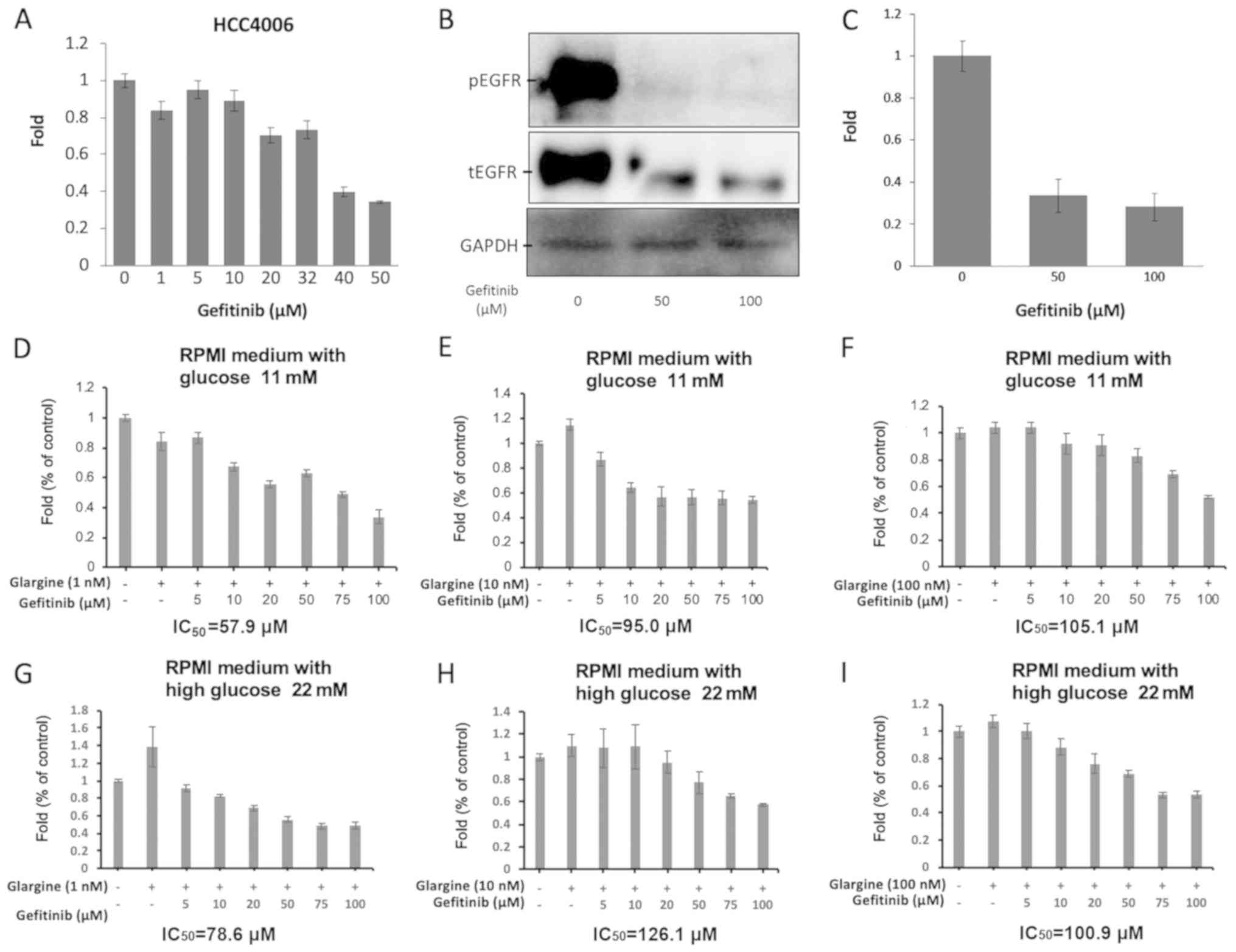
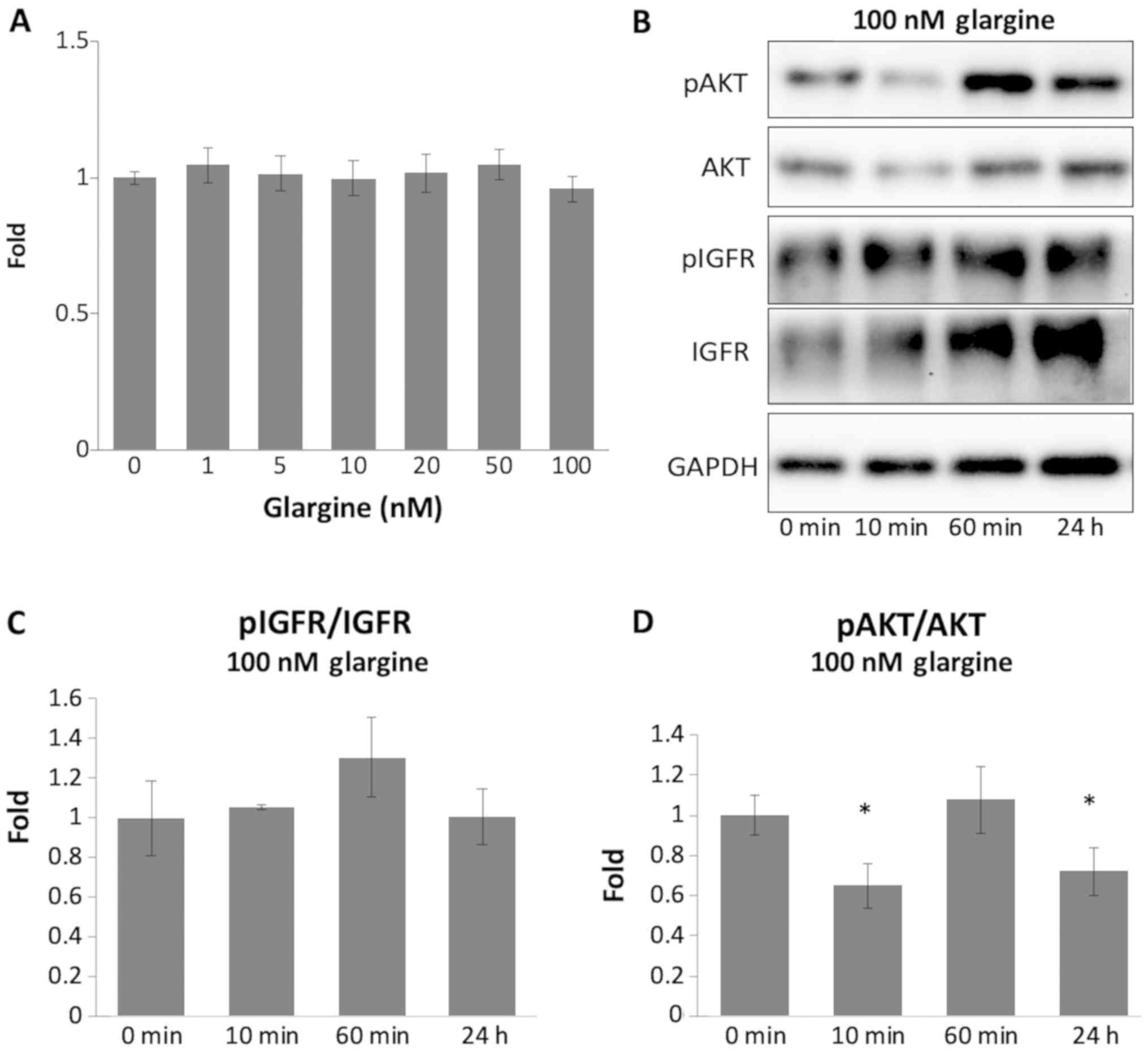
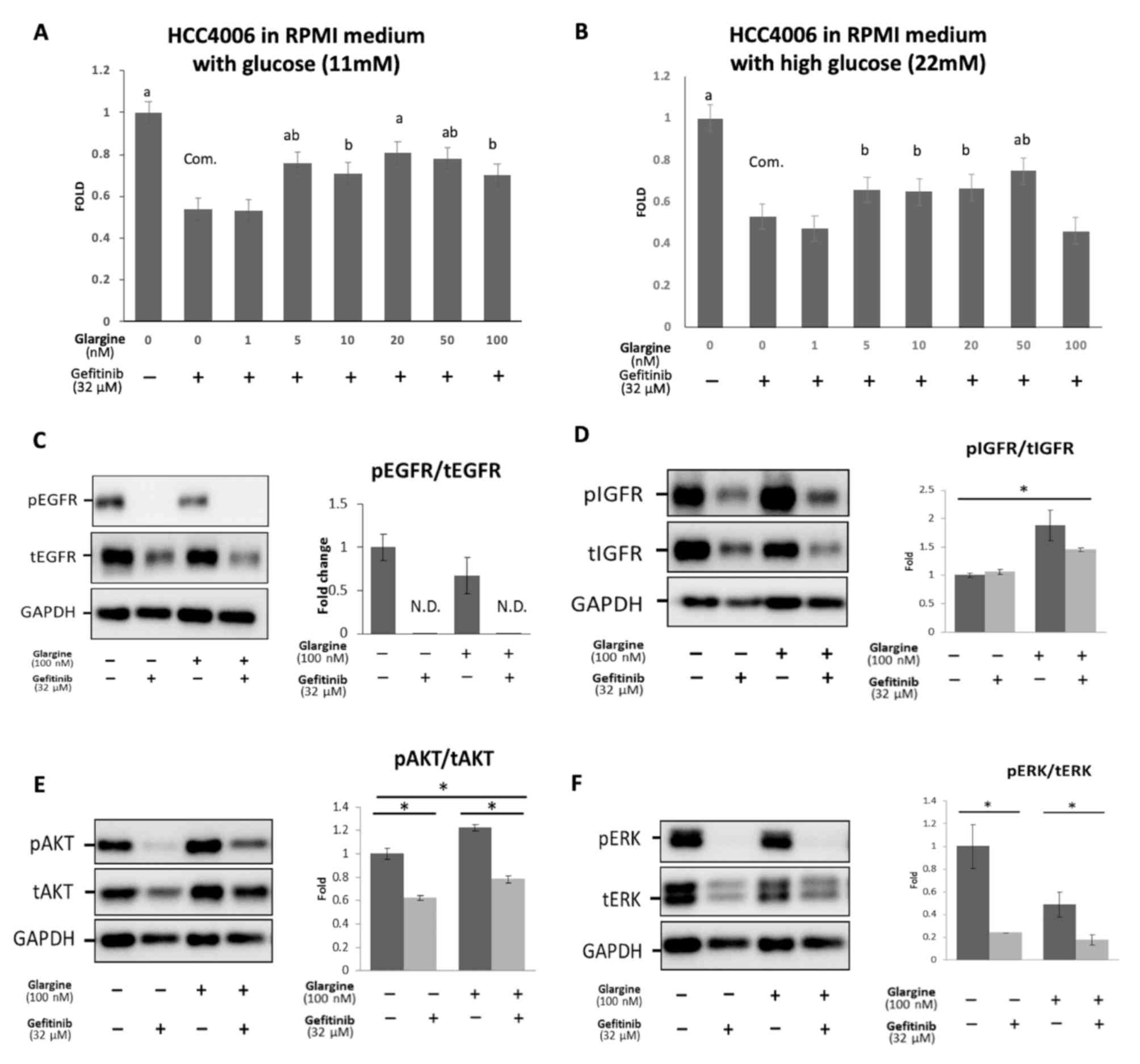
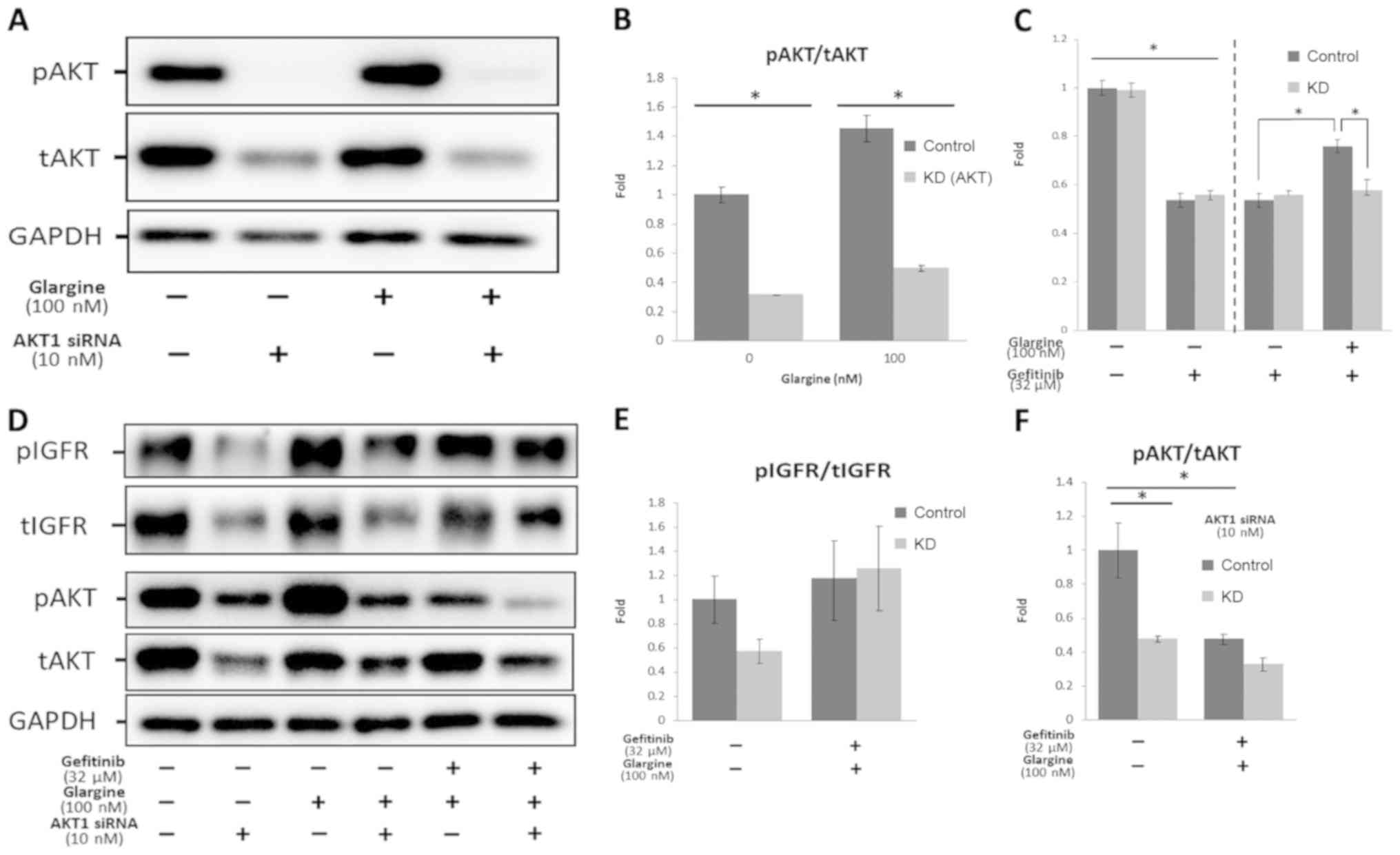
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mok TS, Wu YL, Thongprasert S, Yang CH,
Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et
al: Gefitinib or carboplatin-paclitaxel in pulmonary
adenocarcinoma. N Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Maemondo M, Inoue A, Kobayashi K, Sugawara
S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I,
