Introduction
Lung cancer is the leading cause of cancer
associated mortality worldwide. The prognosis of patients with lung
cancer is poor, and the overall 5-year survival rate is only 16%
(1). One of the most crucial recent
advances in the treatment of non-small cell lung cancer (NSCLC) has
been the development of therapies targeting the epidermal growth
factor receptor (EGFR). Tumors with activating EGFR mutations have
been reported to be particularly sensitive to EGFR-tyrosine kinase
inhibitors (TKIs) (2). EGFR-specific
TKIs have been reported to be associated with improved
progression-free survival rates in patients with EGFR
mutation-positive NSCLC compared with standard chemotherapies
(3–5). However, acquired resistance to
EGFR-specific TKIs may occur within 9–13 months (6); therefore, the mechanisms underlying
acquired resistance warrant further investigation.
Patients with cancer who also have pre-existing
diabetes mellitus (DM) exhibit higher mortality and recurrence
rates following EGFR-TKI treatment compared with those without DM
(7–9). DM is characterized by hyperglycemia,
hyperinsulinemia, or insulin resistance. Additionally, a previous
study reported that patients with lung cancer who harbored
activating EGFR mutations and who had pre-existing DM, in
conjunction with insulin-like growth factor 1 receptor (IGFR)
overexpression, exhibited poor responses to first-line EGFR-TKI
therapy (10). Furthermore,
Ramalingam et al (11)
demonstrated that cross-talk between IGFR and EGFR fulfills a
critical role in tumorigenesis and in developing resistance to
EGFR-TKIs in patients with NSCLC.
Insulin is able to activate insulin receptors and
IGFR since they are structurally and functionally related
heterotetrametric receptors that exhibit cross-talk (12). Exogenous insulin has been identified
to mediate oncogenic effects. Hemkens et al (13) demonstrated that the insulin dose and
long-acting insulin, for example glargine, are associated with a
high risk of malignancy. Baglia et al (14) reported that insulin use may be
associated with poor survival rates among patients with lung,
breast, colorectal, or gastric cancer. However, to date, only a
limited number of studies have been published on the effects of
insulin on EGFR-TKI resistance in NSCLC.
In the present study, whether exogenous insulin
mediates EGFR-TKI resistance in a selected NSCLC cell line
harboring activating EGFR mutations, for example the HCC4006 cell
line, was investigated, and the possible mechanisms underpinning
these actions were elucidated. In addition, whether AKT inhibition
is able to overcome EGFR-TKI resistance induced by long-acting
insulin was also explored.
Materials and methods
Cell lines and cell cultures
The human NSCLC HCC4006 cell line, harboring the
EGFR L747-E749 in-frame deletion in exon 19, was obtained from the
American Type Culture Collection. The cells were cultured in
Roswell Park Memorial Institute (RPMI)-1640 medium supplemented
with 10% FBS, 1% penicillin-streptomycin, HEPES, and 1.5 g/l sodium
bicarbonate under an atmosphere of 5% CO2 at 37°C.
Reagents
Gefitinib (ZD 1839) was purchased from
Sigma-Aldrich; Merck KGaA, and a 25 mM stock solution was prepared
in dimethyl sulfoxide. Insulin glargine is a long-acting basal
insulin analog. Glargine was purchased in its commercially packed
form Sanofi S.A. at a concentration of 100 IU/ml, and a 600 µM
stock solution was prepared in phosphate-buffered saline (PBS).
Antibodies against phosphorylated (p)EGFR, total (t)EGFR, pIGFR,
tIGFR, pAKT, tAKT, pERK, tERK, GAPDH and β-actin were used in the
present study. The antibodies were diluted with 10% bovine serum
albumin (BSA; Gibco; Thermo Fisher Scientific, Inc.) and stored at
−20°C until use. The catalog numbers and dilutions of the
antibodies used in the present study were as follows: AKT (cat. no.
4691S; 1:1,000); pAKT (cat. no. 4060S; 1:1,000); β-actin (cat. no.
8457S; 1:2,000); pEGFR (cat. no. 3777S; 1:1,000); EGFR (cat. no.
4267S; 1:1,000); pERK (cat. no. 4370S; 1:1,000); ERK (cat. no.
4695S; 1:1,000); GAPDH (cat. no. 2118S; 1:1,000); pIGFR (cat. no.
3024S; 1:300); and IGFR (cat. no. 3027S; 1:1,000). All antibodies
were purchased from Cell Signaling Technology, Inc.
Proliferation assay
To determine the effects of gefitinib and glargine
on NSCLC cell proliferation, HCC4006 NSCLC cells
(7.5×104 cells/well) were plated onto 96-well plates.
Following seeding for 24 h, the cells were treated with either
RPMI-1640 culture medium (control) or with different concentrations
of drugs, namely gefitinib (0–100 µM), glargine (1–100 nM), or
both, in RPMI-1640 medium supplemented with 10% FBS. The cells in
the growth medium were cultured for 24 h, and subsequently the
cells were seeded into fresh media of 4 different compositions,
according to the experimental treatment group concerned. A medium
was prepared for the control group, without drug treatment. The
cells in the other three media were treated with: i) Gefitinib
only; ii) glargine only; and iii) gefitinib + glargine,
respectively. The cells of the control group and the other three
drug treatment groups were subsequently cultured for a further 24
h. At the end of the treatment period, cell proliferation was
measured using the MTS assay (BioVision, Inc.), which is based on
the reduction of the MTS tetrazolium compound by viable cells to
generate a colored formazan product that is soluble in cell culture
media. The drug concentration values required to inhibit cell
proliferation by 50% (IC50) were determined from the
dose-response curves for each treatment by using interpolation.
Between 5 and 8 replicate wells were used for each analysis, and at
least 3 independent experiments were performed. Data from replicate
wells were used to determine the mean number of viable cells. The
mean number of viable cells in each group were reported with 95%
confidence intervals. To determine the effect of the combined drug
treatments, any potentiation was estimated by multiplying the
percentages of the cells remaining (% proliferation) in each
treatment.
