Introduction
Lung squamous cell carcinoma (SqCC) accounts for
~30% of non-small cell lung cancer (NSCLC) (1). In recent years, the treatment of lung
SqCC has been less successful compared with lung adenocarcinoma
(2). Although complex biological
studies have improved our understanding of the pathological
mechanisms of SqCC, for example identifying PI3K catalytic subunit
α (PIK3CA), hepatocyte growth factor receptor and fibroblast growth
factor receptor as important driver mutations, there are currently
no effective agents for its treatment (3). In the era of immunotherapy,
pembrolizumab has been listed as the first-line treatment for
patients with NSCLC with programmed death-ligand 1 tumor proportion
score (TPS) ≥50%, according to the 2019 National Comprehensive
Cancer Network (NCCN) guidelines (4). However, the median overall survival
(OS) time of the patients with NSCLC treated with pembrolizumab
alone is only 20 months (5), which
is poor compared with targeted therapy in adenocarcinomas (6). In the most notable clinical trial of
SqCC, CheckMate-017, the median OS time of patients treated with
nivolumab as the second-line treatment for advanced lung SqCC was
prolonged by only 3 months compared with those treated with
docetaxel (7). In the KEYNOTE-010
clinical trial, the OS time in the 2 and 10 mg/kg pembrolizumab
groups was prolonged by 2 and 4 months, respectively, compared with
that in patients treated with docetaxel (8). At present, chemotherapy and
radiotherapy remain the main treatment options for advanced lung
SqCC.
Since epithelial growth factor receptor-tyrosine
kinase inhibitors (EGFR-TKIs) inhibit the phosphorylation of EGFR
tyrosine kinase, they are supposed to exhibit high efficacy in the
tumors with high expression levels of EGFR (9). However, this is not the case in lung
SqCC. Previous studies have demonstrated that ≤84% of patients with
lung SqCC express EGFR, which is significantly higher than the
percentage of patients with EGFR-positive adenocarcinoma (~44%)
(10,11); however, only patients with
adenocarcinoma with sensitizing EGFR mutations respond to EGFR-TKI
treatment, whereas for the other patients with adenocarcinomas and
SqCCs, the efficacy of EGFR-TKIs is not satisfactory (9). In addition, EGFR mutations, in terms of
their predictive value for the response to EGFR-TKI treatment, are
different between SqCC and adenocarcinoma. For example, EGFR
mutations are not valid predictors for EGFR-TKI response in lung
SqCC, with the response rate <30% in patients with lung SqCC
with sensitizing EGFR mutations, as well as the median
progression-free survival time of only 2–3 months (12–14),
which is shorter compared with that in patients with adenocarcinoma
with sensitizing EGFR mutations (10–12 months) (9). Therefore, the intrinsic resistance to
EGFR-TKIs in patients with lung SqCC has become an urgent problem.
The present study was based on the hypothesis that certain genes
may be associated with intrinsic EGFR-TKIs resistance.
In the past decade, the application of next
generation sequencing to investigate the genomic characterization
of lung SqCC has provided a further understanding of the possible
treatment targets. Filipits (15)
have reported that the potential targeted therapies in lung SqCC
include EGFR, fibroblast growth factor receptor 1, Met
proto-oncogene, PI3K, discoidin domain receptor tyrosine kinase 2,
BRAF, AKT, cytotoxic T lymphocyte-associated protein and programmed
cell death 1. Additionally, Schwaederle et al (16) have identified some mutations in TP53
(64.5% of analyzed patients) that are not targeted by any existing
agents, PIK3CA (28.5%), cyclin-dependent kinase inhibitor 2A
(24.4%), SOX2 (17.7%) and cyclin D1 (15.8%). EGFR is highly
expressed in the majority of SqCC tumor tissues, but the clinical
benefits of EGFR-TKIs are modest (17,18). At
present, no effective targeted agents for SqCC have been approved
for use in clinical practice. Therefore, it is crucial to explore
the possible mechanisms of intrinsic EGFR-TKI resistance in lung
SqCC.
The high-throughput RNA interference (RNAi)
technology is the combination of high-throughput chromatin
immunoprecipitation and small interference RNA library that makes
RNAi no longer limited to one gene being silenced, but can be used
for simultaneous large-scale screening of genes and their
functions. In the present study, the most effective gene inhibitors
that can overcome EGFR-TKI resistance of lung SqCC were screened
using high-throughput RNAi technology. The results may bring aid
the development of novel treatments for patients with SqCC.
Materials and methods
Cell lines and culture
The human lung squamous carcinoma cell lines
SK-MES-1, H1688, H1299 and H226 were provided by Cancer Institute
of Tongji University Medical School, (Shanghai, China). The cells
were cultured at 37°C with 5% CO2 in Dulbecco's modified
Eagle's medium (DMEM) supplemented with 10% fetal bovine serum
(FBS) (Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin
and 100 mg/ml streptomycin.
Tumor samples
Tumor samples were collected from 50 patients
recruited at the Shanghai Pulmonary Hospital between July 2012 and
June 2015. The patients were newly diagnosed with histologically
confirmed lung SqCC and adenocarcinoma by biopsy. The mean age was
63.77 (49–74) years, 63.44 (47–71) years and 62.9 (49–72) years in
SqCC, wide-type EGFR adenocarcinoma and mutated-EGFR adenocarcinoma
groups, respectively. There was no females in the SqCC group, but
three (33.3%) females in the wide-type EGFR adenocarcinoma group,
and seven (70%) female in the mutated-EGFR adenocarcinoma group.
The rest of the patients were male. Patients with a previous
medical history of cancer, radiotherapy or chemotherapy were
excluded. This study was approved by the Ethics Committee of Tongji
University (approval no. 2012-FK-14), and all patients provided
signed informed consent.
High content screening (HCS)
In total, 18 target shRNAs (Table I) were selected using the following
criteria according to Genechem database based on the NIH Cancer
Genome Project database, OMIM, MalaCards and UniProtKB database
(https://www.genechem.com.cn/index/index/index.html).
Firstly, the genes were highly correlated with EGFR-TKIs and
certified by pathway and functional network analysis from Genechem,
and the gene function annotations should be relatively clear.
Subsequently, the genes that were reported in PubMed <100 times,
or those whose function were obviously inconsistent with their
expected function, were removed. Lentiviruses carrying the green
fluorescent protein (GFP) gene and expressing short hairpin (sh)RNA
against ILK, phosphatase and tensin homolog (PTEN),
mitogen-activated protein kinase kinase kinase 14 (MAP3K14),
myeloid differentiation primary response gene 88 (MYD88), serum
response factor (SRF), interleukin-1 receptor associated kinase 1
(IRAK1), baculoviral IAP repeat containing 5 (BIRC5),
phosphoinositide-3-kinase, class 2, α polypeptide (PIK3C2A),
protein tyrosine phosphatase type IVA, member 3 (PTP4A3), Bruton's
tyrosine kinase (BTK), nemo-like kinase (NLK), Raf-1 proto-oncogene
(RAF1), signal transducer and activator of transcription 3 (STAT3),
sarcoma gene (SRC), aurora kinase A (AURKA), interleukin-1
receptor-associated kinase 4 (IRAK4), erbb2-interacting protein
(ERBB2IP) or Cadherin (CDH), as well as negative control (NC)
shRNA, were purchased from Shanghai GeneChem Co., Ltd. SK-MES-1
cells were transfected with shRNA lentiviruses (non-targeting
shRNA, PSC1369, PSC1446, PSC14867 mix, PSC14872, PSC3584, PSC1489 6
mix, PSC14359, PSC14907, PSC1675, PSC14821 mix, PSC1786, PSC1809,
PSC8012, PSC4913, PSC2260, PSC14817 mix, PSC4899, and PSC3306 that
target the NC, ILK, PTEN, MAP3K14, MYD88, SRF, IRAK1, BIRC5,
PIK3C2A, PTP4A3, BTK, NLK, RAF1, STAT3, SRC, AURKA, IRAK4, ERBB2IP,
and CDH genes, respectively; Table
I) using Polybrene (Shanghai GeneChem Co., Ltd.) at 37°C for 12
h, and MOI was 10. Following 2 or 3 days of transfection, when the
fluorescence rate reached 80%, the cells were collected for
subsequent cell proliferation experiments. In the HCS experiment, 2
µM erlotinib was used to screen the combined effects of erlotinib
and shRNAs after 72 h treatment. The high-throughput RNAi
experiments were performed by Shanghai GeneChem Co., Ltd.
 | Table I.Target and lentiviral shRNA and
fold-changes of cell proliferation after 72-h treatment with shRNA
and erlotinib. |
Table I.
