Introduction
Mucinous cystic neoplasms (MCNs) are one of the most
common types of cystic neoplasms of the pancreas that specifically
occur in the pancreatic body or tail of perimenopausal women
(1). With the continuous application
and progress of cross-sectional imaging technology, the number of
patients diagnosed with pancreatic MCNs has increased over time
(2). In the 1970s, Compagno and
Oertel (3) proposed that pancreatic
MCNs eventually become malignant as the disease progresses. A
multicenter retrospective analysis reported that MCC or high-grade
dysplasia is present in 14.9% of resected pancreatic MCNs (4). The 5-year survival rate of invasive
pancreatic MCC is <20%, which is similar to that of ductal
adenocarcinoma of the pancreas (5).
Due to its malignant potential, a number of studies have focused on
pancreatic MCNs (5–7). However, few studies have explored the
molecular mechanism underlying the development of invasive
pancreatic MCC.
Autophagy, which is a highly conserved catabolic
process, is critical for cellular homeostasis under normal
physiological conditions, as well as for cell viability under
stress. The mammalian target of rapamycin (mTOR)/AMP-activated
protein kinase pathway serves an essential role in regulating this
process (8,9). Under physiological conditions,
autophagy is considered to inhibit the malignant transformation of
cells by maintaining cell homeostasis and normal metabolism
(10). With the exception of
autophagy-mediated suppression in malignant transformation of
normal cells, autophagy can facilitate tumorigenesis and generate
resistance to anti-tumor therapy under adverse microenvironment
conditions (10). Previous studies
have suggested that autophagy regulates tumor metastasis by
affecting local adhesion, anti-anoikis and epithelial-mesenchymal
transition (EMT) (11). However, the
function of autophagy in pancreatic MCC metastasis remains
unclear.
Considering the poor survival associated with
aggressive malignancy, it is necessary to study the potential
regulators of pancreatic MCC metastasis. Based on the widespread
involvement of autophagy in tumor development, its potential for
targeted therapy has been suggested. The purpose of this study was
to investigate the effect of autophagy on the malignant migration
and invasion of MCC1 cells.
Materials and methods
Clinical samples
A total of 4 paired paraffin-embedded pancreatic
mucinous cystadenocarcinoma samples and matched adjacent tissues
were obtained retrospectively from the Department of Pathology,
Changhai Hospital (Shanghai, China) (Table I). The distance between the tumor and
the adjacent tissue sampling point site was 15 mm. All four
patients were female, with a mean age of 58 years, ranging from 47
to 76 years. Tissue samples were rare and all available samples
were used for this study. Informed consent was obtained from the
subjects prior to the collection of the specimens.
 | Table I.Clinicopathological data of four
cases of pancreatic mucinous cystadenocarcinoma. |
Table I.
Clinicopathological data of four
cases of pancreatic mucinous cystadenocarcinoma.
| Patient number | Sex | Age (years) | Pathological
diagnosis | Tumor location | Lymphatic
metastasis |
|---|
| 1 | Female | 76 | Mucinous
cystadenoma of the pancreas with high-grade intraepithelial
neoplasia, carcinogenesis and local infiltration of the stroma | Pancreatic
tail | 0/1 |
| 2 | Female | 47 | Invasive mucinous
cystadenocarcinoma of the pancreas | Pancreatic body and
tail | 0/1 |
| 3 | Female | 47 | Invasive mucinous
cystadenocarcinoma of the pancreas | Pancreatic body and
tail |
|
| 4 | Female | 62 | Pancreatic mucinous
cystic tumor with invasive carcinoma (mucinous
cystadenocarcinoma) | Pancreatic body and
tail | 0/7 |
Cell culture and reagents
Human pancreatic ductal epithelial (HPDE) cells were
obtained from the Type Culture Collection of the Chinese Academy of
Sciences. The cells were cultured with DMEM (Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum
(HyClone; GE Healthcare Life Sciences) and 1%
penicillin/streptomycin (Thermo Fisher Scientific, Inc.). MCC1
cells were kindly provided by Professor Claudio Sorio from the
University of Verona, Italy. The cells were incubated in RPMI-1640
medium (HyClone; GE Healthcare Life Sciences) supplemented with 10%
fetal bovine serum (HyClone; Cyvita), 2 mM glutamine, 80 µg/ml
gentamicin sulfate and 2.5 µg/ml amphotericin B (Sigma-Aldrich;
Merck KGaA). All the cells were incubated at 37°C in a humidified
atmosphere with 5% CO2. Rapamycin and Chloroquine (CQ)
were obtained from Sigma-Aldrich; Merck KGaA.
Transwell migration and invasion
assays
The Transwell migration assay was performed in
24-well inserts with 8-µm pores (Corning, Inc.), whereas the
invasion assay was performed in 24-well inserts with 8-µm pores
coated with Matrigel (Corning, Inc.) according to the
manufacturer's protocols. Cells (8×104 cells/200 µl of
RPMI-1640 medium containing 0.2% bovine serum albumin
(Sigma-Aldrich; Merck KGaA), 2 mM glutamine (Thermo Fisher
Scientific, Inc.), 80 µg/ml gentamicin sulfate (Sigma-Aldrich;
Merck KGaA) and 2.5 µg/ml amphotericin B (Sigma-Aldrich; Merck
KGaA) per insert were seeded into the upper chamber; the bottom
side was filled with culture RPMI-1640 medium containing 10% foetal
bovine serum, 2 mM glutamine, 80 µg/ml gentamicin sulfate and 2.5
µg/ml amphotericin B. Following incubation for 6 h, the inserts
from the migration and invasion assays were fixed with 4%
paraformaldehyde for 30 min at room temperature (25°C) and stained
with 0.1% crystal violet solution for 30 min at room temperature.
Non-migrated cells were removed from the upper membrane using a
cotton swab; the migrated cells on the lower membrane were dried
and imaged using an Olympus IX51 microscope (Olympus Corporation).
The cells were counted using ImageJ software (version 1.44p;
National Institutes of Health).
Small interfering (si)RNA and cell
transfection
siRNA against BCL2 (siBCL2) was purchased from
Guangzhou RiboBio Co., Ltd. The target sequence of BCL2 was as
follows: 5′-GGAGAACAGGGTACGATAA-3′. The sequences of the negative
control primers were forward, 5′-GGCUCUAGAAAAGCCUAUGCdTdT-3′ and
reverse, 3′-dTdTCCGAGAUCUUUUCGGAUACG-5′. The plasmids and siRNAs
were transfected in miR-224-5p inhibitor and negative control MCC1
cells (5×104) using Lipofectamine® 2000
reagent (Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol at room temperature for 25 min. The ratio
of plasmid or siRNA to lipo is 1 µg: 2 µl. Cells were harvested 48
h after transfection. Cells were harvested 48 h after
transfection.
