Introduction
Liver cancer is the third leading causes of cancer
death in the world (1). Early
detection of liver cancer is difficult and most of cases are
diagnosed at an advanced stage. Surgical resection is the primary
treatment for liver cancer, and in some cases, liver cancer is
treated with chemotherapy, transcatheter arterial
chemoembolization, and radiofrequency ablation (2–5). The
prognosis of liver cancer is not favorable even after complete
surgical resection.
There are multiple mechanisms for carcinogenesis of
liver cancer (6). Mechanistic target
of rapamycin (mTOR) is a serine/threonine kinase, which regulates
cell proliferation, cell death, metabolism and expression of growth
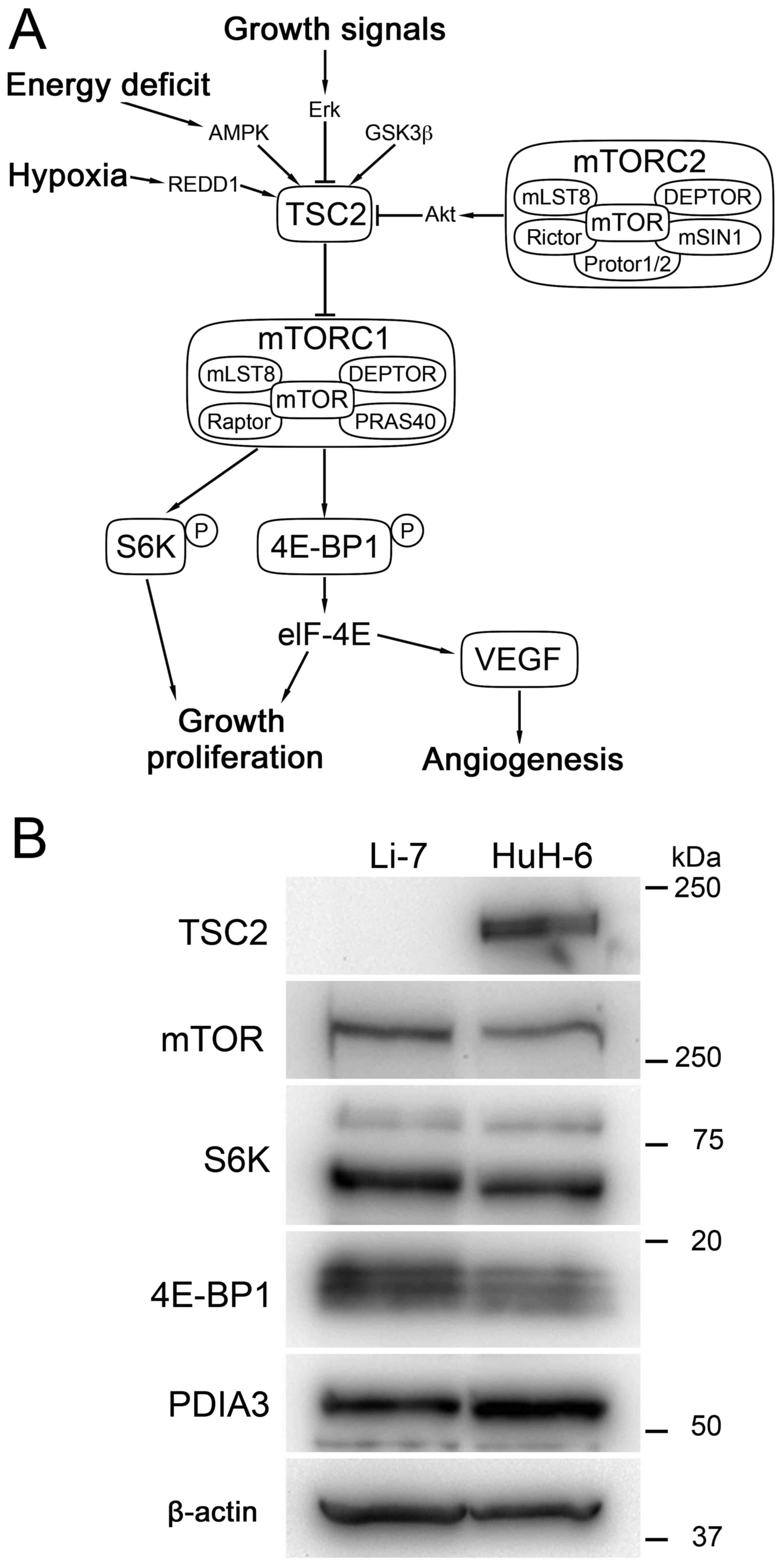
factors (Fig. 1A) (7–10). mTOR
is overexpressed in approximately 40% of liver cancer, and liver
cancer with overexpression of mTOR follows an unfavorable clinical
course (11). Preclinical studies
showed that inhibition of the mTOR pathway suppress the development
of liver cancer (12–16). However, clinical trials of mTOR
inhibitors in liver cancer did not demonstrate significant
improvement of survival (17).