Western blot analysis
Following each treatment, the cells were washed 3
times with ice-cold PBS. Subsequently, cells were directly lysed
with 1X Laemmli buffer [comprising 2% sodium dodecyl sulfate (SDS),
5 mM dithiothreitol (DTT), 10% glycerol, 0.002% bromophenol blue,
and 63 mM Tris-HCl (pH 6.8)], and heated at 98°C for 5 min. The
total protein concentrations were determined using the BCA protein
assay. Samples containing 20–30 µg protein were then separated on
an SDS-PAGE by using 6–15% gels. The separated proteins were
subsequently transferred onto polyvinylidene difluoride membranes
(cat. no. 1620177; Bio-Rad Laboratories, Inc.). The membranes were
briefly activated in methanol and blocked with 5% milk in TBST
buffer (consisting of 20 mM Tris, 150 mM NaCl, and 0.1% Tween-20)
for 1 h at room temperature, followed by overnight incubation at
4°C with antibodies specific to the target proteins. Next, the
membranes were washed 3 times for 10 min with TBST and hybridized
with goat anti-rabbit IgG-HRP (horseradish peroxidase) conjugated
secondary antibodies (cat. no. sc-2004; Santa Cruz Biotechnology)
with 1:5,000 dilution (in TBST containing 5% milk) at room
temperature for a further 1 h. After washing 3 times for 5 min each
in TBST, the bands on the membranes were visualized by adding an
enhanced chemiluminescence substrate (cat. no. RPN2235; GE
Healthcare). All images were recorded using a ChemiDoc gel imaging
system (Bio-Rad Laboratories, Inc.), and densitometric analysis was
conducted using Image Lab software version 6.0.1 (Bio-Rad
Laboratories, Inc.). Internal controls (GAPDH) were used to
normalize the target protein, and the mean quantities were
represented graphically.
siRNA transfection
To determine whether AKT serine/threonine kinase 1
(AKT1) was mainly responsible for the development of gefitinib
resistance, the proliferation of the HCC4006 cells was examined
following knockdown of AKT1. For siRNA transfection, HCC4006 cells
were transfected with siRNA oligo (5′-AAAUCCAGACUCUUUCGAU-3′)
targeting EGFR 3′-UTR with the Lipofectamine RNAiMAX Transfection
Reagent (Thermo Fisher Scientific, Inc.) and used for experiments
48 h after transfection. siRNAs against Akt1
(5′-GACAAGGACGGGCACAUUA-3′) were purchased from GE Healthcare
Dharmacon. Briefly, prior to cell seeding, the final concentration
of human AKT1 siRNA was calculated to be 10 nM in a mixture
containing 2X Lipofectamine RNAiMAX. The cells were incubated at
room temperature for 15 min. Next, the cells were trypsinized,
counted, and mixed with either the siRNA knockdown group solution
or control group solution and seeded 10,000 cells per well on a
96-well plate for the MTS assay or 24-well plate for western blot
analysis, as aforementioned.
Statistical analysis
Each experiment was replicated at least three times.
Data were expressed as mean ± SEM. Data were analyzed using a
one-way analysis of variance (ANOVA), and Tukey's post hoc test.
Two-sided P<0.05 were considered to indicate a statistically
significant difference. All data were analyzed using SigmaPlot 13
(v13.0.0.83; Systat Software, Inc.).
Results
Effect of gefitinib treatment on NSCLC
cells harboring EGFR mutations
The effects of gefitinib on cell proliferation were
assessed using an MTS assay and the basal levels of EGFR and pEGFR
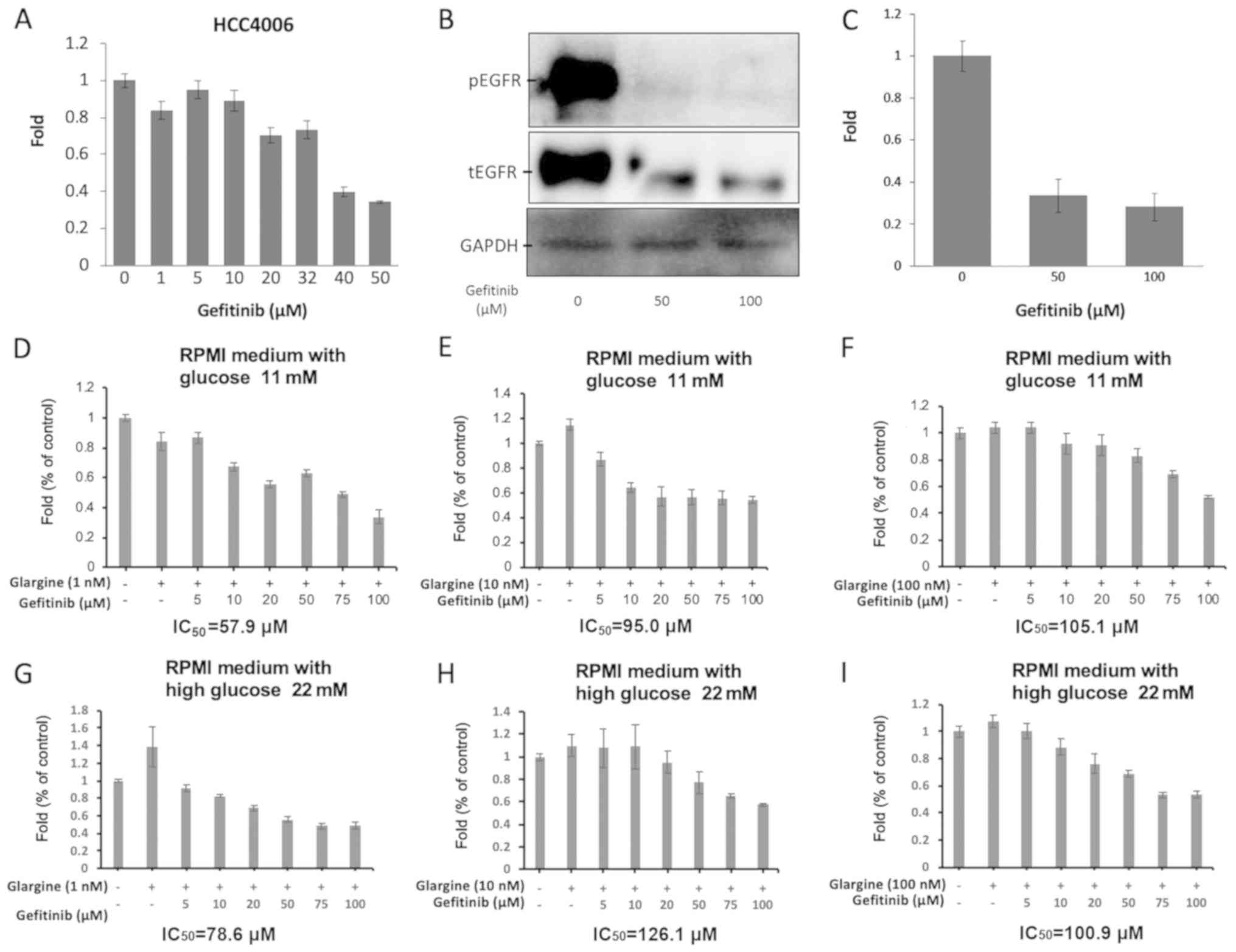
in HCC4006 cells were measured. As shown in Fig. 1, NSCLC cells harboring the EGFR
mutation in exon 19 responded to treatment with gefitinib. HCC4006
cells were sensitive to gefitinib, with an IC50 value of
39.42 µM (Fig. 1A). Subsequently,
the tyrosine kinase activity of EGFR was demonstrated to be
inhibited in a dose-dependent manner, depending on the dose of
EGFR-TKI, in the HCC4006 cell line. Specifically, in the western
blot analysis assay, the protein level of pEGFR in HCC4006 cells
decreased considerably following treatment with gefitinib (Fig. 1B). The HCC4006 cells expressed high
levels of both EGFR and pEGFR. However, the expression levels of
tEGFR and pEGFR were suppressed in HCC4006 cells treated with 50
and 100 µM gefitinib for 72 h in a dose-dependent manner (Fig. 1C). When HCC4006 cells were treated
with gefitinib and different concentrations of glargine in RPMI
medium with 11 mM glucose, the IC50 values of gefitinib
were 57.9 µM with 1 nM glargine (Fig.
