
both cell lines (Fig. 2B and C).
These results demonstrate that DHA induces autophagy in EC
cells.
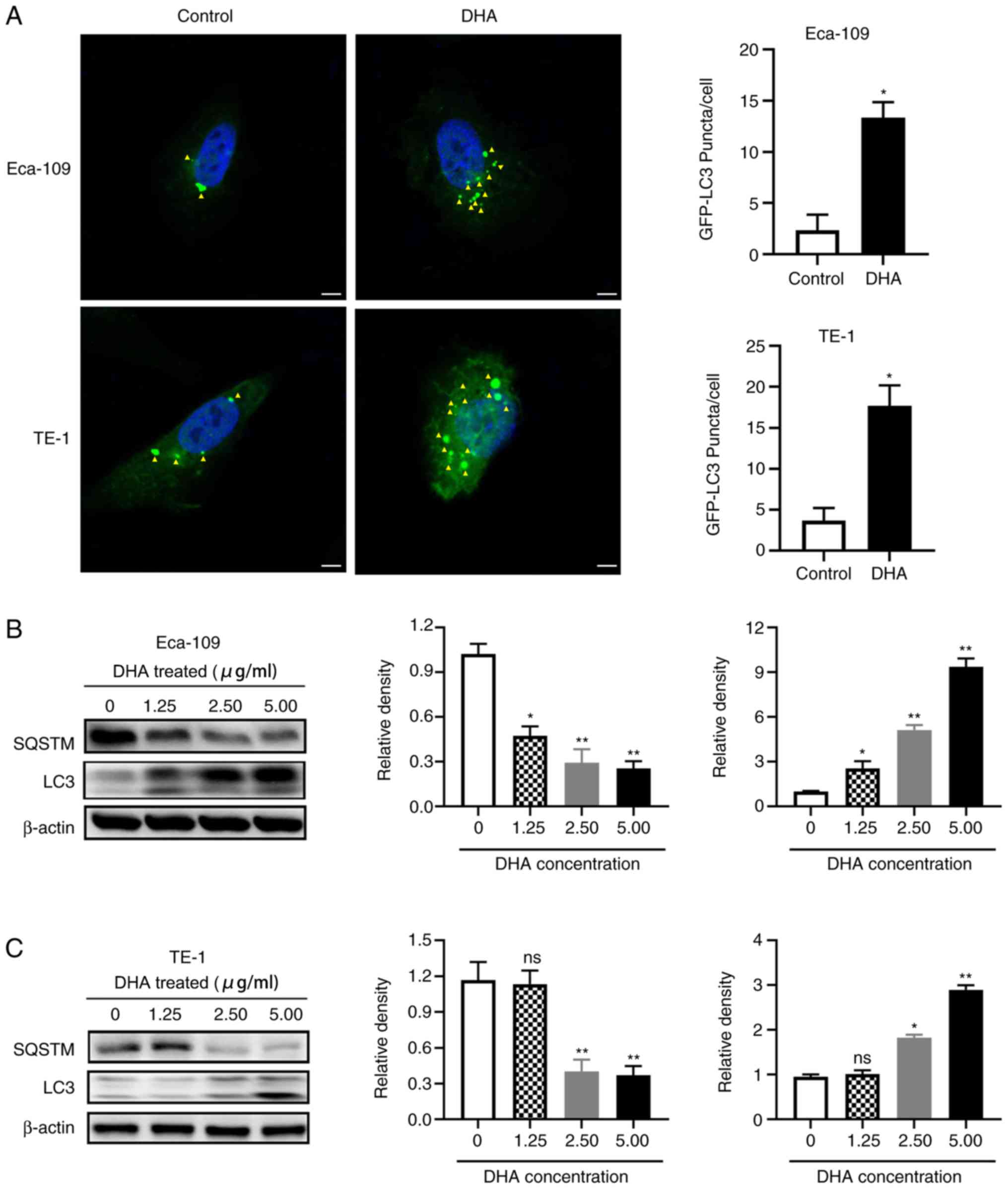 | Figure 2.DHA induces autophagy in TE-1 and
Eca-109 cells. (A) A total of 1×105 cells were seeded
into a 24-well plates and incubated overnight, and then transfected
with an GFP-LC3 expression plasmid for 6 h. The cells were treated
with 2 µg/ml DHA for 24 h, following which the cells were fixed and
stained with DAPI. LC3 expression was analyzed using fluorescence
microscopy, and the number of GFP-LC3 puncta per cell was
determined using Image J. Yellow arrows indicate LC3 protein. Scale
bar, 5 µm. A total of 1×106 (B) Eca-109 and (C) TE-1
cells were seeded into 6-well plates and incubated overnight, and
then treated with 2 µg/ml DHA for 24 h. Levels of
autophagy-associated proteins were detected using western blot
analysis, and the relative density was determined using ImageJ
software. Data are presented as the mean ± standard deviation of 3
independent experiments. *P<0.05 and **P<0.01 vs. respective
control. DHA, dihydroartemisinin; GFP, green fluorescent protein;
LC3, microtubule-associated protein 1A/1B-light chain 3; SQSTM,
sequestosome 1; ns, not significant. |
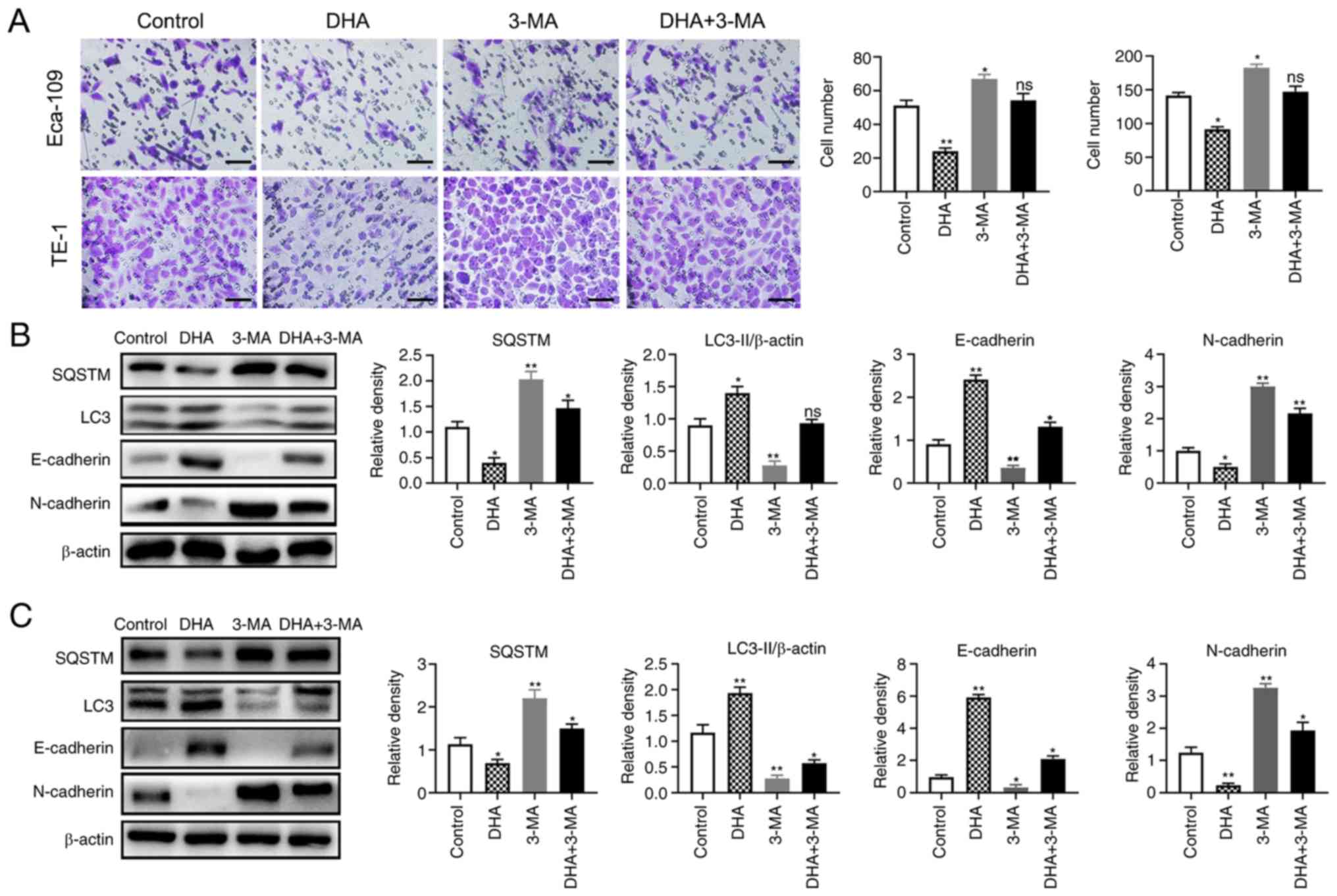
DHA inhibits the migration of EC cells
by inducing autophagy
To determine whether DHA inhibited cell migration by
activating autophagy, Transwell assays and western blot analysis
