Introduction
Marginal zone lymphomatas (MZLs) are a group of
indolent B-cell lymphomata that arise from marginal zone B
lymphocytes (1,2). MZLs comprises ~10% of all non-Hodgkins
lymphomas (NHLs) (1,3). They include splenic marginal zone
lymphoma (SMZL) with or without villous lymphocytes, nodal marginal
zone lymphoma (NMZL) and extranodal marginal-zone B-cell lymphoma
of mucosal-associated lymphoid tissue (MALT) (4). MALT lymphoma is the most common
subtype, comprising 7–8% of NHLs, with NMZL comprising <2% and
SMZL <1% (3). MALT lymphoma
arises from numerous epithelial tissues, including stomach
(60-70%), lung (15%), ocular adnexa (10%) and other less frequent
sites (thyroid, salivary glands, intestine, skin and liver)
(5–7). These organs are usually almost devoid
of lymphoid tissue; however, they accumulate B lymphocytes in
response to persistent inflammation due to chronic infections, such
as by Helicobacter pylori in the stomach (8). Anatomical sites may have prognostic
relevance due to organ-specific clinical problems and different
sites may have distinct natural history (9). In a previous study, long-term outcomes
were investigated in patients with localized MALT lymphoma and the
results identified a significantly improved outcome in gastric and
thyroid lymphoma (10). In general,
despite frequent relapses, MALT lymphoma most often maintains an
indolent course (11).
Chronic inflammatory and immunological responses
promote the acquisition of genetic abnormalities, malignant
transformation and clonal expansion of transformed neoplastic cells
(8,12). Acquired genetic abnormalities serve
critical roles in the development of MALT lymphoma by modulating
similar molecular processes (12).
To the best of our knowledge, genetic abnormalities in MALT
lymphoma have not yet been investigated using whole exome or genome
sequencing. Furthermore, various repeat genetic abnormalities,
including chromosomal translocations, somatic cell mutations and
changes in the number of copies, have been reported to alter
signaling pathways that modulate NF-κB activity through standard
pathways (13).
The etiology of MALT lymphoma involves various
chromosomal translocations (14).
The t(11;18)(q21;q21) chromosome translocation has been
demonstrated to be the most common translocation that results in a
functional chimeric fusion product between the N-terminal of
baculoviral IAP repeat containing 3 (API2) and the C-terminal of
mucosa-associated lymphoid tissue lymphoma translocation protein 1
(MALT1) (15–17). The API2-MALT1 fusion product exhibits
functional gain and activates both the canonical and non-canonical
NF-κB signaling pathways (13). The
fusion product is often detected in stomach and lung MALT lymphoma
(18). The t(1:14) translocation
upregulates B-cell lymphoma/leukemia 10 (BCL10) proteins in 1–2% of
MALT lymphoma cases, including in MALT lymphoma in the stomach,
lungs and skin (12). The t(14;18)
translocation results in the deregulation of MALT1 expression and
it has been reported in 15–20% of MALT lymphoma cases (19). This translocation is most frequently
detected in the MALT lymphoma of the liver, skin, ocular adnexa and
salivary gland (19). The t(3;14)
translocation results in forkhead box protein B1 upregulation
(20). It is associated with MALT
lymphoma of the thyroid, ocular adnexa and skin (20). Numerical chromosomal aberrations,
including trisomy 3, 12 and/or 18 are present as sole abnormalities
in 22% of cases (21).
In MALT lymphoma, TNF-α inducible protein 3
(TNFAIP3) deletions and mutations are found mainly in tumors of the
ocular adnexa, salivary gland and thyroid (22–24).
TNFAIP3 inactivation by deletion and/or mutation abolishes negative
regulations of several signaling pathways, thus activating the
canonical NF-κB signaling pathway (25). The oncogenic activities of this
inactivation depend on the presence of antigenic and inflammatory
stimulations (12). The MYD88 innate
immune signal transduction adaptor (MYD88) L265P somatic mutation
can be observed in ocular adnexal MALT lymphoma (~5% of cases),
although it appears to be infrequent at other anatomic sites
(26). However, it has not yet been
fully investigated (27–29). Somatic mutation in the MYD88 gene
occurs at a single nucleotide (3p22.2) (26). This mutation leads to an amino acid
change from leucine to proline (L265P), which activates
MYD88-dependent signaling in Toll-like receptor signaling pathways,
leading to NF-κB activation (27,30).
Extranodal MZL (EMZL) usually has an indolent
disease course (2,31). Patients with EMZL have a median
survival time of >12 years (32).
Gastric MALT lymphoma tends to be localized to primary tissue sites
for a long time (33). The 10-year
survival and disease-free survival rates for gastric MALT lymphoma
are close to 90 and ~70%, respectively (33,34).
However, in rare instances, aggressive high-grade tumors can arise
or be transformed from MALT lymphoma (35). Extranodal diffuse large B cell
lymphoma (DLBCL) causes a drop in the 10-year survival rate to ~42%
(33). EMZL is a clinically
heterogeneous low-grade B cell lymphoma (1,3,5). Molecular pathways responsible for the
pathogenesis, prognosis and drug resistance of EMZL have not been
fully elucidated. Further research on genetic aberrations is
required to identify the genetic abnormalities of MALT
lymphoma.
Next-generation sequencing (NGS) provides a means of
performing high-throughput, comprehensive analyses of landscapes of
genetic aberrations, copy number alterations and transcript
expression patterns, thus leading to improved understanding of
various genetic abnormalities (29).
The present study aimed to identify mutations that may be
responsible for gastrointestinal MALT lymphoma, including 4 cases
of MALT lymphoma in the small bowel, which is an uncommon
anatomical site, by targeted sequencing using a panel (HemaScan™)
of hematologic malignancy-related genes (Fig. S1). Genes that may be associated with
the pathogenesis and progression of gastrointestinal MALT lymphoma
were suggested.
Materials and methods
Patient characteristics
Patients who were diagnosed with gastrointestinal
MALT lymphoma through endoscopy or surgery at Dong-A University
Hospital were included in the analysis. Patient samples were
excluded if they did not match the following criteria: Subjects
should have enough FFPE samples for NGS and DNA preparation samples
from the FFPE should qualify quality control (QC) criteria. A total
of 5 patients (age, 49–72 years; 2 males and 3 females) underwent
surgical biopsies. All patients were diagnosed histologically with
MALT lymphoma. A total of 4 patients had small intestine lesions,
and 1 patient had a stomach lesion. According to the Stage Lugano
modification of the Ann Arbor staging system (36), 1 patient was in stage I, 3 patients
were in stage III and 1 patient was in stage IV. None of the
patients exhibited an increase in lactate dehydrogenase or B
symptoms. All patients underwent bone marrow biopsies and no bone
marrow involvement was noted. According to the International
Prognostic Index (IPI) scoring system (37), 2 patients had an IPI score of 2 and 3
patients had a score of 0. Helicobacter pylori eradication
therapy was performed for the patient with gastric MALT
lymphoma.
All patients underwent surgery and 4 patients
underwent chemotherapy with a regimen of cyclophosphamide,
vincristine and prednisolone (CVP) for ≥4-6 cycles. Each cycle of
CVP chemotherapy regimens was administered intravenously with
cyclophosphamide 750 mg/m2, and vincristine 1.4
mg/m2 (maximum dose of 2 mg) on first day, and orally
with prednisone 100 mg on day 1 to 5. It was repeated every 21 days
and treated up to six times. Additionally, the patient with gastric
MALT lymphoma underwent radiotherapy prior to chemotherapy with the
addition of intravenous Rituximab 375 mg/m2 on day 1 to
the CVP regimen. Intestinal MALT lymphoma recurred in one patient
at 4 years and 6 months following the completion of the initial
treatment. All patients were alive at the time of writing (68.3
months following initial treatment; Table I).
 | Table I.Patient characteristics. |
Table I.
Patient characteristics.
| Variable | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 |
|---|
| Sex | M | F | M | F | F |
| Age, years | 59 | 72 | 53 | 55 | 49 |
| Diagnosis | MALT | MALT | MALT | MALT | MALT |
| Site of lesion | Stomach | Small
intestine | Small
intestine | Small
intestine | Small
intestine |
| Stage | II | II | I | IV | II |
| B symptoms | – | – | – | – | – |
| Elevated LDH | – | – | – | – | – |
| BM involvement | – | – | – | – | – |
| ECOG PS | 1 | 1 | 1 | 1 | 1 |
| IPI score | 0 | 1 | 0 | 1 | 0 |
| H. pylori
eradiation | + | – | – | – | – |
| Chemotherapy | + | + | – | + | + |
| Radiotherapy | + | – | – | – | – |
| Surgery | + | + | + | + | + |
| Recurrence | – | + | – | – | – |
| Death | – | – | – | – | – |
The present study was approved by the Institutional
Review Board of Dong-A University Hospital, Seo-gu, Busan, Republic
of Korea (approval no. DAUHIRB 20-088).
Study design
All patients were diagnosed with gastrointestinal
MALT lymphoma and underwent surgery to obtain tissue samples.
Samples from the patients were obtained at Dong-A University
Hospital (Busan, Korea) between January 2011 and December 2014.