1D), 95.0 µM with 10 nM glargine (Fig. 1E), and 105.1 µM with 100 nM glargine
(Fig. 1F). Meanwhile, the
IC50 values of gefitinib were 78.6 µM with 1 nM glargine
(Fig. 1G), 126.1 µM with 10 nM
glargine (Fig. 1H), and 100.9 µM
with 100 nM glargine (Fig. 1I), when
HCC4006 cells were treated with gefitinib and different
concentrations of glargine in 22 mM high-glucose of RPMI
medium.
Long-acting insulin glargine did not
significantly affect the NSCLC cells
Initially, the degradation of porcine insulin in
HCC4006 cells was investigated. As shown in Fig. S1, the expression of pAKT/tAKT
decreased at 60 min and 24 h after the addition of porcine insulin,
which indicated porcine insulin might not be active for 24 h.
Therefore, glargine (long-acting insulin) was selected in this
study. To determine the roles of glargine in the development of
gefitinib resistance, the effects of glargine alone on the
proliferation of HCC4006 cells were subsequently investigated. As
shown in Fig. 2, glargine treatment
did not exert a significant effect on the proliferation of HCC4006
cells, even though HCC4006 cells were treated with different
concentrations of glargine (from 0 to 100 nM). No significant
differences in cell proliferation were observed following the
application of different doses of glargine (Fig. 2A). Subsequently, in HCC4006 cells
treated with 100 nM glargine for different time intervals, the
levels of pIGFR/tIGFR and pAKT/tAKT were quantified through western
blot analysis (Fig. 2B). The levels
of pIGFR/tIGFR did not change significantly (Fig. 2C), whereas the level of pAKT/tAKT was
significantly decreased following treatment time periods of 10 min
and 24 h (Fig. 2D).
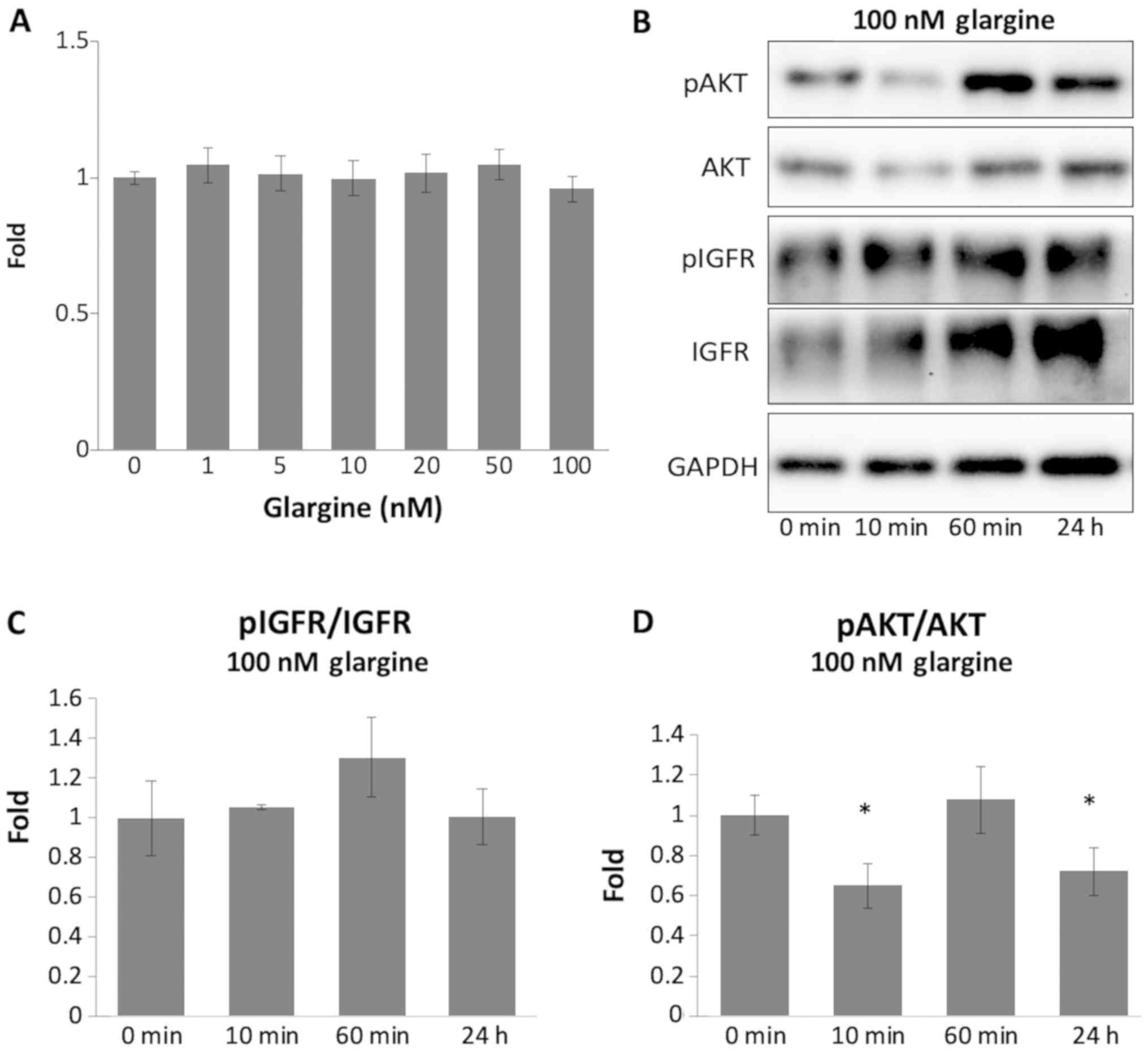 | Figure 2.Long-acting insulin glargine did not
significantly affect NSCLC cells. (A) HCC4006 cells were initially
plated in 96-well plates and grown in experimental medium
containing different concentrations of long-acting insulin glargine
for 24 h. Cell survival was measured using the MTS proliferation
assay. Quantification of the results obtained from the MTS
proliferation assay is shown in the bar graph. The vertical bars
