Introduction
Energy metabolism of cancer cells predominantly
involves aerobic glycolysis, in contrast to normal differentiated
cells, which rely primarily on mitochondrial oxidative
phosphorylation to generate the energy needed for cellular
processes (1,2). To adapt to the hypoxic microenvironment
and compete with surrounding normal cells for limited resources,
cancer cells utilize aerobic glycolysis, converting most glucose to
lactate regardless of whether oxygen is present, to ensure rapid
proliferation (1,3). Although the efficiency of aerobic
glycolysis for energy production is very low, the rate is extremely
high (1). Pyruvate kinase (PK) is a
key enzyme involved in aerobic glycolysis and exists as four
isomers: L, R, M1 and M2. PKL and PKR are expressed in hepatic
cells and red blood cells, respectively. PKM1 is expressed in most
mature cells (4). The M2 isomer of
pyruvate kinase (PKM2) is expressed in cancer cells and tissues,
which has a strong ability to promote cell proliferation and serves
a crucial role in tumor development and survival (4). PKM2 is a driver enzyme of aerobic
glycolysis and a hotspot of present studies in the field of tumor
metabolism (5–7). PKM2 primarily exists in an inactive
dimer form in the tumor, catalyzing the conversion of pyruvic acid.
This accumulates upstream glycolytic intermediates as an anabolic
supply for the synthesis of lipids and nucleic acids, which
promotes the anabolism of bio-macromolecules. The tetramer of PKM2
is activated and catalyzes the conversion of pyruvic acid to ATP.
The subtype of PKM2 is determined by its phosphorylation status,
which mediates glucose conversion (8).
Kress et al (9) found that mRNA levels of PKM2 in
colorectal cancer (CRC) were significantly higher compared with
normal tissues. Subsequently, a meta-analysis by Kumar et al
(10) showed similar results. They
also found that serum PKM2 protein levels in patients with
gastrointestinal tumors and esophageal cancer were higher compared
with healthy patients. PKM2 may be used for tumor diagnosis and
shows similar sensitivity to the classic marker carcinoembryonic
antigen in CRC (11,12), and meta-analyses by Zhang et
al and Hathurusinghe et al (12,13) also
demonstrated that PKM2 was gradually increased with the degree of
malignancy of the disease. Furthermore, the diagnostic and
prognostic value of PKM2 has also been demonstrated in other types
of tumors, including pancreatic cancer (14), cervical cancer (15), lung cancer (16), renal cancer (17), melanoma (18) and breast cancer (19,20).
Shi et al (21) found that PKM2 suppression reduced
tumor growth and also exhibited synergistic effects with docetaxel
in A549 lung cancer cells. In addition, Lin et al (20) revealed high PKM2 expression was
significantly associated with in vitro chemosensitivity to
epirubicin and 5-fluorouracil (5-Fu) in patients with breast
cancer. However, studies on the association between PKM2 and
chemotherapy are limited in metastatic CRC (22,23). A
previous study revealed that the response rate to oxaliplatin was
reduced in the PKM2 downregulated HTOXAR3 cell line of CRC compared
with its parental cell line HT29, which was also validated in
clinical settings (22).
Currently, oxaliplatin-based regimens are the most
effective treatment for CRC (24,25).
Efforts aimed at improving the efficacy of this regimen may result
in improved outcomes (22). The
mechanism of oxaliplatin action is mediated by the formation of DNA
adducts, which induce DNA lesions such as intrastrand crosslinks by
covalently binding the platinum compound to guanine residues.
Oxaliplatin DNA adducts are thought to exert their cytotoxicity by
directly inhibiting DNA and RNA synthesis and inducing apoptosis
(26). The cytotoxic effects of
oxaliplatin in CRC cell lines involve the p53 gene status via
induced activation of the p53-p21 pathway (23). Whether the combination treatment of
PKM2 knockdown and oxaliplatin has a synergistic effect in CRC is
yet to be elucidated. The aim of the present study was to determine
whether PKM2 and oxaliplatin exhibited synergistic effects and to
evaluate the potential mechanism by which PKM2 induced apoptosis.
The results may be valuable in developing novel treatment
approaches targeting PKM2.
Materials and methods
Cell lines and cell culture
The human CRC cell lines HCT116 and DLD1 were
purchased from the Cell Bank of Chinese Academy of Sciences and
cultured in McCoy's 5A and RPMI-1640 medium, respectively, both
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) in humidified conditions with 5% CO2
at 37°C.
Small interfering (si)RNA
transfection
CRC cells (HCT116 and DLD1) were transfected with
siRNA duplex oligonucleotides targeting PKM2 (50 nM) using
Lipofectamine™ RNAiMAX transfection reagent (cat. no. 13778150,
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. Cells were incubated for 24 h at 37°C after transfection.
Western blot analysis was performed to determine the knockdown
efficiency of PKM2-siRNAs. The sequences of the PKM2 siRNAs were:
si155, 5′-GCCAUAAUCGUCCUCACCA-3′; si156, 5′-CCAUAAUCGUCCUCACCAA-3′;
and si27, 5′-AGCAGAGCUGCAUCUA-3′ according to a previous study
(27). Additionally, an siRNA with
no influence on PKM2 function was used as a negative control (NC,
5′-CUUACGCUGAGUACUUCGA-3′).
Western blot analysis of PKM2 and
lactate dehydrogenase (LDH) expression
Transfected cells were washed twice with cold PBS
and harvested in lysis buffer (5% sodium dodecyl sulfate, 10 mM
EDTA, 50 mM NaCl, 10 mM Tris-HCl). A Pierce bicinchoninic acid
protein assay kit (Thermo Fisher Scientific, Inc.) was used to
determine protein concentrations. Subsequently, 50 µg each sample
was electrophoresed using 10% SDS-PAGE and transferred to 0.45 µm
PVDF, sealed using 5% milk at room temperature for 1 h and
incubated with antibodies (EMD Millipore). Membranes were probed
with anti-PKM2, anti-LDH and anti-GAPDH rabbit antibodies (cat.
nos. 4053, 3582 and 5174, respectively; all 1:1,500; Cell Signaling
Technology, Inc.). The primary antibody was incubated at 4°C
overnight, and the secondary antibody was incubated at room
temperature for 2 h. Protein expression was normalized against
β-actin expression (cat. no. 4970; 1:1,000; Cell Signaling
Technology, Inc.) and signal were visualized using an enhanced
chemiluminescence kit (Thermo Fischer Scientific, Inc.).