Protein extraction and western blot
assays
Experimental cells (HPDE and MCC1 cells treated with
NCm, miR-224, NCi, inhibitor-224) and MCC1 treated with rapamycin
were lysed with RIPA buffer (Cell Signaling Technology, Inc.)
containing complete protease inhibitor cocktail (Roche
Diagnostics), phosphatase inhibitors (Roche Diagnostics), 5 mM
dithiothreitol (Sigma-Aldrich; Merck KGaA) and 1 mM
phenylmethylsulfonyl fluoride (Sigma-Aldrich; Merck KGaA) and
incubated on ice for 30 min. The cell lysate was centrifuged at
12,000 × g for 10 min at 4°C. The supernatant was collected and
protein concentrations were determined using the BCA protein assay
kit (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Western blot assays were performed as
previously described (12). The
following antibodies were used: anti-GAPDH (cat. no. M2006M,
1:5,000 dilution) and anti-β-actin (cat. no. M2001M, 1:5,000
dilution) (both from Abmart Pharmaceutical Technology Co., Ltd.),
anti-P62 (cat. no. 5114T, 1:2,000 dilution) and horseradish
peroxidase-conjugated secondary antibody (cat. no. 7074S, 1:2,000
dilution) (both from Cell Signaling Technology, Inc.), anti-LC3
(cat. no. L7543, 1:2,000 dilution; Sigma-Aldrich; Merck KGaA) and
anti-BCL2 (cat. no. L5034901, 1:2,000 dilution; BD Biosciences).
ImageJ (version 1.44p; National Institutes of Health) was used for
densitometry analysis.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was isolated using TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.), cDNA was then
generated by reverse transcription of aliquots of RNA using the
Takara PrimeScript RT Reagent kit (Takara Bio, Inc.) according to
the manufacturer's instructions. The resulting cDNA was used for
qPCR with SYBR® Premix Ex Taq™ kit (Takara Bio, Inc.) in
a StepOne Real-Time PCR Detection System (Thermo Fisher Scientific,
Inc.) (13). For micro (mi)RNA
detection, the total RNA was extracted from cells as
aforementioned, and then miRNA was reverse transcribed and
amplified using a Takara PrimeScript RT Reagent kit according to
the manufacturer's instructions. The amplification reactions were
performed in triplicate in a 96-well plate using the following
thermocycling conditions: 5 min at 95°C, followed by 40 cycles of
10 sec at 95°C and 30 sec at 60°C. The Cq values were calculated
using ABI Sequence Detection System software (version 2.1; Thermo
Fisher Scientific, Inc.). The relative fold-change of each miRNA
was calculated using the comparative 2−ΔΔCq method
(14). The primers used for qPCR are
presented in Table II. The
quantitative miRNA data were normalized to U6 and mRNA expression
of BCL2 of 18S.
| Table II.Primer sequences of primers used in
RT-qPCR. |
Table II.
Primer sequences of primers used in
RT-qPCR.
| Primers | Sequence
(5′→3′) |
|---|
| miR-224-5p | F:
CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCAAGGCAA |
|
| R:
ACACTCCAGCTGGGCAAGTCACTAGTGGT |
| U6 | F:
CTCGCTTCGGCAGCACA |
|
| R:
AACGCTTCACGAATTTGCGT |
| BCL2 | F:
GGTGGGGTCATGTGTGTGG |
|
| R:
CGGTTCAGGTACTCAGTCATCC |
| 18S | F:
ACCGCAGCTAGGAATAATGGA |
|
| R:
CAAATGCTTTCGCTCTGGTC |
Vectors and lentiviral
transduction
All recombinant lentiviruses were obtained from
Shanghai Genechem Co., Ltd. The packaged lentiviruses contained
miR-224 mimic-5p (miR-224), miR-224-5p mimic control (NCm),
miR-224-5p inhibitor (inhibitor-224) and miR-224-5p inhibitor
control (NCi). All negative controls were empty vectors. The
sequences were as follows: Hsa-miR-224-5p-mimic forward,
5′-GAGGATCCCCGGGTACCGGCCAGCTAACCATGGGCCTGCCTC-3′ and reverse,
5′-CACACATTCCACAGGCTAGAGGAGAAAGAAGACCTCTTTTC-3′;
hsa-miR-224-5p-inhibitor forward, 5′-CAAGTCACTAGTGGTTCCGTT-3′ and
reverse, 5′-AACGGAACCACTAGTGACTTG-3′. Lentiviral transduction was
performed following the manufacturer's protocol. The packaged virus
solution was thawed, and the virus stock solution was diluted with
the infection medium (MOI) value equal to 10 using fresh medium
containing the gene transfection enhancer polyamine. After 72 h of
infection at room temperature, the transduction efficiency was
evaluated as the percentage of green fluorescent protein-positive
cells observed under an inverted fluorescence microscope
(magnification ×200; Olympus Corporation). The lentivirus-infected
cells were treated with 2 µg/ml puromycin for 1 week, and the cells
resistant to puromycin were selected.
Immunofluorescence and confocal
imaging
Cells (MCC1 cells transfected with
miR-224-5p/inhibitor-224-5p), and corresponding positive and
negative control were plated on coverslips or in chamber slides.
After reaching a suitable density (8×104/ml), the cells
were fixed with 4% paraformaldehyde overnight at 4°C, permeabilized
in 0.5% Triton X-100 for 8 min at room temperature, blocked with
phosphate-buffered saline containing 10% goat serum for 1 h at room
temperature and incubated overnight with anti-LC3 (cat. no. L7543,
1:500 dilution; Sigma-Aldrich; Merck KGaA) at 4°C. Fixed and
stained coverslips or slides were washed 3 times with
phosphate-buffered saline, and labeled with Alexa Fluor-conjugated
secondary antibodies (cat. no. A11036, 1;200 dilution; Invitrogen;
Thermo Fisher Scientific, Inc.). Anti-LC3 antibody was obtained
from Sigma-Aldrich; Merck KGaA. The coverslips or slides were
mounted with ProLong Gold Antifade Reagent mounting medium
containing DAPI (Thermo Fisher Scientific, Inc.), and the cells
were visualized by confocal laser microscopy (Olympus Corporation).
Three random fields of view of each sample were used for
semi-quantification analysis at 60× magnification. ImageJ software
(version 1.44p; National Institutes of Health) was used for
semi-quantified analysis.