were used to detect the migratory capacity and the levels of
autophagy-associated proteins following treatment with the
autophagy inhibitor 3-MA. In the Transwell assay, the cells of
Eca-109 (P=0.007) and TE-1 (P=0.03) on the underside of the filter
membrane were significantly lower in number compared with that in
the control group, following treatment with DHA (Fig. 3A). However, this phenomenon was
reversed, and the cell number significantly increased with
co-treatment of 3-MA and DHA in both Eca-109 (P=0.003) and TE-1
(P=0.04) cells (Fig. 3A). These
results indicate that the DHA-induced suppression of cell migration
was weakened when autophagy was inhibited. The expression levels of
the EMT markers and autophagy-associated proteins were also
markedly altered; the protein levels of LC3 II and E-cadherin were
significantly increased and SQSTM and N-cadherin levels decreased
after DHA treatment alone. However, the expression levels of
E-cadherin and LC3 II were decreased, while SQSTM and N-cadherin
were increased following co-treatment with 3-MA and DHA (Fig. 3B and C). These results indicate that
DHA inhibits the migration of TE-1 and Eca-109 cells by
upregulating autophagy.
The Akt/mTOR signaling pathway is
involved in the DHA-induced inhibition of EC cell migration
To determine how DHA regulates autophagy and
subsequently inhibits cellular migration, the expression levels of