Immunohistochemistry for gastrointestinal MALT lymphoma diagnosis
with LCA, UCHL-1, CD20, CD5, CD10, Bcl-2, Bcl-6, MUM-1, Cyclin D1
and CD23 was performed using formalin-fixed, paraffin-embedded
(FFPE) tissues. The requirement for informed consent was waived
since the tissues had been donated to the Dong-A University
Hospital tissue bank.
DNA preparation
FFPE tissue samples were cut into 5-µm thick
sections, incubated at 60°C for 30 min, de-paraffinized in three
containers of xylene baths at 60°C (3×5 mins), rehydrated with
decreasing alcohols (100, 95 and 70%, two washes for 5 mins each)
and washed with distilled water two times for 5 mins each.
Subsequently, genomic DNA was extracted from the tissue samples
using the Maxwell 16 CSC DNA FFPE kit (cat. no. AS1350, Promega
Corporation) or the QIAamp DNA FFPE Tissue kit (cat. no. 56404,
Qiagen, Inc.), which provides an automated purification of genomic
DNA from FFPE tissue samples, thereby maximizing simplicity and
convenience. DNA concentration and purity were verified using
Nanodrop 8000 UV–Vis spectrometry (Thermo Fisher Scientific, Inc.)
and Qubit 2.0 Fluorometry (Thermo Fisher Scientific, Inc.). Degrees
of DNA degradation were measured using a 200 TapeStation Instrument
(Agilent Technologies GmbH) and quantitative PCR (Agilent
Technologies GmbH) (38).
Library preparation and
sequencing
DNA was sheared using Covaris S220 (Covaris, Inc.).
Target capture was performed using a SureSelect XT reagent kit, HSQ
(Agilent Technologies GmbH) according to the manufacturers
protocol. A paired-end sequencing library was constructed using a
barcode. Individual ‘barcode’ sequences are added to each DNA
fragment during NGS library preparation so that each read can be
identified and sorted before the final data analysis (39). After checking for library quality,
sequencing was performed on a HiSeq 2500 (Illumina, Inc.) using 100
bp reads. SureSelect XT Human All Exon (version 5; Agilent
Technologies, Inc.) was used for target capture for exome
sequencing and a TruSeq Nano DNA Sample prep kit (Illumina, Inc.)
was used for whole genome sequencing.
Panel design and sequencing
Samples were profiled on a HemaSCAN™, a
targeted-sequencing platform designed by the LabGenomics in the
Samsung Medical Center (81 Irwon-Ro Gangnam-gu. Seoul 06351,
Korea). Samsung medical center participated in the panel design for
the production of customized panels. The platform performs targeted
NGS of cancer-related genes and allows for the detection of a
variety of somatic mutations, including clinically actionable
mutations. HemaSCAN™ analyzed 426 genes, including essential genes
for different blood diseases, as follows: Plasma cell disease [NRAS
proto-oncogene, GTPase (NRAS), KRAS and TP53], acute myeloid
leukemia [CCAAT enhancer binding protein α, fms related receptor
tyrosine kinase 3, Janus kinase 2 (JAK2), KIT proto-oncogene,
receptor tyrosine kinase, nucleophosmin 1, disease resistance
protein RUN1, TP53, isocitrate dehydrogenase (NADP(+)) 1 and
isocitrate dehydrogenase (NADP(+)) 2], acute lymphoblastic leukemia
(TP53, RB transcriptional corepressor 1, JAK2, NRAS and IKAROS
family zinc finger 1) and malignant lymphoma (MYD88, BRAF and
TP53). Additionally, the platform analyzes single nucleotide
variations (SNVs), short insertions and deletions (InDels), copy
number variations (CNVs) and gene rearrangements in 426 target
genes in clinical FFPE specimens. Furthermore, HemaSCAN™ allows
researchers and clinicians to include target genes curated from
literature by request (Fig.
S1).
Purity estimation
Tumor purity is the proportion of cancer cells in a
tumor sample and is a major confounding factor for analyzing cancer
molecular and genomic data with NGS (40). The present study estimated tumor
purity via the computational method that is derived from the
distribution of variant allelic fractions (VAFs) of somatic
single-nucleotide variants (SNVs) within copy-number neutral tumor
segments. Tumor purity estimation using a panel sequencing is a
more delicate process than using the whole genome due to the
limited DNA region in the panel sequencing which could be
insufficient for calculation of the genomic alterations (41). First, copy-neutral regions were
identified by virtual karyotypes using SNP-based arrays. The minor
allele frequencies at known single-nucleotide polymorphisms (SNPs)
in the copy-neutral regions were near 0.5. Since only these SNPs
were considered, their read densities corresponded to the most
prominent peak in the distribution of read coverage at the SNPs,
based on the hypothesis that pure polyploidy tumors with 4N in all
chromosomes are extremely rare (42). The regions of copy number gain and
loss were identified by their adjusted coverage relative to the
copy number-neutral regions. Once the copy-neutral, gain and loss
regions were delineated, the following formula was used to infer
the proportion of each tumor clone: Alternative allele frequency =
[PxY+(1-P)]/[PxX+2(1-P)] where, X and Y are the numbers of all and
alternative alleles at each group of clustered SNPs in the tumor,
respectively, and P is the proportion of tumor clones ranging
between 0 and 1. Tumor purity was inferred from the maximum value
among the Ps estimated at multiple positions, according to the
hypothesis that the largest clone best represents tumor purity
(43). Tumor purity <30% was less
reliable and was not annotated.
Variant detection
Paired-end reads were aligned to the human reference
genome (hg19 downloaded from http://genome.ucsc.edu) using BWA-MEM (ver.0.7.5)
(http://bio-bwa.sourceforge.net/).
SAMTOOLS (ver.0.1.18) (http://samtools.sourceforge.net/), GATK (ver.3.1–1)
(https://gatk.broadinstitute.org/hc/en-us)and Picard
(ver.1.93) (http://picard.sourceforge.net/) were used for file
handling, local realignment and removal of duplicate reads,
respectively. Base quality scores were recalibrated with GATK
BaseRecalibrator, using known SNPs and InDels from SNP database 138
(dbSNP138).
To increase the sensitivity of SNV detection, two
published methods, MuTect (version 1.1.4) (44) and LoFreq (ver.0.6.1) (45), were employed using default
parameters. Unions of variants identified by the two callers (with
high confidence set for MuTect) were used as the candidate set of
variants. Small InDels were identified using Pindel (ver. 0.2.4)
(46). To identify somatic CNVs,
mean read depths at each exon were calculated and normalized to the
coverage of target regions in each sample. Normalized read depth
was further standardized by dividing the expected coverage for a
normal individual (the expected coverage at each exon was taken to
be the median of the read depth at that exon across a set of normal
individuals). These steps addressed variability in capture
efficiencies and GC content at different exons. To infer the
correct copy numbers, amplitudes of copy numbers were adjusted
based on estimated purity. When the adjusted amplitude of a copy
change was >1 or <1 on a log scale, the region concerned was
defined as an amplification or a deletion, respectively.
Furthermore, most fusions involve intronic breakpoints. To identify
fusion using a gene panel, ‘hotspot’ introns that are known to
contain most breakpoints were analyzed for a set of clinically
relevant fusions.
Classification and interpretation of
variants
Variants were classified into three tiers. Tier 1
variants included variants listed as therapeutic targets by the
Korean MFDS or the US FDA, and variants reported to be candidates
in clinical trials. HemaSCAN™ panel covered all positions of tier 1
variants. Tier 2 variants included any mutation reported in COSMIC
(ver. 64) (http://cancer.sanger.ac.uk) and gene
fusions involving a target gene and a known Tier 1 partner (in
COSMIC) or a novel partner. Tier 3 variants included all other
mutations. ‘Actionable’ (that is, potentially responsive to a
targeted therapy) variants were defined as Tier 1 variants, while
‘known’ (but not actionable) variants were defined as Tier 2
variants.
Results
HemaSCAN™ panel
The HemaSCAN™ panel (Fig. S1) is a targeted NGS analysis panel
that identifies clinically relevant genomic variants in tumor DNA
and matches these variants with targeted therapies and/or clinical
implications for diagnostic/prognostic purposes. NGS analysis
revealed the following genomic variants in the five patients
(Figs. 1 and S2): SNVs (Figs.
2 and 3), short InDels (Figs. 2 and 4) and CNVs (Fig.