Immunohistochemical (IHC)
analysis
Immunohistochemical (IHC) analysis was performed as
previously described (13). The
following primary antibodies were used: Anti-LC3 (cat. no. GB11124,
1:500 dilution) antibody and anti-p62 (cat. no. GB11239, 1:500
dilution) antibody from Wuhan Servicebio Technology Co., Ltd. IHC
evaluation of protein expression intensity in normal adjacent
tissues and paired pancreatic mucinous cystadenocarcinoma tissues
was performed independently by two pathologists from the Department
of Pathology, Changhai Hospital. Staining intensity was scored as
previously described (12). Goat
serum (10%; HyClone; Cyvita) was used in the place of primary
antibody as a negative control.
Target gene prediction and luciferase
reporter assay
The online tool TargetScan 7.2 (http://www.targetscan.org) was used to predict the
miR-224-5p targets. BCL2 was selected as a predicted target of
miR-224-5p. A 219-base pair fragment of the BCL2 3′-untranslated
region (UTR) sequence containing one putative miR-224-5p binding
site was cloned into the pmirGV272-luciferase reporter plasmids
(Shanghai GeneChem Co., Ltd.). GV369-miR-224 mimic-transfected MCC1
cells and GV369-miR-224 mimic control-transfected MCC1 cells were
transfected with the luciferase reporters using
Lipofectamine® 2000. A Thermo Scientific, Inc.
Microplate Reader was used to detect firefly luminescence and
Renilla luminescence. Results were evaluated through
normalization of the firefly luciferase activity with
Renilla luciferase activity as previously described
(15).
Statistical analysis
Data are presented as the mean ± SD from at least
three independent experiments. SPSS 21.0 software (IBM Corp.) was
used to conduct statistical analysis. Student's t-test was used to
compare the differences between two groups; one-way ANOVA followed
by the least significant difference post hoc test was used to
compare the differences among multiple groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
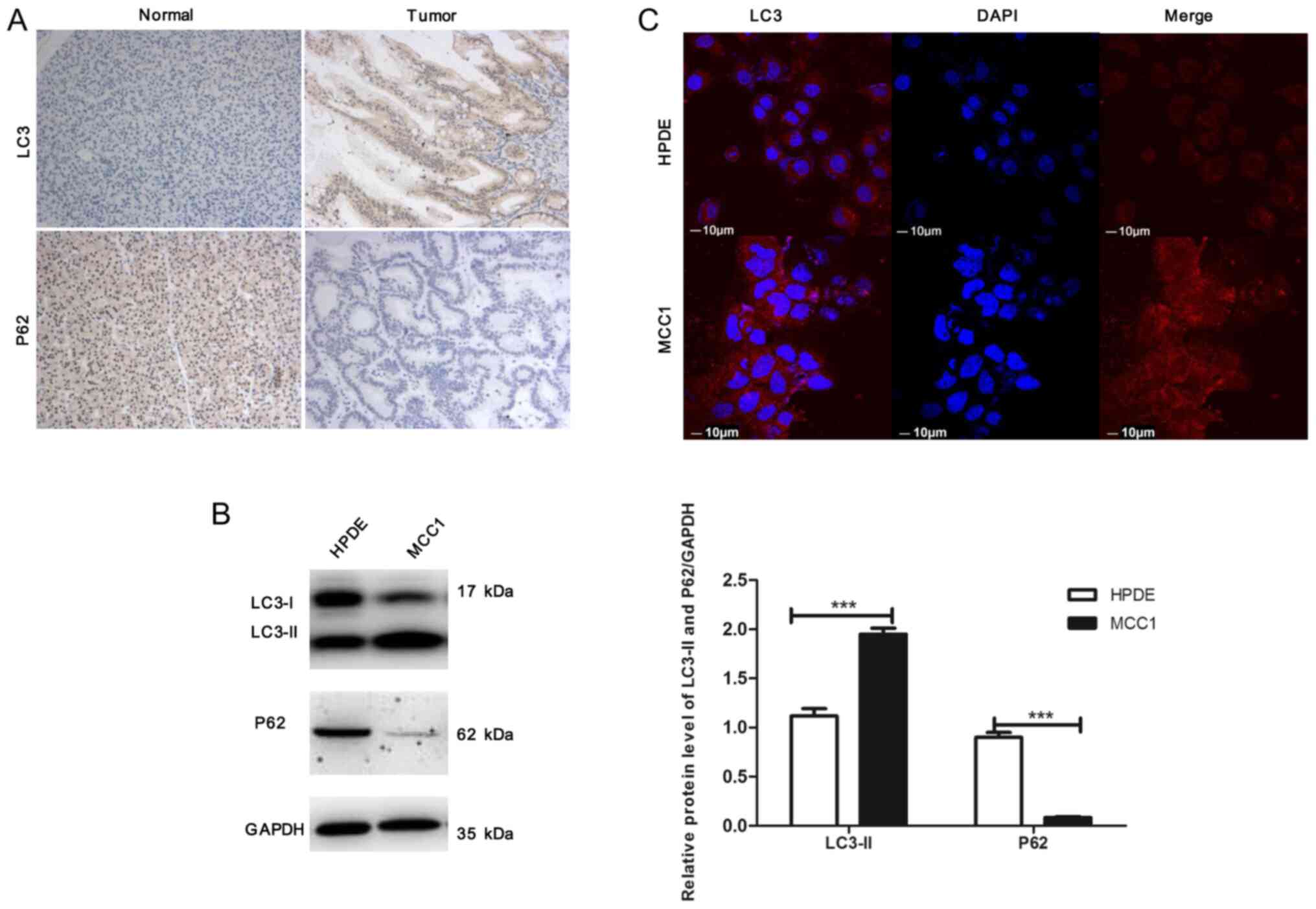
Autophagy is hyperactivated in
pancreatic mucinous cystadenocarcinoma tissues and MCC1
A total of 4 paired pancreatic MCC and adjacent
non-tumor tissues were used for IHC staining with anti-LC3 and
anti-p62 antibodies. The protein expression of LC3 was higher in
the tumors compared with that in adjacent non-tumor tissues,
whereas p62 accumulation was decreased in tumor tissues (Fig. 1A). The level of autophagy signaling
in MCC1 and HPDE cells was determined by western blotting; the
results demonstrated that compared with HPDE cells, the expression
levels of LC3-II were significantly increased in MCC1 cells,
whereas p62 expression levels were notably decreased (Fig. 1B), which was consistent with the
results from clinical samples. Additionally, immunofluorescence
(IF) was performed to detect autophagosomes, which revealed that
the number of LC3 dots (red) in MCC1 cells was increased compared
with HPDE cells (Fig. 1C). These
results indicated that autophagy was hyperactivated in pancreatic
MCC tissues and MCC1 cells.