indicate fold-change compared with the control group. (B) Western
blot analysis of pIGFR, IGFR, pAKT, and AKT was performed in
HCC4006 cells treated with 100 nM glargine for different time
periods (0, 10 and 60 min, and 24 h). (C and D) Quantification of
the data of the western blot analysis of pIGFR, IGFR, pAKT and AKT
is shown using bar graphs. The vertical bars indicate the
fold-change compared with the control group. *P<0.05 vs. control
group using a one-way ANOVA with Tukey's post hoc test. NSCLC,
non-small cell lung cancer; p, phosphorylated; IGFR, insulin-like
growth factor receptor. |
Mechanism of hyperinsulinemia in the
development of gefitinib resistance
Subsequently, whether glargine was able to affect
the efficiency of action of gefitinib, ultimately leading to
gefitinib resistance, was investigated in HCC4006 cells. As shown
in Fig. 3A, HCC4006 cells were
cultured in media containing different concentrations of glargine
and a fixed concentration of gefitinib (at the IC50
concentration of 32 µM) for 24 h. The proliferation of HCC4006
cells decreased significantly following treatment with gefitinib
alone. However, the MTS assay revealed that the proliferation of
HCC4006 cells co-treated with gefitinib and different
concentrations of glargine (0–100 nM) led to a marked increase in
cell proliferation compared with the control cells. Glargine (5–100
nM) caused a significant increase in the fold-change in the MTS
assay (50–80%), which indicated that glargine increased cell
proliferation in a dose-dependent manner. Therefore, we concluded
that the hyperinsulinemia induced by glargine was able to affect
the efficiency of gefitinib action, and furthermore, may induce
gefitinib resistance.
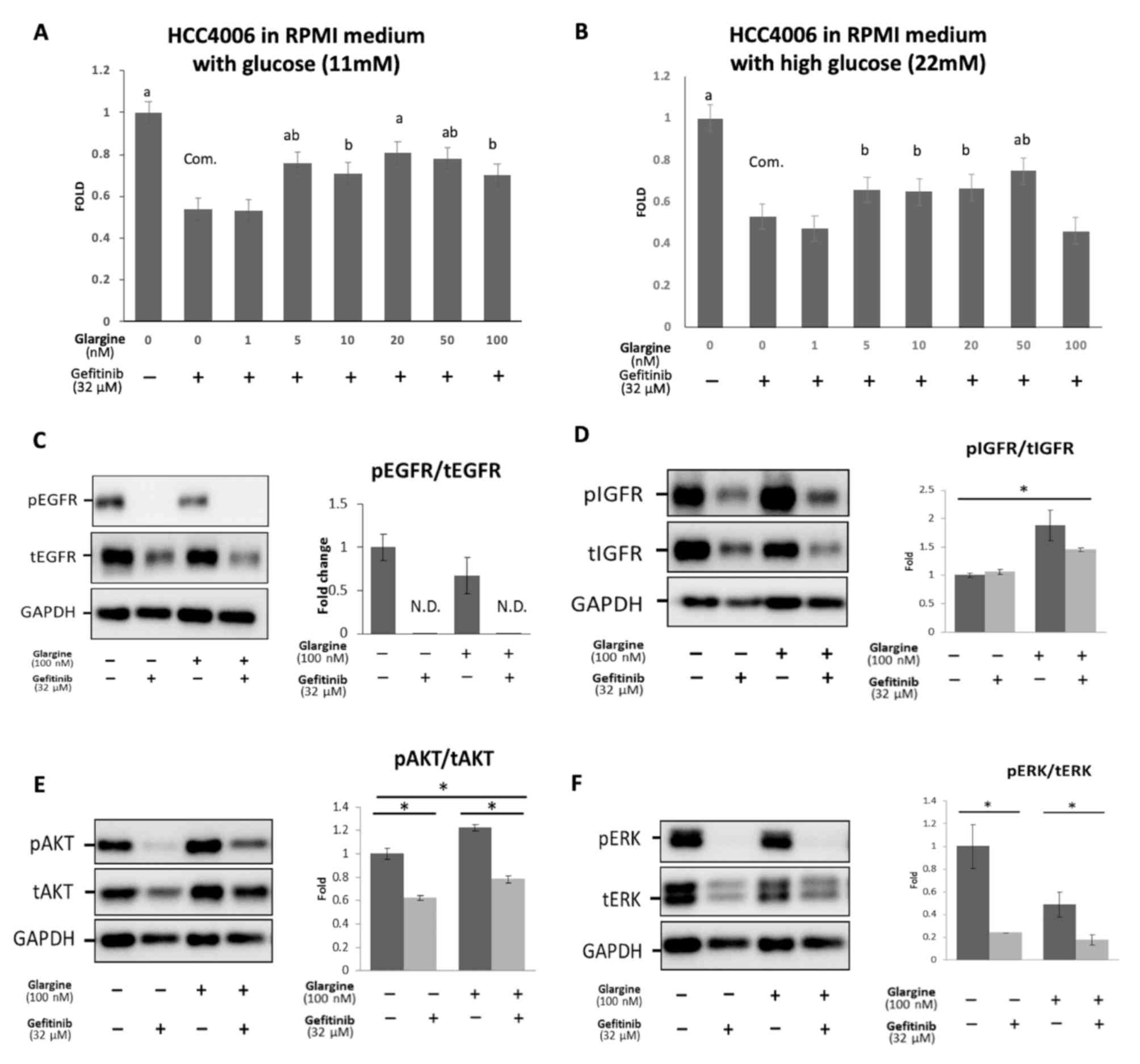 | Figure 3.Mechanism of hyperinsulinemia induced
by glargine in the development of gefitinib resistance. (A) HCC4006
cells were cultured in regular RPMI medium containing 11 mM
glucose. (B) The cells were cultured in RPMI medium containing a
2-fold concentration of glucose (22 mM). HCC4006 cells were treated
with different concentrations of glargine, and a fixed
concentration of gefitinib (IC50 value, 32 µM) for 24 h.