et al: Gefitinib or chemotherapy for non-small-cell lung cancer
with mutated EGFR. N Engl J Med. 362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rosell R, Moran T, Queralt C, Porta R,
Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M,
et al: Screening for epidermal growth factor receptor mutations in
lung cancer. N Engl J Med. 361:958–967. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tamura K, Okamoto I, Kashii T, Negoro S,
Hirashima T, Kudoh S, Ichinose Y, Ebi N, Shibata K, Nishimura T, et
al: Multicentre prospective phase II trial of gefitinib for
advanced non small cell lung cancer with epidermal growth factor
receptor mutations: results of the West Japan Thoracic Oncology
Group trial (WJTOG0403). Br J Cancer. 98:907–914. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, et al: Erlotinib versus
chemotherapy as first-line treatment for patients with advanced
EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG
0802): a multicentre, open-label, randomised, phase 3 study. Lancet
Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Richardson LC and Pollack LA: Therapy
insight: Influence of type 2 diabetes on the development, treatment
and outcomes of cancer. Nat Clin Pract Oncol. 2:48–53. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Barone BB, Yeh HC, Snyder CF, Peairs KS,
Stein KB, Derr RL, Wolff AC and Brancati FL: Long-term all-cause
mortality in cancer patients with preexisting diabetes mellitus: a
systematic review and meta analysis. Jama. 300:2754–2764. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu L, Cao H, Zhang T, Shen H, Dong W,
Wang L and Du J: The effect of diabetes mellitus on lung cancer
prognosis: a PRISMA-compliant meta-analysis of cohort studies.