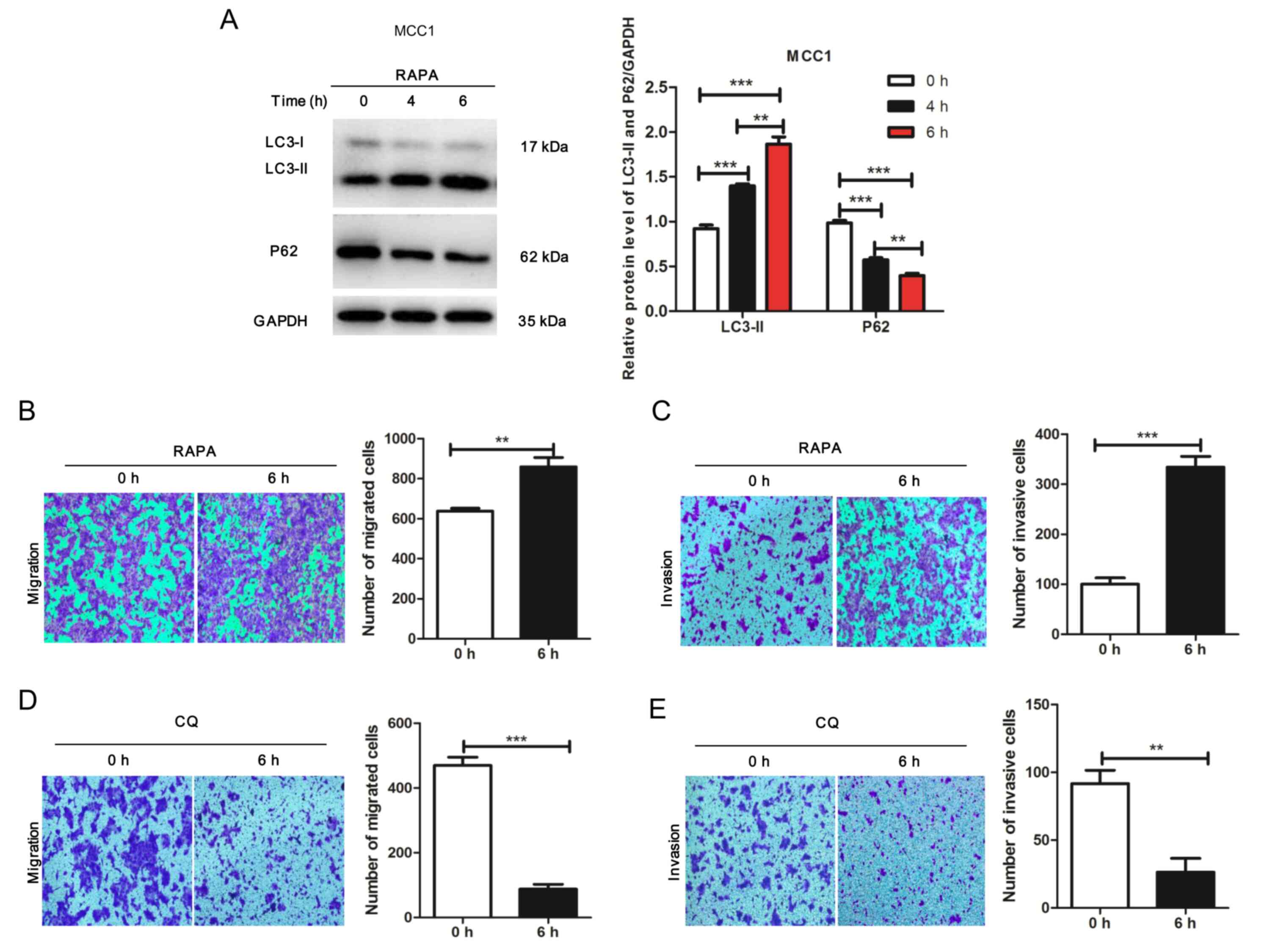
Autophagy promotes the migration and
invasion of MCC1 cells
To investigate the potential effects of autophagy on
the migration and invasion of MCC1 cells, the mTOR inhibitor
rapamycin was used to treat MCC1 for 0, 4 or 6 h. The results
demonstrated that rapamycin induced changes in the expression of
proteins associated with autophagy activity (Fig. 2A). In addition, the results of the
Transwell assay revealed that the migration and invasion of
rapamycin-treated MCC1 cells was significantly enhanced compared
with that in the control groups (Fig. 2B
and C). Additionally, the migration and invasion of MCC1 cells
treated with CQ was significantly decreased compared with that in
the control groups (Fig. 2D and E).
These results demonstrated that autophagy facilitated the migration
and invasion of MCC1 cells.
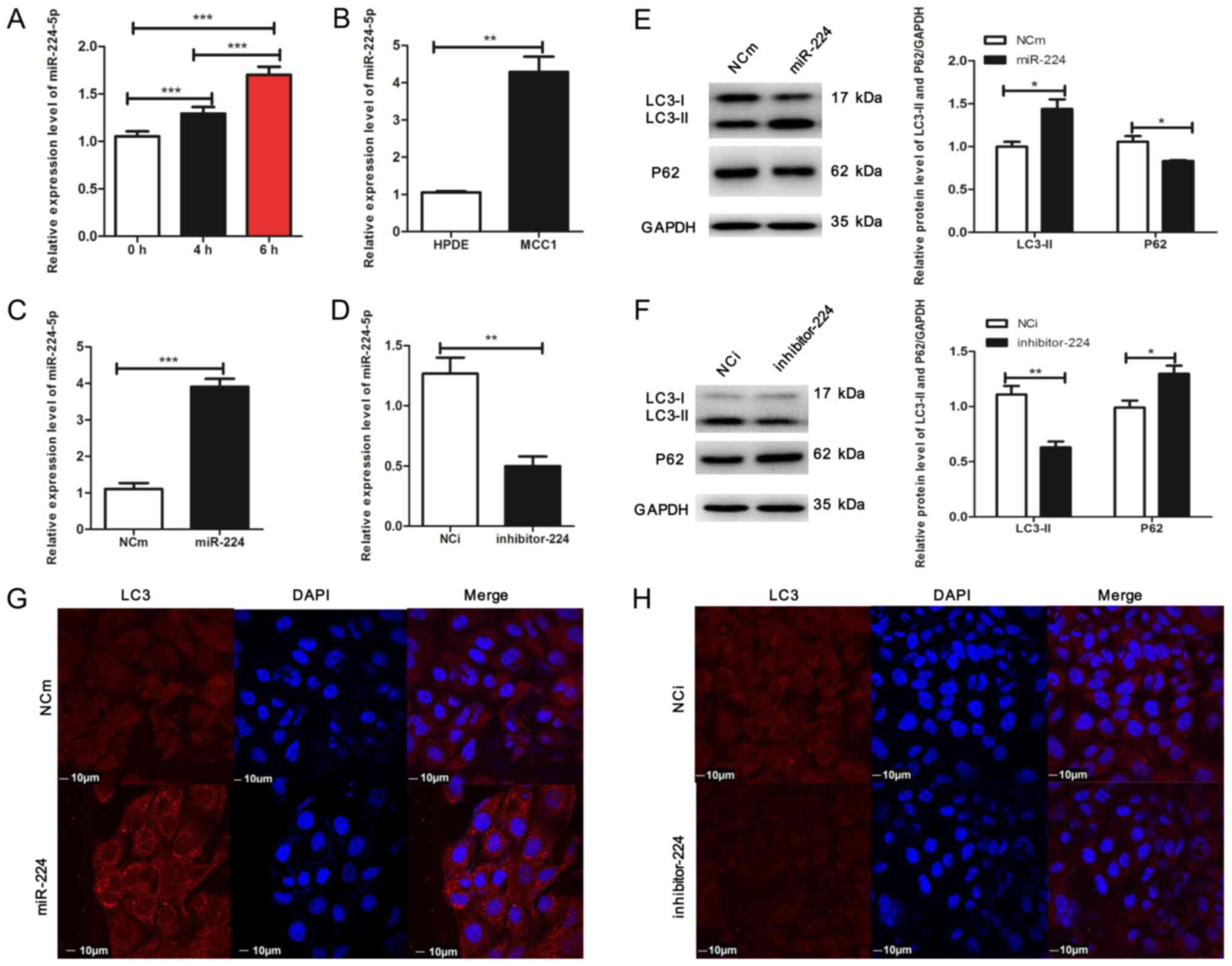
miR-224-5p promotes autophagy in MCC1
cells
The present study examined whether miR-224-5p
expression levels changed in response to stress-induced stimuli in
MCC1. To verify this hypothesis, MCC1 cells were treated with
rapamycin to induce autophagy; the results demonstrated that
miR-224-5p expression levels increased in a time-dependent manner
(Fig. 3A). Additionally, miR-224-5p
expression levels were significantly increased in MCC1 cells
compared with those in HPDE cells (Fig.
3B). Therefore, these results suggested an association between
autophagy activation and miR-224-5p levels in MCC1 cells (Figs. 1B and 3B).
The present study examined whether miR-224-5p
affected autophagy in MCC1 cells. Lentiviruses with a miR-224-5p
mimic or miR-224-5p inhibitor were constructed to interfere with
the level of miR-224-5p in MCC1 cells, and RT-qPCR assay was
performed to assess the expression levels of miR-224-5p in MCC1.
The results revealed overexpression of miR-224-5p in the miR-224-5p
mimic group compared with that in the control group (Fig. 3C). Western blot analysis demonstrated
that compared with the negative control, the expression of LC3-II
was upregulated, whereas that of p62 was reduced in MCC1 cells
overexpressing miR-224-5p (Fig. 3E).
In addition, overexpression of miR-224-5p increased the number of
fluorescent LC3 dots in MCC1 cells compared with that in the
negative control (Fig. 3G), which
indicated the accumulation of autophagosomes. By contrast,
miR-224-5p was downregulated in the presence of the miR-224-5p