Cell survival was measured using an MTS proliferation assay. The
vertical bars indicate a fold-change compared with the control
group. The eight groups were analyzed in a single one-way ANOVA
with Tukey's post hoc test. aP<0.001,
abP<0.01, and bP<0.05 vs. gefitinib 32
µM without glargine group. (C-F) HCC4006 cells were treated with 32
µM gefitinib, 100 nM glargine, or both gefitinib and glargine for
24 h. Western blot analysis was performed to investigate the
protein expression levels of pEGFR, EGFR, pIGFR, IGFR, pAKT, AKT,
pERK and ERK. *P<0.05 vs. control group using a one-way ANOVA
with Tukey's post hoc test. EGFR, epidermal growth factor receptor;
ERK, extracellular signal-regulated kinase; IGFR, insulin-like
growth factor receptor; p, phosphorylated; t, total; com.,
comparison group; N.D., none detected. |
The aim of the present study was not only focused on
the effects of hyperinsulinemia, but the interaction between
hyperglycemia and hyperinsulinemia was also explored, as the role
of glucose in the development of gefitinib resistance could not be
excluded. As shown in Fig. 3B, the
RPMI-1640 medium of altered composition (22 mM), containing a
2-fold concentration of glucose compared with the original
concentration (11 mM), was used. HCC4006 cells were cultured in
high-glucose RPMI-1640 medium (22 mM) for 3 passages. However, the
morphology and proliferation of cells grown in high-glucose
RPMI-1640 medium (22 mM) and those grown in the original medium
containing 11 mM glucose did not differ significantly.
To determine the role of hyperinsulinemia in the
development of gefitinib resistance, the expression of EGFR, pEGFR,
IGFR, and pIGFR, and their downstream factors, such as AKT, pAKT,
extracellular signal-regulated kinase (ERK), and pERK, were
evaluated to elucidate the mechanisms of gefitinib resistance. As
shown in Fig. 3C, HCC4006 cells were
treated for 24 h with 32 µM gefitinib alone, 100 nM glargine alone,
or 32 µM gefitinib + 100 nM glargine. These experiments revealed
that the phosphorylation of EGFR was significantly suppressed by
either gefitinib or co-treatment with gefitinib and glargine. The
ratio of pEGFR/tEGFR was not detectable in the cells treated with
gefitinib or co-treated with gefitinib and glargine. As shown in
Fig. 3D, no significant difference
in the pIGFR level was observed between control and
gefitinib-treated HCC4006 cells, which indicated that the IGFR
pathway could operate independently. The pIGFR level was
significantly increased in the glargine-treated group compared with
the gefitinib-treated group, thereby suggesting that the
hyperinsulinemia induced by glargine was able to switch on the IGFR
pathway, which may represent a potential route for gefitinib
resistance.
To determine downstream pathway factors, AKT, pAKT,
ERK, and pERK were further investigated. As shown in Fig. 3E, pAKT expression was markedly
suppressed in the gefitinib-treated cells. However, among the cells
treated with glargine or glargine and gefitinib, pAKT expression
was notably increased (P<0.01), which suggested that
hyperinsulinemia induced by glargine may switch on the IGFR
pathway, with cross-talk compensation via the AKT pathway.
Therefore, cell proliferation was observed, even when gefitinib
inhibited the EGFR downstream pathway. As shown in Fig. 3F, the pERK expression level was
significantly decreased in the gefitinib treatment group
(P<0.05). However, the pERK level was completely suppressed even
when glargine and gefitinib were added simultaneously, which was
opposite to the effects on pAKT expression. This result indicated
that gefitinib resistance may not have resulted from the
mitogen-activated protein kinase (MAPK)/ERK pathway.
AKT1 knockdown by siRNA rescued
gefitinib resistance induced by glargine
Subsequently, AKT was knocked down by transfecting
the cells with the siRNA of AKT1 to confirm that gefitinib
resistance resulted from the AKT pathway induced by glargine.
First, the efficiency of AKT knockdown was investigated using an
MTS assay and western blot analysis (Fig. 4A and B). Next, 10 nM AKT1 siRNA was
added, and the cells were incubated for 24 h. Then, 100 nM
glargine, show to activate the AKT pathway in Fig. 3E, was added. As shown in Fig. 4A and B, pAKT expression in the
knockdown group was significantly suppressed, and tAKT expression
was successfully knocked down by AKT siRNA. Furthermore, AKT siRNA
did not exert any effect on glargine treatment. When glargine was
subsequently added, pAKT expression was not activated. As shown in
Fig. S2A and B, the expression of
IGFR1, insulin receptor, and pIGFR were not affected by the
knockdown of AKT. When 100 nM glargine was added, pIGFR was
activated (P<0.05) in both control and the AKT knockdown groups.
However, the expression of pIGFR did not significantly change
between the control and the AKT knockdown groups regardless of the
addition of glargine, which indicated that pIGFR activation was
independent of transfection with AKT siRNA. The function of
gefitinib, which exerted appropriate effects in the control and
knockdown groups, was subsequently investigated. Furthermore, the
IC50 values comparing the siRNA knockdown experiment in
Fig. S2C (37 µM) and the control
group in Fig. 1A (39 µM) were not
significantly different. Cell proliferation was not significantly
increased in the knockdown group compared with in the control group
when cells were cotreated with 32 µM gefitinib and 100 nM glargine
(Fig. 4C).