the EMT-associated proteins, E-cadherin (21), N-cadherin (22) and vimentin (23) (important components of epithelial
cell loss, polarization and mesenchymal transformation) were
detected. Autophagy-associated proteins, LC3 and SQSTM were also
detected, as well as the phosphorylation levels of Akt and mTOR
(indicating the activated forms of these proteins), which
negatively regulates autophagy (24–26). As
shown in Fig. 4A and B, Akt and mTOR
phosphorylation were inhibited by DHA treatment, accompanied by
increasing LC3 and E-cadherin expression levels, and decreasing
SQSTM, vimentin and N-cadherin expression, in both cell lines. To
further determine the association between DHA and the Akt/mTOR
signaling pathway, cells were transfected with CA-Akt to restore
DHA-induced Akt/mTOR inhibition, and a wound-healing assay was
conducted. As expected, the migratory abilities of the two EC cell
lines were restored following co-treatment with DHA and CA-Akt,
though migration was significantly decreased by DHA treatment alone
(Fig. 4C and D). The results of
western blot analysis were consistent with those of the
wound-healing assay; in cells transfected with CA-Akt and treated
with DHA, the protein levels of p-Akt, SQSTM and N-cadherin were
significantly increased compared with DHA treatment alone (Fig. 4E and F). These data indicate that DHA
activates autophagy by inhibiting the phosphorylation of Akt, and
subsequently suppressing the migration of TE-1 and Eca-109
cells.