inhibitor compared with the negative control group (Fig. 3D). Additionally, the loss of
miR-224-5p inhibited the expression of LC3II and elevated the
expression of p62 compared with that in the negative control
(Fig. 3F). The results of the IF
assay confirmed that the inhibition of miR-224-5p expression
repressed autophagosome accumulation in MCC1 cells compared with
the negative control (Fig. 3H).
Overall, these results demonstrated that autophagy significantly
increased the expression level of miR-224-5p in MCC1 cells and that
miR-224-5p may facilitate autophagy in the pancreatic mucinous
cystadenocarcinoma cell line MCC1, suggesting that positive
feedback may occur between autophagy and miR-224-5p in MCC1.
miR-224-5p promotes autophagy through
BCL2 in MCC1
To explore the molecular mechanisms of
miR-224-5p-mediated autophagy, TargetScan Human 7.2 was used to
screen potential downstream targets of miR-224-5p in autophagy;
BCL2 was identified as a target gene (Fig. 4A). To confirm whether BCL2 was a
direct target of miR-224-5p, a luciferase reporter assay was
performed. The results demonstrated that overexpression of
miR-224-5p reduced luciferase activity in the BCL2 wild-type 3′-UTR
reporter, but had no effect on the BCL2 mutant 3′-UTR reporter
(Fig. 4B). These results suggested
that BCL2 may be a direct target of miR-224-5p. In addition, the
levels of BCL2 were evaluated in MCC1 cells overexpressing
miR-224-5p, which revealed that the miR-224-5p mimic significantly
decreased the mRNA and protein levels of BCL2 (Fig. 4C and D). Consistently, inhibition of
miR-224-5p increased BCL2 expression at the mRNA and protein level
(Fig. 4E and F). To verify the
effects of BCL2 on miR-224-5p-induced autophagy, MCC1 cells were
transfected with siBCL2, which abolished the expression of BCL2
(Fig. 4G). BCL2 silencing
significantly rescued autophagy in cells transfected with the
miR-224 inhibitor, demonstrating that BCL2 protein knockdown
counteracted the effects of the miR-224-5p inhibitor on autophagy
inhibition (Fig. 4H). The expression
levels of BCL2 were also determined in MCC1 and HPDE cells by
western blotting; the results demonstrated that compared with HPDE
cells, the expression levels of BCL2 were reduced in MCC1 cells
(Fig. 4I). The protein expression of
BCL2 was decreased in tumor tissues compared with that in adjacent
non-tumor tissues (Fig. 4J). These
results suggested that miR-224-5p may functionally promote
autophagy through BCL2.