Cell viability analysis
Cells were seeded at the density of 3×103
cells/well in 100 µl culture medium in a 96-well plate. Following
overnight incubation, culture medium was added to the experimental
(si155, si156, oxaliplatin (3 µmol/l), si155 + oxaliplatin and
si156 + oxaliplatin) or control cells (NC), respectively. Si155 and
si156 groups were added to the same amount of medium as
oxaliplatin. To determine the activity of the cells, cells were
imaged 24 h after oxaliplatin treatment during the logarithmic
growth phase. An MTS assay was performed at 24, 48, 72 and 96 h.
For this assay, 20 µl MTS solution (Promega Corporation) was added
to each well and the cells were incubated at 37°C for 4 h before
the absorbance was determined using a MultiSkan microplate reader
(Thermo Fisher Scientific, Inc.) at a wavelength of 490 nm.
Apoptosis analysis
HCT116 and DLD1 CRC cells were seeded into a 6-well
plate at a density of 1×105 cells/well. A total of 48 h
after transfection, both attached and floating cells were harvested
and washed twice with PBS. Cells were resuspended and stained using
an annexin-V/PI assay kit (Nanjing KeyGen Biotech Co., Ltd.) as
previously described (23). Cell
mortality was determined using a flow cytometer (Beckman Coulter,
Inc.).
Glucose uptake and lactate secretion
assays
Per protein extracellular fluxes, including
glucose/glutamine uptake and lactate/glutamate secretion of HCT116
and DLD1 CRC cells were calculated by subtracting the substrate
concentrations in the final spent medium from those in the initial
medium using a Yellow Springs Instrument (YSI) 2950 biochemistry
analyzer and the YSI 2776 glucose/lactate standard (2.5 g/l
glucose, 0.5 g/l lactate; YSI; Xylem, Inc.) (28). Cells in the logarithmic phase were
seeded into 6-well plates at a density of 2×105
cells/well. Following overnight incubation, culture medium (NC,
si155, si156, oxaliplatin, si155 + oxaliplatin and si156 +
oxaliplatin) were added and the plates were incubated for 24 h and
culture medium collected for detection and normalized to the cell
numbers after 24 h.
Reverse-transcription-quantitative
(RT-q)PCR
Total RNA was extracted from HCT116 CRC cells using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. cDNA was synthesized
using the MLV transcriptase kit (Invitrogen; Thermo Fisher
Scientific, Inc.). Fast SYBR Green master mix was used to determine
the threshold cycle (Cq) value of each sample using a CFX96
real-time quantitative PCR detection system (Bio-Rad Laboratories,
Inc.). GAPDH served as the gene used for normalization. The
fold-changes were calculated using the relative quantification with
2−ΔΔCq (29). All
reactions were performed in a 20 µl reaction volume in triplicate.
The following PCR conditions were used: Initial denaturation at
95°C for 30 sec; followed by 40 cycles of 95°C for 5 sec and 60°C
for 30–60 sec; and stage 3 was dissociation according to the
manufacturer's protocol (cat. no. RR420; Takara Biotechnology Co.,
Ltd.).
The PCR primer sequences used were as follows: GAPDH
forward, 5′-AAGGTCATCCCTGAGCTGAA-3′ and reverse,
5′-TGACAAAGTGGTCGTTGAGG-3′; G6PD forward,
5′-TGCATGAGCCAGATAGGCTG-3′ and reverse, 5′-GGTAGTGGTCGATGCGGTAG-3′;
and PKM2 forward, 5′-ATGCAGCACCTGATAGCTCG-3′ and reverse,
5′-AGGCTCGCACAAGTTCTTCA-3′.
Analysis of cellular glutathione (GSH)
levels
GSH was measured using the GSH-Glo™ glutathione
assay kit (cat. no. V6911; Promega Corporation) according to the
manufacturer's instructions. Briefly, HCT116 cells were seeded at a
density of 2×104 cells/well into 96-well opaque plates
and treated with the indicated siRNA (si155 or si156). After
removing the medium, the cells were incubated in 100 µl mixed
GSH-Glo™ reagent for 35 min at room temperature and subsequently in
100 µl reconstituted Luciferin Detection Reagent for 15 min at room
temperature. Luminescent signals were detected using a Fluoroskan
luminescence scanner (Thermo Fisher Scientific, Inc.).
Analysis of reactive oxygen species
(ROS)
After treatment with PKM2-siRNAs (si155 and si156)
or scrambled siRNAs for 24 h, HCT116 cells were incubated with 10
µM 2′,7′-dichlorofluorescin diacetate (Sigma-Aldrich; Merck KGaA)
for 30 min, followed by flow cytometry analysis using a FACSCalibur
flow cytometer (BD Biosciences).
Statistical analysis
Data were presented as mean ± SD. Differences
between two groups were compared using unpaired Student's t-test
and differences between multiple groups were compared using LSD or
Tukey's post-hoc test following one-way ANOVA test for comparisons
of 3 groups or more respectively. P<0.05 was considered to
indicate a statistically significant difference. GraphPad Prism 7
software (GraphPad Software, Inc.) was used for calculating these
statistics.