5). These genomic variants were also reported as annotated,
known and novel variants. Variant annotations were used to
distinguish ‘real’ variants from sequencing artifacts and to
distinguish potentially pathogenic variants from neutral variants
(47). Estimated tumor purity in
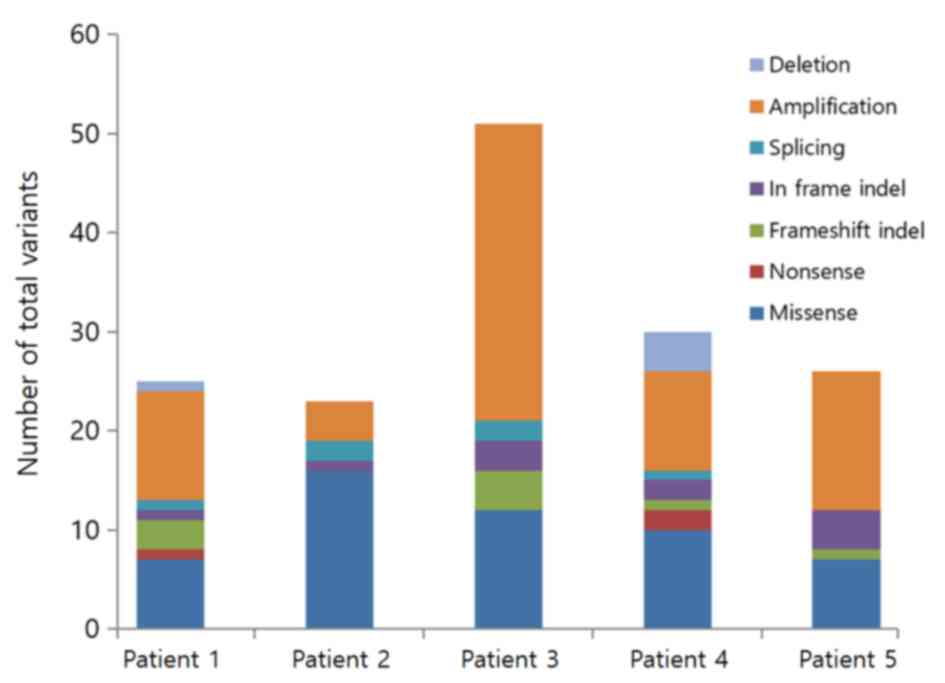
bioinformatics analysis was >90%. HemaSCAN™ analysis results
revealed 25 variants in patient 1, 23 variants in patient 2, 51
variants in patient 3, 30 variants in patient 4 and 26 variants in
patient 5 (Table II).
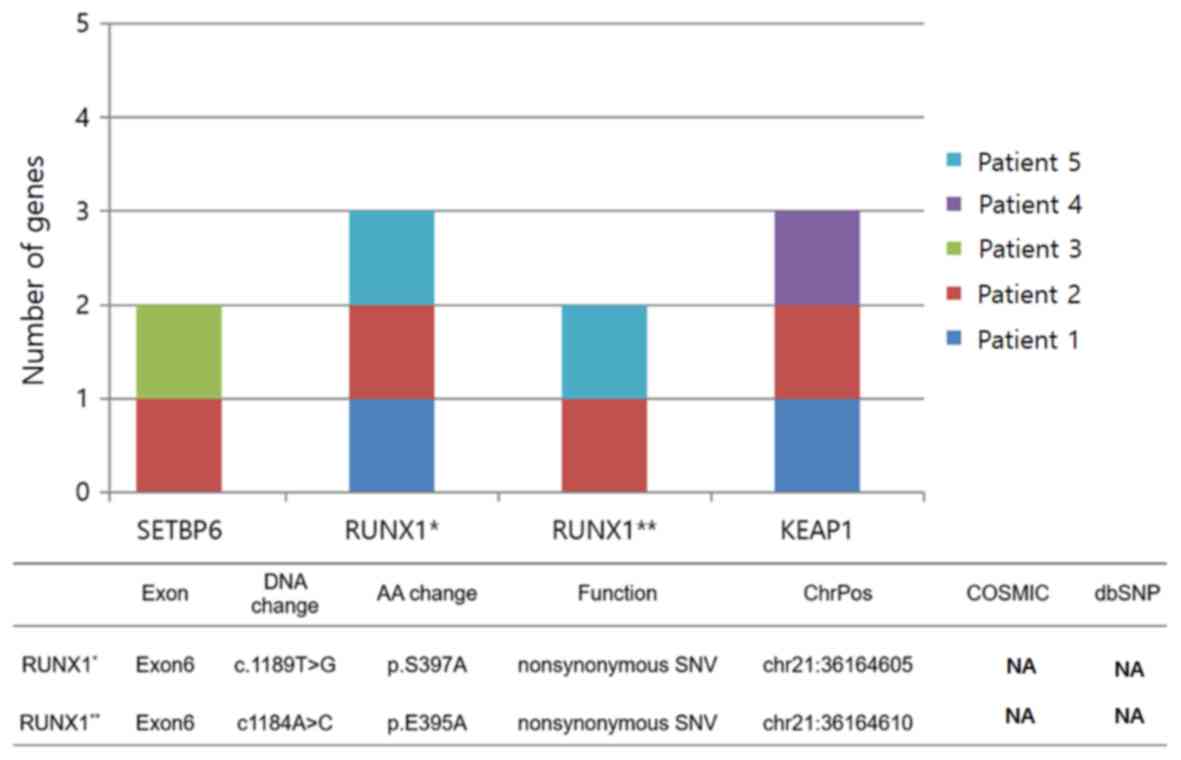 | Figure 3.Genes for which SNVs were identified
in ≥2 patients. SNVs, single nucleotide variations; RUNX1,
runt-related transcription factor 1; KEAP1, Kelch-like
ECH-associated protein 1; T, thymine; G, guanine; A, adenine; C,
cytosine; AA, amino acid; ChrPos, chromosome position; SETBP6, SET
binding protein 6; dbSNP, SNP data base; COSMIC, catalogue of
somatic mutations in cancer; NA, not applicable. |
| Table II.Sum and types of variation in the
five patients. |
Table II.
Sum and types of variation in the
five patients.
| Variation | Patient 1, n | Patient 2, n | Patient 3, n | Patient 4, n | Patient 5, n |
|---|
| SNV |
|
|
|
|
|
|
Missense | 7 | 16 | 12 | 10 | 7 |
|
Nonsense | 1 | – | – | 2 | – |
|
Splicing | 1 | 2 | 2 | 1 | – |
| InDel |
|
|
|
|
|
|
In-frame | 1 | 1 | 3 | 2 | 4 |
|
Frameshift | 3 | – | 4 | 1 | 1 |
| CNV |
|
|
|
|
|
|
Amplification | 11 | 4 | 30 | 10 | 14 |
|
Deletion | 1 | – | – | 4 | – |
| Total | 25 | 23 | 51 | 30 | 26 |
SNVs
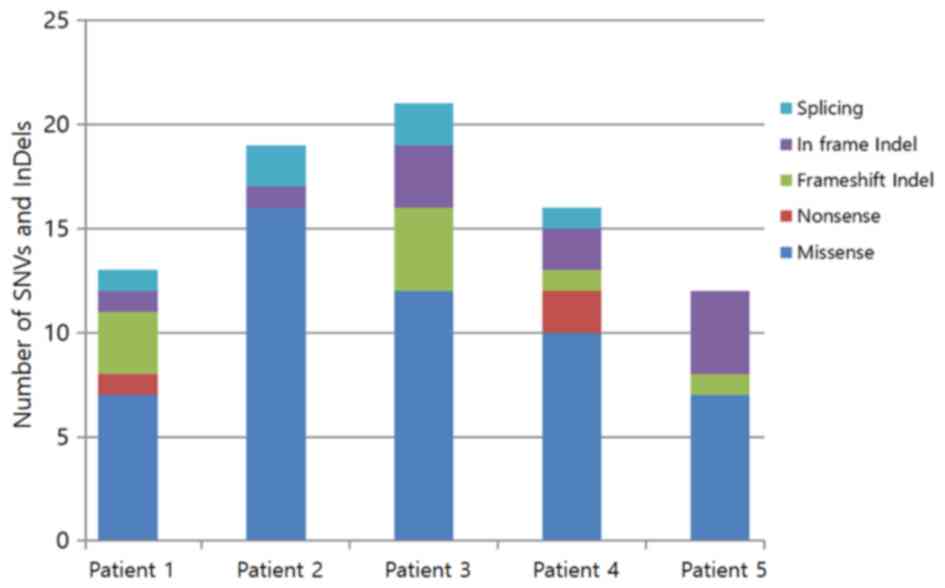
Different numbers of SNVs were identified in the
five patients: Patient 1, 9; patient 2, 18; patient 3, 14; patient
4, 13; and patient 5, 7 SNVs. There were 6 SNVs [Kelch-like
ECH-associated protein 1 (KEAP1), BLM RecQ like helicase, ETS
variant transcription factor 6 (ETV6) and heat shock protein 90α
family class A member 1] at splicing sites, 3 SNVs [CREB binding
protein (CREBBP), TNF receptor superfamily member 14 (TNFRSF14) and
RAD50 double strand break repair protein (RAD50)] that caused stop
codons and 52 nonsynonymous SNVs that altered protein amino acid
sequences. No SNV was identified in the annotated variant. Among
the known variants, the same nonsynonymous SNV was present in the
SET binding protein 6 (SETBP6) gene in patients 2 and 3 (Fig. 3). The SETBP gene is located on
chromosome 18 at 42,643,270 (https://ghr.nlm.nih.gov/gene/SETBP1#conditions). In
exon 6 of the SETBP gene, the 4,398th cDNA nucleotide (guanine) was
replaced by thymine and the amino acid at position 1,466 of the
protein was altered from glutamic acid to aspartic acid (Fig. 3). Among known SNVs, three different
SNVs in the ETV6 gene were identified in patients 2 and 3 (Fig. S2). These SNVs were located in the
same gene; however, they were at different positions in the 12th
chromosome and different exons. Additionally, nucleotide
substitutions and amino acid alterations also differed. Hence, they
were deemed as different mutations.
