Introduction
Colon cancer is one of the most common types of
cancer and the second leading cause of cancer-related mortality
worldwide (1). The tumor-associated
antigen, mucin1 (MUC1), is highly expressed in colon cancer and has
been linked to poor outcomes (2,3). MUC1 is
a transmembrane molecule that is expressed by the glandular
epithelial cells of the prostate, stomach, duodenum, pancreas and
colon, and is also found in hematopoietic cells (4). It has a heavily glycosylated
extracellular domain (4,5). The abnormal glycosylation and
upregulation of MUC1 in human epithelial carcinoma has aroused
great interest as a candidate tumor marker and potential
tumor-killing target (5). Compared
with fully glycosylated MUC1, hypoglycosylated forms and
upregulated MUC1 have pivotal roles in the transcriptional
regulation of genes associated with immune responses, tumor
invasion, apoptosis, angiogenesis, proliferation and inflammation
(6). High expression levels of MUC1
are associated with a poor prognosis in patients with colorectal
cancer (7). In a breast cancer
animal model, intravenous injection of MUC1 induces
immunosuppression and accelerates tumor-bearing mouse death
(8). Serum MUCl expression levels
are also associated with immunosuppression in patients with
metastatic adenocarcinoma treated with active specific
immunotherapy (8–10). In addition, MUC1 causes apoptosis of
activated human T-cells, which inhibits the proliferation of human
T cells and cytotoxic lymphocyte-target cell interactions (11,12).
However, the pro-tumor and immunosuppressive roles of MUC1 in colon
cancer have not been fully elucidated.
The programmed death protein 1 (PD1)-programmed
death ligand 1 (PDL1) signaling pathway is involved in tumor immune
escape, in which the tumor cells evade host immune surveillance by
inhibiting the toxicity of T cells via the PD1-PDL1 signaling
pathway (13). Immunogenic tumor
cells might induce anergy of tumor-specific T cells by expressing
PDL1 on their surfaces (14). A
variety of factors induce PDL1 expression in tumor cells and
antigen-presenting cells, which includes the inflammatory cytokines
IFN-γ and IL-17A (15). MUC1, as a
tumor-associated antigen, recruits white blood cells into the tumor
microenvironment, which results in the secretion of inflammatory
cytokines (16). However, it is
unclear whether the tumor cells upregulate PDL1 expression via MUC1
to escape immune surveillance. In the present study, the mechanisms
underlying PDL1 expression in MUC1-positive tumor tissue were
explored, and the potential of targeting PDL1 to inhibit the
progression of MUC1-overexpressing colon cancer in mice was
demonstrated. A clearer understanding of this process may help to
define the role of MUC1 in promoting tumor immune escape and
provide key information to support immunotherapy through targeting
the PDL1-PD1 signaling pathway in MUC1-positive colon cancer.
Materials and methods
Mice and cell lines
Female BALB/c mice (100 mice; 8 weeks old; 18–21 g)
and female nu/nu mice (40 mice; 8 weeks old; 18–22 g) were
purchased from the Animal Experiment Center of Kunming Medical
University. All animals used in the present study were kept under
specific pathogen-free conditions, in a temperature-controlled
environment (20–23°C) with 40–60% humidity and with a 12-h
light/dark cycle, with food and water freely available. Colon
cancer SW480 (human) and CT26 (mouse) cells, sourced from the
American Type Culture Collection, were cultured in DMEM
supplemented with 10% FBS and with 2 mM L-glutamine (all Gibco;
Thermo Fisher Scientific, Inc.) at 37°C with 5% CO2.
Patients and sample collection
To study the association between the survival time
of patients with colorectal cancer and MUC1 expression in tumor
tissues, a discovery cohort of 230 patients with colorectal cancer
(dataset: Colon and Rectal Cancer) was obtained from The Cancer
Genome Atlas (TCGA) database available from University of
California Santa Cruz Xena (https://xenabrowser.net/) (17,18). To
study the role of immune cells in colorectal cancer tissues, colon
cancer specimens were collected from 33 male patients (Table I) who underwent open surgery at The
First Affiliated Hospital of Kunming Medical University (Kunming,
China) between April 2017 and August 2018. The mean age of the
patients was 56 years old. Individuals with primary colon cancer
who had not received any treatment were deemed eligible for the
present study. The clinical diagnosis was confirmed by
histopathological detection. MUC1 expression was detected using
immunohistochemistry (IHC). The present study was approved by the
Ethics Committee of The First Affiliated Hospital of Kunming
Medical University (approval no. KMU20160901) and conformed with
the ethical standards of the World Medical Association Declaration
of Helsinki. All patients provided written informed consent.
 | Table I.Demographic data of recruited
patients with colorectal cancer. |
Table I.
Demographic data of recruited
patients with colorectal cancer.
| Characteristic |
MUC1high |
MUC1low |
|---|
| Age range,
years | 45-72 | 47-76 |
| Metastasis | 10
(30.3)a | 9
(27.3)a |
| Stage |
|
|
| I | 8 | 6 |
| II | 10 | 9 |
Establishment of a stable cell line
expressing MUC1
The plasm-id
pRP[Exp]-EGFP/Puro-CAG>hMUC1[NM_001371720.1] was purchased from
VectorBuilder, Inc. The human MUC1 gene was amplified and
subcloned in-frame between the NheI and HindIII
restriction sites of pcDNA3.1(−)/Myc-His A (cat. no.
V855-20; Invitrogen; Thermo Fisher Scientific, Inc.). A total of
5×105 CT26 or SW480 cells/well in 500 µl DMEM without
antibiotics were plated in 6-well plates, and then cells were
transfected for 24 h with pcDNA3.1(−)/Myc-His-muc1 (2 µg) or
pcDNA3.1(−)/Myc-His (2 µg) using Lipofectamine™ 2000
transfection reagent (Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocols. The transfected cells were cultured
in DMEM with 10% FBS containing G418 (1 mg/ml; cat. no. 10131027;
Thermo Fisher Scientific, Inc.) to establish stable cell lines
expressing hMUC1. MUC1+CT26 and MUC1+SW480
tumor cells were sorted using flow cytometry and maintained in
medium containing the antibiotic G418 (10% FBS; 1 mg/ml G418). The
CT26 and SW480 cells, which were transfected with
pcDNA3.1(−)/Myc-His as aforementioned, were also maintained
in the same medium containing G418 (10% FBS; 1 mg/ml G418). Human
MUC1 expression was confirmed by flow cytometry and western
blotting one month after cells were transfected.
Western blotting
The cells were lysed in RIPA buffer (Beyotime
Institute of Biotechnology) for 10 min on ice, and then the
supernatant was collected by centrifugation (250 × g; 4°C; 5 min)
to remove cell debris. The protein concentration was measured using
a Bradford assay (Bio-Rad Laboratories, Inc.). Protein samples (20
µg/well) were loaded on a 10% (w/v) tris-HCl SDS-PAGE gel for
electrophoresis and transferred to PVDF membranes (cat. no.
3010040001; Sigma-Aldrich; Merck KGaA), which were blocked in 5%
bovine serum albumin (Sigma-Aldrich; Merck KGaA) for 45 min at room
temperature. Subsequently, the membrane was probed with anti-MUC1
(1:3,000; cat. no. ab36690; Abcam), anti-PDL1 (1:1,000; cat. no.
ab238697; Abcam) or anti-β-actin (1:1,000; cat. no. ab8226; Abcam)
primary antibodies at 4°C overnight. The signal was detected using
a goat anti-mouse secondary antibody (1:3,000; cat. no. ab205719;
Abcam) conjugated to horseradish peroxidase at room temperature for
1 h. Band signals were visualized using the Immobilon Western
Chemiluminescent substrate (Merck KGaA) and detected using the
Odyssey CLx (LI-COR Biosciences). Furthermore, PDL1 expression in
tumor tissues was analyzed using Gel-Pro Analyzer 3.0 (Media
Cybernetics, Inc.).
IHC
The expression levels of MUC1 in colon cancer
tissues were determined using IHC. Briefly, 10% formalin-fixed (8 h
at room temperature), paraffin-embedded sections (4-µm-thick) were
cut using a microtome, mounted on silanized glass slides and dried
overnight at 37°C. A graded alcohol series (100 and 70%) and
distilled water were used to deparaffinize and rehydrate the
slides. Slides containing tumor sections were incubated for 30 min
in citrate buffer (pH 6.0) at 96°C, followed by a 1-h incubation at
room temperature with blocking buffer (10% FBS in PBS). Slides were
then incubated in a humidified chamber at room temperature for 1 h
with rabbit anti-human MUC1 monoclonal antibody (1:1,000; cat. no.
ab109185; Abcam), followed by incubation in a humidified chamber at
room temperature for 30 min with a horseradish peroxidase-labeled
anti-rabbit secondary antibody (1:3,000; cat. no. ab7090; Abcam).
The sections were developed using 3,3′-diaminobenzidine
tetrahydrochloride. Antibody staining in the tissue sections was
observed using light microscopy (Olympus Corporation). For
quantification, cells stained with MUC1 were manually counted at a
magnification of ×400 in five high-power fields of each tumor
tissue. The average number of cells stained with MUC1 in each tumor
sample was counted to reflect the number of MUC1-expressing cells
in the tumor. Similarly to PDL1 expression in tumor tissues of a
previous study (15), tumors with
>90 and ≤90 MUC1-expressing cells were considered to have high
and low expression, respectively.