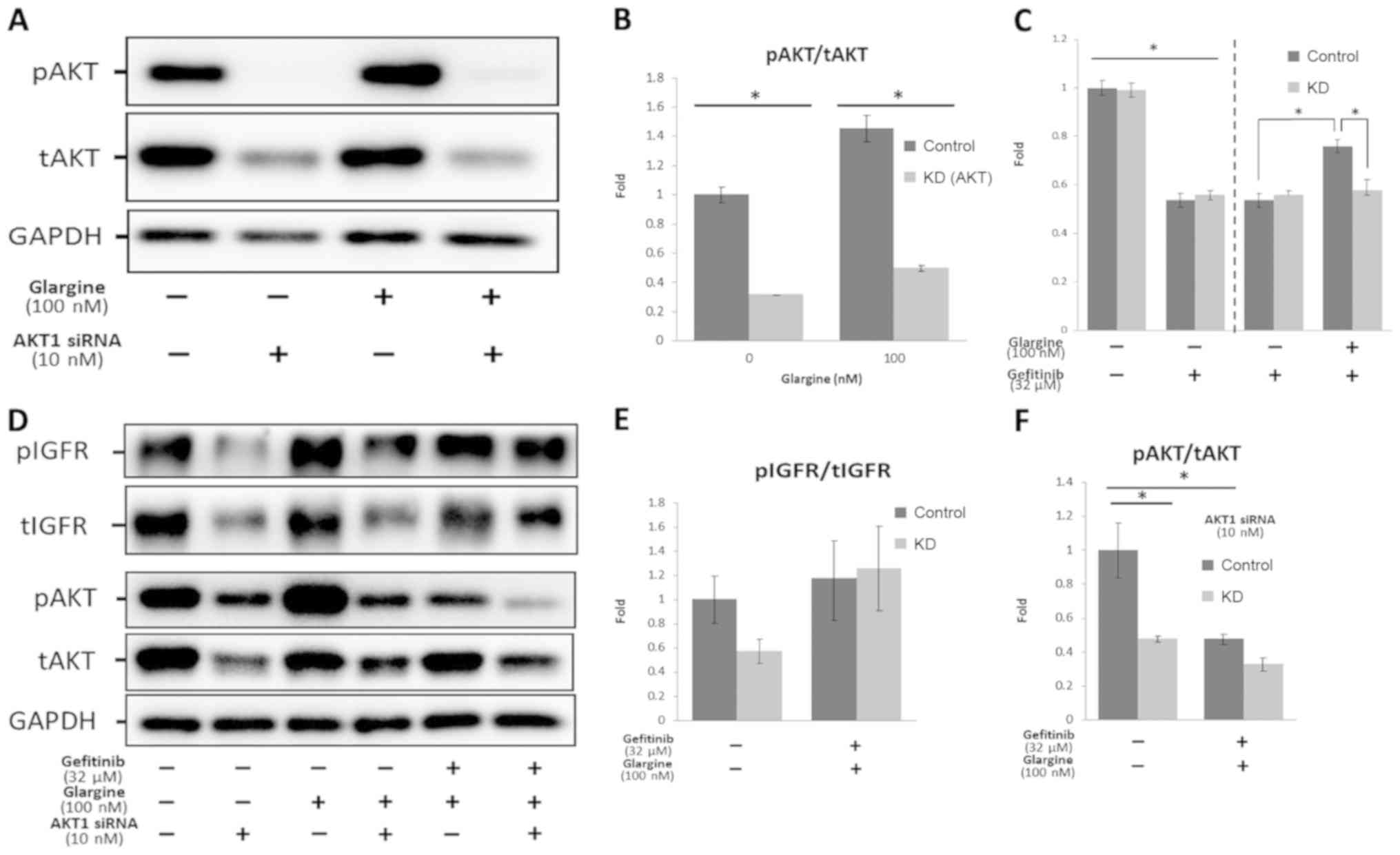 | Figure 4.AKT1 knockdown through siRNA
transfection was able to rescue gefitinib resistance induced by
glargine. (A and B) HCC4006 cells were treated with or without 10
nM AKT1 siRNA, and then treated with or without 100 nM glargine for
24 h, and the results are shown in the representative western blots
and in the bar graphs. (C-F) HCC4006 cells were treated with or
without 10 nM AKT1 siRNA, and subsequently treated with 32 µM
gefitinib, 100 nM glargine, or both. Results are shown in the
representative western blots and the bar graphs. (C) Cell survival
was measured using an MTS proliferation assay. The vertical bars
indicate the fold-change compared with the control group. (D-F)
Quantification of the results of the western blot analysis of pAKT,
AKT, pIGFR and IGFR is shown. The vertical bars indicate the
fold-change compared with the control group. *P<0.05 vs. control
group using a one-way ANOVA with Tukey's post hoc test. IGFR,
insulin-like growth factor receptor; p, phosphorylated; t, total;
siRNA, small interfering RNA; KD, knockdown; IC50,
half-maximal inhibitory concentration. |
To examine the pathway markers, the expression of
pIGFR/IGFR, pAKT/AKT, and pERK/ERK was then investigated. As shown
in Fig. 4D and F, the expression
levels of pAKT and tAKT were significantly decreased in the
knockdown group compared with the control group, and no differences
were observed between the knockdown and the co-treatment (gefitinib
and glargine) groups. When HCC4006 cells were co-treated with
gefitinib and glargine, the expression level of pAKT remained
unchanged. No significant differences were observed between the
control and knockout groups co-treated with gefitinib and glargine,
suggesting that AKT had been successfully knocked down so that the
effects of hyperinsulinemia induced by glargine could not be
mediated via the AKT pathway.
As shown in Fig. 4D and
E, no significant differences in the expression levels of
pIGFR/IGFR were observed between the control and knockdown group,
irrespective of whether co-treatment with gefitinib and glargine
was performed (P=0.134) or not (P=0.516). This result may indicate
that the IGFR pathway, which is normally activated, was not
suppressed by gefitinib.
As shown in Fig. S2D and
E, the levels of pERK were significantly inhibited in the AKT
knockdown group (P<0.01), whereas no inhibition was observed in
the cells co-treated with gefitinib and glargine. However, no
significant differences were observed between the AKT knockdown
group (co-treated with gefitinib and glargine) and the control
group. This could indicate that gefitinib resistance induced by
hyperinsulinemia is not directly affected by the ERK pathway.
Discussion
In the present study, HCC4006 cells were
demonstrated to be sensitive to gefitinib. Co-treatment with
glargine (0–100 nM) and gefitinib induced gefitinib resistance in
HCC4006 cells in a dose-dependent manner. To elucidate the
mechanism underlying gefitinib resistance induced by glargine,
western blot analysis was performed to investigate the
participating signaling pathways. According to the western blot
analysis results, insulin (glargine) increased the phosphorylation
levels of IGFR and AKT excluding ERK, suggesting that glargine may
primarily reactivate the PI3K/AKT pathway, which is associated with
cell proliferation, instead of the MAPK/ERK pathway, which directly
affects DNA transcription in the nucleus. Furthermore, via knocking
down the expression of AKT1 through siRNA transfection, these
experiments were able to further establish the role of AKT in the
development of gefitinib resistance. The MTS assay results revealed
that levels of cell proliferation were decreased in the AKT
knockdown cells compared with the cells that were not transfected,
suggesting that AKT is essential for the development of
resistance.