Nonsynonymous SNVs were identified in two different
Runt-related transcription factor 1 (RUNX1) genes (Fig. 3). One occurred in patients 1, 2 and 5
and the other in patients 2 and 5. Both were identified in exon 6,
although nucleotide changes and the resulting amino acid changes
differed. In the former, thymine at position 1,189 of cDNA was
replaced by guanine, causing a change from serine to alanine at
position 397. In the latter, adenine located at position 1,184 of
the cDNA sequence was replaced by cytosine, causing a change from
glutamic acid to alanine at position 395. SNVs in KEAP1 occurred at
the splicing site and were identified in patients 1, 2 and 4. SNVs
in CREBBP, TNFRSF1 and RAD50 caused stop codons (Fig. S2).
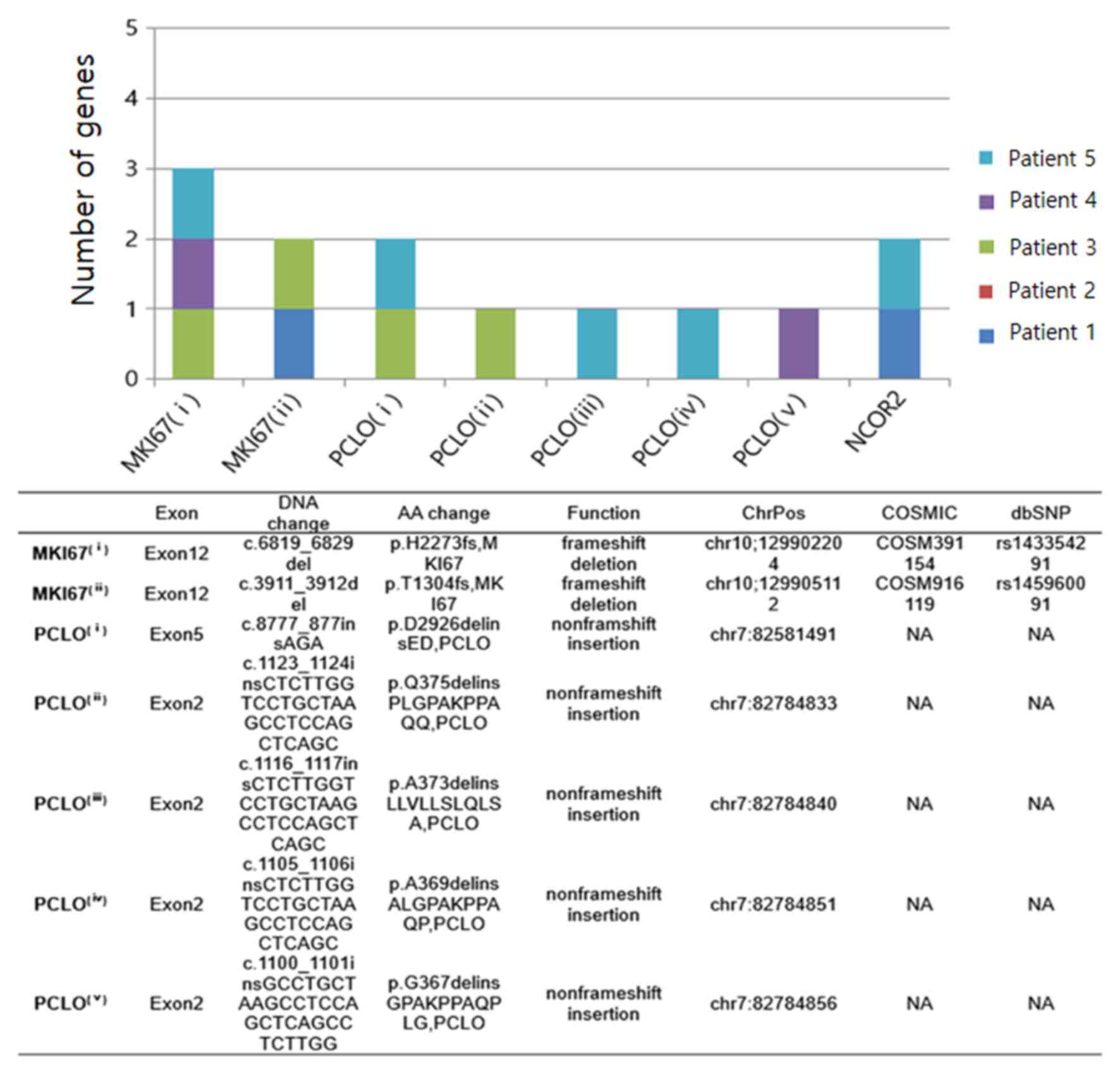
InDels
Among the three variant types, InDels exhibited the
lowest frequency and were not identified in the annotated variant.
Among the known variants, two single-base deletions were identified
in the same marker of proliferation Ki-67 (MKI67) gene; however,
these were at different chromosomal positions, resulting in a
frameshift deletion (Fig. 4). This
deletion occurred at amino acid 1,304 (threonine) of the protein in
patients 1 and 3, and at position 2,273 (histidine) in patients 3,
4 and 5. Furthermore, additional frameshift insertions involving
known variants were confirmed in the forkhead box O3, mucin 2,
oligomeric mucus/gel-forming and BCL10 genes (Fig. S2). Among novel variants, five
single-base insertions were identified in piccolo presynaptic
cytomatrix protein (PCLO) at different chromosomal positions,
resulting in non-frameshift insertions. Non-frameshift mutations
occurred at five different positions of the protein: i) At position
2,926 (aspartic acid) in patients 3 and 5; ii) at 375 (glutamine)
in patient 3; iii) at 373 (alanine) in patient 5; iv) at 369
(alanine) in patient 5; and v) at position 367 (glycin) in patient
4. Additionally, non-frameshift insertions in nuclear receptor
corepressor 2 (NCOR2) were detected in patients 1 and 5.
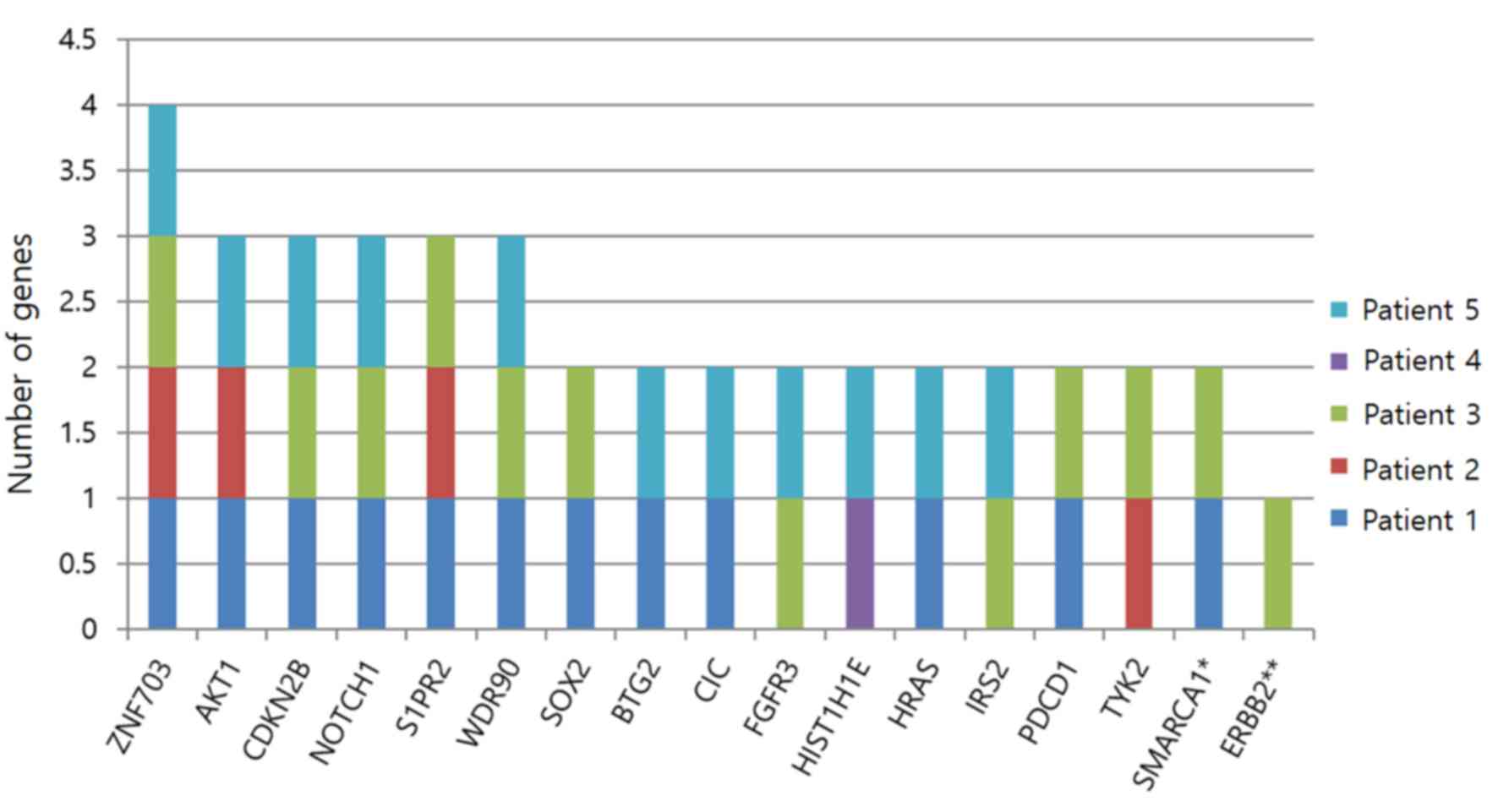
CNVs
According to the results, CNVs were the most common
variant. The largest number of CNVs was found in patient 3. Two
types of CNV were identified: Amplification and deletion.