Cytokine-specific ELISA
ELISA assays were used to analyze IL-10, IL-17A,
TNF-α, TGF-β and IFN-γ levels in the tumor microenvironment of mice
and patients with colon cancer. Fresh tumor tissues were isolated
from patients with colon cancer and tumor-bearing mice. The tumor
tissues were homogenized in PBS (0.1 g tissue/ml). The supernatants
were collected by centrifugation (1,200 × g; 4°C; 10 min) for
analysis of cytokine expression levels using mouse IL-10 (cat. no.
88-7105-22), IL-17A (cat. no. 88-7371-22), TNF-α (cat. no.
88-7324-22), TGF-β (cat. no. 88-8350-22) and IFN-γ (cat. no.
88-7314-22), or human IL-10 (cat. no. KAC1321), IL-17A (cat. no.
BMS2017), TNF-α (cat. no. 88-7346-22), TGF-β (cat. no. 88-8350-22)
and IFN-γ (cat. no. 88-7316-22) ELISA kits (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocols.
Cell proliferation assay
To assess the number of living cells,
1–2×104 CT26/MUC1 or SW480/MUC1 cells were plated and
cultured with 10% FBS-supplemented DMEM containing G418 (1 mg/ml)
for 7 days. CT26/vector or SW480/vector cells were cultured as
negative controls. Proliferation was measured using trypan blue
exclusion. In brief, the cells were centrifuged at 4°C for 5 min at
100 × g and then resuspended in 1 ml PBS. One part 0.4% trypan blue
was mixed with one part cell suspension and incubated for ~3 min at
room temperature. A drop of the trypan blue/cell mixture was
applied to a hemocytometer and the unstained (viable) and stained
(nonviable) cells were counted separately with a binocular
microscope using the hemocytometer. The number of living cells was
counted, and the proliferation of cells was analyzed.
Tumor implantation
CT26/MUC1 or CT26/vector cells (1×106
cells/200 µl PBS/body) were transplanted subcutaneously into the
right flank of BALB/c mice; SW480-MUC1 or SW480/vector cells
(2×106 cells/200 µl PBS/body) were transplanted
subcutaneously into the right flank of nu/nu mice to establish a
mouse model of MUC1-positive and MUC1-negative colon cancer. The
tumor diameters were measured from day 7 after tumor implantation.
The treatment of mice was approved by the Institutional Animal Care
and Use Committee of Kunming Medical University (approval no.
KMU20170121).
For the analysis of survival time of tumor-bearing
mice (the experiments were conducted between January 2017 and
November 2019), tumor size (using microcalipers) and bodyweight
were measured every 2–3 days. The volumes were calculated as:
(length × width × depth)/2. Mice in cages were euthanized by
CO2 exposure for 5 min (flow rate, 3 l/min; 23% chamber
vol/min) when tumor diameters reached 12 mm, according to the
Animal Care and Use Committee Guidelines (19), to minimize the suffering of the
tumor-bearing mice. The mice were considered dead when the
respiration, heartbeat and reflex activity had ceased. The survival
rate of tumor-bearing mice was then evaluated. For surface
molecules and intracellular cytokine analysis (the experiments were
conducted between March 2017 and June 2017), the CT26 tumor-bearing
mice were sacrificed on day 26 after tumor cell inoculation and
surface and intracellular cytokine staining patterns of
myeloid-derived suppressor cells (MDSCs) and T cells were analyzed
using flow cytometry.
For the treatment (the experiments were conducted
between April 2018 and August 2018), the tumor-bearing mice were
anesthetized with 2% pentobarbital (45 mg/kg) via intraperitoneal
injection, and then injected intravenously with an anti-mouse PDL1
antibody (200 µg/mouse; BE0101; Bio X Cell) four times at 3-day
intervals, after palpable tumors (3–5 mm in diameter) had formed.
The tumor volumes and survival rates of tumor-bearing mice were
evaluated. In another experimental set (the experiments were
conducted between April 2018 and June 2018), the tumor-bearing mice
were euthanized on the third day after the end of PD1 antibody
treatment. Surface and intracellular staining of cells was
performed, and cytokine-producing T cells, MDSCs and
tumor-associated macrophages (TAMs) were analyzed using flow
cytometry.
In some experiments (conducted between April 2018
and November 2018) a rat anti-mouse CD8 antibody (500 µg/mouse;
cat. no. BE0223; Bio X Cell) or isotype control rat IgG1 (250
µg/mouse; cat. no. BE0088; Bio X Cell) was injected into the
abdominal cavity of the mouse 3 days before the PDL1 antibody
treatment, followed by evaluation of tumor volumes and survival
rates of tumor-bearing mice.
Surface and intracellular molecular
staining
Transfected CT26 and CT26/MUC1 cells
(2×106 cells/well) were stimulated using IL-17A (10
ng/ml; R&D Systems China Co., Ltd.) and IFN-γ (10 ng/ml;
R&D Systems China Co., Ltd.) at 37°C for 48 h, the cells were
then harvested via centrifugation (250 × g; 4°C; 10 min) and
counted. Subsequently, the cells were stained with PE anti-PDL1 at
4°C for 30 min (1:100; cat. no. 124308; clone 10F.9G2; BioLegend,
Inc.). Fluorescence data were acquired using a CytoFLEX flow
cytometer (Beckman Coulter, Inc.) and analyzed using Kaluza
software v2.1 (Beckman Coulter, Inc.).
For the surface staining of tumor cells and immune
cells, the tumor tissues from tumor-bearing mice or patients with
colon cancer were weighed, homogenized and digested with
hyaluronidase (2 mg/ml), collagenase type IV (2 mg/ml), and
deoxyribonuclease (25 µg/ml; Sigma-Aldrich; Merck KGaA) for 50 min
at 37°C. Immune cells were enriched with Ficoll reagent and stained
with propidium iodide at 4°C for 5 min. After counting viable
cells, the samples were incubated with an anti-CD16/CD32-blocking
Ab (1:100; cat. no. Ab00123-23.0-BT; Absolute Antibody) at 4°C for
10 min and then stained at 4°C for 30 min with Brilliant Violet
510™ anti-mouse CD45 (1:100; cat. no. 103138; clone 30-F11;
BioLegend, Inc.), Brilliant Violet 510™ anti-human CD45 (1:100;
cat. no. 304036; clone HI30; BioLegend, Inc.), FITC anti-mouse CD4
(1:100; cat. no. 100509; clone RM4-5; BioLegend, Inc.), FITC
anti-human CD4 (1:100; cat. no. 317408; clone OKT4; BioLegend,
Inc.), FITC anti-mouse CD8a (1:100; cat. no. 100706; clone 53-6.7;
BioLegend, Inc.), FITC anti-human CD8a (1:100; cat. no. 301050;
clone RPA-T8; BioLegend, Inc.), APC anti-mouse CD279 (PD-1; 1:100;
cat. no. 109112; clone RMP1-30; BioLegend, Inc.), APC anti-human
CD279 (PD-1; 1:100; cat. no. 329908; clone EH12.2H7; BioLegend,
Inc.), PE anti-mouse/human CD11b (1:100; cat. no. 101207; clone
M1/70; BioLegend, Inc.), FITC anti-mouse Ly-6G/Ly-6C (Gr-1; 1:100;
cat. no. 108406; clone RB6-8C5; BioLegend, Inc.), FITC anti-human
CD66b (1:100; cat. no. 305104; clone G10F5; BioLegend, Inc.),
PerCP/Cyanine5.5 anti-mouse F4/80 (1:100; cat. no. 123128; clone
BM8; BioLegend, Inc.), PerCP/Cyanine5.5 anti-human CD68 (1:100;
cat. no. 333813; clone Y1/82A; BioLegend, Inc.), APC anti-mouse
CD274 (B7-H1, PD-L1; 1:100; cat. no. 124311; clone 10F.9G2;
BioLegend, Inc.), APC anti-human CD274 (B7-H1, PD-L1; 1:100; cat.
no. 329708; clone 29E.2A3; BioLegend, Inc.) or PE anti-human MUC1
(1:1,000, cat. no. ab213337; clone EP1024Y; Abcam). The live cells
were gated. Fluorescence data were acquired using a CytoFLEX flow
cytometer and analyzed using Kaluza software v2.1.
For the intracellular staining of immune cells,
1×106 cells/ml were treated with Cell Activation
Cocktail containing Brefeldin A (1:1,000; cat. no. 423304;
BioLegend, Inc.). After 4 h, the cells were collected and stained
with the aforementioned Brilliant Violet 510™- or FITC-conjugated
mAbs against mouse or human CD45, CD4 or CD8 mAbs (BioLegend, Inc)
at 4°C for 30 min. The cells were then fixed with Fixation Buffer
(cat. no. 420801; BioLegend, Inc.) in the dark at room temperature
for 20 min. Next, the cells were permeabilized with 1X
intracellular Staining Permeabilization Wash Buffer (cat. no.
