Introduction
Endometrial cancer (EC) is the sixth most commonly
occurring cancer in women, with an incidence rate rising to 3.5% of
all new cancer cases and representing 2% of all cancer deaths in
the USA (1). While the 5-year
survival rate for patients with EC in economically advantaged
countries is high (~80%) compared with those for patients with
other female-associated types of cancer, such as breast and ovarian
cancer, the prognosis for women with EC is dismal in other parts of
the world where treatment options are limited. The metabolic
disorders type 2 diabetes and obesity are increasingly linked to an
increased risk of cancer, including EC (2–4). The
estimated type 2 diabetes incidence rate for 2019 is 9.3% globally
(5). Furthermore, 35–40% of adults
are obese in the USA and the worldwide prevalence of obesity has
tripled to 13% over a timespan of 40 years (6). The staggering financial and personal
burdens of cancer underscore the need for the development of novel
therapeutic and preventative strategies that are clinically
effective and financially deliverable to the general
population.
Metformin (1,1-dimethylbiguanide hydrochloride; MET)
constitutes the first-line treatment for type 2 diabetes. Previous
studies have highlighted the therapeutic value of MET as an
adjuvant treatment in breast cancer (7). EC is pathologically akin to breast
cancer since it is steroid hormone-driven, tightly linked to
abnormal glucose metabolism and insulin signaling, and exhibits a
complex, multi-factorial origin. In breast cancer cells, MET has
been shown to inhibit cell proliferation, invasion and
inflammation, promote apoptosis and exert a cytotoxic effect on
cancer stem cells (8,9). Nevertheless, the anti-breast cancer
benefits of MET have been inconsistent and appear to be dependent
on context and the duration of exposure. In triple negative breast
cancer cell lines for example, glucose levels in the diabetic range
diminished the effects of MET on cell proliferation, cell death and
cell cycle arrest (10). Conversely,
glucose starvation enhanced the inhibitory effect of MET on the
mTOR pathway, a downstream target of AMP-activated protein kinase
(AMPK) and an activator of growth factor signaling in breast cancer
cells (11). Moreover, breast cancer
cells chronically exposed to MET acquired resistance to the drug,
which was accompanied by changes in the expression of key genes and
resulted in the generation of a metastatic stem-cell like phenotype
(12).
In a previous study, it was shown that the
short-term treatment of non-diabetic and obese women with MET
during the pre-surgical window between diagnosis and hysterectomy
promoted the expression of antitumor biomarkers; this effect was
recapitulated in Ishikawa human endometrial cancer cells treated
with a physiological dose of MET under normal glucose conditions
(13). While these results support
the ability of MET to reduce EC risk and development, meta-analyses
of current clinical data indicate conflicting MET outcomes, with
some showing no effect (14) and
others showing inhibitory effects (15). In light of the findings for breast
cancer, a strong rationale exists for clarifying the ambiguities
regarding the context and dosage at which MET exerts its optimal
therapeutic effects in EC to achieve its potential for clinical
use.
In humans treated with a single 500-mg dose of MET,
the blood concentration of MET 2.4 h post-oral intake was 4 µM, as
measured by high-pressure liquid chromatography (16). In mice, the oral administration of
MET at 50 mg/kg body weight resulted in a MET concentration of
10–70 µM in the portal vein and 10–40 µM in the plasma, as measured
using 14C-radiolabeled MET (17).
Thus, studies that use mM concentrations of MET may not be
physiologically relevant. In the present study, estrogen-treated
Ishikawa EC cells were characterized for their response to 100 µM
MET and glucose concentrations typifying the non-diabetic and
diabetic ranges, using progesterone receptor (PGR) gene
expression as the primary outcome. The present findings demonstrate
the glucose-dependency of the MET-mediated effects on PGR
isoform B (PGR-B) expression, which may be relevant when
assessing the response to progestin-based therapy of diabetic and
non-diabetic patients with EC and other gynecological
pathologies.
Materials and methods
Cell culture and treatments
The Ishikawa human endometrial epithelial carcinoma
cell line (a gift from Dr Bruce Lessey, Greenville Health System;
Prisma Health) was authenticated and propagated as previously
described (13). Cells were
initially grown in Minimal Essential Media (MEM; Gibco; Thermo
Fisher Scientific, Inc.), supplemented with 10% (v/v) fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and containing
phenol red and 1% antibiotic-antimycotic solution (Invitrogen;
Thermo Fisher Scientific, Inc.) in a humidified incubator (5%
CO2/95% air) at 37°C. In subsequent experiments
incorporating various treatments, cells were incubated in either
phenol red-free MEM or phenol red-free Dulbecco's modified Eagle's
medium (DMEM)/Nutrient Mixture F-12 (DMEM/F-12), each supplemented
with charcoal-stripped 10% FBS and 1% antibiotic-antimycotic
solution and referred to hereinafter as MEM-FBS and DMEM-FBS,
respectively. MEM which contains 5.5 mM glucose represents the
normal glucose environment while DMEM with 17.5 mM glucose,
typifies a high glucose environment.
MET (Sigma-Aldrich; Merck KGaA) was dissolved in
phosphate-buffered saline (PBS; Gibco; Thermo Fisher Scientific,
Inc.) and initially evaluated at final concentrations of 10 or 100
µM, which approximate the physiological range found in patients
treated with MET for type 2 diabetes (18). 17β-estradiol (E2; Sigma-Aldrich;
Merck KGaA) was dissolved in dimethyl sulfoxide (DMSO), further
diluted in PBS and used at final concentrations of 0.1 or 10 nM.
Medroxyprogesterone acetate (MPA; Sigma-Aldrich; Merck KGaA) was
dissolved in DMSO, further diluted in PBS and used at a final
concentration of 1 µM. The AMPK activator,
5-aminoimidazole-4-carboxamide ribonucleotide (AICAR;
Sigma-Aldrich; Merck KGaA) was dissolved in PBS and used at a final
concentration of 500 µM. Compound C (dorsomorphin; Sigma-Aldrich;
Merck KGaA), an AMPK inhibitor, was dissolved in DMSO, further
diluted in PBS and used at a final concentration of 5 µM. In all
treatment protocols where DMSO was added to solubilize E2, MPA and
compound C, the same amount of DMSO was added to the control
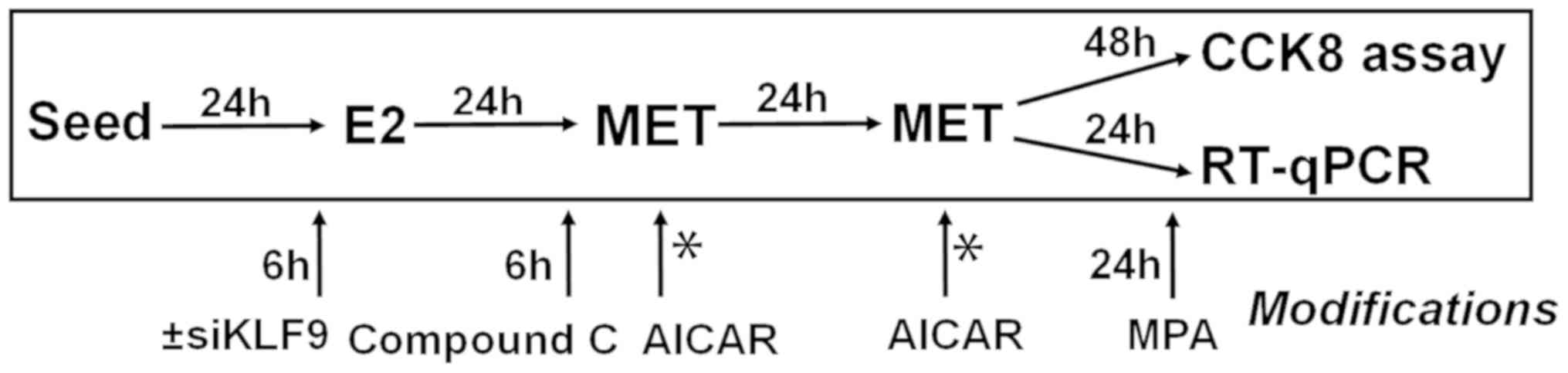
treatment. The various treatment strategies are summarized in
Fig. 1.
Cell proliferation assay
Cells were seeded onto 96-well culture dishes at a
density of 1×104 cells/well in MEM-FBS or DMEM-FBS. After 24 h,
cells were pre-treated with E2 (0.1 or 10 nM) or vehicle control
(DMSO-PBS) and incubated for another 24 h at 37°C. E2 was removed
with a change of media and cells were then treated with MET (10 or
100 µM) or vehicle control (PBS) twice at 24-h intervals at 37°C.
Cells were collected 24 h after the final MET treatment. Cell
viability was evaluated by the Cell Counting Kit-8 (Dojindo
Molecular Technologies, Inc.) metabolic assay according to the
manufacturer's instructions. Absorbance at 450 nm was assessed
using a CLARIOstar plate reader (BMG Labtech GmbH) and the mean
optical density from n=4 wells/treatment group was calculated. The
cells treated with and without E2 (10 nM) and in the presence or
absence of MET (100 µM) were collected and their cyclin D1 (CCND1)
transcript levels were measured by reverse
transcription-quantitative (q)PCR.