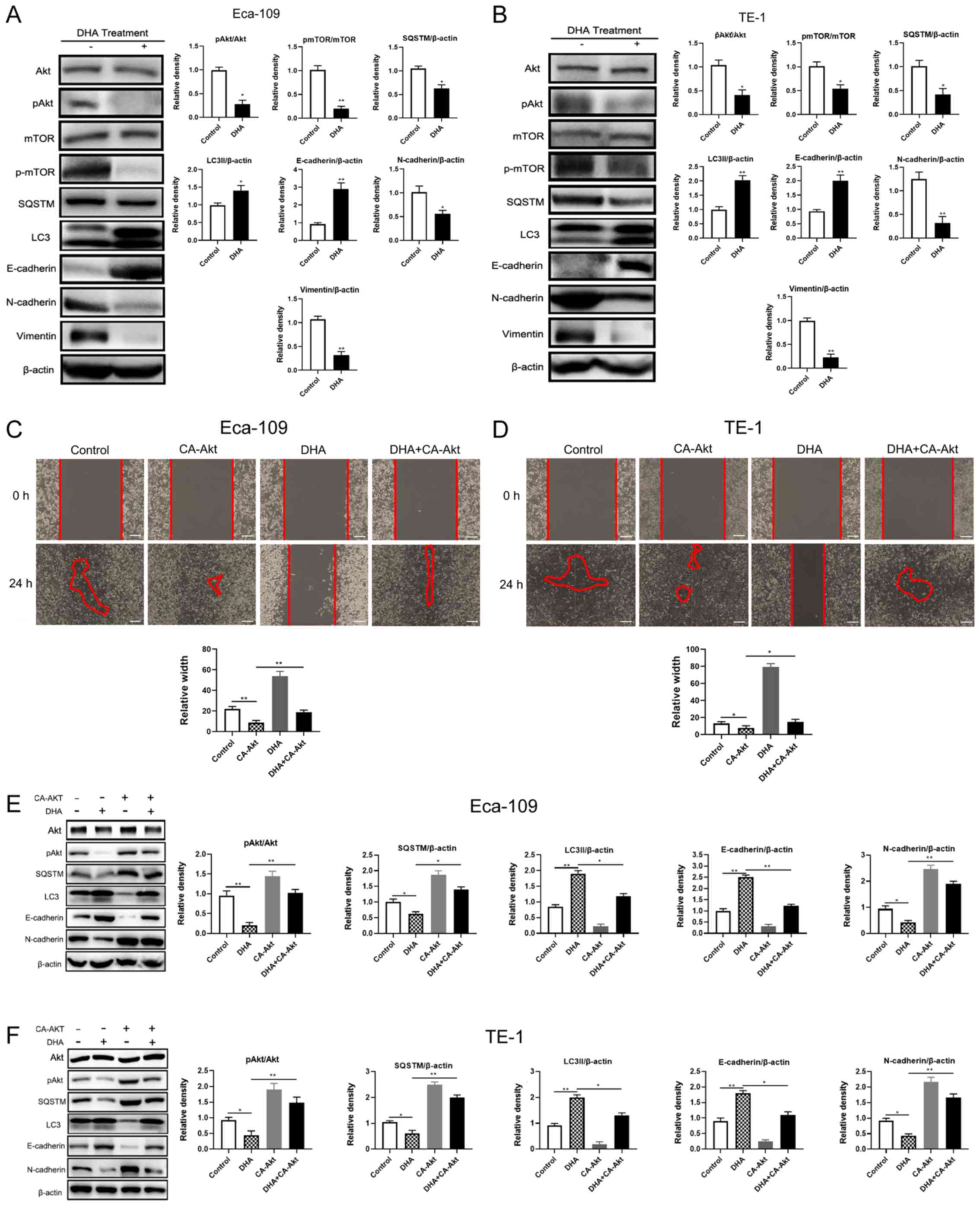 | Figure 4.DHA induces autophagy via the
Akt-mTOR signaling pathway. A total of 1×106 (A) Eca-109
and (B) TE-1 cells were seeded into 6-well plates to adhere
overnight, and then treated with 2 µg/ml DHA for 24 h. Levels of
autophagy- and EMT-associated proteins were detected using western
blot analysis and the relative density was analyzed using ImageJ
software. Furthermore, 1×106 (C) Eca-109 and (D) TE-1
cells were seeded into 6-well plates overnight and then transfected
with CA-Akt. The cell monolayers were scratched with 1-ml tips
after 12 h, subsequently cultured with medium containing 3% FBS,
and then treated with 2 µg/ml DHA. Images of wound closure
distances were captured after DHA treatment at 0 and 24 h. Scale
bar, 100 µm. Following 12 h of transfection with CA-Akt, (E)
Eca-109 and (F) TE-1 cells were treated with 2 µg/ml DHA for 24 h,
and the levels of autophagy- and EMT-associated proteins were
determined using western blot analysis and the relative densities
were analyzed using ImageJ software. Data are presented as the mean
± standard deviation of 3 independent experiments. *P<0.05 and
**P<0.01. DHA, dihydroartemisinin; p, phosphorylated, LC3,
microtubule-associated protein 1A/1B-light chain 3; SQSTM,
sequestosome 1; CA, constitutively active; EMT,
epithelial-mesenchymal transition; -, without; +, with. |
Discussion
DHA, one of the primary derivatives of artemisinin,
has well established anti-malarial effects and has recently been
reported as a potential antitumor compound (27). An increasing amount of evidence has
suggested that DHA can inhibit proliferation by promoting cell
cycle arrest and apoptosis in human hepatic carcinoma (28), cholangiocarcinoma (29), cervical cancer (30) and tongue squamous carcinoma cells
(31). Moreover, DHA can also
prevent the migration of malignant tumor cells, such as lung cancer
(32) and ovarian carcinoma cells
(33). The antitumor activity of DHA
in EC cells was also been shown to affect apoptosis, the cell cycle
and glycolysis (29,34–38);
however, to the best of our knowledge, its inhibitory effects on
cell migration have not previously been reported.
EC is a common malignancy of the digestive tract,
which ranks 7th among 36 cancer types in occurrence worldwide in
2018. The prognosis of patients with EC is poor following
esophagectomy, due to the metastasis and invasion of EC cells
(8). EMT is a precursor of tumor
metastasis, which comprises in the loss of cell polarity and the
acquiring of mesenchymal cell characteristics (39). In the present study, DHA was shown to