421002; BioLegend, Inc.) and incubated with PE-conjugated
anti-IFN-γ anti-IL-17 or anti-granzyme B (BioLegend, Inc.) for 30
min at room temperature. For forkhead box P3 (Foxp3) staining, the
fixed cells were treated using the True-Nuclear™ Transcription
Factor Buffer Set (BioLegend, Inc.). The tumor-infiltrating
lymphocytes (TILs) were gated. Fluorescence data were acquired on a
Beckman CytoFLEX and analyzed using Kaluza software 2.1.
Statistical analysis
The standard two-sample unpaired t-test was used to
compare outcome differences in two-group experiments. One-way ANOVA
followed by Tukey's multiple comparison test was applied to assess
the statistical significance of differences among multiple
treatment groups. The survival analysis of tumor-bearing mice was
performed with Kaplan-Meier survival analysis. The log-rank
(Mantel-Cox) test was used to obtain the P-values. Data are
representative of three experiments and are presented as the mean.
Error bars represent standard error of the mean. P<0.05 was
considered to indicate a statistically significant difference.
Results
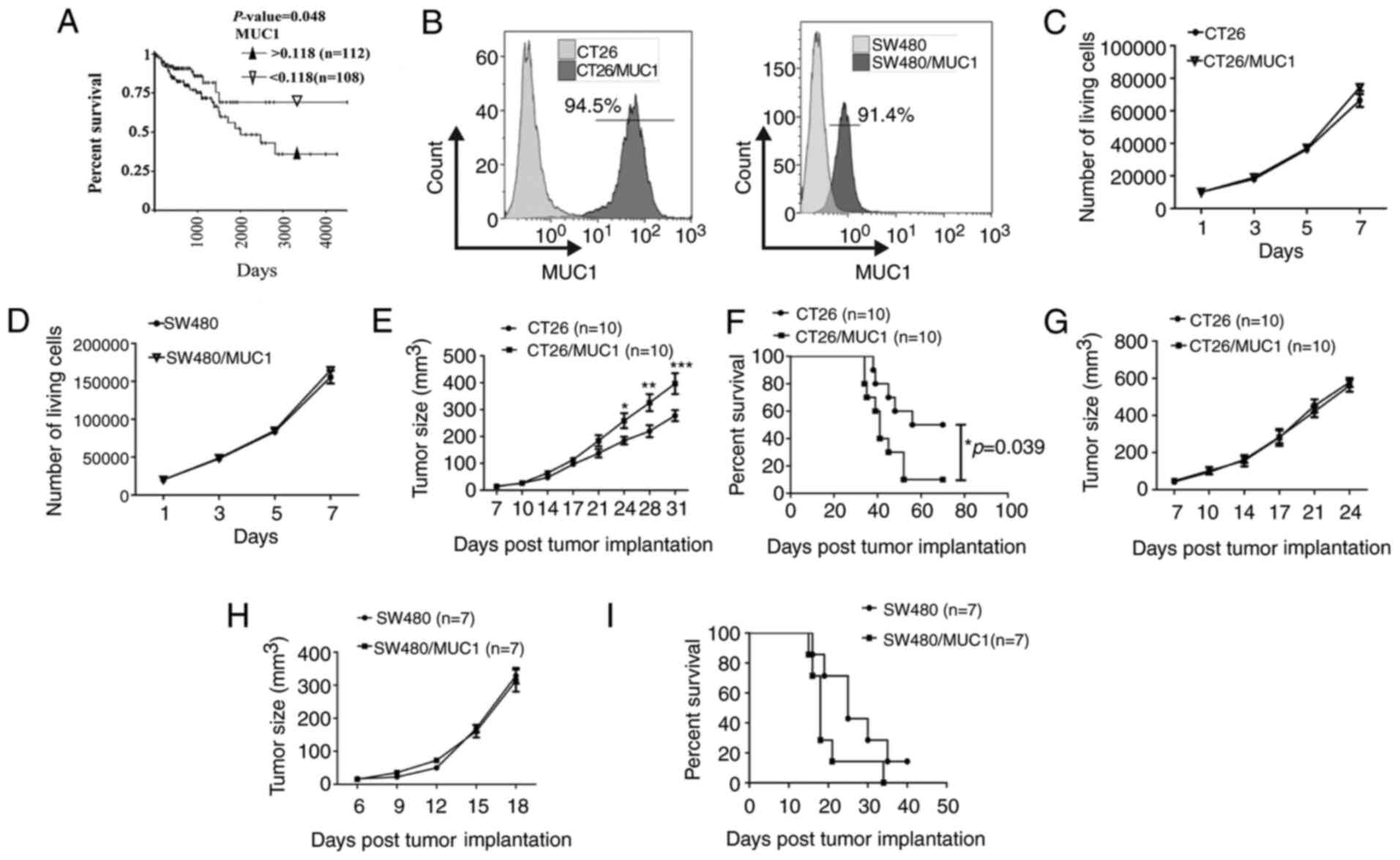
Lymphocytes mediating the immune
response serve a pro-tumor role in MUC1-positive colon cancer
High expression levels of MUC1 in tumor tissues are
associated with poor prognosis in patients with breast cancer
(5,9). However, to the best of our knowledge,
it is unclear whether this association exists in colon cancer. The
relationship between colon cancer survival time and MUC1 expression
was analyzed. In the TCGA discovery dataset, the copy number of
MUC1 was used to determine the cut-off value of expression
in tumor tissues as 0.118 (mean value of copy number). Patients
with high and low expression levels of MUC1 were identified
for further analysis as ≤0.1188 for the low-MUC1 group or
>0.118 for the high-MUC1 group. Kaplan-Meier survival
analysis was performed to compare overall survival according to
MUC1 expression. The data suggested that the survival time
in the low-MUC1 group was improved compared with the
high-MUC1 group (Fig. 1A). To
determine whether MUC1 promotes tumor cell proliferation, mouse
CT26 and human SW480 colon cancer cell lines expressing human MUC1
were prepared. The MUC1+ tumor cells were sorted by flow
cytometry, and MUC1 expression in tumor cells was confirmed
(Fig. 1B). Cell counting assays
suggested that MUC1 itself did not significantly promote cell
proliferation over 7 days (Fig. 1C and
D). In a colon cancer animal model, MUC1-positive CT26 cells
grew faster than MUC1-negative CT26 cells in murine models with
intact immune competence (Figs. 1E;
S1A and B). Additionally, it was
found that the survival time in the MUC1-negative tumor-bearing
BALB/c mice was improved compared with the MUC1-positive group
(Fig. 1F). MUC1 expression on tumor
cells that were isolated from the tumor-bearing mice was confirmed
again by flow cytometry (Fig. S1C).
However, there was no significant difference between the growth of
MUC1-positive and MUC1-negative CT26 tumor cells in nu/nu mice
(Fig. 1G). Furthermore, there was no
significant difference between the growth of MUC1-positive and
MUC1-negative SW480 tumor cells in murine models with lymphocyte
deficiency (Figs. 1H; S1D and E). Additionally, there was no
significant difference in the survival time between MUC1-positive
and MUC1-negative tumor-bearing mice with lymphocyte deficiency
(Fig. 1I). MUC1 expression on SW480
tumor cells that were isolated from the tumor-bearing mice was also
confirmed by flow cytometry (Fig.
S1F). This indicated that MUC1 might elicit a pro-tumor immune
response. Additionally, no signs of weight loss were observed in
any of the treatment groups (Fig.
S1G-I).