Patients with NSCLC and pre-existing DM have been
demonstrated to exhibit shorter survival rates compared with those
without DM (15). DM is a metabolic
disorder characterized by hyperglycemia and hyperinsulinemia. IGFR
has been reported to be a negative biomarker of resistance to the
TKI gefitinib in NSCLC cell lines and patients with NSCLC (16). Therefore, it was possible to
hypothesize that insulin may serve an essential role in the
development of gefitinib resistance. The present study has revealed
that long-acting insulin glargine was able to decrease the
efficiency of gefitinib action in HCC4006 cells co-treated with
gefitinib and glargine. Furthermore, the reason why the long-acting
insulin glargine was selected in preference to porcine insulin was
due to the difference in their duration of action. The western blot
analysis data indicated that hyperinsulinemia induced by glargine
led to a continuation of the activation of the insulin pathway and
suppression of the level of pAKT for >24 h. Conversely, the
activity of porcine insulin lasted only for 2–4 h in Fig. S1. Therefore, the long-acting insulin
glargine was selected for the present study.
Hyperinsulinemia and hyperglycemia are 2 essential
features of DM. Therefore, the present study aimed to clarify the
role not only of high insulin concentrations but also of
high-glucose concentrations in terms of the development of
gefitinib resistance. High-glucose RPMI-1640 medium (22 mM)
contained twice the glucose concentration of regular RPMI-1640
medium (11 mM). HCC4006 cells were passaged 3 times in high-glucose
medium (22 mM), and it was confirmed that the morphology and
proliferation of the cells were unaffected by high-glucose medium
compared with that of HCC4006 cells cultured in regular RPMI-1640
medium (11 mM). The results of the present study demonstrated that
the development of gefitinib resistance may be independent of
high-glucose concentrations and may predominantly be affected by
the activation of insulin.
In the present study, hyperinsulinemia induced
gefitinib resistance in HCC4006 cells, and this resistance was
primarily affected by activation of the PI3K/AKT pathway rather
than the MAPK/ERK pathway. Tyrosine kinase receptors, such as IGFR
or EGFR, were shown to activate the PI3K/AKT and MAPK/ERK pathways.
The dysregulation of the PI3K/AKT pathway has been reported to
frequently occur in NSCLC (17).
Cross-talk between EGFR and IGFR can occur through interaction
between shared downstream components of the activated receptors.
Therefore, multilayered cross-talk and involvement of shared
components of the signaling pathways may lead to acquired
resistance to EGFR-TKI therapy in the treatment of cancer (18). The PI3K/AKT pathway fulfills an
integral part in intracellular signal transduction to promote
metabolism, proliferation, apoptosis, and angiogenesis (19–21).
Activation of the PI3K/AKT pathway has been reported to cause
resistance to various anticancer therapies, including chemotherapy
and TKIs (22–24). Certain studies have shown that the
PI3K/AKT pathway exerts a critical role in the development of
resistance to TKIs (24,25). In the present study, the low
expression levels of pERK and tERK, despite simultaneous glargine
treatment, may have resulted from the high dose of gefitinib (32
µM) used.
The gefitinib dose employed in the present study was
32 µM, which was increased compared with that used in other studies
(26,27). As the duration of glargine action was
an important factor in these experiments, HCC4006 cells were
exposed to drug treatment for only 24 h prior to subsequent
harvesting of the cells. This duration of treatment was shorter
compared with that employed in other studies (72 h) (26,27).
Owing to the short treatment time in the present study (only 24 h),
it was necessary to use a high gefitinib concentration to achieve
the appropriate IC50 value. However, to exclude the
possibility that the increased dose of gefitinib might have exerted
adverse effects on the morphology of the HCC4006 cells, the cells
were treated with the higher concentration of gefitinib for 72 h,
and at the end of the treatment period, normal cell morphology was
still observed.
Acquired resistance to EGFR-TKI is a ‘Gordian knot’
to be tackled, especially in patients with NSCLC accompanied by DM.
The present study investigated whether exogenous insulin or a
high-glucose concentration could facilitate the development of
resistance to EGFR-TKI in the NSCLC cell line (HCC4006). The
results obtained revealed that treatment with long-acting exogenous
insulin, instead of a high concentration of glucose, could lead to
gefitinib resistance in NSCLC cells with activating EGFR mutations.
In addition, the mechanism of gefitinib resistance involved
reactivation of the PI3K/AKT pathway, rather than the MAPK/ERK
pathway. Furthermore, it was observed that the gefitinib resistance
could be circumvented by knocking down the expression of AKT1
through siRNA transfection. However, in spite of these advances in
our knowledge, additional research is warranted, both to
investigate the mechanism in a xenograft mouse model and to examine
the effects of co-targeting exposure to exogenous insulin and the
reactivation of the PI3K/AKT pathway to overcome EGFR-TKI
resistance in NSCLC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grant
106-2320-B-002-040-MY3 (to Dr CHC, National Taiwan University) from
the Ministry of Science and Technology, Taiwan and by The Professor
Lu KS Lung Cancer Foundation (grant no. 2017 to Dr HCL). The
funding agencies were not involved in designing the study,
collecting, analyzing, and interpreting data or writing the
manuscript.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HYC, CMC, SPY, DSJ, LSW, HCL and CHC conceived and
designed the study. HYC, CMC and SPY performed the experiments and
the statistical analysis. HYC, CMC and SPY acquired, analyzed and
interpreted the data. HYC and CMC wrote the original manuscript.