markedly inhibit EC cell migration, with reduced N-cadherin and
increased E-cadherin protein expression. Wound-healing and
Transwell assays consistently demonstrated that the migration of
TE-1 and Eca-109 cells was inhibited following DHA treatment.
Further investigations into the underlying mechanism of DHA
revealed that migration inhibition was mediated by autophagy, and
this was reversed following co-treatment with DHA and the autophagy
inhibitor, 3-MA.
As a ‘self-eating’ process, autophagy serves a
crucial role in cell survival via the degradation of aging
organelles or misfolded proteins (40). An increasing number of studies have
demonstrated that autophagy is associated with EMT (41,42);
however, the means by which autophagy regulates EMT are not
completely clear. Li et al (43) found that autophagy was induced in
hepatoma cells treated with serum-free Hank's medium for 6 h, and
that the expression of EMT and mesenchymal markers were decreased
and increased, respectively. On the other hand, Park et al
(44) demonstrated that SQSTM formed
large aggresome-like induced structures (ALISs). Ubiquitination
protease aggregates are generally rapidly degraded, whereas
proteins bound by ALISs have a longer half-life. Transcription
factors mediating EMT can also be protected in this way (45,46). In
the present study, DHA induced autophagy, subsequently suppressing
EMT and cellular migration via the Akt/mTOR signaling pathway. DHA
treatment in TE-1 and Eca-109 cells also inhibited the
phosphorylation of Akt. In addition, the levels of p-Akt were
significantly increased in the two cell lines, in cells transfected
with a constitutively active form of Akt and active Akt restored
the decreased expression level of SQSTM in DHA-treated cells.
However, the underlying regulation mechanisms of DHA on AKT/mTOR
pathway remains unclear, elucidating the molecular mechanisms of
these processes requires further investigation.
This study was aimed to investigate the antitumor
activity of DHA in esophagus cancer cells. The results demonstrated
that DHA could inhibit the migration capacity of Eca109 and TE-1
cells through inducing autophagy. Exploring the underling mechanism
provides new perspectives for clinical cancer therapy. However,
some limitations remain in the present study. The cells were
cultured with medium containing 3% FBS to maintain cell survival
rather than without FBS in wound healing assay, which may influence
the accuracy of this result. Furthermore, the detailed molecular
mechanisms between DHA and AKT/mTOR pathway are still unknown.