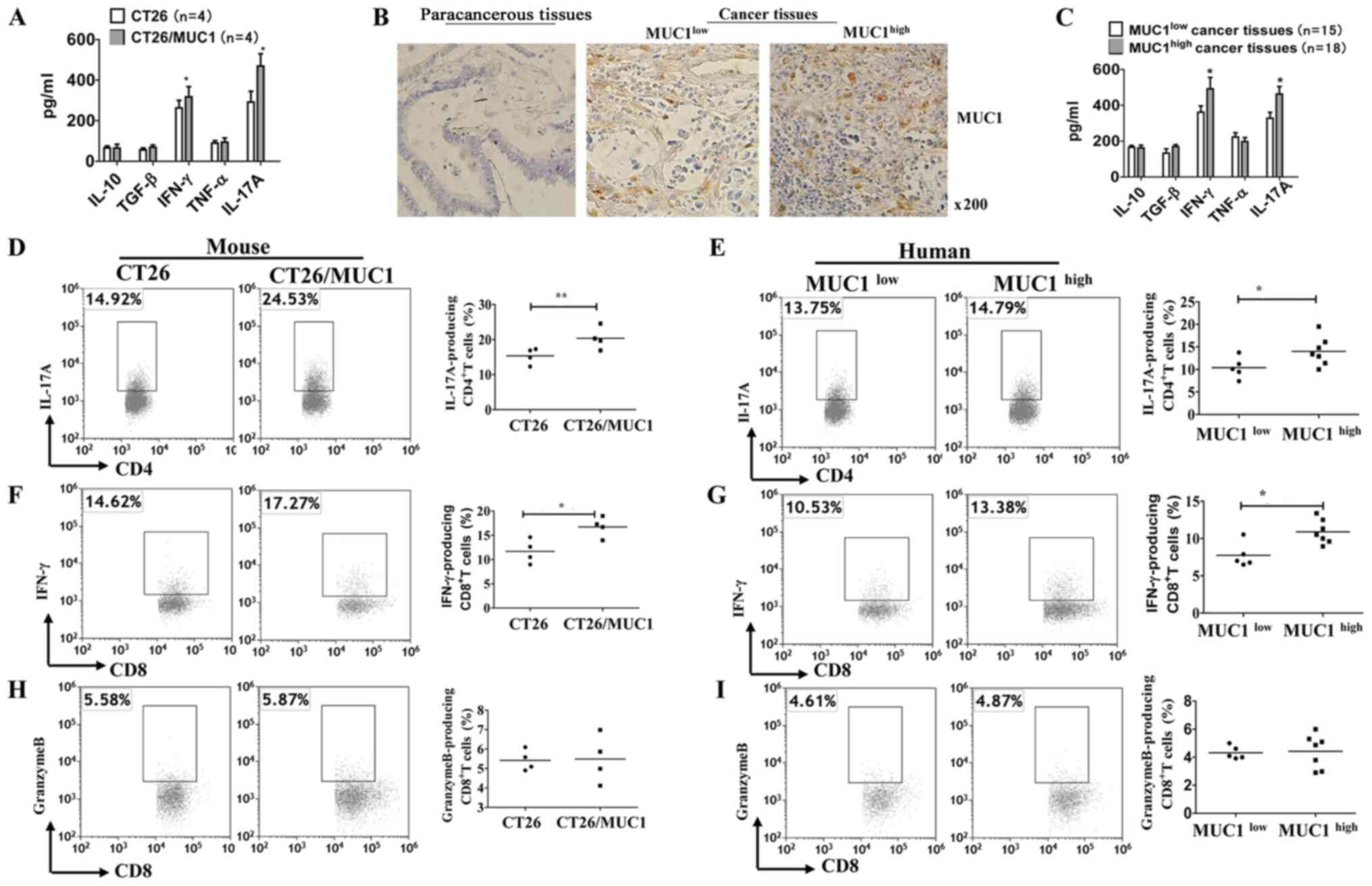
Increased accumulation of inflammatory
cytokines in MUC1-positive tumor tissues
Inflammation is known to be associated with the
development of colon cancer (20,21).
ELISA results measuring inflammatory cytokines in tumor-bearing
mice suggested that IL-17A and IFN-γ levels, but not IL-10, TGF-β
and TNF-α levels, were significantly increased in MUC1-positive
tissues compared with in MUC1-negative tissues (Fig. 2A). Subsequently, the expression
levels of MUC1 in tumor tissue samples were assessed using IHC
(Table I; Fig. 2B). High levels of IL-17A and IFN-γ
were observed in MUC1high tumor tissues, while the
levels of IL-10, TGF-β and TNF-α in MUC1high tumor
tissues were not increased compared with those in
MUC1low tumor tissues (Fig.
2C). To determine whether MUC1 augments or inhibits anti-MUC1
T-cell responses, tumor tissues from different groups of
tumor-bearing mice or patients were harvested, weighed and analyzed
by flow cytometry. CD45+CD3+CD4+
T-cells, CD45+ CD3+CD8+ T-cells,
CD45+CD11b+F4/80+ cells and
CD45+CD11b+GR1+ cells were gated
(Fig. S2). Intracellular cytokine
staining analysis demonstrated that MUC1-positive and
MUC1high tumor cells induced the production of
intracellular IL-17A in CD4+ T-cells (Fig. 2D and E) and IFN-γ in CD8+
T-cells (Fig. 2F and G). Since the
killing of tumor cells by CD8+ T cells is the principal
mechanism of immune protection against tumors (22), the present study detected the number
of granzyme B-producing CD8+ T cells in tumor tissues.
Notably, there was no significant increase in granzyme B-producing
CD8+ T cells in MUC1-positive and MUC1high
tumor tissues (Fig. 2H and I). These
results suggested that MUC1, as a tumor-associated antigen, does
not enhance the cytotoxicity of cytotoxic T cells (CTLs), although
it promotes an inflammatory response in the tumor
microenvironment.
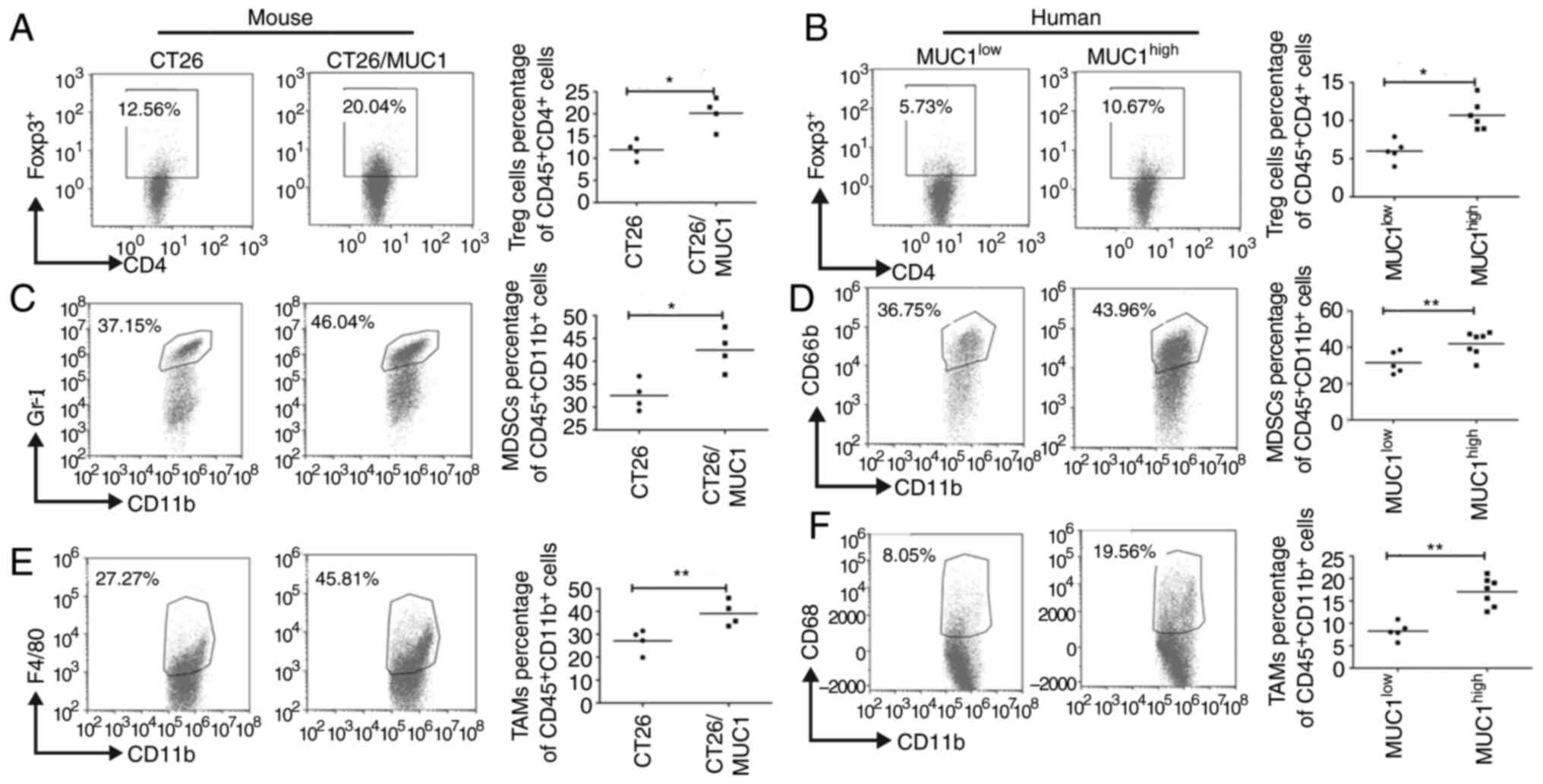
Increased accumulation of
immune-suppressive cells in MUC1-positive tumor tissues
Tumor-infiltrating regulatory T cells (Treg cells)
are a major immune cell population that contribute to the
establishment of an immunosuppressive tumor microenvironment, to
hamper the development of effective antitumor immunity, and are
often associated with poor prognosis (23). Significantly more CD4+
Foxp3+ Tregs were observed to infiltrate MUC1-positive
and MUC1high tumor tissues compared with MUC1-negative
and MUC1low tumor tissues (P<0.05; Fig. 3A and B). Furthermore, tumor growth is
associated with the accumulation of MDSCs and TAMs, where these
cells facilitate tumor immune escape by inhibiting antitumor immune
responses (22). The present results
indicated that significantly more MDSCs (Fig. 3C and D) and TAMs (Fig. 3E and F) accumulated in MUC1-positive
and MUC1high tumor tissues (P<0.05).