Spheroid-formation assay
Ishikawa cells were plated in 24-well ultra-low
attachment plates (Corning Inc.) at a seeding density of 8×103
cells/well. The plating medium consisted of phenol red-free MEM or
phenol red-free DMEM-F12 and was supplemented with B27 (Invitrogen;
Thermo Fisher Scientific, Inc.), human basic fibroblast growth
factor (20 ng/ml; Invitrogen; Thermo Fisher Scientific, Inc.),
human epidermal growth factor (20 ng/ml; Invitrogen; Thermo Fisher
Scientific, Inc.), heparin (10 µg/ml; Sigma-Aldrich; Merck KGaA),
antibiotic-antimycotic solution (1% v/v) and gentamicin (100 µg/ml;
Sigma-Aldrich; Merck KGaA). Cells were treated with PBS alone or
with MET (100 µM) in the presence or absence of E2 (10 nM) at
seeding and incubated in a humidified incubator (5%
CO2/95% air) at 37°C. Culture media, as appropriate for
each treatment group, were replenished with 62.5 µl medium
representing 12.5% of the initial plating volume, on incubation day
3. On day 5, non-adherent spheroids, designated as endospheres,
with diameters >60 µm were counted manually using an Olympus
IMT-2 inverted microscope (Olympus Corporation). In order to assess
the self-renewal capacity of the primary endospheres, non-adherent
spheroids from the first plating (passage 1, P1) were collected,
dissociated into single-cell suspensions with 0.05% trypsin
(Invitrogen; Thermo Fisher Scientific, Inc.), filtered using a
40-µm sieve, and re-plated in MEM or DMEM-F12 supplemented as
described for the plating medium, in ultra-low attachment plates
with no further E2 or MET treatments. Endospheres with diameters
>60 µm, designated as passage 2 (P2), were counted after
incubation for 5 days, with addition of fresh media (MEM or DMEM)
on day 3.
RNA isolation and gene expression
assay
Ishikawa cells were plated in 6-well plates (Falcon)
at a density of 1.5×105 cells/well in MEM-FBS or DMEM-FBS for 24 h
and treated with various reagents, following the timeline
summarized in Fig. 1. For all RNA
analyses, cells were collected 24 h after the final MET treatment.
RNA isolation, preparation of cDNAs and qPCR analyses were
performed as previously described (19), using the geometric mean of β-actin
and TATA-binding protein mRNAs as normalization controls. Primers
(Table I), designed to span introns,
were synthesized by Integrated DNA Technologies, Inc. qPCR was
conducted using the CFX96™ Real-Time PCR System (Bio-Rad
Laboratories, Inc.) under the following thermocycling conditions:
30 sec at 95°C, followed by 39 cycles of 5 sec at 95°C and 30 sec
at 60°C, and a melt curve protocol of 5 sec at 65°C with a gradual
increase in temperature to 95°C by 0.5°C increments. Normalized
mRNA expression was calculated with CFX Manager Software version
3.1.1517.0823 (Bio-Rad Laboratories, Inc.) using the
2−ΔΔCq method (20).
 | Table I.Primer sequences used for reverse
transcription-quantitative PCR. |
Table I.
Primer sequences used for reverse
transcription-quantitative PCR.
| Gene | Primer sequences
(5′-3′) | PCR product size,
bp |
|---|
| ACTB | F:
TCACCAACTGGGACGACATG | 244 |
|
| R:
TCACCGGAGTCCATCACGAT |
|
| CCND1 | F:
CTGGCCATGAACTACCTG | 483 |
|
| R:
GTCACACTTGATCACTCTGG |
|
| TBP | F:
TCCACAGTGAATCTTGGTTGTAAAC | 102 |
|
| R:
CCTCATGATTACCGCAGCAAA |
|
| PGR | F:
CCTTTGGAAGGGCTACGAAGT | 110 |
|
| R:
GAGCTCGACACAACTCCTTTTTG |
|
| PGR-B | F:
CGACCCAGGAGGTGGAGAT | 105 |
|
| R:
GAGGGAAAAGGGAAGGAGGAG |
|
| ESR1 | F:
CGGCATTCTACAGGCCAAATT | 111 |
|
| R:
AGCGAGTCTCCTTGGCAGATT |
|
| ESR2 | F:
CGATTACGCATCGGGATATCA | 136 |
|
| R:
GCGCCGGTTTTTATCGATT |
|
| KLF9 | F:
TGGCTGTGGGAAAGTCTATGG | 124 |
|
| R:
CTCGTCTGAGCGGGAGAACT |
|
| KLF4 | F:
TTCCCATCTCAAGGCACACCT | 120 |
|
| R:
TGTTTACGGTAGTGCCTGGTCA |
|
Transient transfection with small
interfering (si)RNAs
Ishikawa cells were transfected with human
Krüppel-like factor 9 (KLF9) siRNAs (siKLF9), following
previously described protocols (21). Briefly, cells were seeded in 6-well
plates at a density of 1.5×105 cells/well in MEM-FBS or DMEM-FBS
without added antibiotic for 24 h. Cells were then transfected with
Lipofectamine® 2000/OPTI-MEM I (Invitrogen; Thermo
Fisher Scientific, Inc.) containing 50 µM siKLF9 RNA pools
(cat. no. L-011223-00-0005) or non-targeting (scrambled control,
cat. no. D-001810-01-05) siRNAs (GE Healthcare Dharmacon, Inc.).
After 6 h of transfection, cells were incubated for 24 h in MEM-FBS
or DMEM-FBS containing 1% antibiotic-antimycotic solution, and then
treated with E2 (10 nM) for 24 h, followed by MET (100 µM) twice at
24-h intervals at 37°C. RNA isolation and gene expression analysis
of cells collected 24 h after the last MET treatment were performed
by the aforementioned method.
Statistical analysis
Data are presented as the mean ± SEM and were
analyzed for statistical differences between two groups using
Student's t-test, and among three or more groups using two-way
analysis of variance followed by Tukey's post hoc test. Analyses
were performed using SigmaStat (version 3.5; Systat Software,
Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
MET inhibits proliferation of Ishikawa
cells in normal but not high glucose conditions
Previous studies showed that MET decreased the
viability of EC cells exposed to E2 (22,23);
however, those studies used supra-physiological MET concentrations
of >5 mM, which do not reflect the systemic µM concentrations
detected in patients receiving MET (18). In order to evaluate whether
physiologically relevant concentrations of MET act directly on
E2-responsive EC cells to influence their viability, the
well-differentiated Ishikawa endometrial carcinoma cell line was
treated with E2 and exposed to MET in normal and high glucose
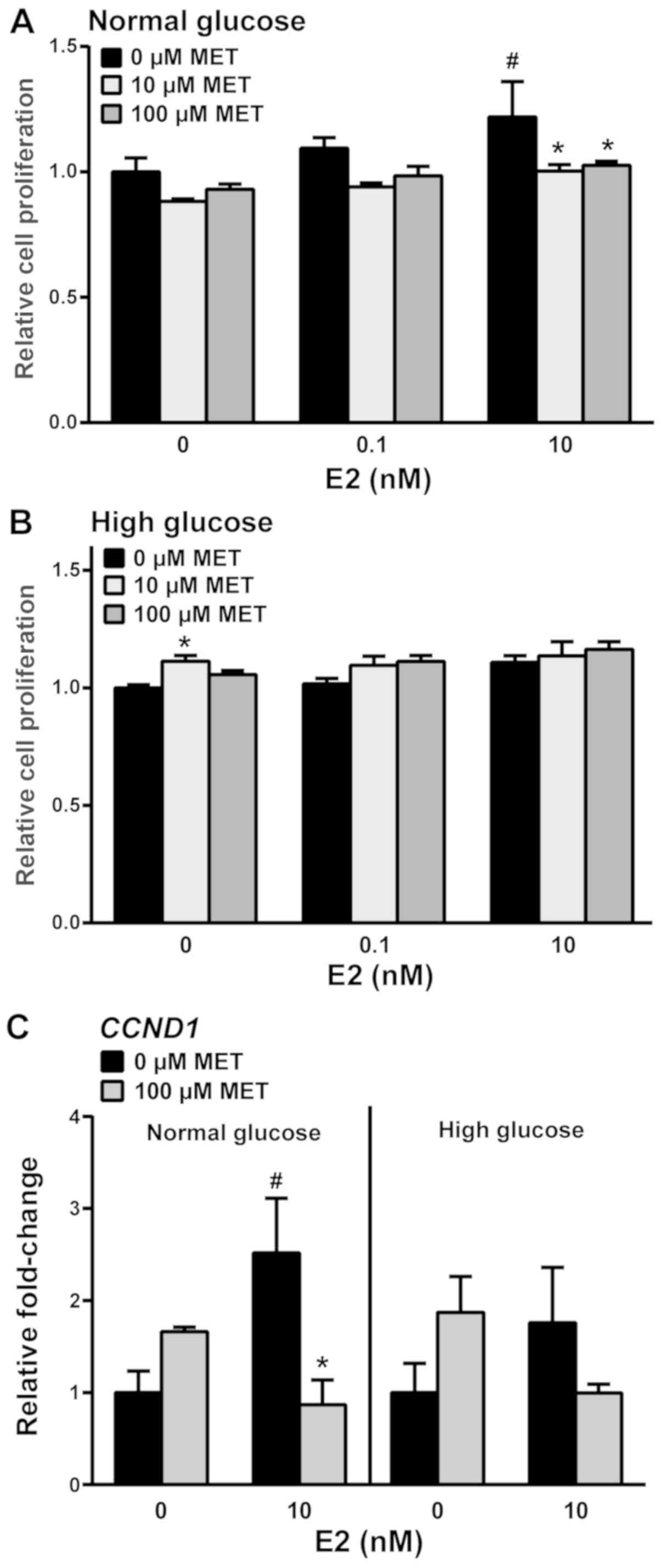
media. Without MET, 10 nM E2-treated cells under normal glucose
conditions exhibited increased cell viability (Fig. 2A); this was decreased to basal levels
when 10 or 100 µM MET was added. By contrast, the viability of
E2-treated cells was not influenced by MET in the high glucose
medium (Fig. 2B). Notably, treatment
with 10 µM MET in the absence of E2 increased cell viability in the
presence of a high concentration of glucose. Gene expression
analyses for the proliferation marker cyclin D1 showed that E2
induced CCND1 transcript levels in the absence of MET and that MET
reduced these E2-induced effects under normal glucose conditions.