SPY, DSJ, LSW, HCL and CHC revised the manuscript for important
intellectual content. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
NSCLC
|
non-small cell lung cancer
|
|
EGFR
|
epidermal growth factor receptor
|
|
TKI
|
tyrosine kinase inhibitor
|
|
DM
|
diabetes mellitus
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mok TS, Wu YL, Thongprasert S, Yang CH,
Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et
al: Gefitinib or carboplatin-paclitaxel in pulmonary
adenocarcinoma. N Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Maemondo M, Inoue A, Kobayashi K, Sugawara
S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I,
et al: Gefitinib or chemotherapy for non-small-cell lung cancer
with mutated EGFR. N Engl J Med. 362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rosell R, Moran T, Queralt C, Porta R,
Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M,
et al: Screening for epidermal growth factor receptor mutations in
lung cancer. N Engl J Med. 361:958–967. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tamura K, Okamoto I, Kashii T, Negoro S,
Hirashima T, Kudoh S, Ichinose Y, Ebi N, Shibata K, Nishimura T, et
al: Multicentre prospective phase II trial of gefitinib for
advanced non small cell lung cancer with epidermal growth factor
receptor mutations: results of the West Japan Thoracic Oncology
Group trial (WJTOG0403). Br J Cancer. 98:907–914. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, et al: Erlotinib versus
chemotherapy as first-line treatment for patients with advanced
EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG
0802): a multicentre, open-label, randomised, phase 3 study. Lancet
Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Richardson LC and Pollack LA: Therapy
insight: Influence of type 2 diabetes on the development, treatment
and outcomes of cancer. Nat Clin Pract Oncol. 2:48–53. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Barone BB, Yeh HC, Snyder CF, Peairs KS,
Stein KB, Derr RL, Wolff AC and Brancati FL: Long-term all-cause
mortality in cancer patients with preexisting diabetes mellitus: a
systematic review and meta analysis. Jama. 300:2754–2764. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu L, Cao H, Zhang T, Shen H, Dong W,
Wang L and Du J: The effect of diabetes mellitus on lung cancer
prognosis: a PRISMA-compliant meta-analysis of cohort studies.
Medicine (Baltimore). 95:e35282016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yeo CD, Park KH, Park CK, Lee SH, Kim SJ,
Yoon HK, Lee YS, Lee EJ, Lee KY and Kim TJ: Expression of
insulin-like growth factor 1 receptor (IGF-1R) predicts poor
responses to epidermal growth factor receptor (EGFR) tyrosine
kinase inhibitors in non-small cell lung cancer patients harboring
activating EGFR mutations. Lung Cancer. 87:311–317. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ramalingam SS, Spigel DR, Chen D, Steins
MB, Engelman JA, Schneider CP, Novello S, Eberhardt WE, Crino L,
Habben K, et al: Randomized phase II study of erlotinib in
combination with placebo or R1507, a monoclonal antibody to
insulin-like growth factor-1 receptor, for advanced stage
non-small-cell lung cancer. J Clin Oncol. 29:4574–4580. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang H, Pelzer AM, Kiang DT and Yee D:
Down-regulation of type I insulin-like growth factor receptor
increases sensitivity of breast cancer cells to insulin. Cancer
Res. 67:391–397. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hemkens LG, Grouven U, Bender R, Günster
C, Gutschmidt S, Selke GW and Sawicki PT: Risk of malignancies in
patients with diabetes treated with human insulin or insulin
analogues: a cohort study. Diabetologia. 52:1732–1744. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baglia ML, Cui Y, Zheng T, Yang G, Li H,
You M, Xu L, Murff H, Gao YT, Zheng W, et al: Diabetes medication
use in association with survival among patients of breast,
colorectal, lung, or gastric cancer. Cancer Res Treat. 51:538–546.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Iams WT and Lovly CM: Molecular pathways:
clinical applications and future direction of insulin-like growth
factor-1 receptor pathway blockade. Clin Cancer Res. 21:4270–4277.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Peled N, Wynes MW, Ikeda N, Ohira T,
Yoshida K, Qian J, Ilouze M, Brenner R, Kato Y, Mascaux C and
Hirsch FR: Insulin-like growth factor-1 receptor (IGF-1R) as a
biomarker for resistance to the tyrosine kinase inhibitor gefitinib
in non-small cell lung cancer. Cell Oncol (Dordr). 36:277–288.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dobashi Y, Suzuki S, Kimura M, Matsubara
H, Tsubochi H, Imoto I and Ooi A: Paradigm of kinase-driven pathway
downstream of epidermal growth factor receptor/Akt in human lung
carcinomas. Hum Pathol. 42:214–226. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oliveira S, Schiffelers R, Storm G,
Henegouwen P and Roovers R: Crosstalk between epidermal growth
factor receptor- and insulin-like growth factor-1 receptor
signaling: implications for cancer therapy. Curr Cancer Drug
Targets. 9:748–760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Osaki M, Oshimura Ma and Ito H: PI3K-Akt
pathway: its functions and alterations in human cancer. Apoptosis.
9:667–676. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Luo J, Manning BD and Cantley LC:
Targeting the PI3K-Akt pathway in human cancer: rationale and
promise. Cancer Cell. 4:257–262. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hennessy BT, Smith DL, Ram PT, Lu Y and
Mills GB: Exploiting the PI3K/AKT pathway for cancer drug
discovery. Nat Rev Drug Discov. 4:988–1004. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Burris HA 3rd: Overcoming acquired
resistance to anticancer therapy: focus on the PI3K/AKT/mTOR
pathway. Cancer Chemother Pharmacol. 71:829–842. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Papadimitrakopoulou V: Development of
PI3K/AKT/mTOR pathway inhibitors and their application in
personalized therapy for non-small-cell lung cancer. J Thorac
Oncol. 7:1315–1326. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gadgeel SM and Wozniak A: Preclinical
rationale for PI3K/Akt/mTOR pathway inhibitors as therapy for
epidermal growth factor receptor inhibitor-resistant non-small-cell
lung cancer. Clin Lung Cancer. 14:322–332. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yamasaki F, Johansen MJ, Zhang D,
Krishnamurthy S, Felix E, Bartholomeusz C, Aguilar RJ, Kurisu K,
Mills GB, Hortobagyi GN and Ueno NT: Acquired resistance to
erlotinib in A-431 epidermoid cancer cells requires down-regulation
of MMAC1/PTEN and up-regulation of phosphorylated Akt. Cancer Res.
67:5779–5788. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Morgillo F, Kim WY, Kim ES, Ciardiello F,
Hong WK and Lee HY: Implication of the insulin-like growth
factor-IR pathway in the resistance of non-small cell lung cancer
cells to treatment with gefitinib. Clin Cancer Res. 13:2795–2803.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim JG, Kang MJ, Yoon YK, Kim HP, Park J,
Song SH, Han SW, Park JW, Kang GH, Kang KW, et al:
Heterodimerization of glycosylated insulin-like growth factor-1
receptors and insulin receptors in cancer cells sensitive to
anti-IGF1R antibody. PLoS One. 7:e333222012. View Article : Google Scholar : PubMed/NCBI
|