In conclusion, the results of the present study
demonstrated that DHA inhibits the migration of EC cells by
inducing Akt/mTOR axis-mediated cytostatic autophagy. These
findings may provide novel insights into the migration inhibiting
activity of DHA and provide evidence to support the potential use
of DHA in the clinical treatment of EC.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed in this study are
included in this published article.
Authors' contributions
YH conceived and designed the study. XC and LYH
prepared the experimental materials and equipment. XC, LYH and SL
performed the experiments and acquired the data. XC and LYH
analyzed and interpreted the data. XC, LYH and SL drafted the
manuscript and revised it critically for important intellectual
content. SL and YH summarized and analyzed the final data for
paper. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Arnold M, Soerjomataram I, Ferlay J and
Forman D: Global incidence of oesophageal cancer by histological
subtype in 2012. GUT. 64:381–387. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Abbasi BA, Iqbal J, Ahmad R, Bibi S,
Mahmood T, Kanwal S, Bashir S, Gul F and Hameed S: Potential
phytochemicals in the prevention and treatment of esophagus cancer:
A green therapeutic approach. Pharmacol Rep. 71:644–652. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang Y: Epidemiology of esophageal
cancer. World J Gastroenterol. 19:5598–5606. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Napier KJ, Scheerer M and Misra S:
Esophageal cancer: A Review of epidemiology, pathogenesis, staging
workup and treatment modalities. World J Gastrointest Oncol.
6:112–120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mir MM and Dar NA: Esophageal cancer in
kashmir (India): An enigma for researchers. Int J Health Sci
(Qassim). 3:71–85. 2009.PubMed/NCBI
|
|
7
|
Wong M, Hamilton W, Whiteman DC, Jiang JY,
Qiao Y, Fung F, Wang H, Chiu P, Ng E, Wu J, et al: Global Incidence
and mortality of oesophageal cancer and their correlation with
socioeconomic indicators temporal patterns and trends in 41
countries. Sci Rep. 8:45222018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nan L, Wei J, Jacko AM, Culley MK, Zhao J,
Natarajan V, Ma H and Zhao Y: Cross-talk between lysophosphatidic
acid receptor 1 and tropomyosin receptor kinase A promotes lung
epithelial cell migration. Biochim Biophys Acta. 1863:229–235.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tripathi V, Shin JH, Stuelten CH and Zhang
YE: TGF-β-induced alternative splicing of TAK1 promotes EMT and
drug resistance. Oncogene. 38:3185–3200. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ota I, Masui T, Kurihara M, Yook JI,
Mikami S, Kimura T, Shimada K, Konishi N, Yane K, Yamanaka T and
Kitahara T: Snail-induced EMT promotes cancer stem cell-like
properties in head and neck cancer cells. Oncol Rep. 35:261–266.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mooney SM, Talebian V, Jolly MK, Jia D,
Gromala M, Levine H and McConkey BJ: The GRHL2/ZEB feedback loop-a
key axis in the regulation of EMT in breast cancer. J Cell Biochem.
118:2559–2570. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cano A and Portillo F: An emerging role
for class I bHLH E2-2 proteins in EMT regulation and tumor
progression. Cell Adh Migr. 4:56–60. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gao FX, Wu J and Ren DL: Effect of
epithelial-to-mesenchymal transition on biological activity of NK
cells in esophageal squamous cell carcinoma. Sichuan Da Xue Xue Bao
Yi Xue Ban. 50:40–47. 2019.(In Chinese). PubMed/NCBI
|
|
14
|
Yokozaki H, Koma YI, Shigeoka M and Nishio
M: Cancer as a tissue: The significance of cancer-stromal
interactions in the development, morphogenesis and progression of
human upper digestive tract cancer. Pathol Int. 68:334–352. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mladinich M, Ruan D and Chan CH: Tackling
cancer stem cells via inhibition of EMT transcription factors. Stem
Cells Int. 2016:52858922016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pattabiraman DR, Bierie B, Kober KI, Thiru
P, Krall JA, Zill C, Reinhardt F, Tam WL and Weinberg RA:
Activation of PKA leads to mesenchymal-to-epithelial transition and
loss of tumor-initiating ability. Science. 351:d36802016.