Similar effects of MET were not observed in high glucose conditions
(Fig. 2C).
MET inhibits Ishikawa sphere formation
in normal but not high glucose conditions
Spheres formed from Ishikawa cells grown under
non-adherent conditions are considered to constitute EC stem-like
cells, based on their characterized genotype and phenotype
(24,25). In other types of tumor, MET has been
shown to selectively target cancer stem cells by decreasing sphere
formation in vitro and tumor formation in vivo
(26). To assess whether MET
influences the formation of endometrial spheres under normal or
high glucose conditions, Ishikawa cells that normally grow as
monolayers in plastic culture dishes (Fig. 3A, top panel) were plated in
low-attachment culture dishes to enable the formation of
endospheres (Fig. 3A, bottom panel)
and treated with 100 µM MET in the presence or absence of 10 nM E2.
Without E2, MET significantly inhibited the growth of primary
endospheres (P1; >60 µm diameter) under normal glucose
conditions; however, this effect of MET was lost with the addition
of E2 (Fig. 3B). By contrast, MET
with and without E2, showed no inhibitory effect on basal
P1-endosphere formation under high glucose conditions (Fig. 3C). P1-endospheres were collected and
re-plated in low attachment plates in normal or high glucose
conditions without further MET or E2 treatments. P2-endospheres
exhibited increased formation efficiency (10–12 vs. 2–3% for P1)
when normalized to initial seeding density, indicating self-renewal
(Fig. 3D and E). Moreover,
P2-endospheres grown in normal and high glucose conditions
exhibited responses to MET without and with added E2 that were
comparable with those observed for the respective
P1-endospheres.
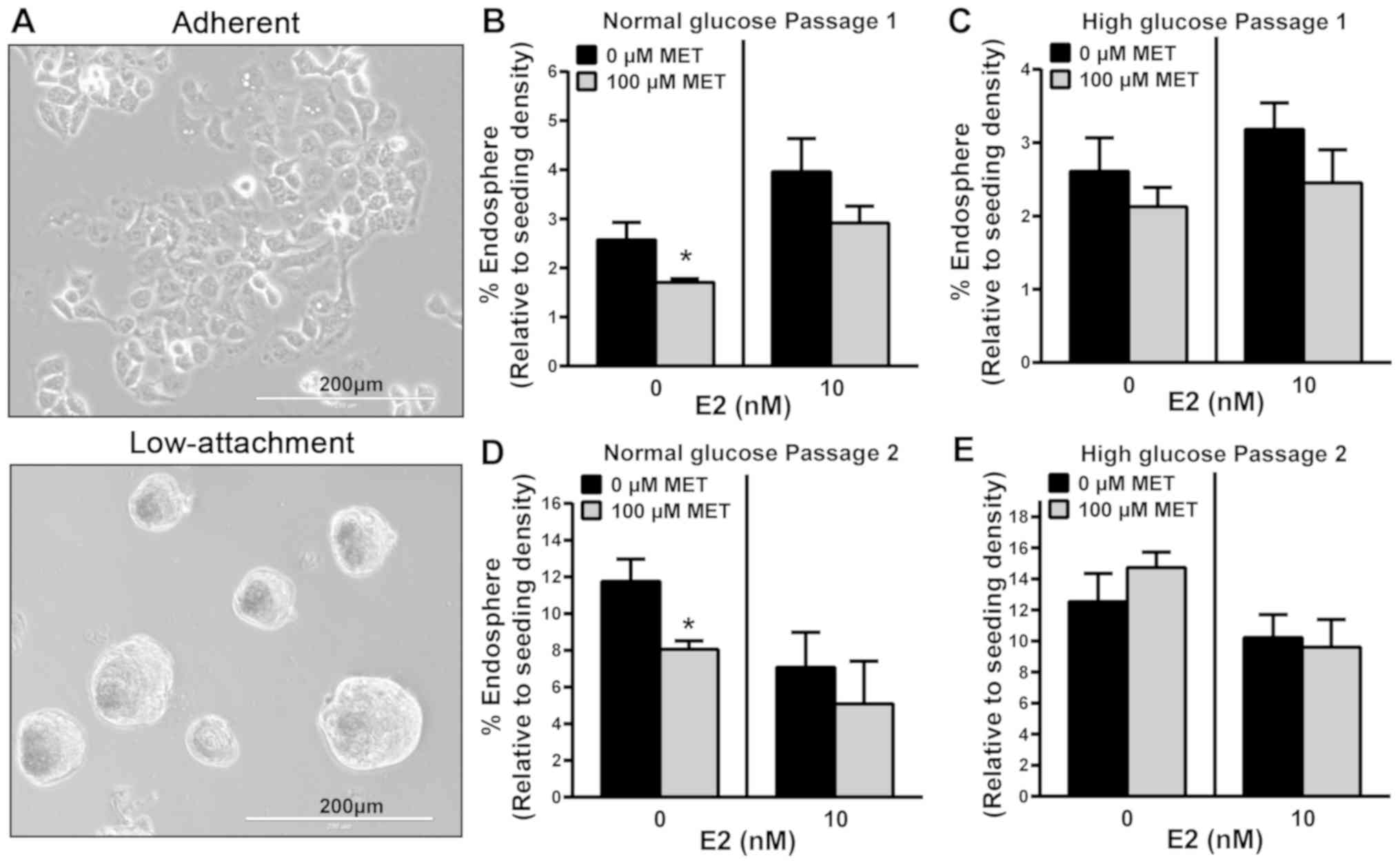 | Figure 3.MET inhibits the sphere formation of
Ishikawa cells under normal but not high glucose conditions. (A)
Representative images show 2-dimensional (top panel) and
3-dimensional (bottom panel) growth of Ishikawa cells, when
cultured in adherent and low-attachment culture plates,
respectively. Ishikawa cells plated in low-attachment plates were
treated with MET (100 µM) in the presence or absence of E2 (10 nM)
at day 1, under (B) normal or (C) high glucose conditions. At day
5, endometrial spheroids from the initial plating with a diameter
of >60 µm were counted (designated passage 1). Non-adherent
spheroids from passage 1 were collected, dissociated into
single-cell suspensions with 0.05% trypsin, filtered using a 40-µm
sieve and re-plated in (D) normal or (E) high glucose conditions in
ultra-low attachment plates with no further treatments. Endometrial
spheroids with a diameter of >60 µm were counted and designated
passage 2. Data are presented as mean ± SEM (n=6) and expressed as
% endosphere, calculated as the ratio of spheroids with a diameter
of >60 µm, relative to the total number of initial plated
Ishikawa cells ×100. *P<0.05 vs. 0 µM MET, as determined by
Student's t-test. MET, metformin; E2, 17β-estradiol. |
MET-induced changes in total PGR and
PGR-B transcript levels are dependent on glucose
We previously showed that MET inhibited the
expression of the antitumor biomarker PGR in EC cells in
vivo (protein) and in vitro (transcript) (13). Transcript levels of total PGR
and its antiproliferative isoform PGR-B were evaluated in
Ishikawa cells in response to 100 µM MET, following a 24-h
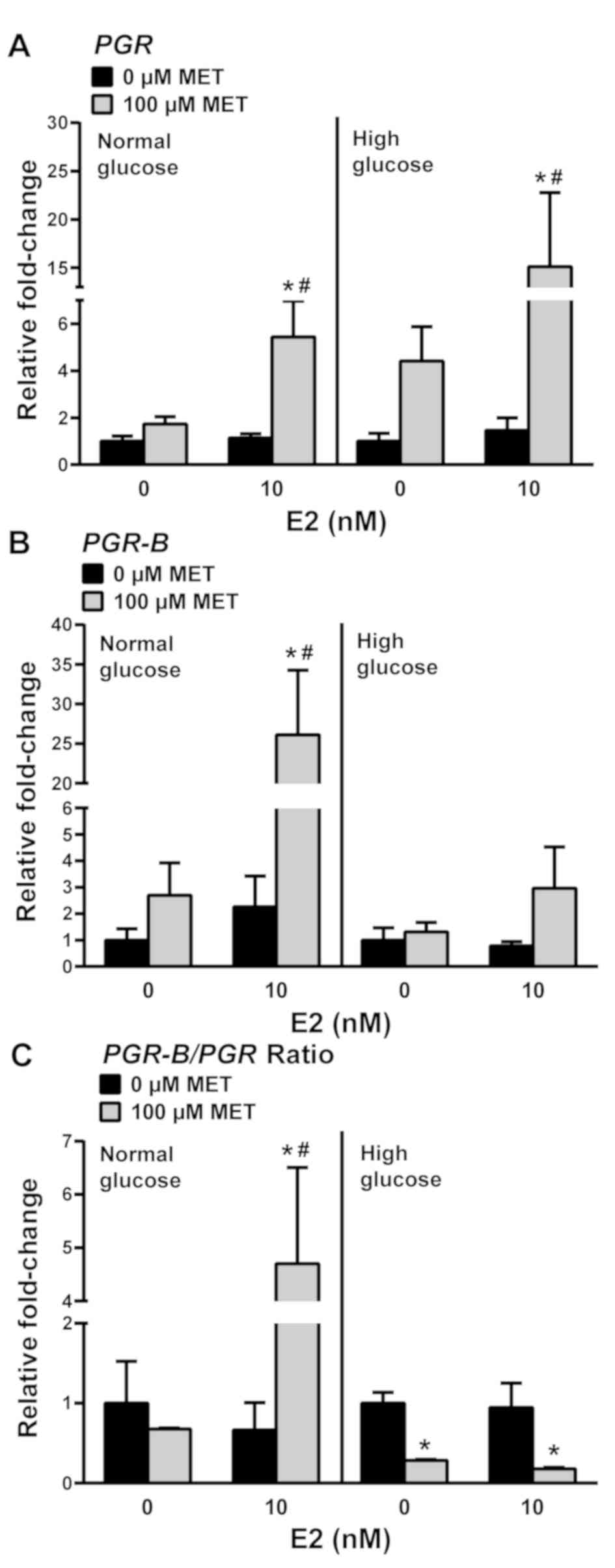
pre-treatment with E2. MET significantly increased total PGR
transcript levels in normal and high glucose conditions by 5- and
15-fold, respectively, relative to the corresponding non-MET
treated levels (Fig. 4A).