PDL1 expression on MDSCs, TAMs and
tumor cells is greater in MUC1high tumor tissues from
mice and patients
PDL1 has previously been demonstrated to exhaust
CTLs by binding PD1 (24,25). PDL1 expression was upregulated on
tumor cells from MUC1-positive and MUC1high tumor
tissues (Fig. 4A). However, in CT26
and SW480 cells transfected with the MUC1-expressing plasmid, MUC1
did not appear to promote PDL1 expression on tumor cells (Fig. 4B and C). These data indicated that
PDL1 expression in MUC1-positive and MUC1high tumor
tissues was upregulated due to other mechanisms. Since the present
data also indicated that inflammatory cytokines were elevated in
MUC1high tumor tissues, it is unclear whether high PDL1
expression on tumor cells was caused by these inflammatory
cytokines. IL-17A and IFN-γ stimulation promoted PDL1 expression on
CT26 cells, as well as CT26/MUC1 cells (Fig. 4D), which suggests that increased
inflammatory cytokine levels could promote PDL1 expression on
CT26/MUC1 cells. Since PDL1 is expressed on tumor cells and
antigen-presenting cells, the present study assessed whether PDL1
expression was increased on MDSCs and TAMs. PDL1 expression on
MDSCs and TAMs was significantly elevated in MUC1high
tumor tissues from mice and humans (Fig.
4E-H). Since PD1 expression on T-cells is similarly critical
for activation of the PDL1-PD1 signaling pathway (25), PD1 expression on CD8+
T-cells was also measured. PD1 staining was analyzed by FACS, and
the results revealed that PDL1 expression, in both mouse and human
CD8+ T-cells, was significantly increased (Fig. 4I and J). This suggested that more
inhibitory immune cells and checkpoint molecules accumulated in
MUC1high tumor tissues from mice and patients.
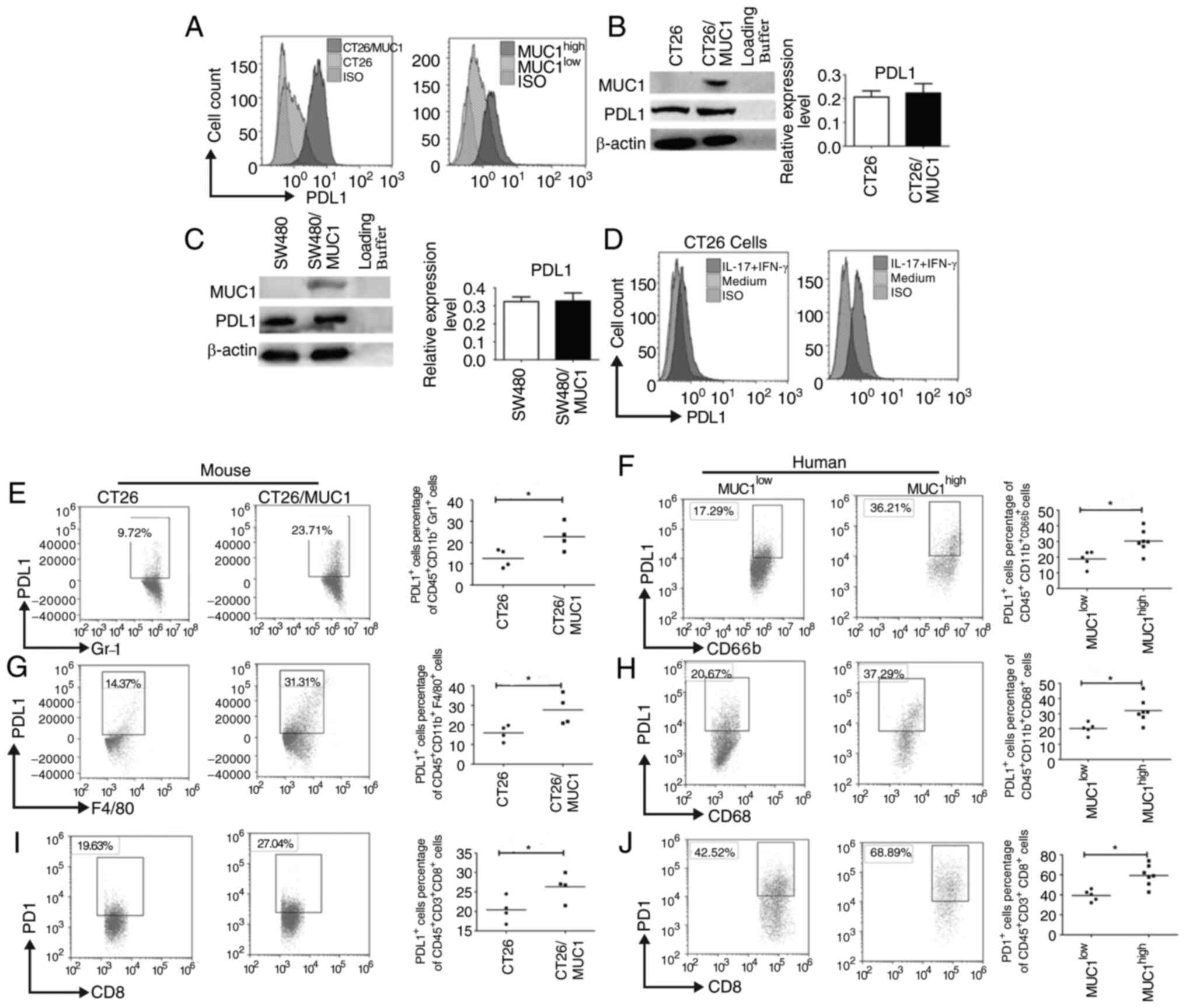 | Figure 4.PDL1 expression on MDSCs, TAMs and
tumor cells is greater in MUC1high tumor tissues from
mice and patients. (A) Tumor cells from tumor-bearing mice or
patients with colon cancer were isolated and PDL1 expression on the
surface of tumor cells was assessed using flow cytometry. (B) CT26
and (C) SW480 tumor cells were transfected with
pcDNA3.1(−)/Myc-His-MUC1. Semi-quantitative analysis of
western blots of PDL1 expression in tumor cells after the
transfection of pcDNA3.1(−)/Myc-His-MUC1 using an anti-PDL1
mAb was performed. β-actin was used as an internal control. (D)
CT26/vector and CT26/MUC1 cells were stimulated with mouse IFN-γ
(20 ng/ml) and IL-17A (10 ng/ml) for 48 h and surface PDL1
expression was assessed. PDL1 expression on the surface of (E)
mouse and (F) human MDSCs and (G) mouse and (H) human TAMs in tumor
tissues. PD1 expression on the surface of (I) mouse and (J) human
CD8+T cells in tumor tissues from tumor-bearing mice
(n=8) or patients with colon cancer (n=12). Data are representative
of four experiments. Error bars represent the standard error of the