Results
Determining the validity of candidate
PKM2-siRNAs
To explore the chemosensitization effects of
targeting PKM2 in CRC cells, the PKM2 gene was initially knocked
down using siRNAs. The si-PKM2 sequences were specifically
synthesized as described by Goldberg et al (27). The most efficient sequences, si27,
si155, and si156, targeting PKM2 were selected, and si-NC was used
the negative control. The knockdown efficiency of PKM2-siRNAs
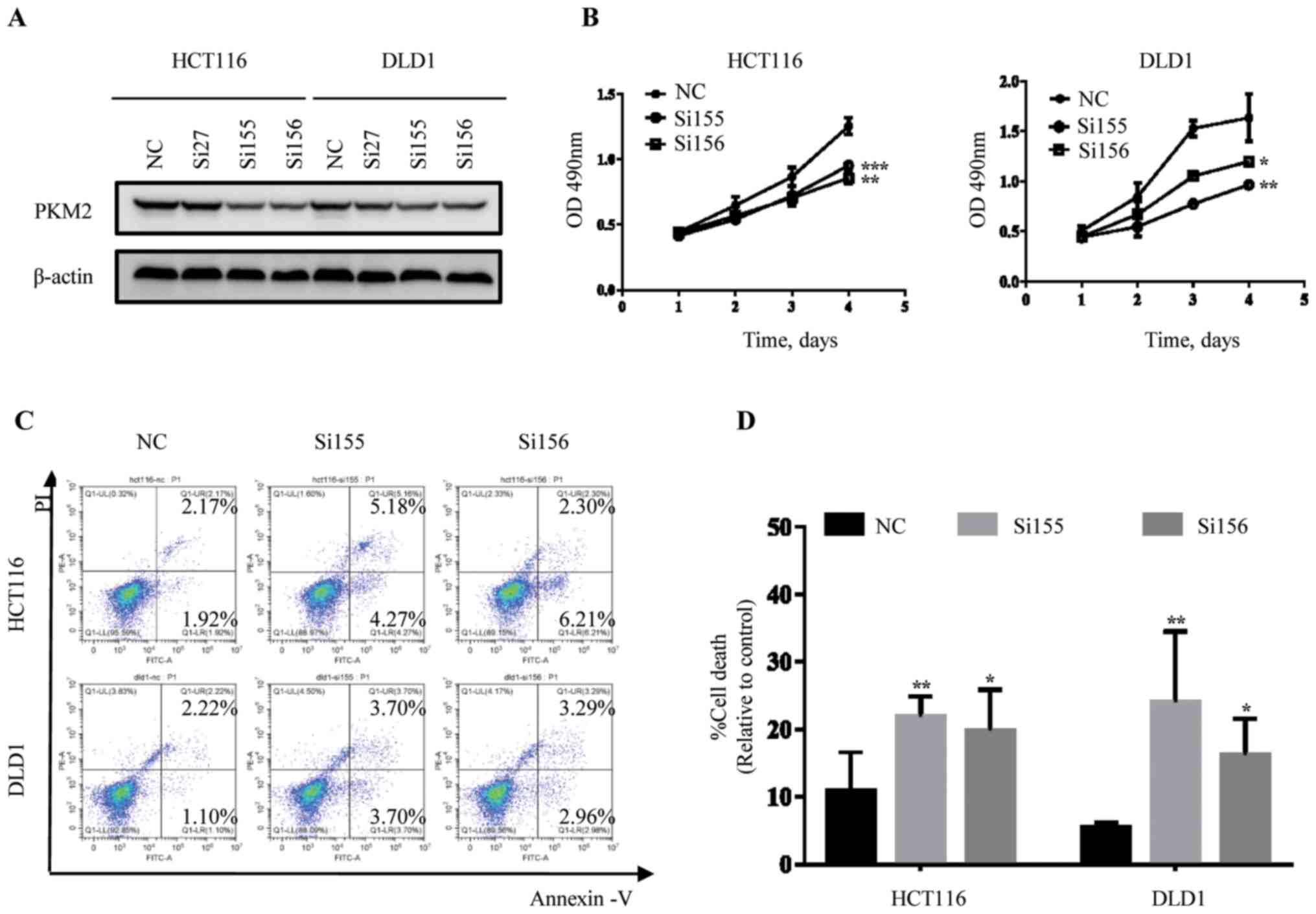
(si27, si155, si156) was verified using western blot analysis.
Protein level of PKM2 was analyzed in two colorectal cancer (CRC)
cell lines (HCT116 and DLD1). PKM2 protein expression was reduced
after transfection of si155 and si156 in CRC cells (HCT116 and
DLD1) compared with si-NC (Fig. 1A).
The results indicated that si155 and si156 were the most effective
siRNAs for knockdown of PKM2 in CRC cells, and thus these siRNAs
were used to downregulate PKM2 in subsequent analyses.
Effects of PKM2 knockdown on
proliferation and apoptosis of CRC cells
si155 and si156 were transfected into HCT116 and
DLD1 cell lines and the effects on cell-proliferation was evaluated
using an MTS assay at different time points (24, 48, 72 and 96 h).
The results showed that both siRNAs significantly attenuated the
proliferative ability of CRC cells (Fig.
1B). Consistently, similar results were also observed with the
apoptosis assay. Transfection of siRNAs resulted in increased
apoptosis in CRC cells compared with the NC group (Fig. 1C and D).
In order to examine the effect of PKM2-siRNAs on
cell viability of HCT116 and DLD1, the logarithmic growth of HCT116
and DLD1 cells was observed in different experimental groups (NC,
PKM2-siRNAs and PKM2-siRNAs + oxaliplatin; Fig. 2).