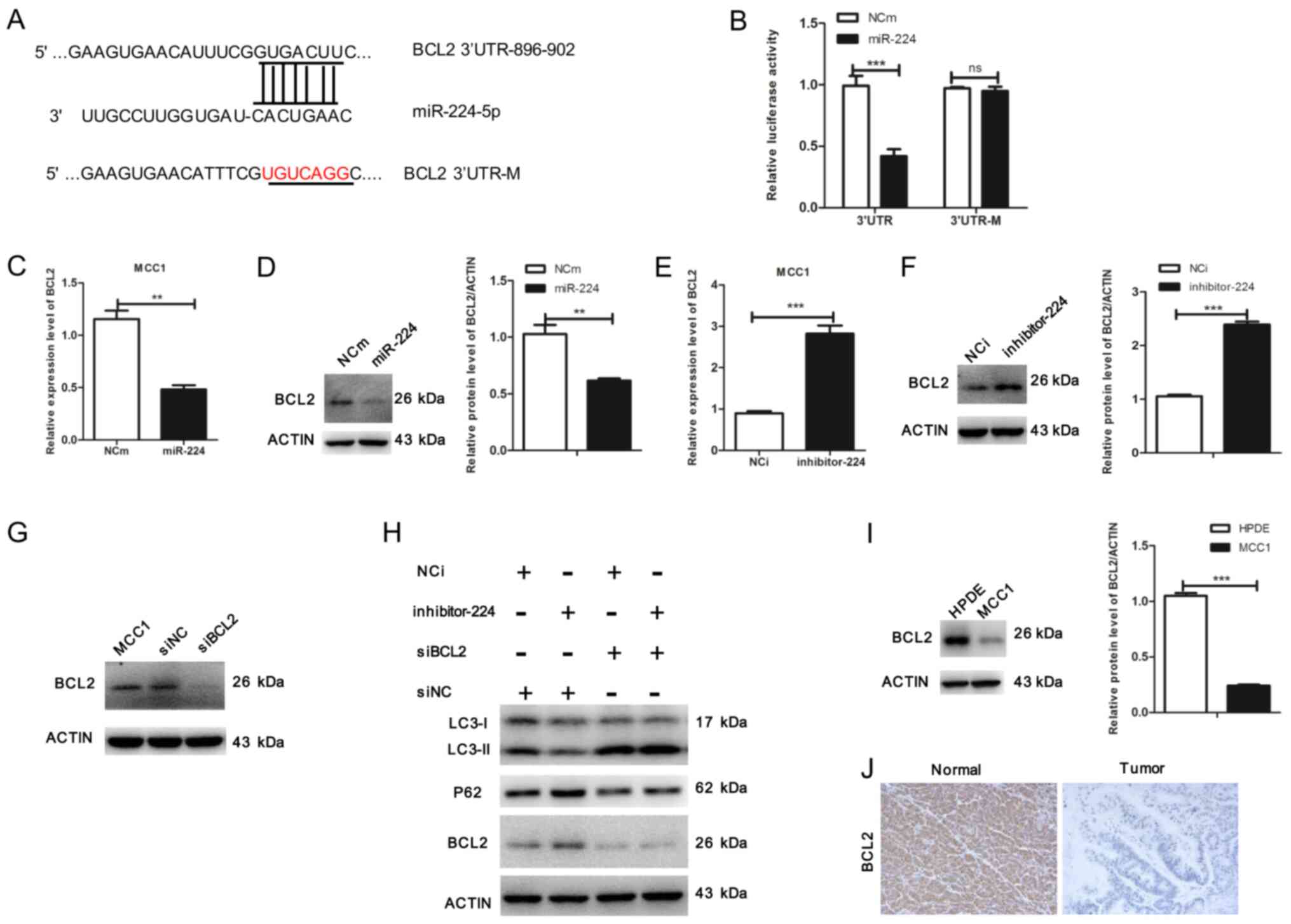 | Figure 4.miR-224-5p promotes autophagy through
BCL2 in MCC1 cells. (A) Predicted binding sequences for miR-224-5p
in the BCL2 3′-UTR. (B) Luciferase reporter vectors containing
wild-type or mutated 3′-UTR fragments of BCL2 cloned into
pmirGV272-luciferase plasmids. Luciferase activity was assayed 48 h
following co-transfection with either wild-type or mutated 3′-UTR
containing plasmids and miR-224-5p mimic or miR-224-5p mimic
control in MCC1 cells. (C) Overexpression of miR-224-5p affected
the expression level of BCL2 in MCC1 determined by reverse
transcription-quantitative PCR. (D) Overexpression of miR-224-5p
affected the protein expression level of BCL2 in MCC1 cells.
Relative protein expression of BCL2 was normalized to the level of
β-actin. (E) Inhibition of miR-224-5p affected the mRNA expression
level of BCL2 in MCC1 cells. (F) Inhibition of miR-224-5p affected
the protein expression level of BCL2 in MCC1 cells. Relative
protein expression of BCL2 was normalized to β-actin. (G) MCC1
cells were transfected with BCL2 siRNA. Western blotting was used
to detect the expression of BCL2. (H) miR-224-5p inhibitor and
miR-224-5p inhibitor control-treated MCC1 cells were transfected
with BCL2 siRNA, and western blot analysis was performed to detect
the expression levels of LC3, P62 and BCL2. (I) Expression of BCL2
was determined by western blotting in HPDE and MCC1 cells. Actin
was used as an internal control. (J) Representative
immunohistochemistry images of pancreatic mucinous
cystadenocarcinoma and matched adjacent tissue sections
demonstrating the staining of BCL2 (magnification ×20). Data are
presented as the mean ± SD (n=3) from three separate experiments.
**P<0.01 and ***P<0.001, Student's t-test. miR, microRNA;
NCm, miR-224-5p mimic control; miR-224, miR-224-5p mimic; NCi,
miR-224-5p inhibitor control; inhibitor-224, miR-224-5p inhibitor;
siNC, siBCL2 control; UTR, untranslated region; M, mutated; siRNA,
small interfering RNA; Normal, matched adjacent tissue; Tumor,
pancreatic mucinous cystadenocarcinoma tissue. |
Discussion
Although autophagy serves an important role in
cancer (16), the function of
autophagy in regulating pancreatic MCC is unknown. The results of
the present study demonstrated that autophagy was increased in
pancreatic MCC tissues and cells compared with the respective
control groups and promoted the migration and invasion of the
pancreatic mucinous cystadenocarcinoma cell line MCC1. In addition,
the expression levels of miR-224-5p were significantly increased in
MCC1 cells when autophagy was activated. A previous study has
suggested that BCL2 inhibits autophagy by directly interacting with
the BH3 domain of beclin 1 in the endoplasmic reticulum (17). Further experiments demonstrated that
miR-224-5p facilitated autophagy in MCC1 by directly targeting
BCL2. These results suggested that a positive feedback loop between
autophagy and miR-224-5p may promote the migration and invasion of
MCC1 cells.
Metastasis is one of the leading causes of
cancer-related death. The present study demonstrated that autophagy
promoted the migration and invasion of MCC1 cells. The association
between autophagy and tumors has been studied previously; findings
have suggested that autophagy also influences tumor metastasis.