Target and lentiviral shRNA and
fold-changes of cell proliferation after 72-h treatment with shRNA
and erlotinib.
| Target | Identifier of
shRNA | Fold-change |
|---|
| NC | Non-targeting
shRNA | 1.00 |
| ILK | PSC1369 | 1.29a |
| PTEN | PSC1446 | 1.14 |
| MAP3K14 | PSC14867mix | 1.09 |
| MYD88 | PSC14872 | 1.05 |
| SRF | PSC3584 | 1.04 |
| IRAK1 | PSC14896mix | 1.03 |
| BIRC5 | PSC14359 | 1.03 |
| PIK3C2A | PSC14907 | 1.00 |
| PTP4A3 | PSC1675 | 1.00 |
| BTK | PSC14821mix | 0.97 |
| NLK | PSC1786 | 0.97 |
| RAF1 | PSC1809 | 0.93 |
| STAT3 | PSC8012 | 0.92 |
| SRC | PSC4913 | 0.92 |
| AURKA | PSC2260 | 0.89 |
| IRAK4 | PSC14817mix | 0.87 |
| ERBB2IP | PSC4899 | 0.86 |
| CDH | PSC3306 | 0.83 |
Celigo, a high-throughput screening system based on
automatic image acquisition and image data analysis, was used for
the HCS test. The target in this test was the SK-MES-1 cells
(growing in a 96-well plate) expressing GFP following lentiviral
infection. Celigo was used to identify the cells with green
fluorescence, capture, analyze and process images and calculate the
number of cells in the various groups in the plate. After 5 days of
continuous reading, the cell growth curves were produced to
determine the cell proliferation.
Cell proliferation analysis
The cells were seeded into 96-well plates
(3×103 cells/well) in triplicate and exposed to various
concentrations (2, 4 and 10 µmol) of erlotinib. After 72 h, 20 µl
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
solution (Abcam) (5 mg/ml) was added to each well and incubated for
4 h at 37°C. Subsequently, the formazan crystal was dissolved with
dimethyl sulfoxide, and the absorbance at 530 nm was read using a
microplate reader. The percentage of surviving cells was calculated
as follows: Survival rate=(mean absorbance of the replicate wells
containing drugs-mean absorbance of the replicate background
wells)/(mean absorbance of the replicate drug-free wells-mean
absorbance of the replicate background wells) ×100%.
Fold-change=[(NC + drug)/NC)/(target shRNA + drug)/target shRNA].
The test was performed independently three times.
Apoptosis analysis
Annexin V-APC Apoptosis Detection kit (eBioscience;
Thermo Fisher Scientific, Inc.) and a transferase-mediated
deoxyuridine triphosphate nick-end labeling (TUNEL) kit (Promega
Corporation) were used to determine apoptosis levels. In the flow
cytometry assay, SK-MES-1 cells (1.2×106 cells/well)
were plated in 6-well plates. Following 24 h incubation at 37°C, 2
µM erlotinib was added into the experimental wells and incubated
for another 72 h at 37°C. The cells were harvested, washed with PBS
and resuspended in 500 µl binding buffer (from the aforementioned
kit). The cells were stained with 5 µl Annexin V-PE and incubated
for 5 min at room temperature in the dark. Quantification of
apoptosis was determined by flow cytometer (FACSCalibur; BD
Bioscience). Fluorescence microscope (micropublisher 3.3RTV,
Olympus Corporation) was used to count the cells with Annexin
V-APC. BD CellQuest software was used for analysis.
For the TUNEL assay, SK-MES-1 cells
(5×105 cells/well) were seeded in 24-well plates and
exposed to 2 µM erlotinib for 72 h. Apoptosis was assessed by the
TUNEL assay kit according to the manufacturer's protocol. The
apoptotic index (%) was calculated using the following formula:
Apoptotic index=positive for Annexin V- or PE-stained cells/total
cells ×100%. The apoptotic index for Annexin V and PE-stained cells
were calculated separately.
Western blotting
Cells were washed twice with ice-cold PBS and lysed
in 0.1 ml lysis buffer (HyClone; Cyvita) on ice for 30 min.
Insoluble debris was removed by centrifuging at 12,000 × g for 15
min at 4°C. BCA Protein Assay kit (HyClone; Cyvita) was used for
protein detection, and 20 µg protein was loaded per lane. The
percentage of the SDS-PAGE gel was 10% and then the proteins were
transferred to PVDF membrane. PVDF membrane was sealed with
blocking solution (TBST solution containing 5% skimmed milk) at
room temperature for 1 h or 4°C overnight. Mouse Anti-Flag
(1:3,000; cat. no. F1804; Sigma-Aldrich; Merck KGaA) was diluted
with blocking solution, and then incubated with the closed PVDF
membrane at room temperature for 2 h or 4°C overnight. Mouse
anti-GAPDH (1:5,000; cat. no. sc-32233; Santa Cruz Biotechnology,
Inc.) was for the reference. Goat anti-mouse IgG (1:5,000; cat. no.
sc-2005; Santa Cruz Biotechnology, Inc.) was diluted with blocking
solution, and then incubated with the closed PVDF membrane at room
temperature for 2 h. Antibodies against human ILK (1:3,000; cat.
no. MAB4266-SP), EGFR (1:1,000; cat. no. MAB3570-SP), β-actin
(1:1,000; cat. no. MAB8929-SP) and GAPDH (1:1,000; cat. no.
AF5718-SP) (all from R&D Systems, Inc.) were used according to
the manufacturer's instructions. The ECL + Plus™ Substrate
(Amersham) was used for color development. Densitometry analysis
was performed using an Odyssey® Infrared Imaging system
(LI-COR Biosciences).
Quantitative (q)PCR
Total RNA was extracted from tumors using TRIzol
(Shanghai Perfectics Co., Ltd), and reverse transcribed to cDNA
using M-MLV Reverse Transcriptase kit (Promega Corporation).
Reverse transcription was carried out at 37°C for 10 min. In total,
1 µg cDNA was used for qPCR analysis using a LightCycler PCR
instrument (Roche Diagnostics) according to the manufacturer's
instructions. The upstream primer of ILK and GAPDH was
5′-GACGACATTTTCACTCAGTGCC-3′ and 5′-TGACTTCAACAGCGACACCCA-3′
respectively, and the downstream primer was
5′-ACGGTTCATTACATTGATCCGTG-3′ and 5′-CACCCTGTTGCTGTAGCCAAA-3′,
respectively. Amplifications were carried out in 20-µl reaction
mixtures under the following conditions: Initial denaturation at
95°C for 2 min; followed by 40 cycles of 95°C for 20 sec, 55°C for
20 sec and 72°C for 35 sec; and a final extension at 72°C for 3
min. GAPDH was used as the internal control. SYBR Master Mixture
(DRR041B, Takara) was used for your gene expression analysis. The
copy numbers of the ILK gene were determined as follows: Target
gene copy number 2−∆∆Cq=(Cqtarget
gene-Cqreference gene) experimental
group-(Cqtarget gene-Cqreference gene)
control group (19).