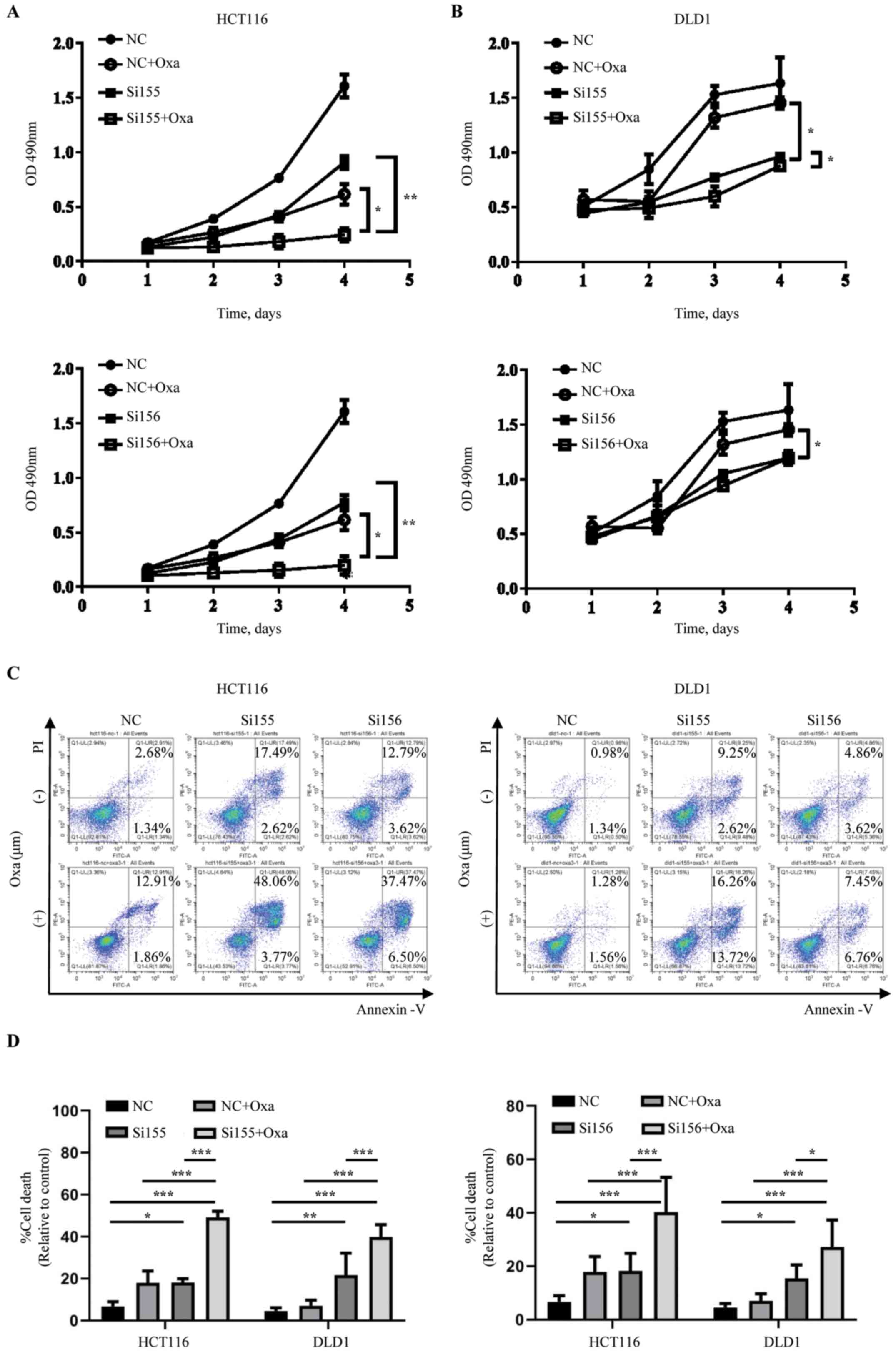
Synergistic anti-proliferative and
apoptotic effects of PKM2-siRNAs and oxaliplatin in vitro
As oxaliplatin-based chemotherapy is not only a
traditional but also an effective regimen in CRC (30), to investigate whether PKM2-siRNAs
augmented the antitumor effects of oxaliplatin, proliferation and
apoptosis assays were performed.
Cell proliferation was evaluated using an MTS assay
in the PKM2-siRNAs + oxaliplatin, PKM2-siRNAs only, oxaliplatin
only and NC groups. Cellular proliferation was significantly
inhibited in the PKM2-siRNAs + oxaliplatin group compared with the
PKM2-siRNAs or the oxaliplatin only group. Additionally, treatment
with oxaliplatin or PKM2-siRNAs alone showed cytotoxic effects
compared to in the NC group (Fig. 3A and
B).
The number of apoptotic cells was analyzed using
flow cytometry. As shown in Fig. 3C and
D, treatment with PKM2-siRNAs + oxaliplatin resulted in a
synergistic increase in apoptosis compared with oxaliplatin or
PKM2-siRNAs alone.
Key factors relating to apoptosis
induced by PKM2 knockdown
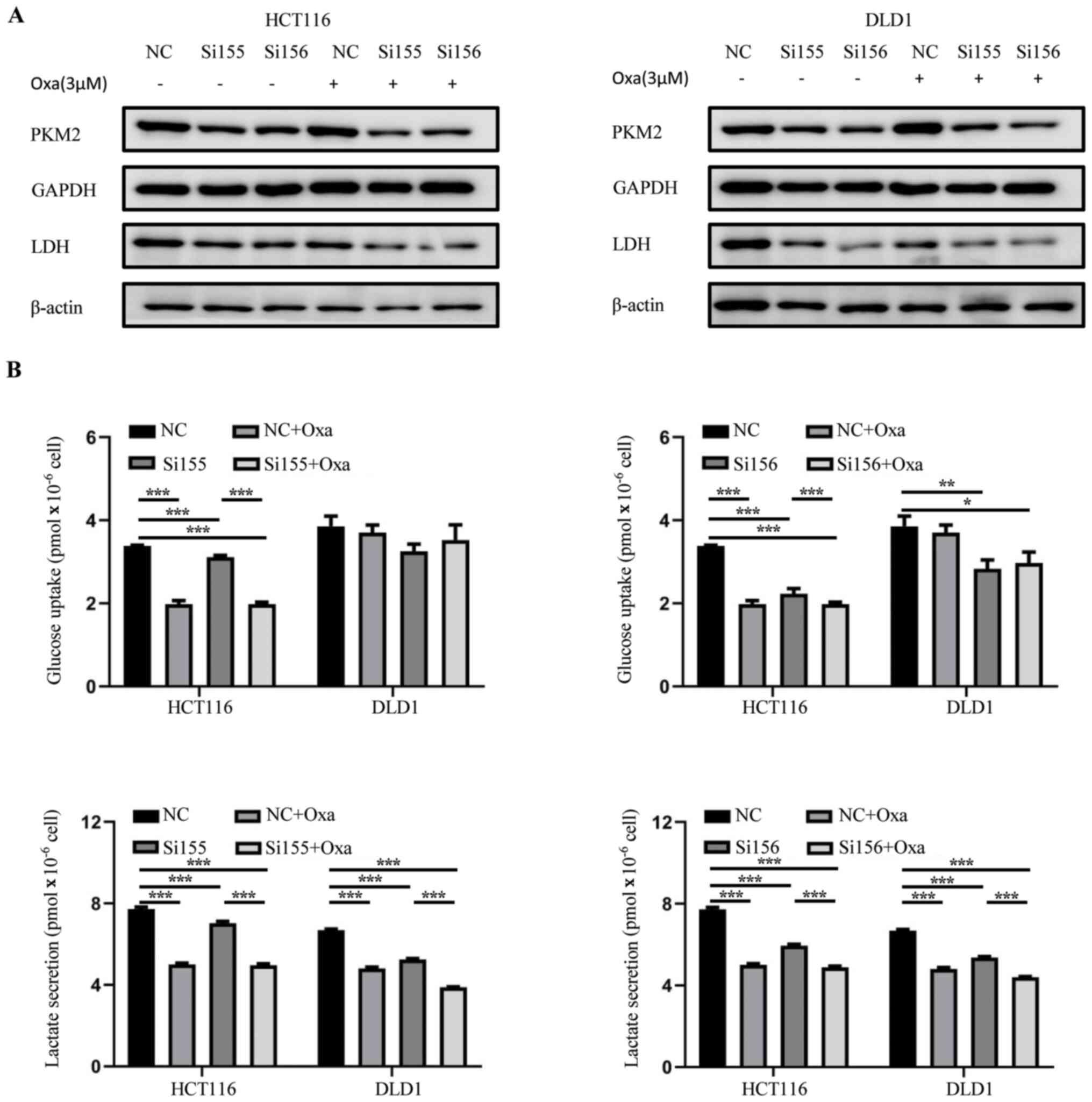
Cancer cells obtain energy and also maintain a
stable state through glycolysis (1,3). Glucose
uptake and lactate secretion fluxes of the NC, PKM2-siRNAs and
PKM2-siRNAs + oxaliplatin treated HCT116 and DLD1 cells were
measured. PKM2 knockdown reduced the protein expression of lactate
dehydrogenase (LDH), glucose uptake and lactate secretion in the
PKM2-siRNAs and PKM2-siRNAs + oxaliplatin groups (Fig. 4), demonstrating that glycolysis was
suppressed. The results indicated that PKM2-siRNAs had greater
effect on HCT116 cells.