Medicine (Baltimore). 95:e35282016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yeo CD, Park KH, Park CK, Lee SH, Kim SJ,
Yoon HK, Lee YS, Lee EJ, Lee KY and Kim TJ: Expression of
insulin-like growth factor 1 receptor (IGF-1R) predicts poor
responses to epidermal growth factor receptor (EGFR) tyrosine
kinase inhibitors in non-small cell lung cancer patients harboring
activating EGFR mutations. Lung Cancer. 87:311–317. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ramalingam SS, Spigel DR, Chen D, Steins
MB, Engelman JA, Schneider CP, Novello S, Eberhardt WE, Crino L,
Habben K, et al: Randomized phase II study of erlotinib in
combination with placebo or R1507, a monoclonal antibody to
insulin-like growth factor-1 receptor, for advanced stage
non-small-cell lung cancer. J Clin Oncol. 29:4574–4580. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang H, Pelzer AM, Kiang DT and Yee D:
Down-regulation of type I insulin-like growth factor receptor
increases sensitivity of breast cancer cells to insulin. Cancer
Res. 67:391–397. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hemkens LG, Grouven U, Bender R, Günster
C, Gutschmidt S, Selke GW and Sawicki PT: Risk of malignancies in
patients with diabetes treated with human insulin or insulin
analogues: a cohort study. Diabetologia. 52:1732–1744. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baglia ML, Cui Y, Zheng T, Yang G, Li H,
You M, Xu L, Murff H, Gao YT, Zheng W, et al: Diabetes medication
use in association with survival among patients of breast,
colorectal, lung, or gastric cancer. Cancer Res Treat. 51:538–546.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Iams WT and Lovly CM: Molecular pathways:
clinical applications and future direction of insulin-like growth
factor-1 receptor pathway blockade. Clin Cancer Res. 21:4270–4277.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Peled N, Wynes MW, Ikeda N, Ohira T,
Yoshida K, Qian J, Ilouze M, Brenner R, Kato Y, Mascaux C and
Hirsch FR: Insulin-like growth factor-1 receptor (IGF-1R) as a
biomarker for resistance to the tyrosine kinase inhibitor gefitinib
in non-small cell lung cancer. Cell Oncol (Dordr). 36:277–288.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dobashi Y, Suzuki S, Kimura M, Matsubara
H, Tsubochi H, Imoto I and Ooi A: Paradigm of kinase-driven pathway
downstream of epidermal growth factor receptor/Akt in human lung
carcinomas. Hum Pathol. 42:214–226. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oliveira S, Schiffelers R, Storm G,
Henegouwen P and Roovers R: Crosstalk between epidermal growth
factor receptor- and insulin-like growth factor-1 receptor
signaling: implications for cancer therapy. Curr Cancer Drug
Targets. 9:748–760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Osaki M, Oshimura Ma and Ito H: PI3K-Akt
pathway: its functions and alterations in human cancer. Apoptosis.
9:667–676. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Luo J, Manning BD and Cantley LC:
Targeting the PI3K-Akt pathway in human cancer: rationale and
promise. Cancer Cell. 4:257–262. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hennessy BT, Smith DL, Ram PT, Lu Y and
Mills GB: Exploiting the PI3K/AKT pathway for cancer drug
discovery. Nat Rev Drug Discov. 4:988–1004. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Burris HA 3rd: Overcoming acquired
resistance to anticancer therapy: focus on the PI3K/AKT/mTOR
pathway. Cancer Chemother Pharmacol. 71:829–842. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Papadimitrakopoulou V: Development of
PI3K/AKT/mTOR pathway inhibitors and their application in
personalized therapy for non-small-cell lung cancer. J Thorac
Oncol. 7:1315–1326. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gadgeel SM and Wozniak A: Preclinical
rationale for PI3K/Akt/mTOR pathway inhibitors as therapy for
epidermal growth factor receptor inhibitor-resistant non-small-cell
lung cancer. Clin Lung Cancer. 14:322–332. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yamasaki F, Johansen MJ, Zhang D,
Krishnamurthy S, Felix E, Bartholomeusz C, Aguilar RJ, Kurisu K,
Mills GB, Hortobagyi GN and Ueno NT: Acquired resistance to
erlotinib in A-431 epidermoid cancer cells requires down-regulation
of MMAC1/PTEN and up-regulation of phosphorylated Akt. Cancer Res.
67:5779–5788. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Morgillo F, Kim WY, Kim ES, Ciardiello F,
Hong WK and Lee HY: Implication of the insulin-like growth
factor-IR pathway in the resistance of non-small cell lung cancer
cells to treatment with gefitinib. Clin Cancer Res. 13:2795–2803.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim JG, Kang MJ, Yoon YK, Kim HP, Park J,
Song SH, Han SW, Park JW, Kang GH, Kang KW, et al:
Heterodimerization of glycosylated insulin-like growth factor-1
receptors and insulin receptors in cancer cells sensitive to
anti-IGF1R antibody. PLoS One. 7:e333222012. View Article : Google Scholar : PubMed/NCBI
|