Cell cycle analysis
SK-MES-1 cells (1.2×106 cells/well) were
incubated in 6-well plates at 37°C for 24 h, and the cell culture
medium was replaced with fresh medium containing 10% FBS with or
without erlotinib at 37°C for another 72 h. The cells were
trypsinized, fixed in ice-cold 70% ethanol overnight and stained
with propidium iodide containing 1 mg/ml RNase (Sigma-Aldrich;
Merck KGaA) according to the instructions of the Cell Cycle Phase
Determination kit (Cayman Chemical Company). The samples were
analyzed using a flow cytometer (FACSCalibur; BD Bioscience). The
cell cycle parameters from 10,000 events were analyzed using
multi-cycle software (version 3.1.1, Phoenix Flow Systems).
Clone formation analysis
Cells (200 cells/well) were seeded in 24-well plates
and treated with erlotinib at 37°C after 12 h. After 2 weeks of
culture, the cells were stained with GIEMSA at room temperature for
20 min, and the number of visible colonies were counted manually.
The relative clone formation ability was calculated by clone
number/well.
Signaling pathway microarray
analysis
The lung SqCC cells were separated into two groups:
i) NC (SK-MES-1 cells treated with erlotinib); and ii) knockdown
(KD; SK-MES-1 cells treated with erlotinib after ILK knockdown by
shRNA). Total RNA was extracted from cells by TRIzol (Shanghai
Perfectics Co., Ltd), and RNA probes were prepared and hybridized
to the GeneChip primeview human 100 format (cat. no. 901838;
Affymetrix GeneChip System; Affymetrix; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. For each
sample, three biological replicates were performed. All arrays were
washed, stained, and read by a GeneChip Scanner 3000 (Affymetrix;
Thermo Fisher Scientific, Inc.). The fluorescence signal was
excited at 532 nm and detected at 570 nm. Data were analyzed using
GeneChip Operating Software 1.4 (Affymetrix; Thermo Fisher
Scientific, Inc.)
Bioinformatics analysis
To analyze differentially expressed genes (DEGs)
between NC and KD groups, the GEO2R tool (http://www.ncbi.nlm.nih.gov/geo/geo2r/) were used. To
control errors, the false discovery rate (FDR) determination
feature automatically included in the GEO2R tool was employed.
Significant DEGs were those that remained significant after FDR
correction. DEGs were selected based on |fold-change|>2 and
P<0.05 for GeneChip analysis (GeneChip Hybridization Wash and
Stain kit; Thermo Fisher Scientific, Inc.). Based on the Kyoto
Encyclopedia of Genes and Genomes (KEGG) database (https://www.kegg.jp/kegg/pathway.html),
significantly changed pathways were identified and connected in a
pathway network (Path-net) to show the association between these
pathways. GO analysis (http://geneontology.org/) was used to organize DEGs
into hierarchical categories. The list of DEGs, containing gene
identifiers and corresponding expression values, was also uploaded
into the IPA software 2012 (Ingenuity Systems, Inc.). The ‘core
analysis’ function included in the software was used to interpret
the differentially expressed genes between NC and KD groups, which
included biological processes, canonical pathways, upstream
transcriptional regulators and gene networks. Each gene identifier
was mapped to its corresponding gene target in the Ingenuity
Pathway Knowledge Base (www.ingenuity.com).
Statistical analysis
Data are presented as the mean ± SEM. Statistical
analysis was performed using SPSS Statistics 23 software (IBM
Corp.). Data were analyzed using unpaired Student's t-test or
one-way ANOVA with Tukey's post hoc test, and P<0.05 was
considered to indicate a statistically significant difference.
Results
ILK knockdown improves the effects of
erlotinib in lung SqCC
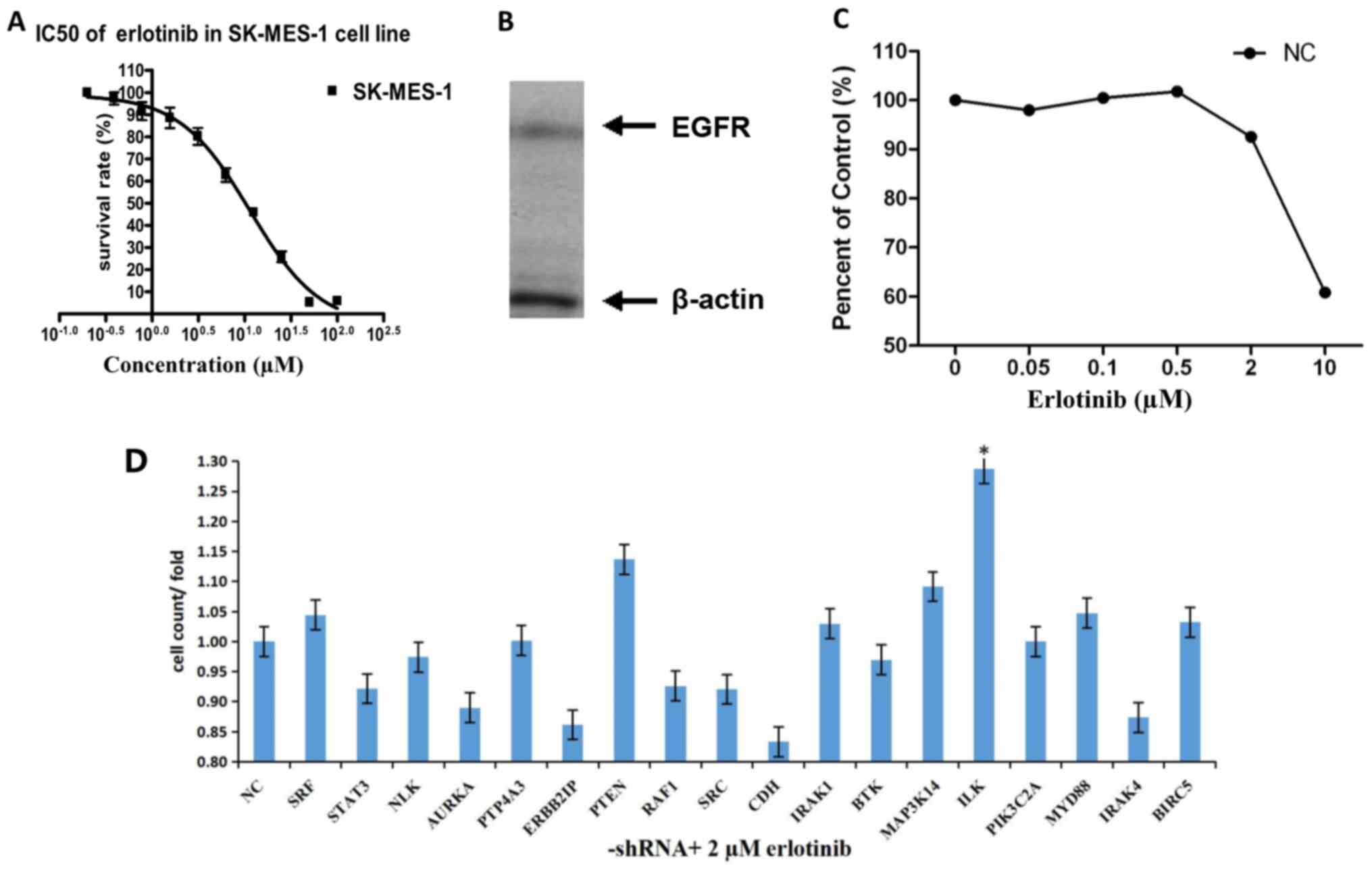
The MTT assay results demonstrated that the
IC50 of SK-MES-1 cells was 11.35 µM, the results of
western blotting revealed that this cell line expressed EGFR
(Fig. 1A and B). The SK-MES-1 cells
were treated with a gradient of different concentrations of
erlotinib following transfection with lentiviral NC shRNA. Cell
proliferation was inhibited by 2 µM erlotinib; thus, 2 µM was used
as the screening concentration in subsequent experiments (Fig. 1C).