mean. *P<0.05 vs. model control. GR-1, Ly-6G/Ly-6C; F4/80
(EMR1), mucin-like receptor 1; PDL1, programmed death ligand 1;
PD1, programmed cell death 1; MDSC, myeloid-derived suppressor
cells; TAM, tumor-associated macrophages; mAb, monoclonal antibody;
MUC1, mucin1; ISO, isotype control. |
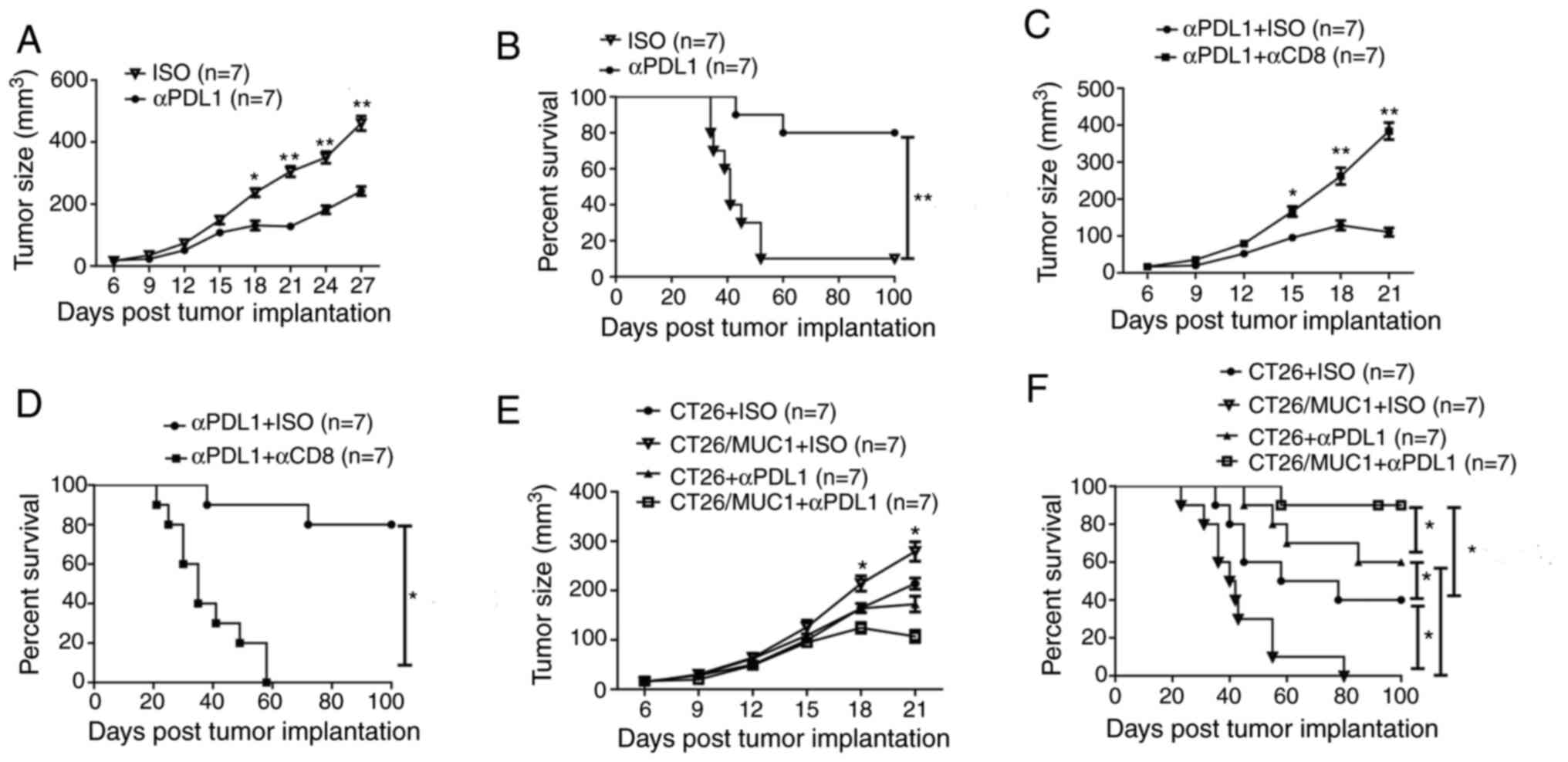
Targeting PDL1 induces an antitumor
immune response
Flow cytometry analysis of TILs in anti-PDL1
antibody-treated tumors indicated that the ratio of CD4/CD8 T cells
was significantly decreased after antibody treatment compared with
that in isotype antibody-treated tumors (Fig. 5A). Furthermore, the present study
revealed that the proportions of MDSCs and TAMs were significantly
lower in mice treated with anti-PDL1 antibodies than in
control-treated mice (Fig. 5B and
C). Notably, the percentage of myeloid-derived cells
(CD11b+ cells) was also significantly lower in mice
treated with anti-PDL1 antibodies compared with the control-treated
mice (Fig. 5D). Using the PDL1
antibody to target PDL1 led to significantly greater proportions of
IFN-γ-producing CD8+ T cells, granzyme B-producing
CD8+ T cells and IFN-γ-producing CD4+ T cells
compared with those in the controls (Fig. 5E-G). The percentage of
PD1+ CD8+ T cells was also significantly
greater in mice injected with the PDL1 antibody (Fig. 5H). These results indicated that
targeting PDL1 reduces the percentage of inhibitory immune cells
and enhances the activity and cytotoxicity of T cells in
MUC1-positive colon cancer models; therefore, targeting PDL1 in
MUC1-positive tumor has the potential to change the tumor
immunosuppressive microenvironment.
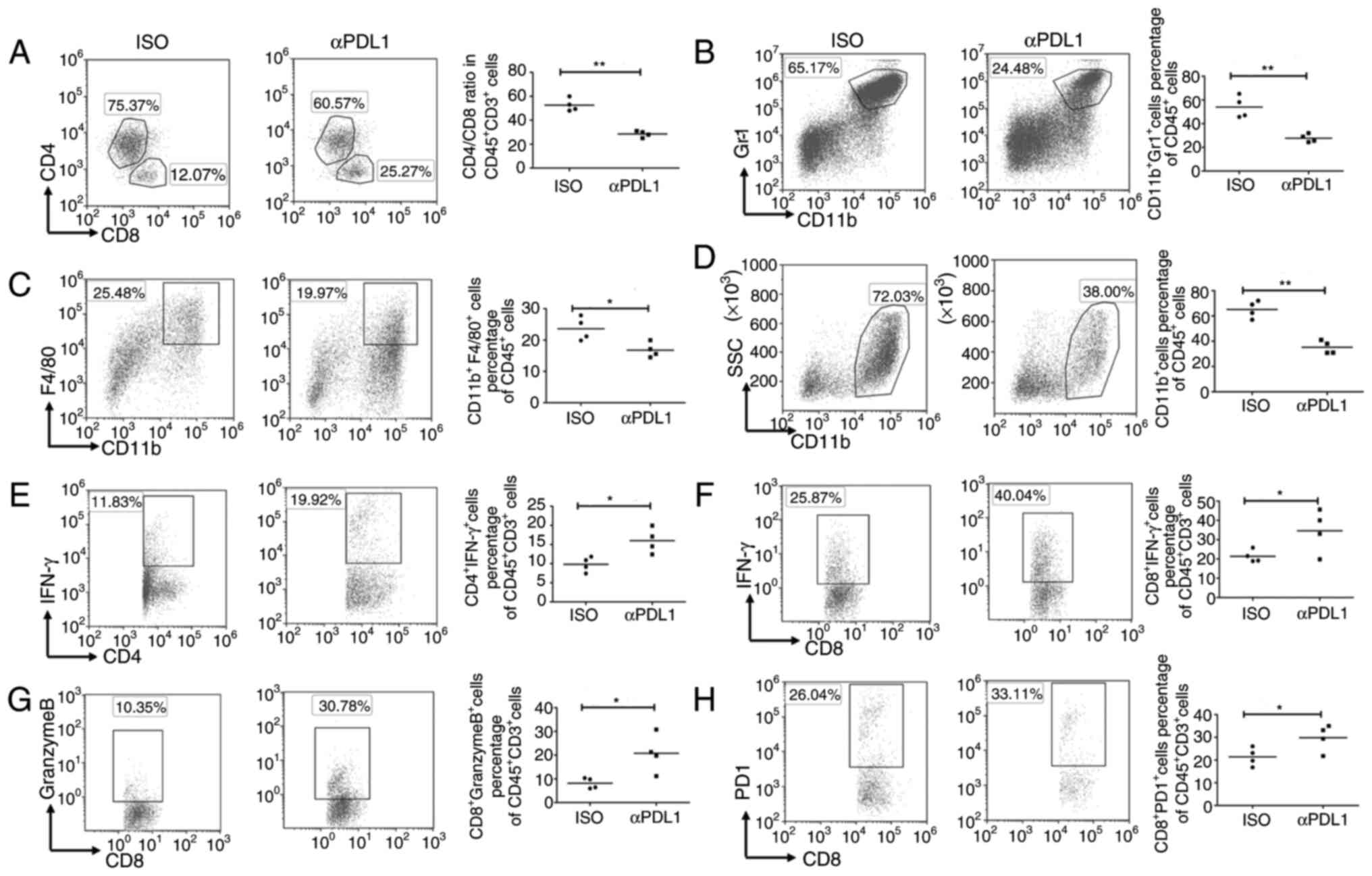 | Figure 5.Targeting PDL1 influences the
immunogenic tumor microenvironment. Mice were subcutaneously
challenged with 1×106 CT26/MUC1 tumor cells. After 7
days, the mice were injected intraperitoneally with an anti-PDL1
antibody (200 µg/mouse; n=4), or control-IgG (200 µg/mouse; n=4).
Antibody treatments were administered an additional three times
every 3 days after CT26/MUC1 inoculation. Surface staining patterns
of (A) T-cells (CD45+CD4+,
CD45+CD8+), (B) myeloid-derived suppressor
cells (CD45+CD11b+Gr1+), (C)
tumor-associated macrophages
(CD45+CD11b+F4/80+) and (D)
myeloid-derived cells (CD45+CD11b+) in tumor
tissues were analyzed using flow cytometry. TILs in tumor tissues
from tumor-bearing mice were stimulated with
phorbol-12-myristate-13-acetate and ionomycin in the presence of
brefeldin A for 4 h. (E) Percentages of IFN-γ+ T-cells
among CD4+CD45+ TILs. (F) Percentages of
IFN-γ+ T-cells among CD8+CD45+
TILs. (G) Percentages of granzyme B+ T-cells among
CD8+CD45+ TILs. (H) PD1 expression on the
surface of CD8+ T-cells in tumor tissues from
tumor-bearing mice. Data are representative of four experiments.