Interestingly, while MET increased the levels of PGR-B
transcripts by 25-fold in cells grown in normal glucose + E2, no
comparable effects on PGR-B were elicited by MET in cells
incubated under high glucose + E2 (Fig.
4B). Given that total PGR transcript levels are the sum
of those for PGR-A + PGR-B isoforms, the relative
changes in PGR-A and PGR-B expression under normal
glucose ± E2 and high glucose ± E2 conditions were determined from
the PGR-B/PGR transcript expression ratios. Under normal
glucose conditions, the increase in PGR expression with MET
+ E2 was due to increased PGR-B transcript levels (Fig. 4C, left panel). Under high glucose
conditions, MET decreased the ratio of PGR-B/PGR transcripts
in the presence or absence of E2, suggesting that levels of
PGR-A transcripts are increased by treatment with MET under
high glucose conditions (Fig. 4C,
right panel).
Reduced KLF9 expression promotes PGR-A
transcription in E2/MET-treated cells independent of glucose
environment
Our previous study showed that human endometrial
tumors display lower levels of the tumor suppressor protein KLF9
compared with those in the adjacent non-tumor endometrial tissue
(21). These findings suggest that
loss of KLF9 expression is a feature of EC cells and that the
antiproliferative effects of MET in the context of normal glucose
(Fig. 2A and C) may be partly due to
the induction of KLF9 expression. To test the latter
hypothesis, KLF9 transcript levels and those of its family
member Krüppel-like factor 4 (KLF4) were measured in
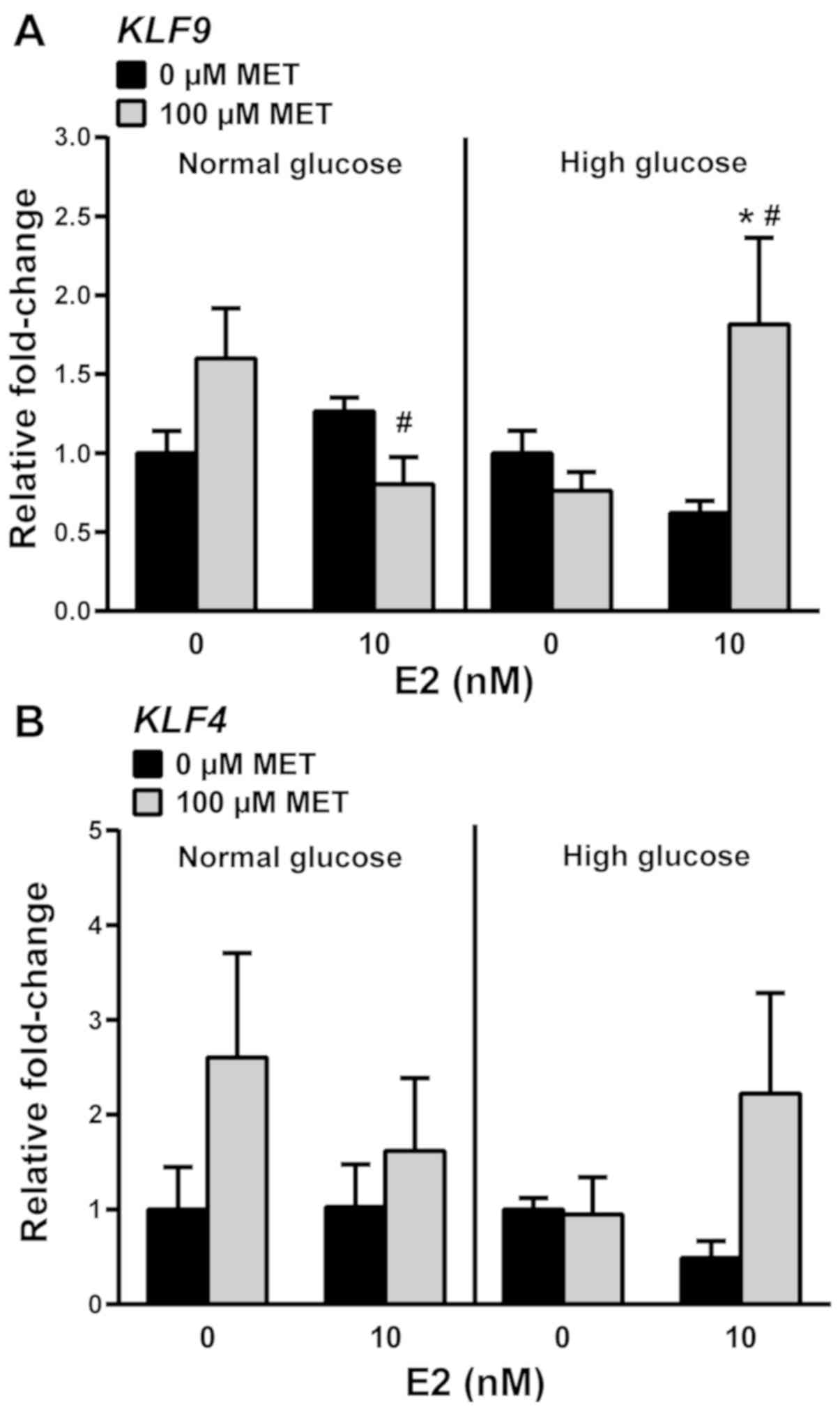
E2/MET-treated cells. Under normal glucose conditions, MET or E2
alone had no effect on KLF9 transcript levels (Fig. 5A). With MET-treatment, KLF9
mRNA levels were modestly decreased by E2, which contrasted with
the increase noted under high glucose conditions (Fig. 5A). KLF4 mRNA levels were
unresponsive to MET, alone or with E2, in normal and high glucose
conditions (Fig. 5B).
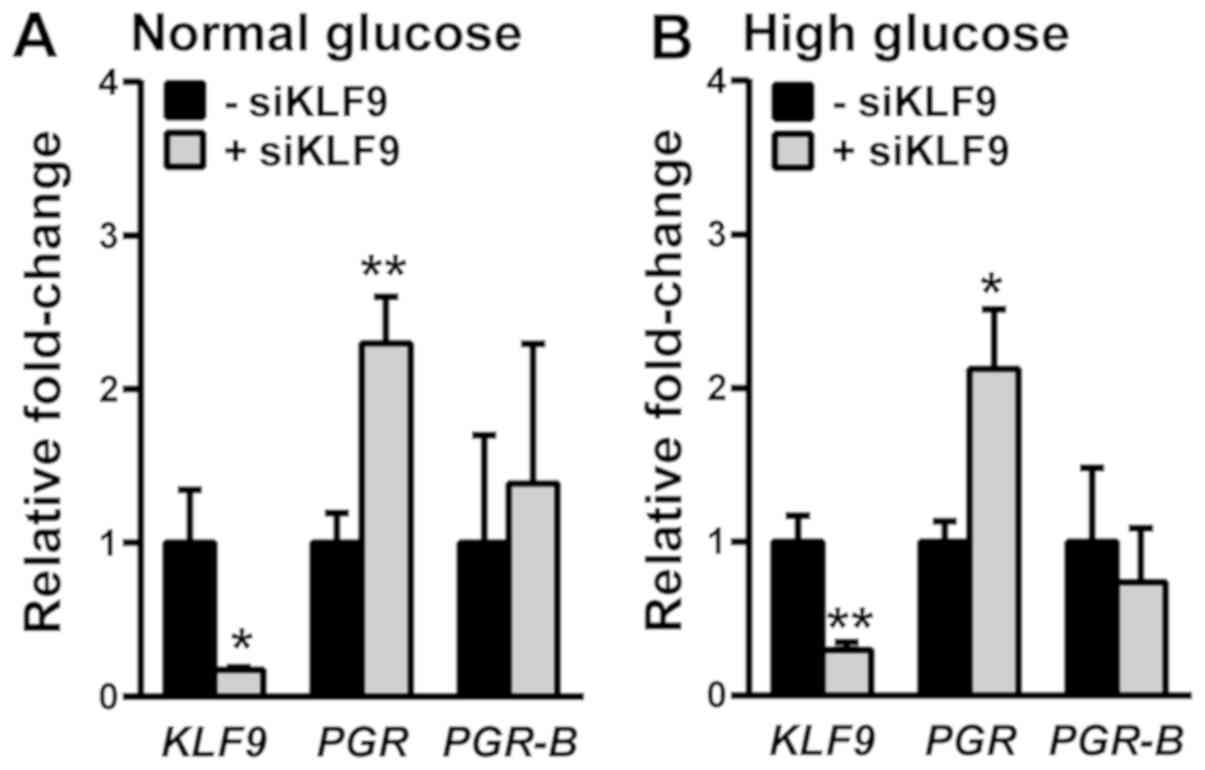
We previously reported that in E2-treated Ishikawa
cells, KLF9 siRNA increased total PGR transcript
levels (27). In the context of
E2/MET treatments (Fig. 1), the
transfection of Ishikawa cells with KLF9 siRNA promoted
total PGR expression in both normal and high glucose culture
conditions (Fig. 6). Parallel
changes in PGR-B transcript levels were not observed when
KLF9 expression was reduced, indicating increased levels of
PGR-A transcripts.