Autophagy promotes the invasion of the mutant KRAS-transformed
human breast cancer cell line MDA-MB-231 by secreting the cytokine
interleukin-6 (18), and autophagy
inhibition substantially reduces tumor metastasis in mice (19). Previous studies have also suggested
that the role of autophagy in tumor metastasis is multifaceted and
depends on the cell type, tumor microenvironment and genomic status
(11,20). Autophagy inhibits metastasis by
blocking the epithelial-mesenchymal transition and fibrosis, but
promotes metastasis by promoting local adhesion, anti-anoikis and
coupling of tumor interstitial metabolism (11). Defects in autophagy can promote the
development of pancreatitis and thus affect the risk of pancreatic
cancer (21,22). Additionally, a previous study has
demonstrated that continuously activated autophagy can lead to
autophagy-induced death of pancreatic cancer cells, thus,
inhibiting tumorigenesis (23).
These findings suggested that the variable effects of autophagy on
the development of pancreatic cancer may be associated with the
stage at which it is detected. By IHC analysis of pancreatic cancer
tissues of 71 patients who underwent pancreaticoduodenectomy,
compared with adjacent non-tumor tissue, autophagy activity in
pancreatic cancer tissue was increased compared with adjacent
non-tumor tissue and significantly associated with poor prognosis
(24). Overexpression and
constitutive activation of the microphthalmia/transcription factor
E family transcription factors in pancreatic cancer further mediate
lysosomal amplification and hyperactivate autophagy to promote the
development of pancreatic cancer (25). The results of the present study
demonstrated that autophagy promoted the migration and invasion of
MCC1 cells, supporting a role for autophagy in the metastasis of
pancreatic mucinous cystadenocarcinoma. It is speculated that
inhibition of autophagy may be a potential therapeutic strategy for
pancreatic mucinous cystadenocarcinoma. Considering the crucial
role of autophagy in tumor metabolism and immunity (26,27), the
effects of autophagy on metabolism and tumor immunity in pancreatic
mucinous cystadenocarcinoma will be investigated further.
Previous studies have suggested that autophagy
regulates miRNA expression or activity (28,29).
Inhibition of autophagy leads to the inhibition of miRNA function
(30). In our previous study,
miR-224 was decreased in pancreatic mucinous cystadenoma (31), whereas the results of the present
study revealed that miR-224-5p was significantly increased in MCC1,
which appears contradictory. Compared with pancreatic duct
epithelial cells in a normal pancreas, mild and moderate pancreatic
intraepithelial neoplasia, moderate or severe LC3 staining was
identified in 81% of pancreatic duct epithelial cells and the
pancreatic cancer cell cytoplasm with severe intraepithelial
neoplasia (32), suggesting that the
autophagic activity of tumor cells is related to tumor stages; the
autophagy activity increased with tumor progression. Pancreatic
mucinous cystadenoma and cystadenocarcinoma are different stages of
tumor progression. However, autophagy serves an important role in
regulating miRNAs and the relatively high autophagy activation also
upregulates the expression of miR-224-5p in MCC1. miRNAs have been
widely reported to be involved in the process of autophagy
(33–36). The results of the present study
revealed that miR-224-5p may facilitate autophagy in MCC1.
In conclusion, the results of the present study
demonstrated that autophagy promoted the migration and invasion of
pancreatic mucinous cystadenocarcinoma MCC1 cells and that
autophagy elevated the expression levels of miR-224-5p in MCC1.
Additionally, miR-224-5p promoted autophagy by targeting BCL2.
These results suggested that pancreatic mucinous cystadenocarcinoma
may exhibit hyperactivated autophagy. The results also revealed
that an autophagy/miR-224-5p positive feedback loop may promote the
migration and invasion of MCC1 cells and provided insight into the
association between autophagy and tumor metastasis in pancreatic
mucinous cystadenocarcinoma.
Acknowledgements
The authors would like to thank Professor Claudio
Sorio (University of Verona, Italy) for the MCC1 cell line.
Funding
The present study was supported by grants from the
National Key R&D Program of China (grant no. 2017YFC1600100),
the Strategic Priority Research Program of the Chinese Academy of
Sciences (grant no. XDA12010100) and the National Natural Science
Foundation of China (grant no. 81672892).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZ and LZ conceived the study. XP and CG acquired
the data. LS and YW analyzed the data. XZ acquired funding. CG, LS
and JL performed the experiments. RC and DF developed the
methodology. CG supervised the study. CG, MY and XP analyzed the
data and interpreted the results. CG wrote the manuscript. XZ and
LZ revised the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Shanghai
Changhai Hospital Ethics Committee (Shanghai, China) (approval no.
CHEC2016-8167111578). Informed consent was obtained from the
subjects prior to the collection of the specimens.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MCNs
|
mucinous cystic neoplasms
|
|
MCC
|
mucinous cystadenocarcinoma
|
|
mTOR
|
mammalian target of rapamycin
|
|
miRNA
|
microRNA
|
|
IHC
|
immunohistochemistry
|
|
RT-qPCR
|
reverse transcription-quantitative
PCR
|
|
HPDE
|
human pancreatic epithelial cell
|
|
DMSO
|
dimethyl sulfoxide
|
References
|
1
|
Farrell JJ: Pancreatic cysts and
guidelines. Dig Dis Sci. 62:1827–1839. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Canto MI and Hruban RH: Managing
pancreatic cysts: Less is more? Gastroenterology. 148:688–691.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Compagno J and Oertel JE: Mucinous cystic
neoplasms of the pancreas with overt and latent malignancy
(cystadenocarcinoma and cystadenoma). A clinicopathologic study of
41 cases. Am J Clin Pathol. 69:573–580. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Postlewait LM, Ethun CG, McInnis MR,
Merchant N, Parikh A, Idrees K, Isom CA, Hawkins W, Fields RC,
Strand M, et al: Association of preoperative risk factors with
malignancy in pancreatic mucinous cystic neoplasms: A multicenter
study. JAMA Surg. 152:19–25. 2016. View Article : Google Scholar
|
|
5
|
Sarr MG, Carpenter HA, Prabhakar LP,
Orchard TF, Hughes S, van Heerden JA and DiMagno EP: Clinical and
pathologic correlation of 84 mucinous cystic neoplasms of the
pancreas: Can one reliably differentiate benign from malignant (or
premalignant) neoplasms? Ann Surg. 231:205–212. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Del Chiaro M, Verbeke C, Salvia R, Klöppel
G, Werner J, McKay C, Friess H, Manfredi R, Van Cutsem E, Löhr M,
et al: European experts consensus statement on cystic tumours of
the pancreas. Dig Liver Dis. 45:703–711. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tanaka M, Fernández-del Castillo C, Adsay
V, Chari S, Falconi M, Jang JY, Kimura W, Levy P, Pitman MB,
Schmidt CM, et al: International consensus guidelines 2012 for the
management of IPMN and MCN of the pancreas. Pancreatology.