Amplifications were encountered more frequently than deletions.
Among the 74 CNVs identified, one was an annotated variant and six
were known variants, while the others were novel variants.
Among the novel variants, a CNV in the zinc finger
protein 703 (ZNF703) gene was the most common and was identified in
patients 1, 2, 3 and 5. A total of 3 patients had CNVs in the AKT1,
cyclin dependent kinase inhibitor 2B (CDKN2B), NOTCH1,
sphingosine-1-phosphate receptor 2 and WD repeat domain 90 genes,
two patients had CNVs in SRY-box transcription factor 2 (SOX2), BTG
anti-proliferation factor 2, capicua transcriptional repressor,
fibroblast growth factor receptor 3, H1.4 linker histone, cluster
member (HIST1H1E), HRas proto-oncogene, GTPase (HRAS), insulin
receptor substrate 2, programmed cell death 1, tyrosine kinase 2
and SWI/SNF related, matrix associated, actin dependent regulator
of chromatin, subfamily a, member 1 and one patient had a CNV in
erb-b2 receptor tyrosine kinase 2 (ERBB2). CNVs were present in
oncogenes (AKT1, ERBB2, SOX2 and ZNF703), a proto-oncogene (HRAS)
and a tumor-suppressor gene (CDKN2B; Fig. 5).
Discussion
The present study performed a comprehensive analysis
using an NGS customized panel for MALT lymphoma. NGS technology has
been applied to lymphoid neoplasms to provide early insights into
the mutational landscapes of several lymphoid cancers, including
DLBCL (48), follicular lymphoma
(FL) (48), chronic lymphocytic
leukemia (CLL) (49), NMZL (50), SMZL (51), mantle cell lymphoma (MCL) (52) and peripheral T-cell lymphoma (PTCL)
(53). Although the analyzed sample
size was small, genetic mutations for rare gastrointestinal MALT
lymphoma were identified. Various mutations or genetic alterations
identified in previous studies using NGS of MALT lymphoma (54,55) were
confirmed, and several variations affecting the development of MALT
lymphoma for which the exact mechanisms have not yet been
elucidated were identified. Hyeon et al (54) analyzed 19 cases of gastric MALT
lymphoma using targeted sequencing and the majority of genes
affected by genetic alterations were involved in NF-κB pathway
activation and included MALT1 rearrangement and somatic mutations
of TNF receptor associated factor 3, TNFAIP3 and NOTCH1. Cascione
et al (55) analyzed 72 cases
of MALT lymphoma derived from different anatomic sites using a
large panel of previously known genes and reported genetic
mutations or alterations (SNVs and CNVs) that code for proteins
involved in chromatin remodeling and transcription regulation, the
breakpoint cluster region protein/NF-κB signaling pathway, immune
escape and the NOTCH singaling pathway [trisomy 3, TNFAIP3, CREBBP,
lysine methyltransferase 2C, tet methylcytosine dioxygenase 2, spen
family transcriptional repressor (SPEN), trisomy 18, lysine
methyltransferase 2D, LDL receptor related protein 1B, PR/SET
domain 1, baculoviral IAP repeat containing 3-MALT, E1A binding
protein p300 (EP300), TNFRSF14, NOTCH1/NOTCH2, β-2-microglobulin
and 6p gains, in common order]. In the present study, genetic
alteration was confirmed in NOTCH1 (80%). CNV was patients 1, 3 and
5 (Fig. 5). InDel/SNV was patient 1
and 2 (Fig. S2). And various other
genetic alterations [caspase recruitment domain family member 11,
SPEN, TNFRSF14, EP300, CREBBP, APC regulator of WNT signaling
pathway, phosphoinositide-3-kinase regulatory subunit 2, SET
binding protein 1 (SETBP1), HIST1HC, HIST1HD and HIST1H1E]
(Fig. S2) were observed.
A total of three patients exhibited an SNV in the
RUNX1 gene. This gene encodes a Runt-related transcription factor
of the RUNX gene family. The protein encoded by RUNX1 constitutes
the α subunit of core binding factor (56). RUNX1 exhibits nuclear localization
and is widely expressed in hematopoietic cells (57). It serves an essential role in the
development and maintenance of hematopoiesis (58). In adults, RUNX1 disruption by
intragenic mutation leads to a pre-leukemic state that predisposes
acute myeloid leukemia (59). In the
present study, the RUNX1 mutation was observed at exon 6.
KEAP1 was identified in three patients at its
splicing site. Oxidative stress is associated with cancer
development (60). Therefore,
oxidative stress rescuing mechanisms serve critical roles, as they
can potentially cause chemo- and radio-resistance and affect
outcomes (61). It has previously
been reported that a genetic polymorphism in the KEAP1 gene is
associated with breast cancer risk and survival outcomes (62).
In two patients, an SNV was identified in the SETBP6
gene. The SETBP1 gene provides instructions for making SET binding
protein 1 (63). However, the
function of SETBP1 protein and the effect of SET binding remain
unclear. Further studies are required to determine if this genetic
change influences disease progression.
InDels are the second most common type of human
genetic variation (64). According
to the Human Gene Mutation database (2008 update), most cases of
severe disease are due to missense mutations (44%) and small coding
InDels (23%), worldwide (65). In
the present study, the most commonly detected gene mutation among
frameshift InDels was detected for MKI67, and was a frameshift
deletion. The MKI67 gene encodes the antigen pKI-67 in humans. The
best-studied examples are carcinomas of the prostate (66), brain (67) and breast (68), as well as nephroblastoma (69) and neuroendocrine tumors (70). Furthermore, the role and significance
of pKi-67 have been studied in NHL (71). Since pKi-67 appears to be crucial for
cell proliferation, mutations in its gene may cause dysfunction in
the cell cycle and cell division to influence the development of
uterine cervix, colon and lung cancer (72). The present study did not clarify how
frameshift deletions in the pKi-67 gene affect the development or
progression of MALT lymphoma. MALT lymphoma is an indolent disease
for which a ‘watch and wait’ strategy is used (73,74).
Therefore, low or no expression of pKi-67 may be associated with
its indolent character.
Patient 1 exhibited a frameshift deletion in the
DEAD-box helicase 3 X-linked (DDX3X) gene (Fig. S2). DDX3X is hypothesized to serve
nuclear and cytoplasmic roles. Dysregulation of DDX3X has been
associated with tumorigenesis caused by loss of function (75). Furthermore, DDX3X has been identified
as a mutational cancer driver in medulloblastoma, cutaneous
melanoma and chronic lymphocytic leukemia by PAN-cancer analysis
(76–78). However, to the best of our knowledge,
the role of DDX3X in MALT lymphoma has not been elucidated and the
relationship between DDX3X mutations and disease occurrence has not
been clarified.
Among the in-frame InDels detected, non-frameshift
insertions were observed in PCLO and NCOR2. PCLO encodes the
piccolo protein in humans, which functions as part of the
presynaptic cytoskeletal matrix and is hypothesized to be involved
in the regulation of neurotransmitter release (79). Mutations in the PCLO gene in DLBCL
have been reported to be extremely common, although their role in
cancer has not been clarified (80).
NCOR2 encodes a nuclear receptor co-repressor that mediates
transcriptional silencing of CCND1, VDUP1 and GADD45A
target genes (81). Aberrant
expression of NCOR2 has been associated with acute promyelocytic
leukemia, breast, bladder and prostate cancers (82). In the present study, mutations in
PCLO and NCOR2 were found to alter their protein expression
(Fig. S2).
Chromosomal aberrations are a hallmark of cancer
cells. The term CNV indicates an intermediate-scale genetic change,
defined as segments >1,000 base pairs in length; however, they
are typically <5 megabases (83).
ZNF703 is located on human chromosome 8 in the short arm region,
commonly referred to as chromosome region 8p12 and is ubiquitously
expressed in human tissues (84). It
localizes to the nucleus under nonpathological conditions (85). However, it has been reported that
this gene is amplified and/or upregulated in breast cancer,
colorectal cancer, stomach cancer and pulmonary cancer (86–89).
ZNF703 gene amplification was observed in four cases (80%). To the
best of our knowledge, no previous studies have reported that
ZNF703 acts as an oncogene in lymphoma. Further research is
required to determine whether ZNF703 gene amplification is
associated with the progression of diseases in primary
gastrointestinal lymphoma, including MALT lymphoma.
NOTCH1 translocation-associated signaling is
associated with direct cell-cell communication, thereby controlling
cell differentiation, proliferation and apoptosis (90). Dysregulation of the NOTCH signaling
pathway has been associated with several hematologic (ALL, CLL and
MALT lymphoma) and solid malignancies (melanoma,
cholangiocarcinoma, colorectal cancer, lung adenocarcinoma, renal
cell carcinoma and prostate cancer) (91). Dysregulation of the NOTCH signaling
pathway occurs by a variety of mechanisms, including mutational
activation or inactivation, overexpression, post-translational
modification and epigenetic regulation (91). Previous studies have indicated that
NOTCH activation serves important oncogenic roles in various B-cell
malignancies (92–94). In particular, NOTCH signaling is
important for the generation of marginal zone B-cells (93,94),
indicating that in MALT lymphoma, mutated NOTCH1 activation is not
a common pathogenic mechanism (95).