Error bars represent the standard error of the mean. **P<0.01
and *P<0.05 vs. model control. GR-1, Ly-6G/Ly-6C; F4/80 (EMR1),
mucin-like receptor 1; TIL, tumor-infiltrating lymphocytes; PDL1,
programmed death ligand 1; PD1, programmed cell death 1; ISO,
isotype control; MUC1, mucin1. |
Targeting PDL1 induces antitumor
effects
Since PDL1-targeting in MUC1-positive tumor tissues
was revealed to enhance the T-cell response, the present study next
aimed to determine whether antibody treatment with anti-PDL1 could
strengthen antitumor immunity. The data suggested that anti-PDL1
antibody treatment could significantly inhibit the growth of tumors
on day 18, 21, 24 and 27 after tumor implantation, and prolong the
survival time of tumor-bearing mice compared with the control
antibody (Figs. 6A and B; S3A and B). To explore the cellular
mechanisms underlying antitumor immunity, CD8+ T cells
in the mice were depleted during PDL1 antibody treatment. Mice with
depleted CD8+ cells lost the protective antitumor
effects induced by the PDL1 antibody (Fig. 6C and D). These findings suggest that
CD8+ T cells are critical for the antitumor immune
responses elicited by PDL1 antibodies. Additionally, no signs of
weight loss were observed in any of the treatment groups (Fig. S3C; data not shown for
anti-PDL1+anti-CD8). As more CD8+ and CD4+ T
cells were attracted to the tumor site in MUC1-positive
tumor-bearing mice (Fig. 2C-G),
despite the antitumor immune response of these immune cells being
diminished by the PD1-PDL1 signaling pathway, the present study
also determined whether stronger antitumor immunity in
MUC1-positive tumor-bearing mice could be elicited by anti-PDL1
antibody treatment, as compared with that in MUC1-negative tumor
bearing mice. Notably, MUC1-positive tumor-bearing mice had larger
tumors and shorter survival times than MUC1-negative tumor-bearing
mice, but MUC1-positive tumor-bearing mice injected with anti-PDL1
antibodies had smaller tumors and significantly longer survival
times than MUC1-negative tumor-bearing mice injected with anti-PDL1
antibodies (Fig. 6E and F). These
data suggest that targeting of PDL1 using PDL1 antibodies in
MUC1-positive tumor-bearing mice is necessary to effectively elicit
an antitumor immune response.
Discussion
The heterodimeric MUC1 protein is aberrantly
upregulated in colon cancer and has been used as a candidate target
antigen in peptide, dendritic cell and whole tumor vaccines
(6). High expression levels of MUC1
have been linked to poor outcomes in colon cancer (5,7).
However, the relationship between MUC1 upregulation and antitumor
immune response in the tumor microenvironment is unclear. The
present study demonstrated that MUC1 has a pro-tumor role in
immune-competent mice, and that Tregs, MDSCs and TAMs accumulate in
MUC1-positive tumor tissues. Furthermore, MUC1-positive tumor cells
were found to promote PDL1 expression on tumor cells, MDSCs and
TAMs by attracting more inflammatory cytokines and suppressing the
antitumor immune response. Finally, targeting PDL1 in MUC1-positive
tumor-bearing mice was demonstrated to transform the tumor
microenvironment, enhance the antitumor immune response and inhibit
tumor growth.
Tumor-infiltrating myeloid cells and
pro-inflammatory cytokines promote the development of colorectal
carcinoma (26). Tumor-associated
inflammation, manifested based on IL-17 expression, is of great
importance for the pathogenesis of colorectal carcinoma (27,28). The
present study observed greater IL-17A production in MUC1-positive
tumor tissues than in MUC1-negative tumor tissues. Since tumor
tissues tend to attract inflammatory cytokines during an immune
response (29), it is feasible that
high expression levels of IL-17A in MUC1-positive tumor tissues
might be specific to the tumor-associated antigen MUC1, but the
mechanisms responsible for this effect have not been addressed.
Notably, the present data also suggested that the expression levels
of IFN-γ and the percentage of IFN-γ-producing CD8+ T
cells were also greater in MUC1-positive tumor tissues compared
with in MUC1-negative tumor tissues, although the percentage of
granzyme B-producing CD8+ T cells was unchanged. IFN-γ
and IFN-γ-producing CD8+ T cells serve a pivotal role in
the killing of tumor cells, but a high level of IFN-γ can also
impair the cytotoxicity of CD8+ T cells and result in
T-cell exhaustion (30).
Immunosuppressive cells promote tumor immune escape by inducing
immunosuppression and accumulate in tumors where they exert T-cell
immunosuppression (31,32). When analyzing Treg cells, MDSCs and
TAMs in tumor tissues, more inhibitory immune cells were found to
accumulate in MUC1-positive tumor tissues than in MUC1-negative
tumor tissues, and thus, it was hypothesized that this might
represent a mechanism by which MUC1-positive and
MUC1high tumor cells escape immune surveillance.
MUC1 is a ligand for sialoadhesin, which is a
macrophage-restricted adhesion molecule, and as such, might be
involved in the recruitment of macrophages to the tumor site
(4). This could be why macrophages
are attracted to tumor sites in colon cancer (5,9). It is
also possible that the large numbers of inhibitory immune cells
neutralized the antitumor immune response of IFN-γ-producing
CD8+ T-cells.
High PDL1 expression on tumor cells or myeloid cells
is associated with poor prognosis compared with PDL1-negative
tumors (33,34). MUC1 can promote PDL1 expression via
the recruitment of MYC and NF-κB p65 to the PDL1 promoter in
certain solid tumors, such as triple-negative breast cancer, where
they contribute to an aggressive pathogenesis (35). Additionally, PDL1 expression is
associated with high expression levels of inflammatory factors,
such as IFN-γ and IL-17A, because these cytokines can stimulate
tumor cells or myeloid cells to produce PDL1 and then suppress the
cytotoxicity of tumor-specific T cells (15,36). The
present data indicates that MUC1 did not directly promote PDL1
expression on colon cancer cells, but IL-17A, IFN-γ and myeloid
cells were more attracted to MUC1-positive tumor tissues than
MUC1-negative tumor tissues, which emphasizes the importance of
exploring the mechanism through which these inhibitory immune cells
and inflammatory cytokines promote the growth of MUCI-positive
tumor cells. IL-17A and IFN-γ were observed to enhance PDL1
expression on CT26/MUC1 and SW480/MUC1 tumor cells more so than
MUC1 itself, and high PDL1 expression was observed on MDSCs and
TAMs. This might be because MUC1-positive tumor cells recruit
robust inflammatory cytokines and enhance PDL1 expression on tumor
cells and myeloid cells. Overall, MUC1 promoted PDL1 expression on
tumor cells through the recruitment of inflammatory cytokines and
then inhibited the antitumor immune response via the PDL1/PD1
signaling pathway, enabling the evasion of immune surveillance.
Targeting PDL1 on tumor cells, MDSCs and macrophages
can suppress the growth of tumor cells (37–40).
While it is possible to restore the tumor microenvironment and
suppress tumor cell growth by blocking the PDL1-PD1 signaling
pathway, it was found that this approach in MUC1-positive
tumor-bearing mice promoted the infiltration of IFN-γ-producing
CD8+ T-cells and inhibited the infiltration of myeloid
cells, MDSCs and TAMs in the tumor microenvironment. This is
pivotal to transform the tumor immunosuppressive microenvironment
and enhance antitumor immune responses in favor of tumor
eradication (41), since the
presence of a high number of CD8+ T lymphocytes and
fewer MDSCs and TAMs in tumor tissues is associated with improved
prognosis (42,43). Interestingly, the present findings
also suggest that targeting PDL1 in MUC1-positive tumor-bearing
mice inhibited tumor growth and elicited a stronger antitumor
effect than that in MUC1-negative tumor-bearing mice. This provides
further in vivo evidence of the tumor suppressor function of
PDL1 (44–46).
Notably, the present study only investigated the
status of immune cells in MUC1-positive and MUC1-negative tumor
tissues, and further studies are required to explore the mechanism
through which MUC1 upregulation promotes the accumulation of
suppressive immune cells in MUC1-positive tumor tissues. In
summary, the present study demonstrated a pro-tumor role of MUC1
with important implications for MUC1-positive colorectal cancer,
and provides a foundation for the application of PDL1 inhibitors to
MUC1-positive colon cancer.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present work was supported by grants from the
Leader Training Program in Medical Subjects of Health and Family
Planning Commission of Yunnan Province (grant no. D-201657) and the
Scientific Research Fund General Projects of Yunnan Provincial
Department of Education (grant no. 2015Y150).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YHZ and XD performed the immunoassays and drafted
the manuscript. LB and XS collected the patient samples and
performed the statistical analysis. YJZ designed the study and
helped to revise the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The treatment of mice was approved by the
Institutional Animal Care and Use Committee of Kunming Medical
University (approval no. KMU20170121). The patient research was
approved by the Ethics Committee of The First Affiliated Hospital
of Kunming Medical University (approval no. KMU20160901) and
conformed with the ethical standards of the World Medical
Association Declaration of Helsinki. All patients provided written
informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MUC1
|
mucin1
|
|
Treg cells
|
regulatory T cells
|
|
MDSCs
|
myeloid-derived suppressor cells
|
|
TAM
|
tumor-associated macrophage
|
|
PDL1
|
programmed death ligand 1
|
|
PD1
|
programmed cell death 1
|
|
TILs
|
tumor-infiltrating lymphocytes
|
|
CTL
|
cytotoxic T cell
|
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ahmad R, Alam M, Hasegawa M, Uchida Y,
Al-Obaid O, Kharbanda S and Kufe D: Targeting MUC1-C inhibits the
AKT-S6K1-elF4A pathway regulating TIGAR translation in colorectal
cancer. Mol Cancer. 16:332017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kasprzak A, Siodła E, Andrzejewska M,
Szmeja J, Seraszek-Jaros A, Cofta S and Szaflarski W: Differential
expression of mucin 1 and mucin 2 in colorectal cancer. World J
Gastroenterol. 24:4164–4177. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Singh R and Bandyopadhyay D: MUC1: A
target molecule for cancer therapy. Cancer Biol Ther. 6:481–486.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Taylor-Papadimitriou J, Burchell J, Miles
DW and Dalziel M: MUC1 and cancer. Biochim Biophys Acta.