View Article : Google Scholar
|
|
17
|
Gugnoni M, Sancisi V, Gandolfi G, Manzotti
G, Ragazzi M, Giordano D, Tamagnini I, Tigano M, Frasoldati A,
Piana S and Ciarrocchi A: Cadherin-6 promotes EMT and cancer
metastasis by restraining autophagy. Oncogene. 36:667–677. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ouyang F, Huang H, Zhang M, Chen M, Huang
H, Huang F and Zhou S: HMGB1 induces apoptosis and EMT in
association with increased autophagy following H/R injury in
cardiomyocytes. Int J Mol Med. 37:679–689. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin R, Zhang Z, Chen L, Zhou Y, Zou P,
Feng C, Wang L and Liang G: Dihydroartemisinin (DHA) induces
ferroptosis and causes cell cycle arrest in head and neck carcinoma
cells. Cancer Lett. 381:165–175. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu T, Guo J, Wang T, Zhang S, Yu X, Hou C
and Guo D: Network pharmacology-based analysis of mechanisms of the
anti-hepatocellular carcinoma effect by dihydroartemisinin. Discov
Med. 28:139–147. 2019.PubMed/NCBI
|
|
21
|
Daugaard I, Sanders KJ, Idica A,
Vittayarukskul K, Hamdorf M, Krog JD, Chow R, Jury D, Hansen LL,
Hager H, et al: miR-151a induces partial EMT by regulating
E-cadherin in NSCLC cells. Oncogenesis. 6:e3662017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang X, Liu G, Kang Y, Dong Z, Qian Q and
Ma X: N-cadherin expression is associated with acquisition of EMT
phenotype and with enhanced invasion in erlotinib-resistant lung
cancer cell lines. PLoS One. 8:e576922013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ivaska J: Vimentin: Central hub in EMT
induction? Small GTPases. 2:51–53. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liang P, Jiang B, Li Y, Liu Z, Zhang P,
Zhang M, Huang X and Xiao X: Autophagy promotes angiogenesis via
AMPK/Akt/mTOR signaling during the recovery of heat-denatured
endothelial cells. Cell Death Dis. 9:11522018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang J, Pi C and Wang G: Inhibition of
PI3K/Akt/mTOR pathway by apigenin induces apoptosis and autophagy
in hepatocellular carcinoma cells. Biomed Pharmacother.
103:699–707. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fan S, Zhang B, Luan P, Gu B, Wan Q, Huang
X, Liao W and Liu J: PI3K/AKT/mTOR/p70S6K pathway is involved in
Aβ25-35-induced autophagy. Biomed Res Int. 2015:1610202015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cao P, Leng D, Li Y, Zhang Z, Liu L and Li
X: Progress on anti-tumor molecular mechanisms of
dihydroartemisinin. Zhejiang Da Xue Xue Bao Yi Xue Ban. 45:501–507.
2016.(In Chinese). PubMed/NCBI
|
|
28
|
Zhang CZ, Zhang H, Yun J, Chen GG and Lai
PB: Dihydroartemisinin exhibits antitumor activity toward
hepatocellular carcinoma in vitro and in vivo.
Biochem Pharmacol. 83:1278–1289. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Thongchot S, Vidoni C, Ferraresi A,
Loilome W, Yongvanit P, Namwat N and Isidoro C: Dihydroartemisinin
induces apoptosis and autophagy-dependent cell death in
cholangiocarcinoma through a DAPK1-BECLIN1 pathway. Mol Carcinog.
57:1735–1750. 2018. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li X, Ba Q, Liu Y, Yue Q, Chen P, Li J,
Zhang H, Ying H, Ding Q, Song H, et al: Dihydroartemisinin
selectively inhibits PDGFRalpha-positive ovarian cancer growth and
metastasis through inducing degradation of PDGFRalpha protein. Cell
Discov. 3:170422017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shi X, Wang L, Li X, Bai J, Li J, Li S,
Wang Z and Zhou M: Dihydroartemisinin induces autophagy-dependent
death in human tongue squamous cell carcinoma cells through DNA
double-strand break-mediated oxidative stress. Oncotarget.