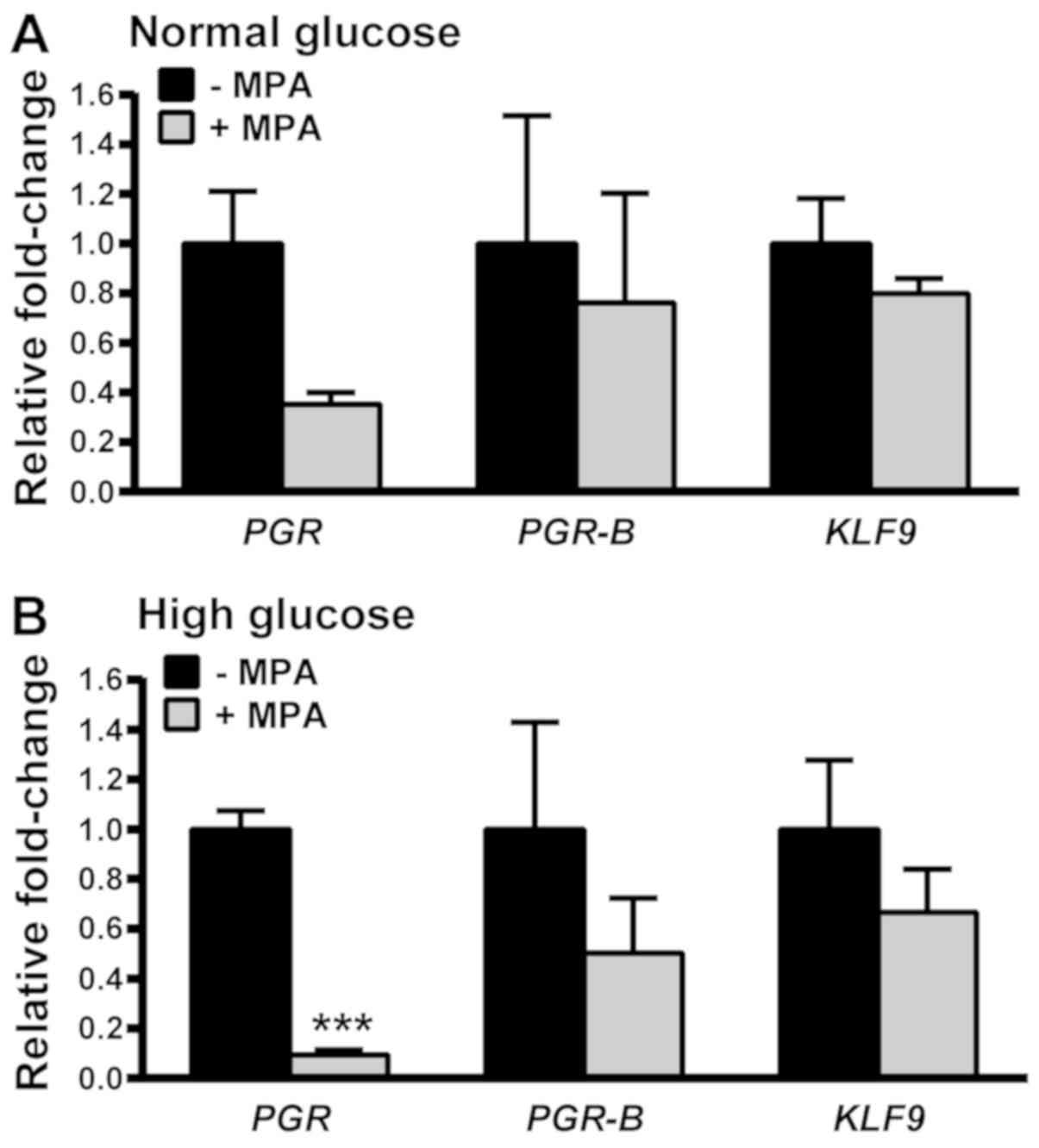
MPA-mediated downregulation of PGR
expression is greater with MET treatment under high compared with
normal glucose conditions
Treatment with progestins can often lead to
resistance over time due to progestin-mediated downregulation
and/or desensitization of its receptor (28,29). To
determine if MET alters the progestin sensitivity of EC cells by
inhibiting PGR downregulation, cells were treated with MPA
after exposure to E2/MET in normal and high glucose environments.
KLF9 mRNA levels were not altered by MPA co-treatment under
normal and high glucose conditions. Total PGR mRNA levels
were significantly decreased by MPA under high glucose but not
normal glucose conditions (Fig. 7).
Notably, no corresponding changes in PGR-B transcript levels
were noted under either glucose condition. These results suggest
that MET promotes the MPA-induced downregulation of PGR-A
mRNA under high glucose conditions.
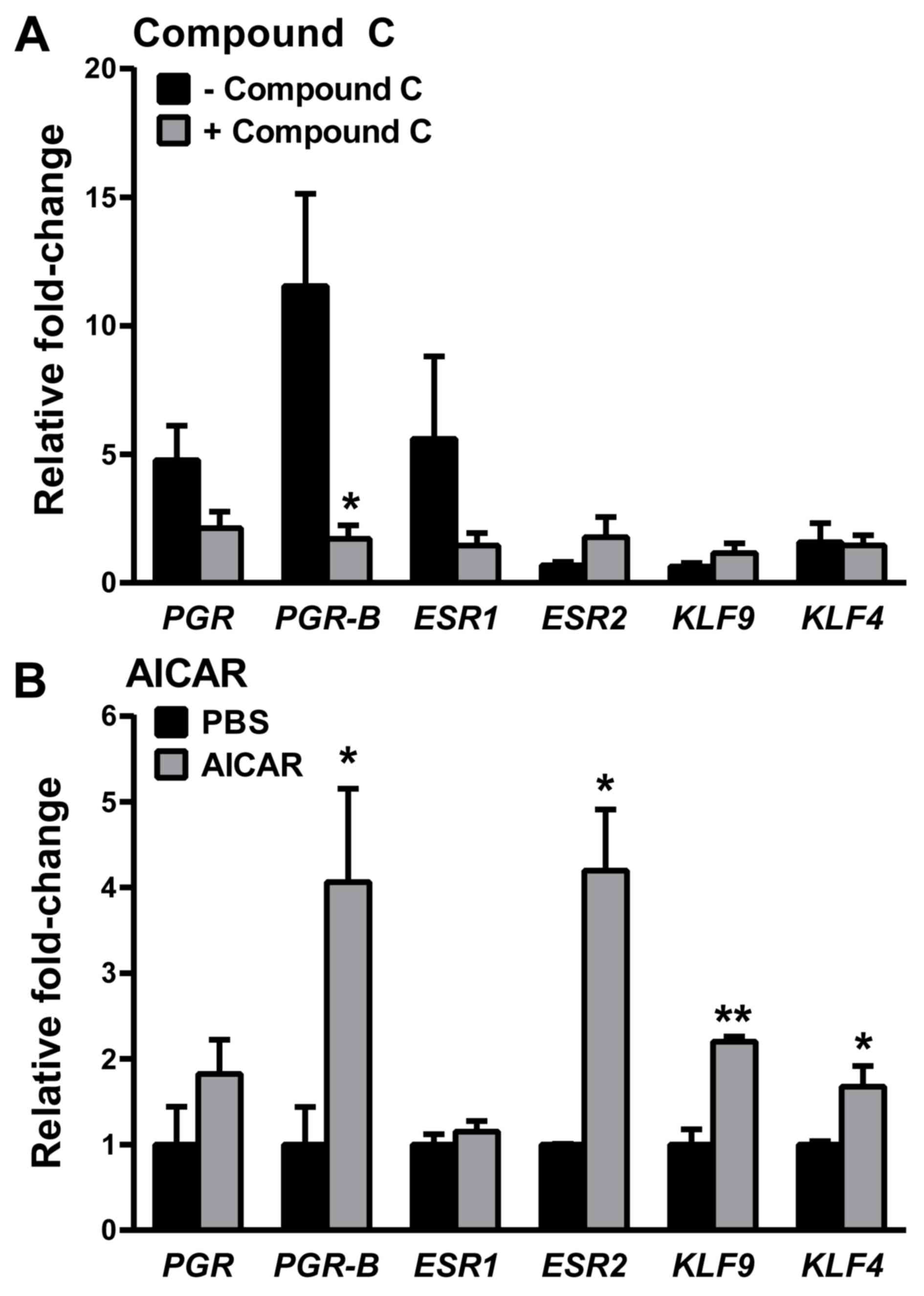
MET effects on PGR gene expression are
partly AMPK-dependent
In EC cells, supra-physiological doses of MET have
been shown to activate AMPK, leading to inhibition of the mTOR
signaling pathway and resulting in decreased cellular protein
synthesis and proliferation (30–32). To
determine whether the effects of physiologically relevant doses of
MET on PGR signaling under normal glucose conditions are mediated
by the AMPK pathway, PGR expression levels in the presence
of the AMPK inhibitor Compound C (33) were evaluated. Consistent with the
aforementioned data (Fig. 4A and B),
the MET-induced increase in transcript levels for total PGR, while
significant, was less robust than that for PGR-B (~5- vs.