However, since the NOTCH signaling pathway serves a crucial role in
MALT lymphoma, further studies on other components of the NOTCH
signaling pathway may elucidate the precise contribution to the
pathogenesis of MALT made by NOTCH signaling. Further larger scale
studies are required to confirm the effect of NOTCH1 mutations on
MALT lymphoma.
The present study had several limitations. As some
of the patients clinical data had been analyzed, there may be other
clinical correlations of genetic variation. In addition, the whole
genome of each patient was not assessed, thus, there is a
possibility that some genetic alterations, which could have
clinical significance, remain unrevealed. Notably, the sample size
was too small, thus a larger scale study is required to
comprehensively examine the clinical relevance of genetic
alternation and MALT lymphoma.
In conclusion, since few NGS studies have been
conducted on MALT lymphoma, various mutations identified in the
present study cannot be regarded as ‘actionable’ (that is,
potentially responsive to a targeted therapy) and whether these
mutations have therapeutic implications could not be confirmed.
Further research is required for the development of treatments by
genetic mutation observed in the present study.
Supplementary Material
Supporting Data
Acknowledgements
The abstract of the present study was presented at
the 44th ESMO congress 2019 in Barcelona [abstract no. 1082P;
Annals of Oncology Volume 30, Supplement 5, Pages v1-v934 (October
2019].
Funding
The present study was supported by the Dong-A
University Research Fund (2020; Seo-gu, Busan, Republic of
Korea).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors contributions
SYO, JHL and HJK conceptualized and designed the
current study and coordinated and supervised data collection. SJH,
SL and SYO drafted and revised the manuscript and analyzed data.
SJH, SHK and MKP conducted the pathological review and performed
NGS data analysis. All authors agreed to be responsible for all
aspects of the study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board of Dong-A University Hospital, Yeonje-gu, Republic of
Korea (approval no. DAUHIRB 20-088). The requirement for informed
consent was waived as the tissues had been donated to the Dong-A
University Hospital tissue bank.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CNV
|
copy number variation
|
|
CLL
|
chronic lymphocytic leukemia
|
|
dbSNP
|
The Single Nucleotide Polymorphism
database
|
|
DLBCL
|
diffuse large B cell lymphoma
|
|
EMZL
|
extranodal marginal zone lymphoma
|
|
FL
|
follicular lymphoma
|
|
InDels
|
short insertions and deletions
|
|
KEAP1
|
Kelch-like ECH-associated protein
1
|
|
MCL
|
mantle cell lymphoma
|
|
MALT
|
mucosal-associated lymphoid tissue
|
|
MZL
|
marginal zone lymphoma
|
|
NGS
|
next-generation sequencing
|
|
NMZL
|
nodal marginal zone lymphoma
|
|
PTCL
|
peripheral T-cell lymphoma
|
|
SETBP
|
SET binding protein
|
|
SMZL
|
splenic marginal zone lymphoma
|
|
SNP
|
single-nucleotide polymorphisms
|
|
SNV
|
single nucleotide variation
|
|
TNFAIP3
|
TNF-α inducible protein 3
|
|
ZNF703
|
zinc finger protein 703
|
References
|
1
|
Kahl B and Yang D: Marginal zone
lymphomas: Management of nodal, splenic, and MALT NHL. Hematology
Am Soc Hematol Educ Program. 359–364. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thieblemont C, Berger F, Dumontet C,
Moullet I, Bouafia F, Felman P, Salles G and Coiffier B:
Mucosa-associated lymphoid tissue lymphoma is a disseminated
disease in one third of 158 patients analyzed. Blood. 95:802–806.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Armitage JO: A clinical evaluation of the
International Lymphoma Study Group classification of non-Hodgkins
lymphoma. Blood. 89:3909–3918. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chan JK, Banks PM, Cleary ML, Delsol G, De
Wolf-Peeters C, Falini B, Gatter KC, Grogan TM, Harris NL, Isaacson
PG, et al: A revised European-American classification of lymphoid
neoplasms proposed by the International Lymphoma Study Group. A
summary version. Am J Clin Pathol. 103:543–560. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Isaacson PG and Du MQ: MALT lymphoma: From
morphology to molecules. Nat Rev Cancer. 4:644–653. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ferreri AJ, Govi S and Ponzoni M: Marginal
zone lymphomas and infectious agents. Semin Cancer Biol.
23:431–440. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thieblemont C, Bertoni F, Copie-Bergman C,
Ferreri AJ and Ponzoni M: Chronic inflammation and extra-nodal
marginal-zone lymphomas of MALT-type. Semin Cancer Biol. 24:33–42.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Witkowska M and Smolewski P: Helicobacter
pylori infection, chronic inflammation, and genomic transformations
in gastric MALT lymphoma. Mediators Inflamm. 2013:5231702013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kwee I, Rancoita PM, Rinaldi A, Ferreri
AJ, Bhagat G, Gascoyne RD, Canzonieri V, Gaidano G, Doglioni C,
Zucca E, et al: Genomic profiles of MALT lymphomas: Variability
across anatomical sites. Haematologica. 96:1064–1066. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Goda JS, Gospodarowicz M, Pintilie M,
Wells W, Hodgson DC, Sun A, Crump M and Tsang RW: Long-term outcome
in localized extranodal mucosa-associated lymphoid tissue lymphomas
treated with radiotherapy. Cancer. 116:3815–3824. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zucca E, Conconi A, Pedrinis E, Cortelazzo
S, Motta T, Gospodarowicz MK, Patterson BJ, Ferreri AJ, Ponzoni M,
Devizzi L, et al: Nongastric marginal zone B-cell lymphoma of
mucosa-associated lymphoid tissue. Blood. 101:2489–2495. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Du MQ: MALT lymphoma: Genetic
abnormalities, immunological stimulation and molecular mechanism.
Best Pract Res Clin Haematol. 30:13–23. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Du MQ: MALT lymphoma: A paradigm of NF-κB
dysregulation. Semin Cancer Biol. 39:49–60. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Farinha P and Gascoyne RD: Molecular
pathogenesis of mucosa-associated lymphoid tissue lymphoma. J Clin
Oncol. 23:6370–6378. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Morgan JA, Yin Y, Borowsky AD, Kuo F,
Nourmand N, Koontz JI, Reynolds C, Soreng L, Griffin CA,
Graeme-Cook F, et al: Breakpoints of the t(11; 18)(q21; q21) in
mucosa-associated lymphoid tissue (MALT) lymphoma lie within or
near the previously undescribed gene MALT1 in chromosome 18. Cancer
Res. 59:6205–6213. 1999.PubMed/NCBI
|
|
16
|
Akagi T, Motegi M, Tamura A, Suzuki R,
Hosokawa Y, Suzuki H, Ota H, Nakamura S, Morishima Y, Taniwaki M
and Seto M: A novel gene, MALT1 at 18q21, is involved in t (11;
18)(q21; q21) found in low-grade B-cell lymphoma of
mucosa-associated lymphoid tissue. Oncogene. 18:5758–5794. 1999.
View Article : Google Scholar
|
|
17
|
Dierlamm J, Baens M, Wlodarska I,
Stefanova-Ouzounova M, Hernandez JM, Hossfeld DK, De Wolf-Peeters
C, Hagemeijer A, Van den Berghe H and Marynen P: The apoptosis
inhibitor gene API2 and a novel 18q gene, MLT, are recurrently
rearranged in the t (11; 18)(q21; q21) associated with
mucosa-associated lymphoid tissue lymphomas. Blood. 93:3601–3609.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Auer I, Gascoyne R, Conners J, Cotter FE,
Greiner TC, Sanger WG and Horsman DE: t (11; 18)(q21; q21) is the
most common translocation in MALT lymphomas. Ann Oncol. 8:979–985.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Streubel B, Lamprecht A, Dierlamm J,
Cerroni L, Stolte M, Ott G, Raderer M and Chott A:
T(14;18)(q32;q21) involving IGH and MALT1 is a frequent chromosomal
aberration in MALT lymphoma. Blood. 101:2335–2339. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Streubel B, Vinatzer U, Lamprecht A,
Raderer M and Chott A: T(3;14)(p14.1;q32) involving IGH and FOXP1
is a novel recurrent chromosomal aberration in MALT lymphoma.
Leukemia. 19:652–658. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Streubel B, Simonitsch-Klupp I, Müllauer
L, Lamprecht A, Huber D, Siebert R, Stolte M, Trautinger F, Lukas
J, Püspök A, et al: Variable frequencies of MALT
lymphoma-associated genetic aberrations in MALT lymphomas of
different sites. Leukemia. 18:1722–1726. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chanudet E, Ye H, Ferry J, Bacon CM, Adam
P, Müller-Hermelink HK, Radford J, Pileri SA, Ichimura K, Collins
VP, et al: A20 deletion is associated with copy number gain at the
TNFA/B/C locus and occurs preferentially in translocation-negative
MALT lymphoma of the ocular adnexa and salivary glands. J Pathol.