12:183–197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mizushima N: A brief history of autophagy
from cell biology to physiology and disease. Nat Cell Biol.
20:521–527. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
ulk1. Nat Cell Biol. 13:132–141. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Levy JM, Towers CG and Thorburn A:
Targeting autophagy in cancer. Nat Rev Cancer. 17:528–542. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dower CM, Wills CA, Frisch SM and Wang HG:
Mechanisms and context underlying the role of autophagy in cancer
metastasis. Autophagy. 14:1110–1128. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu Y, Chang R, Peng Z, Wang Y, Ji W, Guo
J, Song L, Dai C, Wei W, Wu Y, et al: Loss of polarity protein AF6
promotes pancreatic cancer metastasis by inducing snail expression.
Nat Commun. 26:71842015. View Article : Google Scholar
|
|
13
|
Chang R, Song L, Xu Y, Wu Y, Dai C, Wang
X, Sun X, Hou Y, Li W, Zhan X and Zhan L: Loss of Wwox drives
metastasis in triple-negative breast cancer by JAK2/STAT3 axis. Nat
Commun. 28:34862018. View Article : Google Scholar
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu XD, Song XW, Li Q, Wang GK, Jing Q and
Qin YW: Attenuation of microRNA-22 derepressed PTEN to effectively
protect rat cardiomyocytes from hypertrophy. J Cell Physiol.
227:1391–1398. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Maes H, Rubio N, Garg AD and Agostinis P:
Autophagy: Shaping the tumor microenvironment and therapeutic
response. Trends Mol Med. 19:428–446. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
He C and Levine B: The beclin 1
interactome. Curr Opin Cell Biol. 22:140–149. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lock R, Kenific CM, Leidal AM, Salas E and
Debnath J: Autophagy-dependent production of secreted factors
facilitates oncogenic RAS-driven invasion. Cancer Discov.
4:466–479. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sharifi MN, Mowers EE, Drake LE, Collier
C, Chen H, Zamora M, Mui S and Macleod KF: Autophagy promotes focal
adhesion disassembly and cell motility of metastatic tumor cells
through the direct interaction of paxillin with LC3. Cell Rep.
15:1660–1672. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Görgülü K, Diakopoulos KN, Ai J, Schoeps
B, Kabacaoglu D, Karpathaki AF, Ciecielski KJ, Kaya-Aksoy E, Ruess
DA, Berninger A, et al: Levels of the autophagy-related 5 protein
affect progression and metastasis of pancreatic tumors in mice.
Gastroenterology. 156:203–217. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Diakopoulos KN, Lesina M, Wörmann S, Song
L, Aichler M, Schild L, Artati A, Römisch-Margl W, Wartmann T,
Fischer R, et al: Impaired autophagy induces chronic atrophic
pancreatitis in mice via sex- and nutrition-dependent processes.
Gastroenterology. 148:626–638. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Antonucci L, Fagman JB, Kim JY, Todoric J,
Gukovsky I, Mackey M, Ellisman MH and Karin M: Basal autophagy
maintains pancreatic acinar cell homeostasis and protein synthesis
and prevents ER stress. Proc Natl Acad Sci USA. 112:E6166–E6174.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sun L, Hu L, Cogdell D, Lu L, Gao C, Tian
W, Zhang Z, Kang Y, Fleming JB and Zhang W: MIR506 induces
autophagy-related cell death in pancreatic cancer cells by
targeting the STAT3 pathway. Autophagy. 13:703–714. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fujii S, Mitsunaga S, Yamazaki M, Hasebe
T, Ishii G, Kojima M, Kinoshita T, Ueno T, Esumi H and Ochiai A:
Autophagy is activated in pancreatic cancer cells and correlates
with poor patient outcome. Cancer Sci. 99:1813–1819.
2008.PubMed/NCBI
|
|
25
|
Perera RM, Stoykova S, Nicolay BN, Ross
KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK,
Ferrone CR, et al: Transcriptional control of autophagy-lysosome
function drives pancreatic cancer metabolism. Nature. 524:361–365.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kimmelman AC and White E: Autophagy and
tumor metabolism. Cell Metab. 25:1037–1043. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rybstein MD, Bravo-San Pedro JM, Kroemer G
and Galluzzi L: The autophagic network and cancer. Nat Cell Biol.
20:243–251. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lan SH, Wu SY, Zuchini R, Lin XZ, Su IJ,
Tsai TF, Lin YJ, Wu CT and Liu HS: Autophagy suppresses
tumorigenesis of hepatitis B virus-associated hepatocellular
carcinoma through degradation of microRNA-224. Hepatology.
59:505–517. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jing Z, Han W, Sui X, Xie J and Pan H:
Interaction of autophagy with microRNAs and their potential
therapeutic implications in human cancers. Cancer Lett.
356:332–338. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gibbings D, Mostow S, Jay F, Schwab Y,
Cossart P and Voinnet O: Selective autophagy degrades DICER and
AGO2 and regulates miRNA activity. Nat Cell Biol. 14:1314–1321.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang B, Guo X, Zhang J, Liu X, Zhan X and
Li Z: MicroRNA-224 is downregulated in mucinous cystic neoplasms of
the pancreas and may regulate tumorigenesis by targeting jagged1.
Mol Med Rep. 10:3303–3309. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang S, Wang X, Contino G, Liesa M, Sahin
E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al:
Pancreatic cancers require autophagy for tumor growth. Genes Dev.
25:717–729. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Füllgrabe J, Klionsky DJ and Joseph B: The
return of the nucleus: Transcriptional and epigenetic control of
autophagy. Nat Rev Mol Cell Biol. 15:65–74. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Y, Jiang J, Liu W, Wang H, Zhao L, Liu
S, Li P, Zhang S, Sun C, Wu Y, et al: MicroRNA-378 promotes
autophagy and inhibits apoptosis in skeletal muscle. Proc Natl Acad
Sci USA. 115:E10849–E10858. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li Z, Wang G, Feng D, Zu G, Li Y, Shi X,
Zhao Y, Jing H, Ning S, Le W, et al: Targeting the
miR-665-3p-ATG4B-autophagy axis relieves inflammation and apoptosis
in intestinal ischemia/reperfusion. Cell Death Dis. 9:483.2018.
|
|
36
|
Korkmaz G, le Sage C, Tekirdag KA, Agami R
and Gozuacik D: MiR-376b controls starvation and mTOR
inhibition-related autophagy by targeting ATG4C and BECN1.
Autophagy. 8:165–176. 2012. View Article : Google Scholar : PubMed/NCBI
|