PKM2-siRNA induced downregulation of glucose uptake
and lactate production, which suggests a decline in cell
metabolism. While the above results indicated that the difference
in glycolysis metabolism between PKM2-siRNAs + oxaliplatin and
oxaliplatin was not as profound as the difference between cell
proliferation and apoptosis. It was hypothesized that there might
be other glucose metabolic pathways closely associated with
glycolysis which induce apoptosis.
It is known that the production of ROS can cause
cellular damage and severe cytotoxicity, which can induce cell
apoptosis (31). While NADPH, a
metabolite of the PPP (pentose phosphate pathway), converts GSSH to
GSH, which is the main free-radical scavenger neutralizing
intracellular ROS (3) and then
decrease ROS-induced apoptosis (32). Therefore, PKM2-siRNAs were postulated
to increase ROS level through the suppression of PPP.
As expected, ROS levels were increased in the
PKM2-siRNAs groups compared with the NC group (Fig. 5A and B). Subsequently, whether
transfecting HCT116 cell lines with PKM2-siRNA attenuated GSH
levels was investigated. As indicated in Fig. 4C, the GSH level in the PKM2-siRNAs
groups was lower compared with that in the NC group.
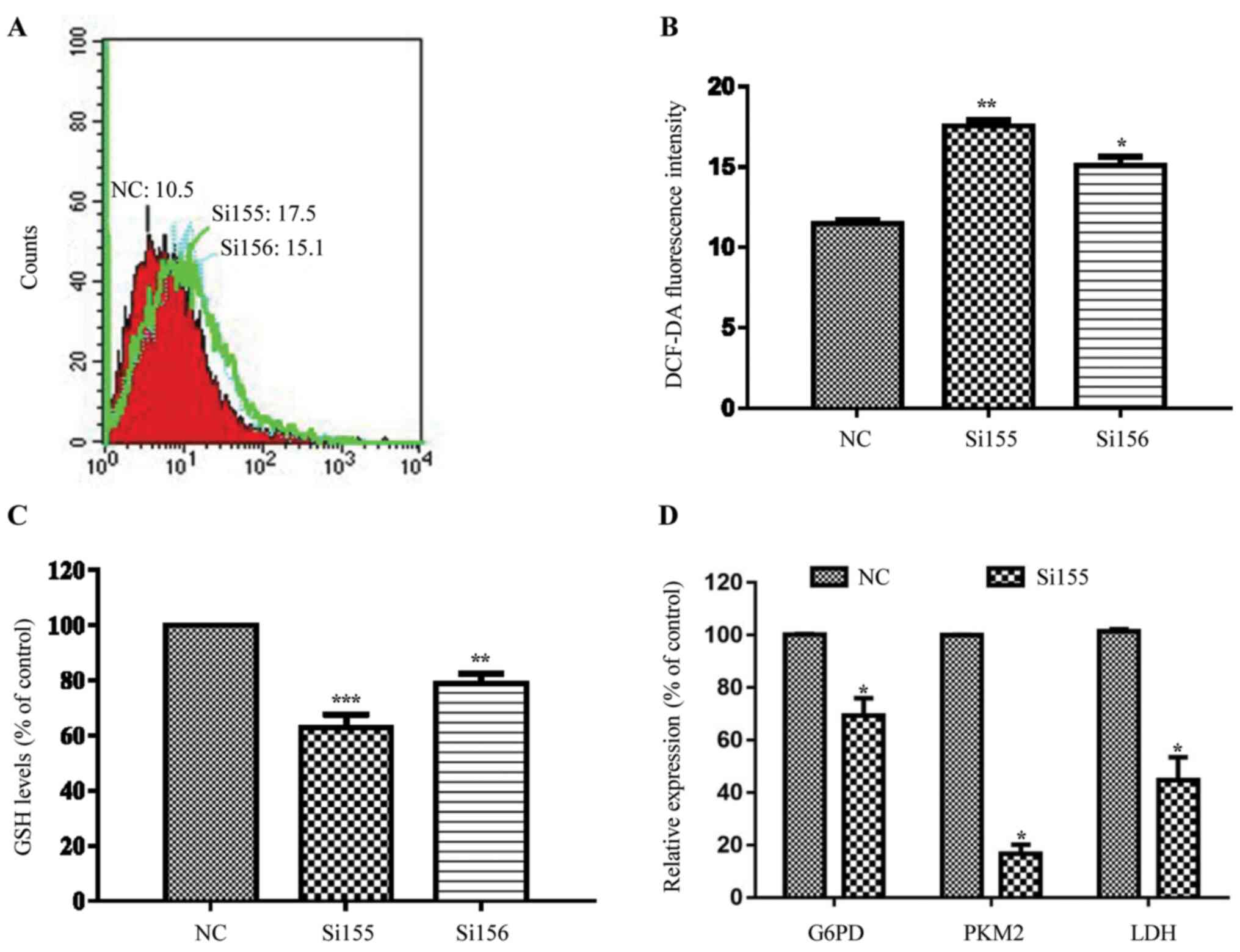 | Figure 5.Changes in the pentose phosphate
pathway redox parameters in PKM2-siRNAs transfected HCT116 cells.