217:420–430. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chanudet E, Huang Y, Ichimura K, Dong G,
Hamoudi RA, Radford J, Wotherspoon AC, Isaacson PG, Ferry J and Du
MQ: A20 is targeted by promoter methylation, deletion and
inactivating mutation in MALT lymphoma. Leukemia. 24:483–487. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Honma K, Tsuzuki S, Nakagawa M, Karnan S,
Aizawa Y, Kim WS, Kim YD, Ko YH and Seto M: TNFAIP3 is the target
gene of chromosome band 6q23. 3-q24. 1 loss in ocular adnexal
marginal zone B cell lymphoma. Genes Chromosomes Cancer. 47:1–7.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Novak U, Rinaldi A, Kwee I, Nandula SV,
Rancoita PM, Compagno M, Cerri M, Rossi D, Murty VV, Zucca E, et
al: The NF-{kappa}B negative regulator TNFAIP3 (A20) is inactivated
by somatic mutations and genomic deletions in marginal zone
lymphomas. Blood. 113:4918–4921. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Martinez-Lopez A, Curiel-Olmo S, Mollejo
M, Cereceda L, Martinez N, Montes-Moreno S, Almaraz C, Revert JB
and Piris MA: MYD88 (L265P) somatic mutation in marginal zone
B-cell lymphoma. Am J Surg Pathol. 39:644–651. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ngo VN, Young RM, Schmitz R, Jhavar S,
Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, et al:
Oncogenically active MYD88 mutations in human lymphoma. Nature.
470:115–119. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li ZM, Rinaldi A, Cavalli A, Mensah AA,
Ponzoni M, Gascoyne RD, Bhagat G, Zucca E and Bertoni F: MYD88
somatic mutations in MALT lymphomas. Br J Haematol. 158:662–664.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao
Y, Sheehy P, Manning RJ, Patterson CJ, Tripsas C, et al: MYD88
L265P somatic mutation in Waldenströms macroglobulinemia. N Engl J
Med. 367:826–833. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu L, Hunter ZR, Yang G, Zhou Y, Cao Y,
Liu X, Morra E, Trojani A, Greco A, Arcaini L, et al: MYD88 L265P
in Waldenström macroglobulinemia, immunoglobulin M monoclonal
gammopathy, and other B-cell lymphoproliferative disorders using
conventional and quantitative allele-specific polymerase chain
reaction. Blood. 121:2051–2058. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Oh SY, Kwon HC, Kim WS, Park YH, Kim K,
Kim HJ, Kwon JM, Lee J, Ko YH, Ahn YC, et al: Nongastric marginal
zone B-cell lymphoma: A prognostic model from a retrospective
multicenter study. Cancer Lett. 258:90–97. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Olszewski AJ and Castillo JJ: Survival of
patients with marginal zone lymphoma. Cancer. 119:629–638. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cogliatti SB, Schmid U, Schumacher U,
Eckert F, Hansmann ML, Hedderich J, Takahashi H and Lennert K:
Primary B-cell gastric lymphoma: A clinicopathological study of 145
patients. Gastroenterology. 101:1159–1170. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fischbach W: Gastric MALT lymphoma-update
on diagnosis and treatment. Best Pract Res Clin Gastroenterol.
28:1069–1077. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Alderuccio JP, Zhao W, Desai A, Ramdial J,
Gallastegui N, Kimble E, de la Fuente MI, Husnain M, Rosenblatt JD,
Alencar AJ, et al: Short survival and frequent transformation in
extranodal marginal zone lymphoma with multiple mucosal sites
presentation. Am J Hematol. 94:585–596. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cheson BD, Fisher RI, Barrington SF,
Cavalli F, Schwartz LH, Zucca E, Lister TA; Alliance Australasian
Leukaemia and Lymphoma Group and Eastern Cooperative Oncology
Group; European Mantle Cell Lymphoma Consortium; Italian Lymphoma
Foundation, ; et al: Recommendations for initial evaluation,
staging, and response assessment of Hodgkin and non-Hodgkin
lymphoma: The Lugano classification. J Clin Oncol. 32:3059–3068.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
International Non-Hodgkins Lymphoma
Prognostic Factors Project, . A predictive model for aggressive
non-Hodgkins lymphoma. N Engl J Med. 329:987–994. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shin HT, Choi YL, Yun JW, Kim NKD, Kim SY,
Jeon HJ, Nam JY, Lee C, Ryu D, Kim SC, et al: Prevalence and
detection of low-allele-fraction variants in clinical cancer
samples. Nat Commun. 8:13772017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Illumina, . Sample Multiplexing Overview.
https://www.illumina.com/techniques/sequencing/ngs-library-prep/multiplexing.htmlAugust
26–2020
|
|
40
|
Locallo A, Prandi D, Fedrizzi T and
Demichelis F: TPES: Tumor purity estimation from SNVs.
Bioinformatics. 35:4433–4435. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Oh BY, Shin HT, Yun JW, Kim KT, Kim J, Bae
JS, Cho YB, Lee WY, Yun SH, Park YA, et al: Intratumor
heterogeneity inferred from targeted deep sequencing as a
prognostic indicator. Sci Rep. 9:45422019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Coward J and Harding A: Size does matter:
Why polyploid tumor cells are critical drug targets in the war on
cancer. Front Oncol. 4:1232014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Andor N, Graham TA, Jansen M, Xia LC,
Aktipis CA, Petritsch C, Ji HP and Maley CC: Pan-cancer analysis of
the extent and consequences of intratumor heterogeneity. Nat Med.
22:105–113. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cibulskis K, Lawrence MS, Carter SL,
Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES
and Getz G: Sensitive detection of somatic point mutations in
impure and heterogeneous cancer samples. Nat Biotechnol.
31:213–219. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wilm A, Aw PP, Bertrand D, Yeo GH, Ong SH,
Wong CH, Khor CC, Petric R, Hibberd ML and Nagarajan N: LoFreq: A
sequence-quality aware, ultra-sensitive variant caller for
uncovering cell-population heterogeneity from high-throughput
sequencing datasets. Nucleic Acids Res. 40:11189–11201. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ye H, Gong L, Liu H, Hamoudi RA, Shirali
S, Ho L, Chott A, Streubel B, Siebert R, Gesk S, et al: MALT
lymphoma with t(14;18)(q32;q21)/IGH-MALT1 is characterized by
strong cytoplasmic MALT1 and BCL10 expression. J Pathol.
205:293–301. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Oetting WS, Brookes AJ, Béroud C and
Taschner PE: Clinical interpretation of variants from
next-generation sequencing: The 2016 scientific meeting of the
human genome variation society. Hum Mutat. 37:1110–1113. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Morin RD, Johnson NA, Severson TM, Mungall
AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et
al: Somatic mutations altering EZH2 (Tyr641) in follicular and
diffuse large B-cell lymphomas of germinal-center origin. Nat
Genet. 42:181–185. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
49
|
Puente XS, Pinyol M, Quesada V, Conde L,
Ordóñez GR, Villamor N, Escaramis G, Jares P, Beà S, González-Díaz
M, Bassaganyas L, et al: Whole-genome sequencing identifies
recurrent mutations in chronic lymphocytic leukaemia. Nature.
475:101–105. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Spina V, Khiabanian H, Messina M, Monti S,
Cascione L, Bruscaggin A, Spaccarotella E, Holmes AB, Arcaini L,
Lucioni M, et al: The genetics of nodal marginal zone lymphoma.
Blood. 128:1392–1373. 2016. View Article : Google Scholar
|
|
51
|
Kiel MJ, Velusamy T, Betz BL, Zhao L,
Weigelin HG, Chiang MY, Huebner-Chan DR, Bailey NG, Yang DT, Bhagat
G, et al: Whole-genome sequencing identifies recurrent somatic
NOTCH2 mutations in splenic marginal zone lymphoma. J Exp Med.
209:1553–1565. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kridel R, Meissner B, Rogic S, Boyle M,
Telenius A, Woolcock B, Gunawardana J, Jenkins C, Cochrane C,
Ben-Neriah S, et al: Whole transcriptome sequencing reveals
recurrent NOTCH1 mutations in mantle cell lymphoma. Blood.
119:1963–1971. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Feldman AL, Dogan A, Smith DI, Law ME,
Ansell SM, Johnson SH, Porcher JC, Ozsan N, Wieben ED, Eckloff BW
and Vasmatzis G: Discovery of recurrent t (6; 7)(p25. 3; q32. 3)
translocations in ALK-negative anaplastic large cell lymphomas by
massively parallel genomic sequencing. Blood. 117:915–919. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hyeon J, Lee B, Shin SH, Yoo HY, Kim SJ,
Kim WS, Park WY and Ko YH: Targeted deep sequencing of gastric
marginal zone lymphoma identified alterations of TRAF3 and TNFAIP3
that were mutually exclusive for MALT1 rearrangement. Mod Pathol.
31:1418–1428. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Cascione L, Rinaldi A, Bruscaggin A,
Tarantelli C, Arribas AJ, Kwee I, Pecciarini L, Mensah AA, Spina V,
Chung EYL, et al: Novel insights into the genetics and epigenetics
of MALT lymphoma unveiled by next generation sequencing analyses.
Haematologica. 104:e558–e561. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Bonifer C, Levantini E, Kouskoff V and
Lacaud G: Runx1 Structure and Function in Blood Cell Development.
Adv Exp Med Biol. 962:65–81. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Maki K, Yamagata T and Mitani K: Role of
the RUNX1-EVI1 fusion gene in leukemogenesis. Cancer Sci.