In HCS, the fold-changes of cell proliferation were
detected by MTT after 72-h treatment with shRNAs and erlotinib.
Transfection with the shRNAs targeting ILK, PTEN, MAP3K14 and
MYD88, increased the effects of erlotinib on SK-MES-1 cells
compared with NC. Following ILK knockdown, the cell proliferation
was significantly inhibited by treatment with erlotinib compared
with NC (fold-change, 1.29; P<0.05; Fig. 1D and Table
I).
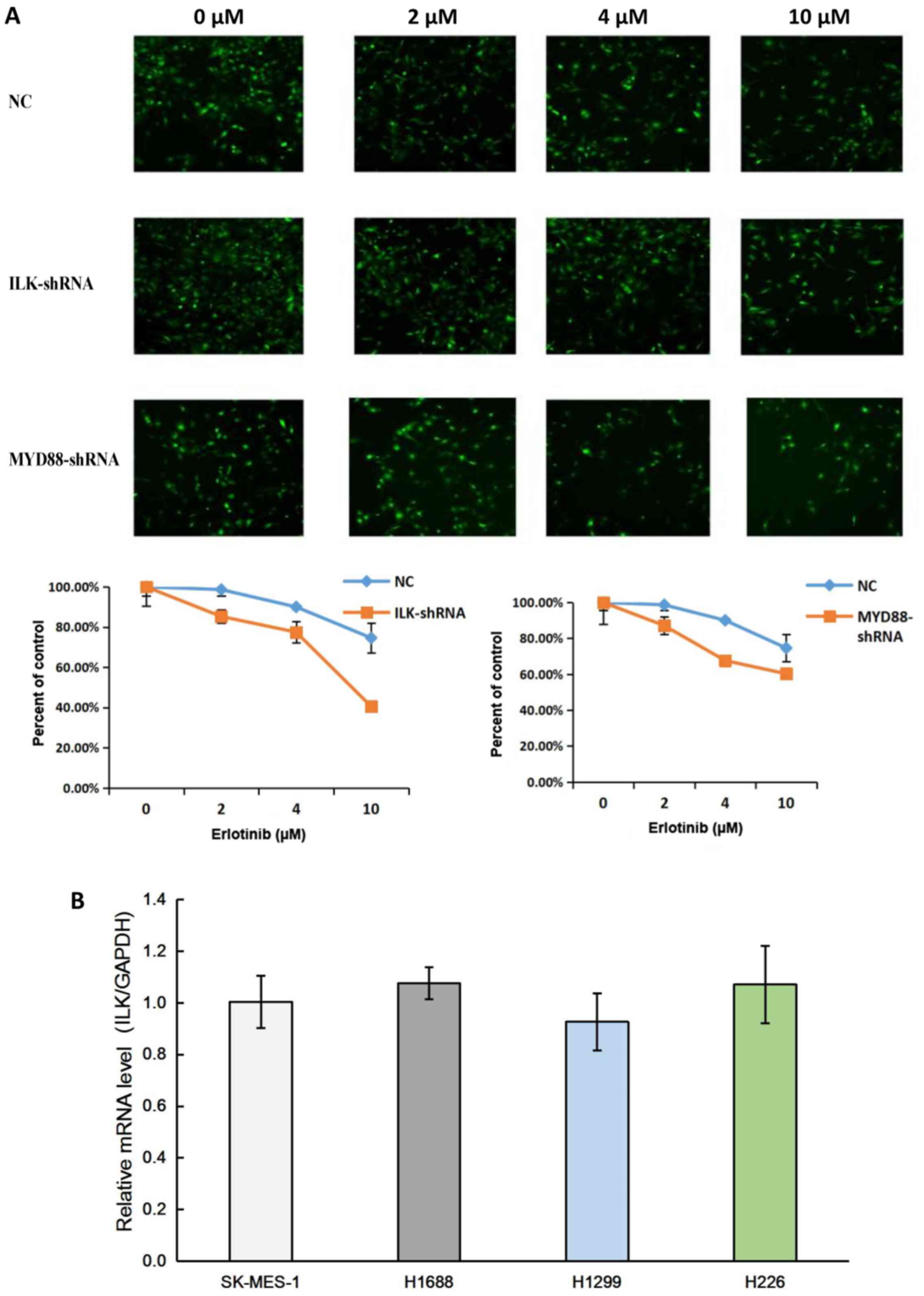
To identify the role of ILK in EGFR-TKI resistance,
the cell survival rates of SK-MES-1 cells transfected with NC and
ILK, MYD88 target shRNAs were measured following 72-h incubation
with 2, 4 or 10 µM erlotinib. The results demonstrated that ILK
shRNA significantly reduced the survival rate of SK-MES-1 cells,
indicating that ILK knockdown improved the effects of erlotinib
(Fig. 2A). The results of qPCR
confirmed that ILK was expressed in lung cancer cell lines,
including the SqCC cell lines SK-MES-1 and H226 (Fig. 2B).
Effects of ILK on erlotinib resistance
in lung SqCC SK-MES-1 cells
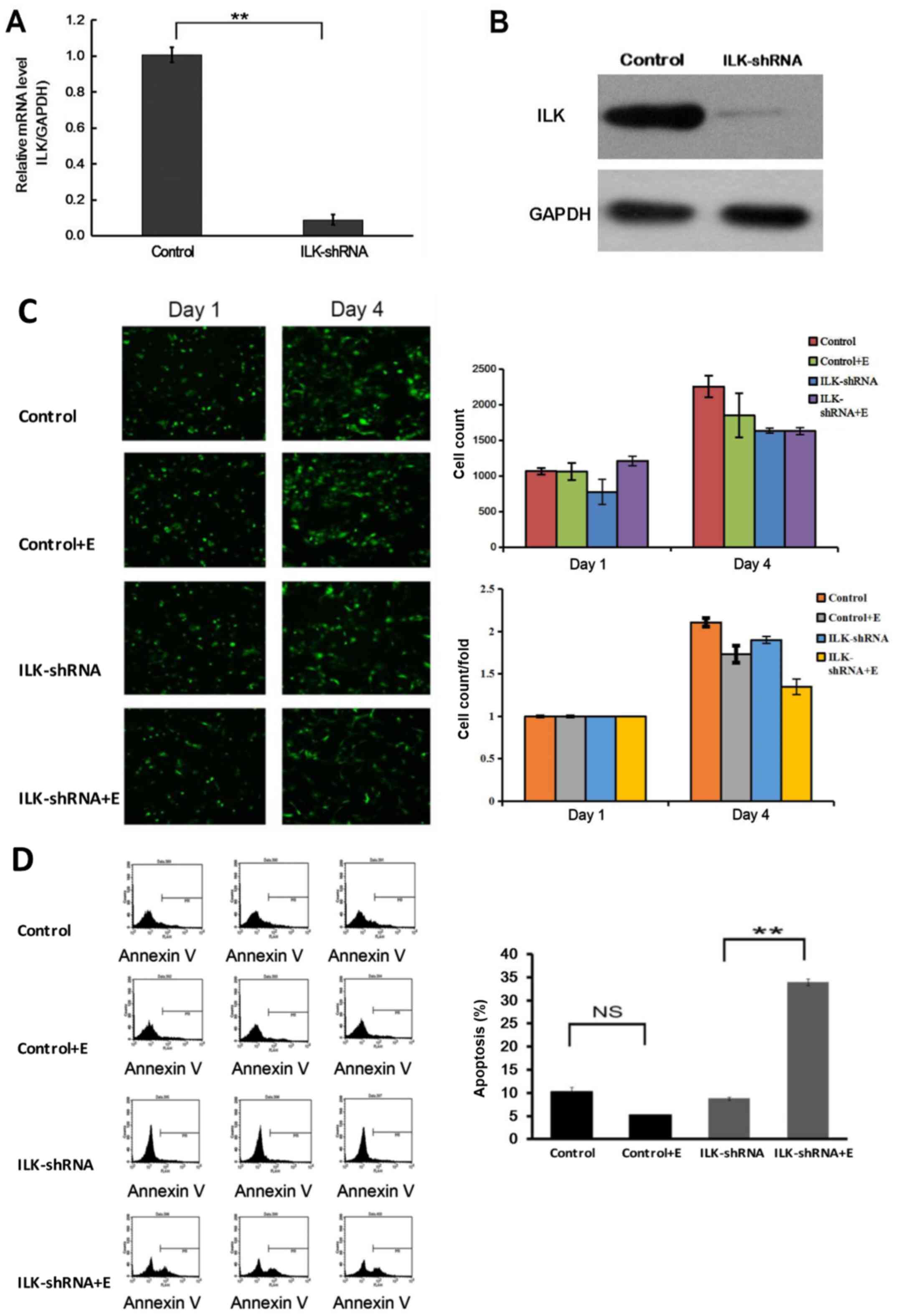
The results of qPCR and western blotting confirmed
that the expression of ILK was significantly inhibited in SK-MES-1
cells after RNA interference (P<0.01; Fig. 3A and B). Treatment with erlotinib
following ILK knockdown significantly inhibited the proliferation
of SK-MES-1 cells (P<0.05; Fig.