8:45981–45993. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jiang J, Geng G, Yu X, Liu H, Gao J, An H,
Cai C, Li N, Shen D, Wu X, et al: Repurposing the anti-malarial
drug dihydroartemisinin suppresses metastasis of non-small-cell
lung cancer via inhibiting NF-kappaB/GLUT1 axis. Oncotarget.
7:87271–87283. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Im E, Yeo C, Lee HJ and Lee EO:
Dihydroartemisinin induced caspase-dependent apoptosis through
inhibiting the specificity protein 1 pathway in hepatocellular
carcinoma SK-Hep-1 cells. Life Sci. 192:286–292. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Y, Sui H, Jiang C, Li S, Han Y, Huang
P, Du X, Du J and Bai Y: Dihydroartemisinin increases the
sensitivity of photodynamic therapy via NF-κB/HIF-1α/VEGF pathway
in esophageal cancer cell in vitro and in vivo. Cell
Physiol Biochem. 48:2035–2045. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiang C, Li S, Li Y and Bai Y: Anticancer
effects of dihydroartemisinin on human esophageal cancer cells
in vivo. Anal Cell Pathol (Amst).
2018:87597452018.PubMed/NCBI
|
|
36
|
Li S, Huang P, Gan J, Ling X, Du X, Liao
Y, Li L, Meng Y, Li Y and Bai Y: Dihydroartemisinin represses
esophageal cancer glycolysis by down-regulating pyruvate kinase M2.
Eur J Pharmacol. 854:232–239. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li YJ, Zhou JH, Du XX, Jia DX, Wu CL,
Huang P, Han Y, Sui H, Wei XL, Liu L, et al: Dihydroartemisinin
accentuates the anti-tumor effects of photodynamic therapy via
inactivation of NF-κB in Eca109 and Ec9706 esophageal cancer cells.
Cell Physiol Biochem. 33:1527–1536. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Du XX, Li YJ, Wu CL, Zhou JH, Han Y, Sui
H, Wei XL, Liu L, Huang P, Yuan HH, et al: Initiation of apoptosis,
cell cycle arrest and autophagy of esophageal cancer cells by
dihydroartemisinin. Biomed Pharmacother. 67:417–424. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Alonso-Alconada L, Eritja N, Muinelo-Romay
L, Barbazan J, Lopez-Lopez R, Matias-Guiu X, Gil-Moreno A, Dolcet X
and Abal M: ETV5 transcription program links BDNF and promotion of
EMT at invasive front of endometrial carcinomas. Carcinogenesis.
35:2679–2686. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhou S, Zhao L, Kuang M, Zhang B, Liang Z,
Yi T, Wei Y and Zhao X: Autophagy in tumorigenesis and cancer
therapy: Dr. Jekyll or Mr. Hyde? Cancer Lett. 323:115–127. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Colella B, Faienza F and Di Bartolomeo S:
EMT regulation by autophagy: A new perspective in glioblastoma
biology. Cancers (Basel). 11:3122019. View Article : Google Scholar
|
|
42
|
Feng H, Zhao X, Guo Q, Feng Y, Ma M, Guo
W, Dong X, Deng C, Li C, Song X, et al: Autophagy resists EMT
process to maintain retinal pigment epithelium homeostasis. Int J
Biol Sci. 15:507–521. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo
Y, Song Z, Zheng Q and Xiong J: Autophagy promotes hepatocellular
carcinoma cell invasion through activation of
epithelial-mesenchymal transition. Carcinogenesis. 34:1343–1351.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Park S, Ha SD, Coleman M, Meshkibaf S and
Kim SO: p62/SQSTM1 enhances NOD2-mediated signaling and cytokine
production through stabilizing NOD2 oligomerization. PLoS One.
8:e571382013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Qiang L, Zhao B, Ming M, Wang N, He TC,
Hwang S, Thorburn A and He YY: Regulation of cell proliferation and
migration by p62 through stabilization of Twist1. Proc Natl Acad
Sci USA. 111:9241–9246. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Qiang L and He YY: Autophagy deficiency
stabilizes TWIST1 to promote epithelial-mesenchymal transition.
Autophagy. 10:1864–1865. 2014. View Article : Google Scholar : PubMed/NCBI
|