~12-fold). Moreover, while there was a marked reduction in
PGR-B expression with Compound C, no significant reduction
was found in total PGR expression (Fig. 8A). When cells were treated with
AICAR, an AMPK-activator (34),
instead of MET, transcript levels of PGR-B but not total
PGR were increased (Fig. 8B),
albeit the fold increase (~4-fold) was lower compared with that
achieved with MET (~25-fold; Fig.
4B). However, AICAR demonstrated a broader range of gene
targets with increased levels of estrogen receptor 2 (ESR2),
KLF9 and KLF4 (Fig.
8B), which were not significantly affected by MET (Fig. 5 for KLF9 and KLF4;
ESR2, data not shown) and by the inhibition of MET-mediated
AMPK signaling (Fig. 8A).
Discussion
MET as an adjuvant in cancer treatments is
increasingly gaining support, based on population studies
demonstrating a reduction in cancer incidence and mortality in
patients with type 2 diabetes taking MET (7). Nevertheless, the benefit of MET in the
management of EC remains largely unknown, given limited clinical
studies to date and the lack of an established causal relationship
between physiologically relevant MET exposure and the EC genotype
and phenotype in patients within normal and diabetic glucose
ranges. The present study compared the proliferation, stem
cell-like growth/self-renewal and expression of selected genes in
the well-differentiated Ishikawa EC cell line treated with E2 and
exposed to physiologically-relevant MET doses (µM) in the presence
of normal and high concentrations of glucose. The results showed
that MET reduced EC cell numbers and spheroid formation under
normal glucose conditions, and that these MET-elicited changes were
lost when glucose levels were elevated. The reduction in
CNND1 transcript levels and the inhibition of stem cell-like
activity with MET in normal but not high glucose conditions,
respectively indicate that MET has inhibitory effects on EC cell
cycle progression and metastatic potential (25,35),
which predict better outcomes with MET in non-diabetic patients
with EC compared with their diabetic counterparts. Moreover, it was
found that while MET increased total PGR transcript levels,
irrespective of the level of glucose exposure, MET preferentially
induced PGR-B transcript levels under normal glucose
conditions, and conversely, those of PGR-A under high
glucose conditions. These findings suggest that in non-diabetic
compared with diabetic patients with EC, MET may elicit distinct
effects on progestin signaling, which differ according to their
mediation by PGR-B and PGR-A isoforms. A role for MET in progestin
signaling is supported by a previous study demonstrating that the
PI3K/AKT/mTOR pathway promotes progestin resistance in EC cells
(22). Furthermore, the present
study showed that the ability of MET to enhance progestin signaling
via the induction of PGR-B transcript levels in EC cells is
partly but not entirely dependent on AMPK activation. The present
results indicate that the direct effects of MET on EC are cellular
context-dependent, and suggest that the potential use of MET as an
adjuvant in EC management involving progestin therapy may involve
its induction of PGR-B expression via AMPK and other yet
unknown signaling pathways.
The present study modeled the effects of MET in
non-diabetic and diabetic patients with EC using MEM (normal
glucose) and DMEM (high glucose), respectively. While MEM and DMEM
have differences in several constituents, including their levels of
vitamins and amino acids, the major difference is their respective
glucose concentrations, which we hypothesize to have more
consequential effects on cellular metabolic status. In a previous
study to evaluate the effects of MET in EC cells, MEM and DMEM were
similarly used to mimic normal and hyper-insulinemic conditions
(36). However, the present results
are likely to be more physiologically relevant, since the MET doses
used were in the µM range rather than the supra-physiological (mM)
doses used in earlier studies (22,23). The
present findings align with those reported in triple-negative
breast cancer cell lines wherein the antiproliferative effects of
MET were enhanced and inhibited in glucose-starved and
glucose-excess conditions, respectively (10). The present study also found that the
formation of EC cell spheroids, referred to as endospheres and
considered to display cancer stem cell-like activity and metastatic
potential (25,35), was differentially responsive to MET,
dependent on cellular glucose. MET alone was shown to inhibit
endosphere growth (P1) and regeneration (P2) under normal glucose
conditions. By contrast, under high glucose conditions, Ishikawa
spheroid formation was unresponsive to MET, paralleling the lack of
MET-elicited antiproliferative response for these cells.
MET-induced inhibition of spheroid growth in the presence of a
normal glucose concentration has been similarly demonstrated in
breast, ovarian and colon cancer cell lines (37). Currently, there is no explanation for
the E2-induced abrogation of the effects of MET on the formation of
spheroids under normal glucose conditions, given that endometrial
epithelial stem/progenitor cells are reported to lack estrogen
receptor expression (38).
The in vivo relevance of PGR signaling as a
MET target is supported by our previous study showing the induction
of total PGR protein levels in the endometrial tumors of
non-diabetic patients with EC following short-term pre-surgical MET
treatment (13). The present study
reports the novel finding that changes in glucose conditions modify
the effects of MET on PGR signaling. While MET increased total
PGR transcript levels, irrespective of the glucose
environment, MET under normal glucose conditions preferentially
induced PGR-B isoform transcript levels, whereas under high
glucose conditions it increased PGR-A isoform transcript
levels. Given the pro-differentiation activity of PGR-B and the
function of PGR-A as a repressor of PGR-B transcriptional activity
(39), changes in their expression
ratios can significantly impact EC phenotype and gene signaling
networks. Thus, the loss of inhibitory effects of MET on cell
proliferation and endosphere formation in the presence of high
concentrations of glucose may reflect the less-differentiated state
of EC cells as a consequence of predominant PGR-A isoform activity.
Another study also reported the upregulation of PGR-B mRNA
and protein levels by MET, albeit at supra-physiological doses, in
Ishikawa and HEC-1A cells under normal glucose conditions (40). Given that MET-responsive Ishikawa
cells express PGR, we postulate that this mechanism of MET will not
be applicable to triple-negative breast cancer cells, further
affirming the pleiotropic context-dependent signaling of MET in
distinct cancer types. While the PGR-A and PGR-B protein levels
corresponding to changes in the PGR-B and PGR-A
isoform transcript levels were not quantified in the present study,
previous studies (27,41,42) have
demonstrated that changes in the respective PGR transcript
levels are closely recapitulated in their protein levels and
transcriptional activity.
The influence of the glucose environment on the
ability of MET to modify EC cell response, based on two parameters
related to PGR signaling, namely KLF9 expression and response to
MPA administration, was evaluated. Our previous study showed that
KLF9 is a PGR-B interacting protein (43,44) and
that human EC tumors demonstrate loss of KLF9 expression (21). The present study found that the
transfection of Ishikawa cells with siKLF9, under conditions
resulting in the reduction of KLF9 protein levels (13), caused an increase in total PGR
transcript levels without a corresponding change in PGR-B
transcript levels, under normal and high glucose conditions. The
indicated increase in PGR-A isoform transcript levels with
reduced KLF9 expression suggests that in more advanced
tumors, in which KLF9 expression is already significantly
attenuated, MET may not positively affect EC outcome, irrespective
of the glucose environment. In the context of MPA administration
and a high glucose environment, MET appeared to amplify the
reduction of PGR-A isoform levels, given the lack of change
in PGR-B isoform transcript levels. Whether the increase in
PGR-B relative to PGR-A expression with MPA/MET
treatments is functionally consequential to EC remains to be
determined. However, in a previous study the treatment of Ishikawa
cells expressing only PGR-B with progestin resulted in complete
growth inhibition, while those expressing only PGR-A showed only
50% growth inhibition (45). Thus,
by favoring the expression of PGR-B over PGR-A under
high glucose conditions, MET may synergize with MPA to increase
progestin-sensitivity and thereby favor cell differentiation at the
expense of cell proliferation, with significant relevance to
progestin therapy for patients with EC.
Using two pharmacological agents, namely Compound C
and AICAR, to inhibit and stimulate AMPK-signaling, respectively, a
substantial reduction (~90%) of PGR-B expression with
Compound C and more modest induction of PGR-B transcript
levels with AICAR were demonstrated. Indeed, the induction of
PGR-B transcript levels by AICAR (~5-fold) is ~2-fold lower
in magnitude than that by MET (12–15-fold), suggesting that MET may
utilize other pathways to promote PGR-B transcript levels.
In this regard, Compound C has been reported to inhibit the
activities of other kinases, in addition to AMPK, suggesting a role
for MET in the regulation of multiple kinase activities in target
cells. The ability of AICAR to increase ESR2, KLF9 and
KLF4 transcript levels in the absence of similar effects by
MET under the same experimental conditions (Fig. 5; data not shown for ESR2) may
be attributable to pathways other than AMPK that have been
implicated in AICAR signaling (46,47).