(A) Flow cytometric plots and (B) respective quantitative analysis
of ROS levels. ROS levels following transfection with different
PKM2-siRNAs were significantly increased compared with the NC
group. (C) GSH levels in the PKM2-siRNAs groups were lower compared
with the NC group. (D) mRNA expression levels of PKM2, G6PD and LDH
were significantly decreased compared with the NC group.
*P<0.05, **P<0.01, ***P<0.001 vs. respective NC. PKM2, M2
isomer of pyruvate kinase; siRNA, small interfering RNA; ROS,
reactive oxygen species; GSH, glutathione; NC, negative control;
LDH, lactate dehydrogenase; DCFDA, 2′,7′-dichlorofluorescin
diacetate. |
Subsequently, the changes in the PPP (pentose
phosphate pathway) were examined. As G6PD is a key enzyme involved
in PPP, which converts glucose-6-phosphate to 6 Gluconolactone 6
phosphate (33), RT-qPCR was used to
detect the change of mRNA level of G6PD following the transfection
of PKM2-si155 into HCT116 cells. The results indicated that the
average mRNA expression level of G6PD was decreased compared with
the NC group, which was consistent with PKM2-si155 and LDH
(Fig. 5D).
Discussion
PKM2 is highly expressed in tumors compared with
normal tissues (2). Aerobic
glycolysis is the primary metabolic pathway used by cancer cells,
and PKM2 is a key enzyme involved in this pathway (1). Additionally, PKM2 promotes tumor growth
through non-metabolic pathways, such as by influencing the cell
cycle protein D1, POU domain, class 5, transcription factor 1, Myc,
mTOR, and Mucin-1 (34–38). PKM2 and hypoxia-inducible factor
(HIF)-1 also interact in tumor cells. HIF-1 upregulates the
expression of PKM2, and PKM2 assists HIF-1 in activating hundreds
of genes in downstream pathways to overcome and adjust to hypoxic
conditions (39,40); HIF-1 target genes include those
encoding: The glucose transporter GLUT1, which increases glucose
uptake; lactate dehydrogenase A (LDHA), which converts pyruvate to
lactate; and pyruvate dehydrogenase kinase 1 (PDK1), which
inactivates pyruvate dehydrogenase, thereby transporting pyruvate
away from the mitochondria and inhibiting O2 consumption
(41). Autophagy suppresses the
function of PKM2 through acetylation (42). Thus, given the crucial role of PKM2
in tumors, targeting PKM2 may be an effective treatment
strategy.
Previous studies have examined whether PKM2 can be
used as a target in cancer treatment. Christofk et al
(43) firstly demonstrated that lung
cancer cell proliferation was significantly inhibited by
suppression of PKM2. Subsequent cell based and nude mice animal
model studies of cholangiocarcinoma (44) revealed that the suppression of PKM2
decreased the proliferation of cancer cells, and also suppressed
the invasion and angiogenesis of tumors. The present study revealed
that PKM2 suppression results in proliferation inhibition and it
also showed that tumor cell apoptosis increased after knockdown of
PKM2, which is in line with previous studies. In the A549 lung
cancer cell line (21), PKM2
targeting or docetaxel-based treatment killed cancer cells, and
their combination showed synergistic effects. In CRC, the PKM2 mRNA
expression levels were associated with oxaliplatin efficacy, tumors
with the lowest PK-M2 levels exhibited the lowest response rates
(22). In previous studies, Ginés
et al (45) reported novel
non-glycolytic roles of PKM2 in response to genotoxic damage and
proposes BMF as a possible target gene of PKM2. The present study
explored the mechanism of PKM2 in oxidative damage induced by the
glycolytic pathway when combined with oxaliplatin. Compared with
the NC and oxaliplatin only groups, cellular proliferation was
inhibited and apoptosis was significantly increased in the
PKM2-siRNAs and PKM2-siRNAs + oxaliplatin groups. PKM2-siRNAs
combined with oxaliplatin showed a synergistic effect in CRC cells.
Oxaliplatin may have reduced proliferation and differentiation of
cells by crosslinking with DNA and suppressing DNA synthesis
(26). PKM2-siRNAs affected the
major metabolic pathway used by cancer cells resulting in
apoptosis. As a result, PKM2 and oxaliplatin serve significant
roles in cell proliferation and metabolism, respectively, avoiding
overlapping effects and explaining the synergistic effects of the
co-treatment.
To examine how siPKM2 initiated apoptosis, glucose
uptake and lactate secretion fluxes of NC, PKM2-siRNAs, and
PKM2-siRNAs + oxaliplatin groups in HCT116 and DLD1 cells were
determined. PKM2 knockdown reduced glucose uptake and lactate
secretion in the PKM2-siRNAs and PKM2-siRNAs + oxaliplatin groups,
demonstrating that glycolysis was suppressed. However, the
difference in glycolysis metabolism between PKM2-siRNAs +
oxaliplatin and oxaliplatin was not as profound as the difference
between cell proliferation and apoptosis. The mechanism may involve
ROS. In an aerobic environment, anoxia (46) and hypoxia-oxidation result in the
production of large amounts of ROS (47), which can induce cell apoptosis
(31). PKM2 enables tumor cells to
escape from ROS damage (3). Anoxia
also upregulates HIF-1, thus increasing PKM2 expression (40). ROS oxidizes PKM2 Cys358, leading
glucose into the PPP and the production of large quantities of
NADPH to neutralize ROS (3). As a
result, intracellular ROS does not increase. Therefore, it may be
hypothesized that once PKM2 was suppressed, the balance between
PKM2 and ROS is disrupted, resulting in increased ROS-induced
apoptosis. A previous study also shows that G6PD knockdown lowers
NADPH levels and increases cellular susceptibility to oxidative
stress (32). The mRNA level of G6PD
was significantly decreased after suppression with PKM2-siRNAs,
indicating that the PPP was inhibited. PKM2-si155 induces
downregulation of glucose uptake and lactate production, which
suggests a decline in cell metabolism (43). As a result, the downregulation of
G6PD mRNA may be the result of changes in cell metabolism. As NADPH
is a metabolite of the PPP and GSH is the downstream product of
NADPH, GSH levels were evaluated. Although NADPH was not examined
in the present study, the levels of GSH were determined. GSH, to
some extent, is a downstream mediator of NADPH, which interacts
with ROS. The results showed that the GSH levels in the PKM2-siRNAs
groups were significantly lower compared with the NC group.