1455:301–313. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nath S and Mukherjee P: MUC1: A
multifaceted oncoprotein with a key role in cancer progression.
Trends Mol Med. 20:332–342. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guo M, You C and Dou J: Role of
transmembrane glycoprotein mucin 1 (MUC1) in various types of
colorectal cancer and therapies: Current research status and
updates. Biomed Pharmacother. 107:1318–1325. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Smith JS, Colon J, Madero-Visbal R, Isley
B, Konduri SD and Baker CH: Blockade of MUC1 expression by glycerol
guaiacolate inhibits proliferation of human breast cancer cells.
Anticancer Agents Med Chem. 10:644–6650. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Apostolopoulos V, Pietersz GA and McKenzie
IF: MUC1 and breast cancer. Curr Opin Mol Ther. 1:98–103.
1999.PubMed/NCBI
|
|
10
|
Yang E, Hu XF and Xing PX: Advances of
MUC1 as a target for breast cancer immunotherapy. Histol
Histopathol. 22:905–922. 2007.PubMed/NCBI
|
|
11
|
Agrawal B, Krantz MJ, Reddish MA and
Longenecker BM: Cancer-associated MUC1 mucin inhibits human T-cell
proliferation, which is reversible by IL-2. Nat Med. 4:43–49. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
van de Wiel-van Kemenade E, Ligtenberg MJ,
de Boer AJ, Buijs F, Vos HL, Melief CJ, Hilkens J and Figdor CG:
Episialin (MUC1) inhibits cytotoxic lymphocyte-target cell
interaction. J Immunol. 151:767–776. 1993.PubMed/NCBI
|
|
13
|
Topalian SL, Drake CG and Pardoll DM:
Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor
immunity. Curr Opin Immunol. 24:207–212. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tang J, Yu JX, Hubbard-Lucey VM,
Neftelinov ST, Hodge JP and Lin Y: Trial watch: The clinical trial
landscape for PD1/PDL1 immune checkpoint inhibitors. Nat Rev Drug
Discov. 17:854–855. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ma YF, Chen C, Li D, Liu M, Lv ZW, Ji Y
and Xu J: Targeting of interleukin (IL)-17A inhibits PDL1
expression in tumor cells and induces anticancer immunity in an
estrogen receptor-negative murine model of breast cancer.
Oncotarget. 5:7614–7624. 2017. View Article : Google Scholar
|
|
16
|
Baldus SE, Engelmann K and Hanisch FG:
MUC1 and the MUCs: A family of human mucins with impact in cancer
biology. Crit Rev Clin Lab Sci. 41:189–231. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Goldman MJ, Craft B, Hastie M, Repečka K,
McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, et al:
Visualizing and interpreting cancer genomics data via the Xena
platform. Nat Biotechnol. 38:675–678. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cancer Genome Atlas Network: Comprehensive
molecular characterization of human colon and rectal cancer.
Nature. 487:330–337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Helfinger V, Henke N, Harenkamp S, Walter
M, Epah J, Penski C, Mittelbronn M and Schröder K: The NADPH
oxidase Nox4 mediates tumour angiogenesis. Acta Physiol (Oxf).
216:435–446. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Koboziev I, Karlsson F and Grisham MB:
Gut-associated lymphoid tissue, T cell trafficking, and chronic
intestinal inflammation. Ann N Y Acad Sci. 1207 (Suppl 1):E86–E93.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Terzić J, Grivennikov S, Karin E and Karin
M: Inflammation and colon cancer. Gastroenterology.
138:2101–2114.e5. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gajewski TF, Schreiber H and Fu YX: Innate
and adaptive immune cells in the tumor microenvironment. Nat
Immunol. 14:1014–1022. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Villarreal DO, L'Huillier A, Armington S,
Mottershead C, Filippova EV, Coder BD, Petit RG and Princiotta MF:
Targeting CCR8 induces protective antitumor immunity and enhances
vaccine-induced responses in colon cancer. Cancer Res.
78:5340–5348. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Amarnath S, Mangus CW, Wang JC, Wei F, He
A, Kapoor V, Foley JE, Massey PR, Felizardo TC, Riley JL, et al:
The PDL1-PD1 axis converts human TH1 cells into regulatory T cells.
Sci Transl Med. 3:111ra1202011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dong H, Strome SE, Salomao DR, Tamura H,
Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al:
Tumor-associated B7-H1 promotes T-cell apoptosis: A potential
mechanism of immune evasion. Nat Med. 8:793–800. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Beatty P, Ranganathan S and Finn OJ:
Prevention of colitis-associated colon cancer using a vaccine to
target abnormal expression of the MUC1 tumor antigen.
Oncoimmunology. 1:263–270. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dmitrieva-Posocco O, Dzutsev A, Posocco
DF, Hou V, Yuan W, Thovarai V, Mufazalov IA, Gunzer M, Shilovskiy
IP, Khaitov MR, et al: Cell-type-specific responses to
interleukin-1 control microbial invasion and tumor-elicited
inflammation in colorectal cancer. Immunity. 50:166–180.e7. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang K and Karin M: Tumor-elicited
inflammation and colorectal cancer. Adv Cancer Res. 128:173–196.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen J, Pitmon E and Wang K: Microbiome,
inflammation and colorectal cancer. Semin Immunol. 32:43–53. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Farhood B, Najafi M and Mortezaee K:
CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A
review. J Cell Physiol. 234:8509–8521. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bianchi G, Borgonovo G, Pistoia V and
Raffaghello L: Immunosuppressive cells and tumour microenvironment:
Focus on mesenchymal stem cells and myeloid derived suppressor
cells. Histol Histopathol. 26:941–951. 2011.PubMed/NCBI
|
|
32
|
Liu Y and Cao X: Immunosuppressive cells
in tumor immune escape and metastasis. J Mol Med (Berl).
94:509–522. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Muenst S, Schaerli AR, Gao F, Däster S,
Trella E, Droeser RA, Muraro MG, Zajac P, Zanetti R, Gillanders WE,
et al: Expression of programmed death ligand 1 (PD-L1) is
associated with poor prognosis in human breast cancer. Breast
Cancer Res Treat. 146:15–24. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sabatier R, Finetti P, Mamessier E,
Adelaide J, Chaffanet M, Ali HR, Viens P, Caldas C, Birnbaum D and
Bertucci F: Prognostic and predictive value of PDL1 expression in
breast cancer. Oncotarget. 6:5449–5464. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Maeda T, Hiraki M, Jin C, Rajabi H, Tagde
A, Alam M, Bouillez A, Hu X, Suzuki Y, Miyo M, et al: MUC1-C
induces PD-L1 and immune evasion in triple-negative breast cancer.
Cancer Res. 78:205–215. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pardoll DM: The blockade of immune
checkpoints in cancer immunotherapy. Nat Rev Cancer. 12:252–264.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Curiel TJ, Wei S, Dong H, Alvarez X, Cheng
P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC, et
al: Blockade of B7-H1 improves myeloid dendritic cell-mediated
antitumor immunity. Nat Med. 9:562–567. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ghebeh H, Tulbah A, Mohammed S, Elkum N,
Bin Amer SM, Al-Tweigeri T and Dermime S: Expression of B7-H1 in
breast cancer patients is strongly associated with high
proliferative Ki-67-expressing tumor cells. Int J Cancer.
121:751–758. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu Y, Zeng B, Zhang Z, Zhang Y and Yang
R: B7-H1 on myeloid-derived suppressor cells in immune suppression
by a mouse model of ovarian cancer. Clin Immunol. 129:471–481.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Parsa AT, Waldron JS, Panner A, Crane CA,
Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, et
al: Loss of tumor suppressor PTEN function increases B7-H1
expression and immunoresistance in glioma. Nat Med. 13:84–88. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xiao W, Ibrahim ML, Redd PS, Klement JD,
Lu C, Yang D, Savage NM and Liu K: Loss of Fas expression and
function is coupled with colon cancer resistance to immune
checkpoint inhibitor immunotherapy. Mol Cancer Res. 17:420–430.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Noy R and Pollard JW: Tumor-associated
macrophages: From mechanisms to therapy. Immunity. 41:49–61. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wherry EJ: T cell exhaustion. Nat Immunol.
12:492–499. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lucas J, Hsieh TC, Halicka HD,
Darzynkiewicz Z and Wu JM: Upregulation of PD-L1 expression by
resveratrol and piceatannol in breast and colorectal cancer cells
occurs via HDAC3/p300-mediated NF-κB signaling. Int J Oncol.
53:1469–1480. 2018.PubMed/NCBI
|
|
45
|
Stenehjem DD, Tran D, Nkrumah MA and Gupta
S: PD1/PDL1 inhibitors for the treatment of advanced urothelial
bladder cancer. Onco Targets Ther. 11:5973–5989. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zheng A, Li F, Chen F, Zuo J, Wang L, Wang
Y, Chen S, Xiao B and Tao Z: PD-L1 promotes head and neck squamous
cell carcinoma cell growth through mTOR signaling. Oncol Rep.
41:2833–2843. 2019.PubMed/NCBI
|