Taken together, the present results are suggestive of
AMPK-dependent and -independent actions of MET on EC cells, as has
been shown for other systems such as T cells, retinal epithelial
cells and mouse embryonic fibroblasts (46,48),
which may contribute to the preferential inductive effect of MET on
PGR-B transcript levels.
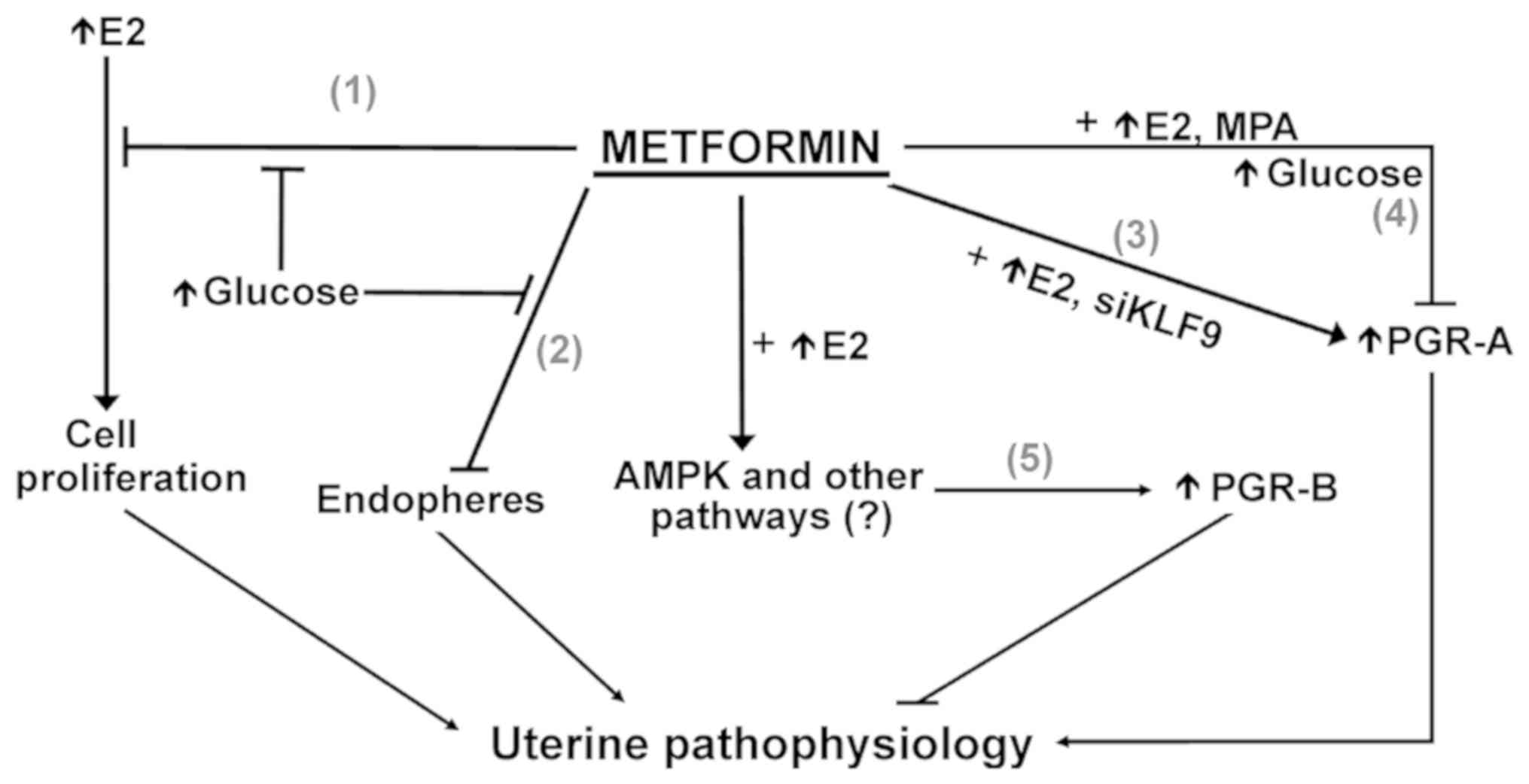
In conclusion, the present study underscores the
context-dependent effects of MET in its selective targeting of
PGR-B isoform transcript levels in EC cells (Fig. 9). Given the current global pandemic
of diabetes and the increasing focus on MET as a viable adjuvant
for cancer management, it is essential to understand the context
underlying the favorable effects of MET on EC. The present data
indicate that the PGR signaling pathway may constitute an important
and novel MET target to increase the progestin sensitivity of EC
cells. This may provide an opportunity for the early prevention of
EC and other progestin-dependent gynecological pathologies in women
with increased risks.
Acknowledgements
Not applicable.
Funding
This study was funded by the University of the
Philippines Office of the Vice-President for Academic
Affairs-Emerging Interdisciplinary Research (grant no.
EIDR-C08-006-MCV/RCMS), the University of Arkansas Barton Endowment
Funds (RCMS) and the National Institutes of Health (RCMS; grant no.
RO1HD21961). Training and internship (SJUS) were sponsored by the
University of the Philippines Office of Internet
Linkages-Continuous Operational and Outcomes-Bases Partnership for
Excellence in Research and Academic Training Enhancement and the
Department of Science and Technology Accelerated Science and
Technology Human Resource Development Program National Science
Consortium, Philippines. IA is a recipient of the 2019 Summer
Fellowship from The Endocrine Society (USA).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
RCMS and MCV supervised the study; SJUS, RCMS and
MCV conceived and designed the experiments; SJUS performed the
experiments; SJUS and IA analyzed the data; SJUS, RCMS and MCV
wrote the manuscript; SJUS, IA, RCMS and MCV revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Howlader N, Noone AM, Krapcho M, Miller D,
Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, et al:
SEER Cancer Statistics Review, 1975–2017, based on November 2019
SEER data submission. National Cancer Institute; Bethesda, MD:
2020, https://seer.cancer.gov/csr/1975_2017/April
15–2020
|
|
2
|
Friberg E, Mantzoros CS and Wolk A:
Diabetes and risk of endometrial cancer: A population-based
prospective cohort study. Cancer Epidemiol Biomarkers Prev.
16:276–280. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kitson SJ, Evans DG and Crosbie EJ:
Identifying high-risk women for endometrial cancer prevention
strategies: Proposal of an endometrial cancer risk prediction
model. Cancer Prev Res (Phila). 10:1–13. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ohkuma T, Peters SAE and Woodward M: Sex
differences in the association between diabetes and cancer: A
systematic review and meta-analysis of 121 cohorts including 20
million individuals and one million events. Diabetologia.
61:2140–2154. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Saeedi P, Petersohn I, Salpea P, Malanda
B, Karuranga S, Unwin N, Colagiuri S, Guariguata L, Motala AA,
Ogurtsova K, et al IDF Diabetes Atlas Committee, : Global and
regional diabetes prevalence estimates for 2019 and projections for
2030 and 2045: Results from the International Diabetes Federation
Diabetes Atlas, 9th edition. Diabetes Res Clin Pract.
157:1078432019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abarca-Gómez L, Abdeen ZA, Hamid ZA,
Abu-Rmeileh NM, Acosta-Cazares B, Acuin C, Adams RJ, Aekplakorn W,
Afsana K, Aguilar-Salinas CA, et al NCD Risk Factor Collaboration
(NCD-RisC), : Worldwide trends in body-mass index, underweight,
overweight, and obesity from 1975 to 2016: A pooled analysis of
2416 population-based measurement studies in 128·9 million
children, adolescents, and adults. Lancet. 390:2627–2642. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Roshan MH, Shing YK and Pace NP: Metformin
as an adjuvant in breast cancer treatment. SAGE Open Med.
7:20503121198651142019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Davies G, Lobanova L, Dawicki W, Groot G,
Gordon JR, Bowen M, Harkness T and Arnason T: Metformin inhibits
the development, and promotes the resensitization, of
treatment-resistant breast cancer. PLoS One. 12:e01871912017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shi P, Liu W, Tala, Wang H, Li F, Zhang H,
Wu Y, Kong Y, Zhou Z, Wang C, et al: Metformin suppresses
triple-negative breast cancer stem cells by targeting KLF5 for
degradation. Cell Discov. 3:170102017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Varghese S, Samuel SM, Varghese E, Kubatka
P and Büsselberg D: High Glucose Represses the Anti-Proliferative
and Pro-Apoptotic Effect of Metformin in Triple Negative Breast
Cancer Cells. Biomolecules. 9:162019. View Article : Google Scholar
|
|
11
|
Ariaans G, Jalving M, Vries EG and Jong S:
Anti-tumor effects of everolimus and metformin are complementary
and glucose-dependent in breast cancer cells. BMC Cancer.
17:2322017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Oliveras-Ferraros C, Vazquez-Martin A,
Cuyàs E, Corominas-Faja B, Rodríguez-Gallego E, Fernández-Arroyo S,
Martin-Castillo B, Joven J and Menendez JA: Acquired resistance to
metformin in breast cancer cells triggers transcriptome
reprogramming toward a degradome-related metastatic stem-like
profile. Cell Cycle. 13:1132–1144. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pabona JMP, Burnett AF, Brown DM, Quick
CM, Simmen FA, Montales MTE, Liu SJ, Rose T, Alhallak I, Siegel ER,
et al: Metformin Promotes Anti-tumor Biomarkers in Human
Endometrial Cancer Cells. Reprod Sci. 27:267–277. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Soliman PT, Zhang Q, Broaddus RR, Westin
SN, Iglesias D, Munsell MF, Schmandt R, Yates M, Ramondetta L and
Lu KH: Prospective evaluation of the molecular effects of metformin
on the endometrium in women with newly diagnosed endometrial
cancer: A window of opportunity study. Gynecol Oncol. 143:466–471.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Laskov I, Drudi L, Beauchamp M-C, Yasmeen
A, Ferenczy A, Pollak M and Gotlieb WH: Anti-diabetic doses of
metformin decrease proliferation markers in tumors of patients with
endometrial cancer. Gynecol Oncol. 134:607–614. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Caillé G, Lacasse Y, Raymond M, Landriault
H, Perrotta M, Picirilli G, Thiffault J and Spénard J:
Bioavailability of metformin in tablet form using a new high
pressure liquid chromatography assay method. Biopharm Drug Dispos.