Furthermore, to explore if there was a negative association between
GSH and ROS levels, ROS levels were detected. The ROS levels in the
si155 group were increased compared with the NC group. Thus,
suppressing PKM2 led to a decrease in GSH, which was closely
associated with increased ROS and cell apoptosis induction.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the Natural
Science Foundation of Guangdong, China (grant no. 2015A030313010),
Science and Technology Program of Guangzhou, China (grant no.
1563000305) and the National Natural Science Foundation of China
(grant nos. 81272641 and 81572409).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LPX and YMH conceived and designed the study. CXY
and WHL designed and performed the experiments. MZM, QY and WZH
analyzed the data. CXY and WHL wrote, revised, and edited the
manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lu Z: Nonmetabolic functions of pyruvate
kinase isoform M2 in controlling cell cycle progression and
tumorigenesis. Chin J Cancer. 31:5–7. 2012.PubMed/NCBI
|
|
3
|
Anastasiou D, Poulogiannis G, Asara JM,
Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW,
Auld DS, et al: Inhibition of pyruvate kinase M2 by reactive oxygen
species contributes to cellular antioxidant responses. Science.
334:1278–1283. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Christofk HR, Vander Heiden MG, Wu N,
Asara JM and Cantley LC: Pyruvate kinase M2 is a
phosphotyrosine-binding protein. Nature. 452:181–186. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gomez-Escudero J, Clemente C, Garcia-Weber
D, Acín-Pérez R, Millán J, Enríquez JA, Bentley K, Carmeliet P and
Arroyo AG: PKM2 regulates endothelial cell junction dynamics and
angiogenesis via ATP production. Sci Rep. 9:150222019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin Y, Zhai H, Ouyang Y, Lu Z, Chu C, He Q
and Cao X: Knockdown of PKM2 enhances radiosensitivity of cervical
cancer cells. Cancer Cell Int. 19:1292019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu VM, Howell AJ, Hosios AM, Li Z,
Israelsen WJ and Vander Heiden MG: Cancer-associated mutations in
human pyruvate kinase M2 impair enzyme activity. FEBS Lett.
594:646–664. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gupta V and Bamezai RN: Human pyruvate
kinase M2: A multifunctional protein. Protein Sci. 19:2031–2044.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kress S, Stein A, Maurer P, Weber B,
Reichert J, Buchmann A, Huppert P and Schwarz M: Expression of
hypoxia-inducible genes in tumor cells. J Cancer Res Clin Oncol.
124:315–320. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kumar Y, Tapuria N, Kirmani N and Davidson
BR: Tumour M2-pyruvate kinase: A gastrointestinal cancer marker.
Eur J Gastroenterol Hepatol. 19:265–276. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schneider J and Schulze G: Comparison of
tumor M2-pyruvate kinase (tumor M2-PK), carcinoembryonic antigen
(CEA), carbohydrate antigens CA 19-9 and CA 72-4 in the diagnosis
of gastrointestinal cancer. Anticancer Res. 23:5089–5093.
2003.PubMed/NCBI
|
|
12
|
Zhang B, Chen JY, Chen DD, Wang GB and
Shen P: Tumor type M2 pyruvate kinase expression in gastric cancer,
colorectal cancer and controls. World J Gastroenterol.
10:1643–1646. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hathurusinghe HR, Goonetilleke KS and
Siriwardena AK: Current status of tumor M2 pyruvate kinase (tumor
M2-PK) as a biomarker of gastrointestinal malignancy. Ann Surg
Oncol. 14:2714–2720. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ogawa H, Nagano H, Konno M, Eguchi H,
Koseki J, Kawamoto K, Nishida N, Colvin H, Tomokuni A, Tomimaru Y,
et al: The combination of the expression of hexokinase 2 and
pyruvate kinase M2 is a prognostic marker in patients with
pancreatic cancer. Mol Clin Oncol. 3:563–571. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao Y, Shen L, Chen X, Qian Y, Zhou Q,
Wang Y, Li K, Liu M, Zhang S and Huang X: High expression of PKM2
as a poor prognosis indicator is associated with radiation
resistance in cervical cancer. Histol Histopathol. 30:1313–1320.
2015.PubMed/NCBI
|
|
16
|
Schneider J, Morr H, Velcovsky HG, Weisse
G and Eigenbrodt E: Quantitative detection of tumor M2-pyruvate
kinase in plasma of patients with lung cancer in comparison to
other lung diseases. Cancer Detect Prev. 24:531–535.
2000.PubMed/NCBI
|
|
17
|
Hegele A, Varga Z, Kosche B, Stief T,
Heidenreich A and Hofmann R: Pyruvate kinase type tumor M2 in
urological malignancies. Urol Int. 70:55–58. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ugurel S, Bell N, Sucker A, Zimpfer A,
Rittgen W and Schadendorf D: Tumor type M2 pyruvate kinase
(TuM2-PK) as a novel plasma tumor marker in melanoma. Int J Cancer.