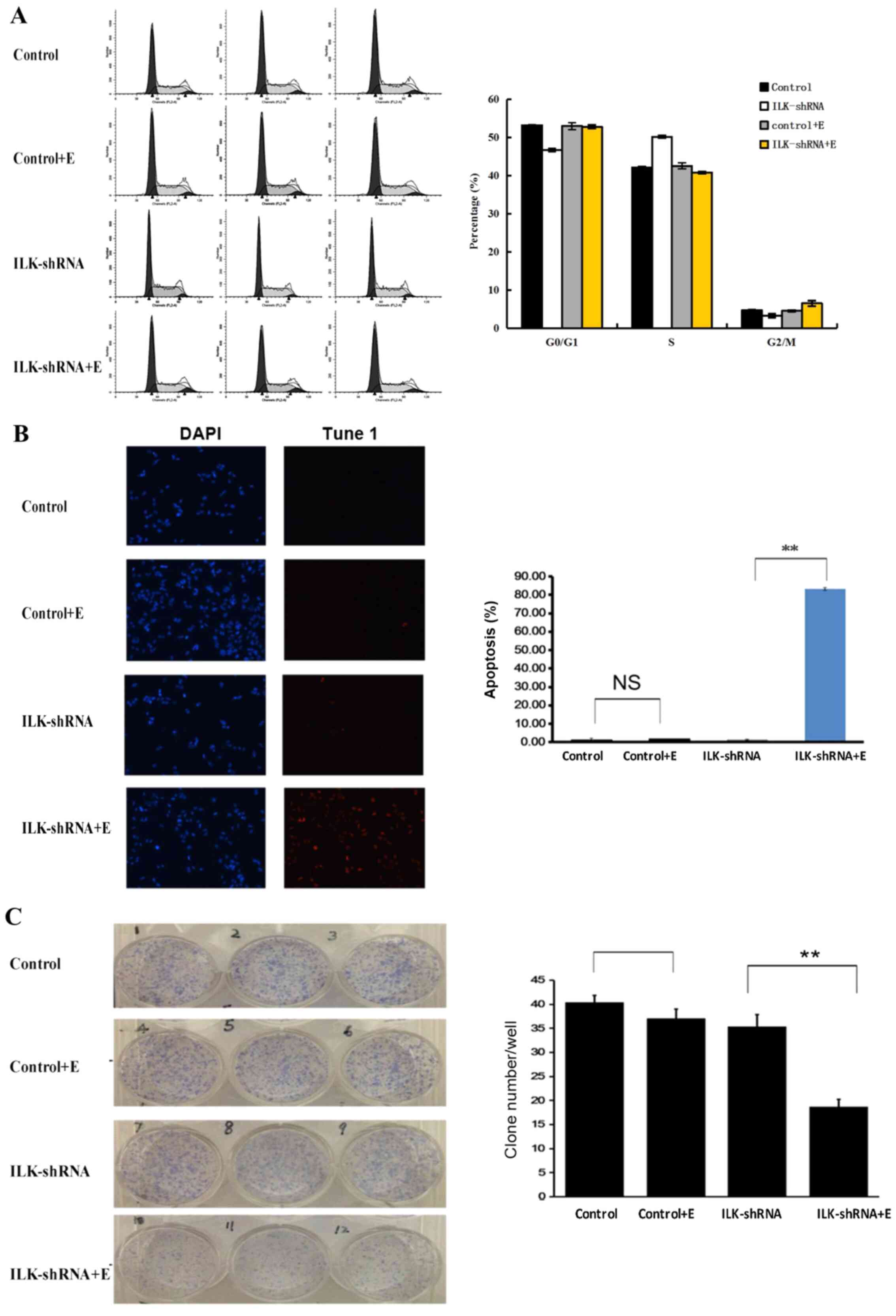
3C), induced apoptosis (P<0.01; Fig. 3D) and cell cycle arrest at the G2/M
and G1 phases (P<0.01; Fig. 4A)
compared with those in the group treated with erlotinib alone. The
flow cytometry apoptosis assay results were verified by TUNEL
assay; the observed apoptotic rate was 83.24% in cells treated with
erlotinib after ILK knockdown, which was notably higher compared
with that of cells treated with erlotinib alone (P<0.01;
Fig. 4B). The results also
demonstrated that the cell clone formation ability decreased
significantly in the group treated with erlotinib after ILK
knockdown compared with that in the group treated with erlotinib
alone (P<0.01; Fig. 4C).
Expression of ILK in patients with
lung SqCC
ILK expression was detected in tumors of patients
with lung squamous cell carcinoma, adenocarcinoma with wild-type
EGFR or with sensitizing EGFR mutations using qPCR. A total of 31
cases of SqCC, 9 cases of wild-type EGFR adenocarcinoma and 10
cases of adenocarcinoma with sensitizing EGFR mutations were
included in the analysis. The results demonstrated that the
expression levels of ILK in patients with adenocarcinoma with
sensitizing EGFR mutations were significantly lower compared with
those in patients with adenocarcinoma with wild-type EGFR or SqCC
(P<0.01; Fig. 5A). No significant
differences were observed in ILK expression levels between patients
with adenocarcinoma with wild-type EGFR and SqCC.