99:1878–1883. 2008.PubMed/NCBI
|
|
58
|
Gaidzik VI, Teleanu V, Papaemmanuil E,
Weber D, Paschka P, Hahn J, Wallrabenstein T, Kolbinger B, Köhne
CH, Horst HA, et al: RUNX1 mutations in acute myeloid leukemia are
associated with distinct clinico-pathologic and genetic features.
Leukemia. 30:22822016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ichikawa M, Yoshimi A, Nakagawa M,
Nishimoto N, Watanabe-Okochi N and Kurokawa M: A role for RUNX1 in
hematopoiesis and myeloid leukemia. Int J Hematol. 97:726–734.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Klaunig JE: Oxidative stress and cancer.
Curr Pharm Des. 24:4771–4778. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Reuter S, Gupta SC, Chaturvedi MM and
Aggarwal BB: Oxidative stress, inflammation, and cancer: How are
they linked? Free Radic Biol Med. 49:1603–1616. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hartikainen JM, Tengström M, Winqvist R,
Jukkola-Vuorinen A, Pylkäs K, Kosma VM, Soini Y and Mannermaa A:
KEAP1 genetic polymorphisms associate with breast cancer risk and
survival outcomes. Clin Cancer Res. 21:1591–1601. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Piazza R, Magistroni V, Redaelli S, Mauri
M, Massimino L, Sessa A, Peronaci M, Lalowski M, Soliymani R,
Mezzatesta C, et al: SETBP1 induces transcription of a network of
development genes by acting as an epigenetic hub. Nat Commun.
9:21922018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Montgomery SB, Goode DL, Kvikstad E,
Albers CA, Zhang ZD, Mu XJ, Ananda G, Howie B, Karczewski KJ, Smith
K, et al: The origin, evolution, and functional impact of short
insertion-deletion variants identified in 179 human genomes. Genome
Res. 23:749–761. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Stenson PD, Mort M, Ball EV, Howells K,
Phillips AD, Thomas NS and Cooper DN: The human gene mutation
database: 2008 update. Genome Med. 1:132009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Fisher G, Yang ZH, Kudahetti S, Møller H,
Scardino P, Cuzick J and Berney DM; Transatlantic Prostate Group, :
Prognostic value of Ki-67 for prostate cancer death in a
conservatively managed cohort. Br J Cancer. 108:271–277. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Niemiec J: Ki-67 labelling index in human
brain tumours. Folia Histochem Cytobiol. 39:259–262.
2001.PubMed/NCBI
|
|
68
|
Inwald EC, Klinkhammer-Schalke M,
Hofstädter F, Zeman F, Koller M, Gerstenhauer M and Ortmann O:
Ki-67 is a prognostic parameter in breast cancer patients: Results
of a large population-based cohort of a cancer registry. Breast
Cancer Res Treat. 139:539–552. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Jurić I, Pogorelić Z, Kuzmić-Prusac I,
Biocić M, Jakovljević G, Stepan J, Zupancić B, Culić S and Kruslin
B: Expression and prognostic value of the Ki-67 in Wilms tumor:
Experience with 48 cases. Pediatr Surg Int. 26:487–493. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Nadler A, Cukier M, Rowsell C, Kamali S,
Feinberg Y, Singh S and Law CH: Ki-67 is a reliable pathological
grading marker for neuroendocrine tumors. Virchows Arch.
462:501–505. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Broyde A, Boycov O, Strenov Y, Okon E,
Shpilberg O and Bairey O: Role and prognostic significance of the
Ki-67 index in non-Hodgkins lymphoma. Am J Hematol. 84:338–343.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Bubán T, Schmidt M, Broll R, Antal-Szalmás
P and Duchrow M: Detection of mutations in the cDNA of the
proliferation marker Ki-67 protein in four tumor cell lines. Cancer
Genet Cytogenet. 149:81–84. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Oh SY, Ryoo BY, Kim WS, Park YH, Kim K,
Kim HJ, Kwon JM, Lee J, Ko YH, Ahn YC, et al: Nongastric marginal
zone B-cell lymphoma: Analysis of 247 cases. Am J Hematol.
82:446–452. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Thieblemont C: Clinical presentation and
management of marginal zone lymphomas. Hematology Am Soc Hematol
Educ Program. 307–313. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Riva V and Maga G: From the magic bullet
to the magic target: Exploiting the diverse roles of DDX3X in viral
infections and tumorigenesis. Future Med Chem. 11:1357–1381. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Ojha J, Secreto CR, Rabe KG, Van Dyke DL,
Kortum KM, Slager SL, Shanafelt TD, Fonseca R, Kay NE and Braggio
E: Identification of recurrent truncated DDX3X mutations in chronic
lymphocytic leukaemia. Br J Haematol. 169:445–448. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Phung B, Cieśla M, Sanna A, Guzzi N,
Beneventi G, Cao Thi Ngoc P, Lauss M, Cabrita R, Cordero E, Bosch
A, et al: The X-Linked DDX3X RNA helicase dictates translation
reprogramming and metastasis in melanoma. Cell Rep. 27:3573–3586
e3577. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Patmore DM, Jassim A, Nathan E, Gilbertson
RJ, Tahan D, Hoffmann N, Tong Y, Smith KS, Kanneganti TD, Suzuki H,
et al: DDX3X suppresses the susceptibility of hindbrain lineages to
medulloblastoma. Dev Cell. S1534-5807(20)30416-0. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Fenster SD and Garner CC: Gene structure
and genetic localization of the PCLO gene encoding the presynaptic
active zone protein Piccolo. Int J Dev Neurosci. 20:161–171. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Lohr JG, Stojanov P, Lawrence MS, Auclair
D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW,
Slager SL, et al: Discovery and prioritization of somatic mutations
in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing.
Proc Natl Acad Sci USA. 109:3879–3884. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Chen JD and Evans RM: A transcriptional
co-repressor that interacts with nuclear hormone receptors. Nature.
377:454–457. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Battaglia S, Maguire O and Campbell MJ:
Transcription factor co-repressors in cancer biology: Roles and
targeting. Int J Cancer. 126:2511–2519. 2010.PubMed/NCBI
|
|
83
|
Rosenfeld JA, Coe BP, Eichler EE, Cuckle H
and Shaffer L: Estimates of penetrance for recurrent pathogenic
copy-number variations. Genet Med. 15:478–481. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Nakamura M, Choe SK, Runko AP, Gardner PD
and Sagerström C: Nlz1/Znf703 acts as a repressor of transcription.
BMC Dev Biol. 8:1082008. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Pereira-Castro I, Costa AM, Oliveira MJ,
Barbosa I, Rocha AS, Azevedo L and da Costa LT: Characterization of
human NLZ1/ZNF703 identifies conserved domains essential for proper
subcellular localization and transcriptional repression. J Cell
Biochem. 114:120–133. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Reynisdottir I, Arason A, Einarsdottir BO,
Gunnarsson H, Staaf J, Vallon-Christersson J, Jonsson G, Ringnér M,
Agnarsson BA, Olafsdottir K, et al: High expression of ZNF703
independent of amplification indicates worse prognosis in patients
with luminal B breast cancer. Cancer Med. 2:437–446. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Yang G, Ma F, Zhong M, Fang L, Peng Y, Xin
X, Zhong J, Yuan F, Gu H, Zhu W and Zhang Y: ZNF703 acts as an
oncogene that promotes progression in gastric cancer. Oncol Rep.
31:1877–1882. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Ma F, Bi L, Yang G, Zhang M, Liu C, Zhao
Y, Wang Y, Wang J, Bai Y and Zhang Y: ZNF703 promotes tumor cell
proliferation and invasion and predicts poor prognosis in patients
with colorectal cancer. Oncol Rep. 32:1071–1077. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Baykara O, Dalay N, Kaynak K and Buyru N:
ZNF703 Overexpression may act as an oncogene in non-small cell lung
cancer. Cancer Med. 5:2873–2878. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Previs RA, Coleman RL, Harris AL and Sood
AK: Molecular pathways: Translational and therapeutic implications
of the Notch signaling pathway in cancer. Clin Cancer Res.
21:955–961. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Ntziachristos P, Lim JS, Sage J and
Aifantis I: From fly wings to targeted cancer therapies: A
centennial for notch signaling. Cancer Cell. 25:318–334. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Mirandola L, Comi P, Cobos E, Kast WM,
Chiriva-Internati M and Chiaramonte R: Notchoing from T-cell to
B-cell lymphoid malignancies. Cancer Lett. 308:1–13. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Hozumi K, Negishi N, Suzuki D, Abe N,
Sotomaru Y, Tamaoki N, Mailhos C, Ish-Horowicz D, Habu S and Owen
MJ: Delta-like 1 is necessary for the generation of marginal zone B
cells but not T cells in vivo. Nat Immunol. 5:638–644. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Santos MA, Sarmento LM, Rebelo M, Doce AA,
Maillard I, Dumortier A, Neves H, Radtke F, Pear WS, Parreira L and
Demengeot J: Notch1 engagement by Delta-like-1 promotes
differentiation of B lymphocytes to antibody-secreting cells. Proc
Natl Acad Sci USA. 104:15454–15459. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Mensah AA, Rinaldi A, Ponzoni M,
Canzonieri V, Uccella S, Rossi D, Bhagat G, Gaidano G, Zucca E and
Bertoni F: Absence of NOTCH1 gene mutations in MALT lymphomas. Br J
Haematol. 157:382–384. 2012. View Article : Google Scholar : PubMed/NCBI
|