117:825–830. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lüftner D, Mesterharm J, Akrivakis C,
Geppert R, Petrides PE, Wernecke KD and Possinger K: Tumor type M2
pyruvate kinase expression in advanced breast cancer. Anticancer
Res. 20((6D)): D5077–D5082. 2000.
|
|
20
|
Lin Y, Lv F, Liu F, Guo X, Fan Y, Gu F, Gu
J and Fu L: High expression of pyruvate kinase M2 is associated
with chemosensitivity to epirubicin and 5-fluorouracil in breast
cancer. J Cancer. 6:1130–1139. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shi HS, Li D, Zhang J, Wang YS, Yang L,
Zhang HL, Wang XH, Mu B, Wang W, Ma Y, et al: Silencing of pkm2
increases the efficacy of docetaxel in human lung cancer xenografts
in mice. Cancer Sci. 101:1447–1453. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Martinez-Balibrea E, Plasencia C, Ginés A,
Martinez-Cardús A, Musulén E, Aguilera R, Manzano JL, Neamati N and
Abad A: A proteomic approach links decreased pyruvate kinase M2
expression to oxaliplatin resistance in patients with colorectal
cancer and in human cell lines. Mol Cancer Ther. 8:771–778. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shiragami R, Murata S, Kosugi C, Tezuka T,
Yamazaki M, Hirano A, Yoshimura Y, Suzuki M, Shuto K and Koda K:
Enhanced antitumor activity of cerulenin combined with oxaliplatin
in human colon cancer cells. Int J Oncol. 43:431–438. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gelibter AJ, Caponnetto S, Urbano F,
Emiliani A, Scagnoli S, Sirgiovanni G, Napoli VM and Cortesi E:
Adjuvant chemotherapy in resected colon cancer: When, how and how
long? Surg Oncol. 30:100–107. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Petrelli F, Ghidini A and Zaniboni A:
Cetuximab in association with an oxaliplatin-based chemotherapy as
first-line treatment of metastatic colorectal cancer. Recenti Prog
Med. 108:128–135. 2017.(In Italian). PubMed/NCBI
|
|
26
|
Woynarowski JM, Faivre S, Herzig MC,
Arnett B, Chapman WG, Trevino AV, Raymond E, Chaney SG, Vaisman A,
Varchenko M and Juniewicz PE: Oxaliplatin-induced damage of
cellular DNA. Mol Pharmacol. 58:920–927. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Goldberg MS and Sharp PA: Pyruvate kinase
M2-specific siRNA induces apoptosis and tumor regression. J Exp
Med. 209:217–224. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang H, Badur MG, Divakaruni AS, Parker
SJ, Jäger C, Hiller K, Murphy AN and Metallo CM: Distinct metabolic
states can support self-renewal and lipogenesis in human
pluripotent stem cells under different culture conditions. Cell
Rep. 16:1536–1547. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(T)(-Delta Delta C) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wils J: Adjuvant treatment of colon
cancer: Past, present and future. J Chemother. 19:115–122. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Deng S, Yang Y, Han Y, Li X, Wang X, Li X,
Zhang Z and Wang Y: UCP2 inhibits ROS-mediated apoptosis in A549
under hypoxic conditions. PLoS One. 7:e307142012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ju HQ, Lu YX, Wu QN, Liu J, Zeng ZL, Mo
HY, Chen Y, Tian T, Wang Y, Kang TB, et al: Disrupting
G6PD-mediated Redox homeostasis enhances chemosensitivity in
colorectal cancer. Oncogene. 36:6282–6292. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stanton RC: Glucose-6-phosphate
dehydrogenase, NADPH, and cell survival. IUBMB Life. 64:362–369.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang W, Xia Y, Ji H, Zheng Y, Liang J,
Huang W, Gao X, Aldape K and Lu Z: Nuclear PKM2 regulates β-catenin
transactivation upon EGFR activation. Nature. 480:118–122. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee J, Kim HK, Han YM and Kim J: Pyruvate
kinase isozyme type M2 (PKM2) interacts and cooperates with Oct-4
in regulating transcription. Int J Biochem Cell Biol. 40:1043–1054.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ohkouchi S, Block GJ, Katsha AM, Kanehira
M, Ebina M, Kikuchi T, Saijo Y, Nukiwa T and Prockop DJ:
Mesenchymal stromal cells protect cancer cells from ROS-induced
apoptosis and enhance the Warburg effect by secreting STC1. Mol
Ther. 20:417–423. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sun Q, Chen X, Ma J, Peng H, Wang F, Zha
X, Wang Y, Jing Y, Yang H, Chen R, et al: Mammalian target of
rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is
critical for aerobic glycolysis and tumor growth. Proc Natl Acad
Sci USA. 108:4129–4134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kosugi M, Ahmad R, Alam M, Uchida Y and
Kufe D: MUC1-C oncoprotein regulates glycolysis and pyruvate kinase
M2 activity in cancer cells. PLoS One. 6:e282342011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Luo W and Semenza GL: Pyruvate kinase M2
regulates glucose metabolism by functioning as a coactivator for
hypoxia-inducible factor 1 in cancer cells. Oncotarget. 2:551–556.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Luo W, Hu H, Chang R, Zhong J, Knabel M,
O'Meally R, Cole RN, Pandey A and Semenza GL: Pyruvate kinase M2 is
a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell.
145:732–744. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wheaton WW and Chandel NS: Hypoxia. 2.
Hypoxia regulates cellular metabolism. Am J Physiol Cell Physiol.
300:C385–C393. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H,
Zha Z, Liu Y, Li Z, Xu Y, et al: Acetylation targets the M2 isoform
of pyruvate kinase for degradation through chaperone-mediated
autophagy and promotes tumor growth. Mol Cell. 42:719–730. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Christofk HR, Vander Heiden MG, Harris MH,
Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL and
Cantley LC: The M2 splice isoform of pyruvate kinase is important
for cancer metabolism and tumour growth. Nature. 452:230–233. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yu G, Yu W, Jin G, Xu D, Chen Y, Xia T, Yu
A, Fang W, Zhang X, Li Z and Xie K: PKM2 regulates neural invasion
of and predicts poor prognosis for human hilar cholangiocarcinoma.
Mol Cancer. 14:1932015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ginés A, Bystrup S, Ruiz de Porras V,
Guardia C, Musulén E, Martínez-Cardús A, Manzano JL, Layos L, Abad
A and Martínez-Balibrea E: PKM2 subcellular localization is
involved in oxaliplatin resistance acquisition in HT29 human
colorectal cancer cell lines. PLoS One. 10:e01238302015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Solaini G, Baracca A, Lenaz G and Sgarbi
G: Hypoxia and mitochondrial oxidative metabolism. Biochim Biophys
Acta. 1797:1171–1177. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Collins P, Jones C, Choudhury S, Damelin L
and Hodgson H: Increased expression of uncoupling protein 2 in
HepG2 cells attenuates oxidative damage and apoptosis. Liver Int.
25:880–887. 2005. View Article : Google Scholar : PubMed/NCBI
|