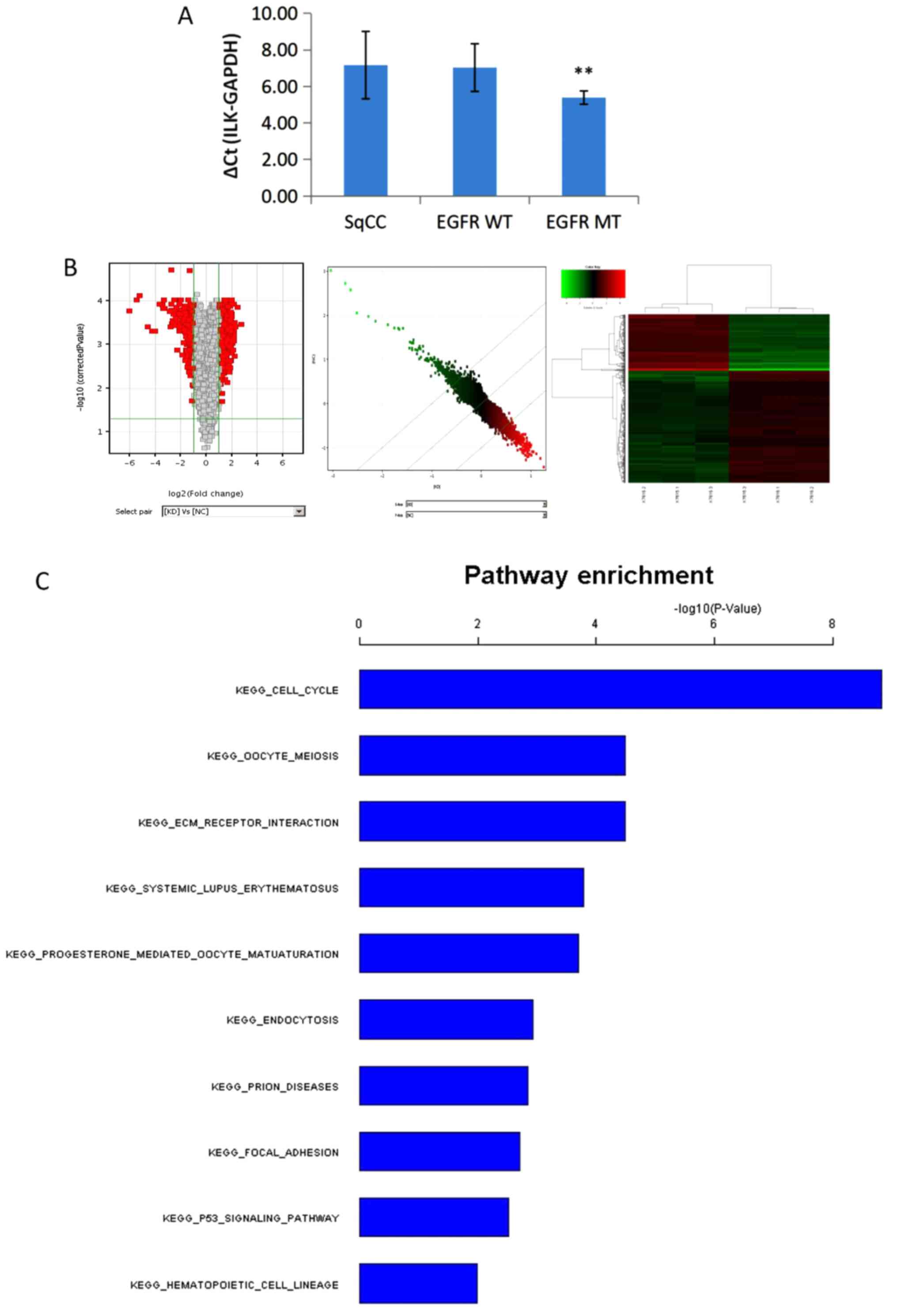 | Figure 5.Expression of ILK and genome-wide
transcriptional analysis of the key pathways of ILK. (A) The
expression of ILK in patients with lung SqCC was significantly
higher compared with that in patients with adenocarcinoma with EGFR
mutation (**P<0.01). No significant differences were observed
between patients with wild-type EGFR adenocarcinoma and SqCC. (B) A
total of 484 differentially expressed transcripts (317 upregulated
and 167 downregulated) in the KD group compared with the NC group,
based on fold-change >2 and P<0.05 threshold, passed the
filtering process and were used for the cluster analysis. In the
volcano map, the x-axis shows the negative log of P-value
calculated by t-test, and the y-axis shows the converted value
after log2 transformation under the comparison of the KD
and NC groups. Scatter plot and heatmap showed the overall
distribution and concentration trend of two groups of data. (C)
Functional analysis of the gene expression profiling using pathways
analysis according to the KEGG and BioCarta Database revealed that
the top three targets enriched by ILK knockdown were the ‘cell
cycle’, ‘ECM receptor interaction’ and ‘oocyte meiosis’. SqCC,
squamous cell carcinoma; ILK, integrin-linked kinase; WT,
wild-type; MT, mutated; KEGG, Kyoto Encyclopedia of Genes and
Genomes; ECM, extracellular matrix KD, SK-MES-1 cells treated with
erlotinib after ILK knockdown; NC, negative control. |
Genome-wide transcriptional analysis
of the key pathways of ILK in EGFR-TKI resistance of the SK-MES-1
cell line
In order to further explore the mechanism of ILK in
intrinsic EGFR-TKI resistance of lung SqCC, genome-wide
transcriptional microarray analysis was carried out to compare the
global gene expression between the control (NC group) and
ILK-knockdown (KD group) SK-MES-1 cells following treatment with
erlotinib using the Affymetrix GeneChip PrimeView Human Gene
Expression Array. A total of 484 transcripts (317 upregulated and
167 downregulated) that were differentially expressed in the KD
group compared with the NC group were selected for the cluster
analysis (Fig. 5B).
The pathway enrichment analysis of the gene
expression profiling based on KEGG and the BioCarta Database
revealed that the top three targets enriched by ILK knockdown were
the ‘cell cycle’, ‘ECM receptor interaction’ and ‘oocyte meiosis’,
as shown in Fig. 5C.
The DEGs were also subjected to GO analysis. Among
the molecular function-associated terms, the DEGs affected ‘protein
kinase activity’ and ‘DNA binding’, which associated with the
action mechanism of EGFR-TKIs (20,21). In
the cellular component analysis, changes occurred in the
‘intracellular non-membrane bound organelle’, ‘spindle’ and
‘cytoplasm’. These parts are closely associated with cell
proliferation and intracellular signal transduction, which are the
important factors for EGFR-TKI efficacy (22,23). In
the biological process analysis, the changes involved ‘mitotic cell
cycle’, ‘mitosis’, ‘response to stress’ and ‘cell proliferation’,
suggesting that ILK knockdown may affect EGFR-TKI resistance via
these cell processes.