14:257–263. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wilcock C and Bailey CJ: Accumulation of
metformin by tissues of the normal and diabetic mouse. Xenobiotica.
24:49–57. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Martin-Castillo B, Vazquez-Martin A,
Oliveras-Ferraros C and Menendez JA: Metformin and cancer: Doses,
mechanisms and the dandelion and hormetic phenomena. Cell Cycle.
9:1057–1064. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pabona JM, Simmen FA, Nikiforov MA, Zhuang
D, Shankar K, Velarde MC, Zelenko Z, Giudice LC and Simmen RC:
Krüppel-like factor 9 and progesterone receptor coregulation of
decidualizing endometrial stromal cells: Implications for the
pathogenesis of endometriosis. J Clin Endocrinol Metab.
97:E376–E392. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Δ Δ C(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Simmons CD, Pabona JM, Heard ME, Friedman
TM, Spataro MT, Godley AL, Simmen FA, Burnett AF and Simmen RC:
Krüppel-like factor 9 loss-of-expression in human endometrial
carcinoma links altered expression of growth-regulatory genes with
aberrant proliferative response to estrogen. Biol Reprod.
85:378–385. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu Z, Qi S, Zhao X, Li M, Ding S, Lu J
and Zhang H: Metformin inhibits 17β-estradiol-induced
epithelial-to-mesenchymal transition via βKlotho-related ERK1/2
signaling and AMPKα signaling in endometrial adenocarcinoma cells.
Oncotarget. 7:21315–21331. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang J, Xu H, Zhou X, Li Y, Liu T, Yin X
and Zhang B: Role of metformin in inhibiting estrogen-induced
proliferation and regulating ERα and ERβ expression in human
endometrial cancer cells. Oncol Lett. 14:4949–4956. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Deane JA, Cousins FL and Gargett CE:
Endometrial organoids: in vitro models for endometrial research and
personalized medicine. Biol Reprod. 97:781–783. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kitson SJ, Rosser M, Fischer DP, Marshall
KM, Clarke RB and Crosbie EJ: Targeting endometrial cancer stem
cell activity with metformin is inhibited by patient-derived
adipocyte-secreted factors. Cancers (Basel). 11:6532019. View Article : Google Scholar
|
|
26
|
Hirsch HA, Iliopoulos D, Tsichlis PN and
Struhl K: Metformin selectively targets cancer stem cells, and acts
together with chemotherapy to block tumor growth and prolong
remission. Cancer Res. 69:7507–7511. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Velarde MC, Zeng Z, McQuown JR, Simmen FA
and Simmen RCM: Kruppel-like factor 9 is a negative regulator of
ligand-dependent estrogen receptor α signaling in Ishikawa
endometrial adenocarcinoma cells. Mol Endocrinol. 21:2988–3001.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim JJ and Chapman-Davis E: Role of
progesterone in endometrial cancer. Semin Reprod Med. 28:81–90.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang Y, Wang Y, Zhang Z, Park JY, Guo D,
Liao H, Yi X, Zheng Y, Zhang D, Chambers SK, et al: Mechanism of
progestin resistance in endometrial precancer/cancer through
Nrf2-AKR1C1 pathway. Oncotarget. 7:10363–10372. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cantrell LA, Zhou C, Mendivil A, Malloy
KM, Gehrig PA and Bae-Jump VL: Metformin is a potent inhibitor of
endometrial cancer cell proliferation--implications for a novel
treatment strategy. Gynecol Oncol. 116:92–98. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hanna RK, Zhou C, Malloy KM, Sun L, Zhong
Y, Gehrig PA and Bae-Jump VL: Metformin potentiates the effects of
paclitaxel in endometrial cancer cells through inhibition of cell
proliferation and modulation of the mTOR pathway. Gynecol Oncol.
125:458–469. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Viollet B, Guigas B, Sanz Garcia N,
Leclerc J, Foretz M and Andreelli F: Cellular and molecular
mechanisms of metformin: An overview. Clin Sci (Lond). 122:253–270.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou G, Myers R, Li Y, Chen Y, Shen X,
Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al: Role of
AMP-activated protein kinase in mechanism of metformin action. J
Clin Invest. 108:1167–1174. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Corton JM, Gillespie JG, Hawley SA and
Hardie DG: 5-aminoimidazole-4-carboxamide ribonucleoside. A
specific method for activating AMP-activated protein kinase in
intact cells? Eur J Biochem. 229:558–565. 1995.PubMed/NCBI
|
|
35
|
Carvalho MJ, Laranjo M, Abrantes AM,
Casalta-Lopes J, Sarmento-Santos D, Costa T, Serambeque B, Almeida
N, Gonçalves T, Mamede C, et al: Endometrial Cancer Spheres Show
Cancer Stem Cells Phenotype and Preference for Oxidative
Metabolism. Pathol Oncol Res. 25:1163–1174. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
de Barros Machado A, Dos Reis V, Weber S,
Jauckus J, Brum IS, von Eye Corleta H, Strowitzki T, Capp E and
Germeyer A: Proliferation and metastatic potential of endometrial
cancer cells in response to metformin treatment in a high versus
normal glucose environment. Oncol Lett. 12:3626–3632. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hu T, Chung YM, Guan M, Ma M, Ma J, Berek
JS and Hu MCT: Reprogramming ovarian and breast cancer cells into
non-cancerous cells by low-dose metformin or SN-38 through FOXO3
activation. Sci Rep. 4:58102014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Janzen DM, Cheng D, Schafenacker AM, Paik
DY, Goldstein AS, Witte ON, Jaroszewicz A, Pellegrini M and
Memarzadeh S: Estrogen and progesterone together expand murine
endometrial epithelial progenitor cells. Stem Cells. 31:808–822.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vegeto E, Shahbaz MM, Wen DX, Goldman ME,
O'Malley BW and McDonnell DP: Human progesterone receptor A form is
a cell- and promoter-specific repressor of human progesterone
receptor B function. Mol Endocrinol. 7:1244–1255. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xie Y, Wang YL, Yu L, Hu Q, Ji L, Zhang Y
and Liao QP: Metformin promotes progesterone receptor expression
via inhibition of mammalian target of rapamycin (mTOR) in
endometrial cancer cells. J Steroid Biochem Mol Biol. 126:113–120.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Graham JD, Yager ML, Hill HD, Byth K,
O'Neill GM and Clarke CL: Altered progesterone receptor isoform
expression remodels progestin responsiveness of breast cancer
cells. Mol Endocrinol. 19:2713–2735. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mouttet D, Laé M, Caly M, Gentien D,
Carpentier S, Peyro-Saint-Paul H, Vincent-Salomon A, Rouzier R,
Sigal-Zafrani B, Sastre-Garau X, et al: Estrogen-Receptor,
Progesterone-Receptor and HER2 Status Determination in Invasive
Breast Cancer. Concordance between Immuno-Histochemistry and
MapQuant™ Microarray Based Assay. PLoS One. 11:e01464742016.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang D, Zhang XL, Michel FJ, Blum JL,
Simmen FA and Simmen RCM: Direct interaction of the Krüppel-like
family (KLF) member, BTEB1, and PR mediates progesterone-responsive
gene expression in endometrial epithelial cells. Endocrinology.
143:62–73. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang XL, Zhang D, Michel FJ, Blum JL,
Simmen FA and Simmen RCM: Selective interactions of Kruppel-like
factor 9/basic transcription element-binding protein with
progesterone receptor isoforms A and B determine transcriptional
activity of progesterone-responsive genes in endometrial epithelial
cells. J Biol Chem. 278:21474–21482. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Smid-Koopman E, Blok LJ, Kühne LCM, Burger
CW, Helmerhorst TJM, Brinkmann AO and Huikeshoven FJ: Distinct
functional differences of human progesterone receptors A and B on
gene expression and growth regulation in two endometrial carcinoma
cell lines. J Soc Gynecol Investig. 10:49–57. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rao E, Zhang Y, Li Q, Hao J, Egilmez NK,
Suttles J and Li B: AMPK-dependent and independent effects of AICAR
and compound C on T-cell responses. Oncotarget. 7:33783–33795.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhao X, Luo G, Cheng Y, Yu W, Chen R, Xiao
B, Xiang Y, Feng C, Fu W, Duan C, et al: Compound C induces
protective autophagy in human cholangiocarcinoma cells via
Akt/mTOR-independent pathway. J Cell Biochem. 119:5538–5550. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Philippe C, Pinson B, Dompierre J,
Pantesco V, Viollet B, Daignan-Fornier B and Moenner M: AICAR
Antiproliferative Properties Involve the AMPK-Independent
Activation of the Tumor Suppressors LATS 1 and 2. Neoplasia.
20:555–562. 2018. View Article : Google Scholar : PubMed/NCBI
|