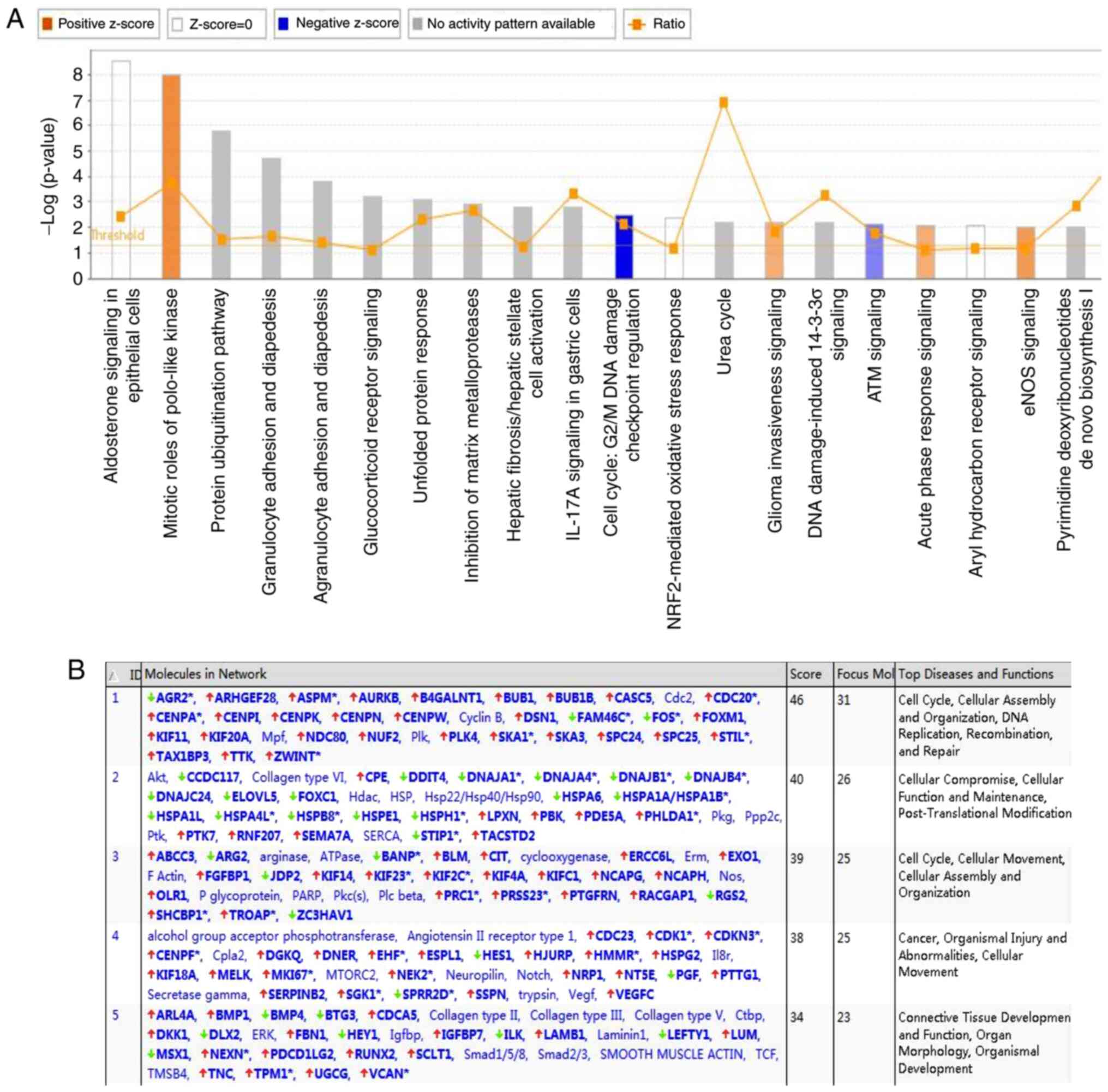
The results of the IPA analysis revealed highly
significant overlap of 207 canonical pathways associated with the
DEGs in the SK-MES-1 cell line, including the ‘mitotic roles of
polo-like kinase’, ‘protein ubiquitination pathway’, ‘inhibition of
matrix metalloproteases’ and ‘cell cycle: G2/M DNA damage
checkpoint regulation’. These pathways were scored based on the
number of genes participating in any particular network. The
results demonstrated that the ‘cell cycle pathway: G2/M DNA damage
checkpoint regulation’ (Z-score, −2), was significantly inhibited
in the Classic Pathway Analysis (Fig.
6A). The IPA-based network analysis demonstrated the
interaction between molecules in the data set; all networks were
sorted according to score values. The network diagram ranked first
in this study was mainly associated with the ‘cell cycle’,
‘cellular assembly organization’, ‘DNA replication’,
‘recombination’ and ‘repair’ (Fig.
6B).
Discussion
Despite recent advances in immunotherapy, the
prognosis of the majority of patients with SqCC is considerably
worse compared with those with adenocarcinoma due to targeted
therapy (24). However, patients
with SqCC express higher levels EGFR compared with those with
adenocarcinoma (9); thus, it is
important to identify the mechanism of intrinsic resistance to
EGFR-TKIs. In the present study, high-throughput RNAi screening
revealed that the knockout of ILK improved the efficacy of
erlotinib in the SqCC cell line SK-MES-1. Based on these
observations, the effects of ILK knockout on the biological
activities of SK-MES-1 cells was investigated. The results
demonstrated that cell proliferation and clone formation was
inhibited, apoptosis was upregulated, and the cells were arrested
at the G1 and G2/M phases following transfection with ILK siRNA
compared with. These results confirmed that inhibition of ILK may
be involved in reversing the resistance to erlotinib.
ILK, an important serine/threonine protein
phosphatase, serves a key role in the regulation of signal
transduction and remodeling of the tumor extracellular matrix (ECM)
(25). High expression of ILK is
associated with the occurrence and development of lung cancer, as
well as with anti-cancer drug resistance, including resistance to
EGFR-TKIs (26–31); thus, it was hypothesized that ILK may
be a target for reversing erlotinib resistance. The results of the
present study confirmed that the expression of ILK was remarkably
higher in patients with lung SqCC compared with that in patients
with adenocarcinoma with sensitizing EGFR mutations, suggesting
that ILK may be a key marker of EGFR-TKI resistance in lung
SqCC.
To gain further insight into the mechanism of ILK in
the resistance of lung SqCC to EGFR-TKIs, KEGG, GO and IPA were
used to explore the associated signaling pathways and gene networks
in the present study. The ‘cell cycle’, ‘oocyte meiosis,
‘ECM-receptor interaction’, ‘protein kinase activity’ and ‘DNA
binding’ were identified to be closely associated with the effects
of ILK on EGFR-TKI resistance. As the cell cycle (32,33),
ECM-receptor interaction (34,35) and
mitosis (36) have been identified
to be activated in cancer drug resistance, ILK may serve a crucial
role in the EGFR-TKI resistance of lung SqCC via these pathways. GO
analysis results also demonstrated that ‘mitotic cell cycle’ and
‘mitosis’ were important signaling pathways involved in EGFR-TKI
resistance in ILK-knockout cells. IPA further confirmed that ‘cell
cycle: G2/M DNA damage checkpoint regulation’ was the
most key signaling pathway regulated by ILK knockout in the
EGFR-TKI resistance of lung SqCC. In conclusion, ILK may improve
our understanding of lung SqCC and may be a future therapeutic
target.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural
Science Foundation of China (grant no. 81207106), the Science and
Technology Commission of Shanghai Municipality (grant nos.
17401932400 and 19401930800) and Shanghai Pulmonary Hospital (grant
no. fkgg1807).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
LJ conceived and designed the study. ZD and JY
acquired, analyzed and interpreted the data and drafted the
manuscript. ML analyzed the data and revised the manuscript
critically for important intellectual content. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by The Ethics Committee of
Shanghai Pulmonary Hospital (Shanghai, China). Signed informed
consents were obtained from the patients and/or guardians.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wahbah M, Boroumand N, Castro C, El-Zeky F
and Eltorky M: Changing trends in the distribution of the
histologic types of lung cancer: A review of 4439 cases. Ann Diagn
Pathol. 11:89–96. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Camidge DR, Doebele RC and Kerr KM:
Comparing and contrasting predictive biomarkers for immunotherapy
and targeted therapy of NSCLC. Nat Rev Clin Oncol. 16:341–355.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sands JM, Nguyen T, Shivdasani P, Sacher
AG, Cheng ML, Alden RS, Jänne PA, Kuo FC, Oxnard GR and Sholl LM:
Next-generation sequencing informs diagnosis and identifies
unexpected therapeutic targets in lung squamous cell carcinomas.
Lung Cancer. 140:35–41. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
National Comprehensive Cancer Network
(NCCN). Clinical Practice Guidelines in Oncology. Plymouth; PA:
2018
|
|
5
|
Mok TSK, Wu YL, Kudaba I, Kowalski DM, Cho
BC, Turna HZ, Castro G Jr, Srimuninnimit V, Laktionov KK,
Bondarenko I, et al: Pembrolizumab versus chemotherapy for
previously untreated, PD-L1-expressing, locally advanced or
metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised,
open-label, controlled, phase 3 trial. Lancet. 393:1819–1830. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hochmair MJ, Morabito A, Hao D, Yang CT,
Soo RA, Yang JC, Gucalp R, Halmos B, Wang L, Märten A and Cufer T:
Sequential afatinib and osimertinib in patients with EGFR
mutation-positive non-small-cell lung cancer: Updated analysis of
the observational GioTag study. Future Oncol. 15:2905–2914. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brahmer J, Reckamp KL, Baas P, Crinò L,
Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE,
Holgado E, et al: Nivolumab versus Docetaxel in advanced
squamous-cell non-small-cell lung cancer. N Engl J Med.
373:123–135. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Herbst RS, Baas P, Kim DW, Felip E,
Pérez-Gracia JL, Han JY, Molina J, Kim JH, Arvis CD, Ahn MJ, et al:
Pembrolizumab versus docetaxel for previously treated,
PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010):
A randomised controlled trial. Lancet. 387:1540–1550. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Solassol I, Pinguet F and Quantin X: FDA-
and EMA-approved tyrosine kinase inhibitors in advanced
EGFR-mutated non-small cell lung cancer: Safety,
tolerability, plasma concentration monitoring, and management.
Biomolecules. 9:6682019. View Article : Google Scholar
|
|
10
|
Hirsch FR, Varella-Garcia M, Bunn PA Jr,
Di Maria MV, Veve R, Bremmes RM, Barón AE, Zeng C and Franklin WA:
Epidermal growth factor receptor in non-small-cell lung carcinomas:
Correlation between gene copy number and protein expression and
impact on prognosis. J Clin Oncol. 21:3798–3807. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cancer Genome Atlas Research Network, .
Comprehensive genomic characterization of squamous cell lung
cancers. Nature. 489:519–525. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Z, Shen Z, Li Z, Duan J, Fu S, Liu Z,
Bai H, Zhang Z, Zhao J, Wang X and Wang J: Activation of the
BMP-BMPR pathway conferred resistance to EGFR-TKIs inlung squamous
cell carcinoma patients with EGFR mutations. Proc Natl Acad Sci
USA. 112:9990–9995. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hata A, Katakami N, Yoshioka H, Kunimasa
K, Fujita S, Kaji R, Notohara K, Imai Y, Tachikawa R, Tomii K, et
al: How sensitive are epidermal growth factor receptor-tyrosine
kinase inhibitors for squamous cell carcinoma of the lung harboring
EGFR gene-sensitive mutations? J Thorac Oncol. 8:89–95. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shukuya T, Takahashi T, Kaira R, Ono A,
Nakamura Y, Tsuya A, Kenmotsu H, Naito T, Kaira K, Murakami H, et
al: Efficacy of gefitinib for non-adenocarcinoma non-small-cell
lung cancer patients harboring epidermal growth factor receptor
mutations: A pooled analysis of published reports. Cancer Sci.
102:1032–1037. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Filipits M: New developments in the
treatment of squamous cell lung cancer. Curr Opin Oncol.
26:152–158. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schwaederle M, Elkin SK, Tomson BN, Carter
JL and Kurzrock R: Squamousness: Next-generation sequencing reveals
shared molecular features across squamous tumor types. Cell Cycle.
14:2355–2361. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Goss GD and Spaans JN: Epidermal growth
factor receptor inhibition in the management of squamous cell
carcinoma of the lung. Oncologist. 21:205–213. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Memon AA, Zhang H, Gu Y, Luo Q, Shi J,
Deng Z, Ma J and Ma W: EGFR with TKI-sensitive mutations in exon 19
is highly expressed and frequently detected in Chinese patients
with lung squamous carcinoma. Onco Targets Ther. 10:4607–4613.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ray P, Raghunathan K, Ahsan A, Allam US,
Shukla S, Basrur V, Veatch S, Lawrence TS, Nyati MK and Ray D:
Ubiquitin ligase SMURF2 enhances epidermal growth factor receptor
stability and tyrosine-kinase inhibitor resistance. J Biol Chem.
295:12661–12673. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lei T, Zhang L, Song Y, Wang B, Shen Y,
Zhang N and Yang M: miR-1262 Transcriptionally modulated by
an enhancer genetic variant improves efficiency of epidermal growth
factor receptor-tyrosine kinase inhibitors in advanced lung
adenocarcinoma. DNA Cell Biol. 39:1111–1118. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nilsson MB, Sun H, Robichaux J, Pfeifer M,
McDermott U, Travers J, Diao L, Xi Y, Tong P, Shen L, et al: A
YAP/FOXM1 axis mediates EMT-associated EGFR inhibitor resistance
and increased expression of spindle assembly checkpoint components.
Sci Transl Med. 12:eaaz45892020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rong X, Liang Y, Han Q, Zhao Y, Jiang G,
Zhang X, Lin X, Liu Y, Zhang Y, Han X, et al: Molecular mechanisms
of tyrosine kinase inhibitor resistance induced by
membranous/cytoplasmic/nuclear translocation of epidermal growth
factor receptor. J Thorac Oncol. 14:1766–1783. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pinheiro FD, Teixeira AF, de Brito BB, da
Silva FAF, Santos MLC and de Melo FF: Immunotherapy-new perspective
in lung cancer. World J Clin Oncol. 11:250–259. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Assi K, Bergstrom K, Vallance B, Owen D
and Salh B: Requirement of epithelial integrin-linked kinase for
facilitation of Citrobacter rodentium-induced colitis. BMC
Gastroenterol. 13:1372013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen D, Zhang Y, Zhang X, Li J, Han B, Liu
S, Wang L, Ling Y, Mao S and Wang X: Overexpression of
integrin-linked kinase correlates with malignant phenotype in
non-small cell lung cancer and promotes lung cancer cell invasion
and migration via regulating epithelial-mesenchymal transition
(EMT)-related genes. Acta Histochem. 115:128–136. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Espinoza I and Miele L: Deadly crosstalk:
Notch signaling at the intersection of EMT and cancer stem cells.
Cancer Lett. 341:41–45. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yu J, Shi R, Zhang D, Wang E and Qiu X:
Expression of integrin-linked kinase in lung squamous cell
carcinoma and adenocarcinoma: Correlation with E-cadherin
expression, tumor microvessel density and clinical outcome.
Virchows Arch. 458:99–107. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Posch F, Setinek U, Flores RM, Bernhard D,
Hannigan GE, Mueller MR and Watzka SB: Serum integrin-linked kinase
(sILK) concentration and survival in non-small cell lung cancer: A
pilot study. Clin Transl Oncol. 16:455–462. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jia Z: Role of integrin-linked kinase in
drug resistance of lung cancer. Onco Targets Ther. 8:1561–1565.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Augustin A, Lamerz J, Meistermann H,
Golling S, Scheiblich S, Hermann JC, Duchateau-Nguyen G, Tzouros M,
Avila DW, Langen H, et al: Quantitative chemical proteomics
profiling differentiates erlotinib from gefitinib in EGFR wild-type
non-small cell lung carcinoma cell lines. Mol Cancer Ther.
12:520–529. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu M, Xu S, Wang Y, Li Y, Li Y, Zhang H,
Liu H and Chen J: PD 0332991, a selective cyclin D kinase 4/6
inhibitor, sensitizes lung cancer cells to treatment with epidermal
growth factor receptor tyrosine kinase inhibitors. Oncotarget.
7:84951–84964. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dermawan JK, Gurova K, Pink J, Dowlati A,
De S, Narla G, Sharma N and Stark GR: Quinacrine overcomes
resistance to erlotinib by inhibiting FACT, NF-κB, and cell-cycle
progression in non-small cell lung cancer. Mol Cancer Ther.
13:2203–2214. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kaylan KB, Gentile SD, Milling LE, Bhinge
KN, Kosari F and Underhill GH: Mapping lung tumor cell drug
responses as a function of matrix context and genotype using cell
microarrays. Integr Biol (Camb). 8:1221–1231. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang J, Wang B, Chu H and Yao Y: Intrinsic
resistance to EGFR tyrosine kinase inhibitors in advanced
non-small-cell lung cancer with activating EGFR mutations. Onco
Targets Ther. 9:3711–3726. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Salmela AL and Kallio MJ: Mitosis as an
anti-cancer drug target. Chromosoma. 122:431–449. 2013. View Article : Google Scholar : PubMed/NCBI
|