Subpopulation analysis of a clinical trial of everolimus (Ev)
suggested that liver cancer without the expression of tuberous
sclerosis complex 2 (TSC2), which suppresses mTOR complex 1
(mTORC1), is associated with susceptibility to Ev, and liver cancer
with TSC2 expression was resistant to Ev (18). Alternatively, the activation of other
signaling pathways such as mitogen-activated protein kinase (MAPK)
and phosphatidylinositol-3 kinase (PI3K) has been suggested
(6). A novel strategy is awaited to
improve the effect of mTOR inhibitor and the prognosis of patients
of liver cancer.
Protein disulfide isomerase (PDI) is a chaperone
protein that supports the folding of synthesized proteins (19). PDIs have also been shown to be
involved in multiple cellular functions such as degradation of
protein, antigen processing, stabilization of receptors and
intracellular signaling, and cell death (20). PDIs are involved in cellular
functions in carcinoma cells. In liver cancer, a molecule of the
PDI family, PDI A member 3 (PDIA3), which is known as GRP58 or
ERp57, is highly expressed (21).
The prognosis of liver cancer with a high PDIA3 expression level is
worse than that of liver cancer with a low expression level.
PDIA3 is involved in the assembly and stability of
signaling molecules such as mTOR and STAT3 (19,22). It
has been shown that PDIA3 forms a complex with mTORC1 and
stabilizes the signaling pathway (23). The knock-down of PDIA3 reduced the
phosphorylation activity of mTORC1, whereas overexpression enhanced
the activity. It is thus plausible that inhibition of PDIA3
function destabilizes mTORC1 and attenuates its signaling activity.
16F16 is a small compound that inhibits the function of PDIs
(24). It is expected that the
suppression of the PDIA3 function by 16F16 destabilizes the
assembly of mTORC1 and increases the effect of mTOR inhibitor
against liver cancer.
The aim of the present study is to explore whether
PDIA3 inhibitor could increase the antiproliferative effect of Ev
in liver cancer. The effect was investigated in 2 cultured liver
cancer cell lines; i.e., Li-7, which lacks TSC2 expression and is
susceptible to Ev, and HuH-6, which expresses TSC2 and is resistant
to Ev (18). Using these cultured
cell lines, the effects of Ev and 16F16 on cell proliferation and
phosphorylation of molecules in the mTOR signaling pathway were
investigated. The expression of vascular endothelial growth factor
(VEGF), which is essential for the formation of blood vessels in
liver cancer (25), was also
examined.
Materials and methods
Cell lines and culture
Cultured human liver cancer cell lines, Li-7 and
HuH-6, were obtained from RIKEN BioResource Center, and Japanese
Collection of Research Bioresources, respectively. Li-7 was derived
from hepatocellular carcinoma (HCC) (26), and HuH-6 was derived from
hepatoblastoma (27). The cells were
cultured in RPMI-1640 (Thermo Fisher Scientific, Inc.) supplemented
with 10% fetal bovine serum (Nichirei Biosciences, Inc.) at
37°C.
Viability assay
Cultured cells were plated in 96-well plates at a
density of 3×103cells/well and cultured at 37°C for 24
h. Ev, an inhibitor of mTOR (Cell Signaling Technology, Inc.), and
16F16, an inhibitor of PDIs (Enzo Life Sciences, Inc.), were then
added to the culture medium, and the cells were cultured at 37°C
for 72 h. Viable cells were determined using Cell Counting Kit-8
(Dojindo Molecular Technologies, Inc.). Ten microliters CCK-8
solution containing
2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium,
monosodium salt was added to each well and incubated at 37°C for 2
h. The absorbance at 450 nm was measured using an iMark Microplate
Reader (Bio-Rad Laboratories, Inc.). Experiments were performed in
triplicate. Cell viability was calculated as the percentage of
viable cells treated with 16F16 and/or Ev compared with untreated
cells.
Proliferation assay
Cultured cells were plated in 96-well plates at a
density of 3×103 cells/well and cultured at 37°C for 24
h. Then, 0.01 µM Ev and 2 µM 16F16 were added to the culture
medium, and the cells were cultured at 37°C. Viable cells were
determined using Cell Counting Kit-8 at 0, 24, 48, and 72 h.
Experiments were performed in triplicate. Viable cells were
indicated by absorbance at 450 nm.
Preparation of protein samples
Cultured cells were plated in 100-mm dishes at a
density of 1.0×106 cells/dish and cultured at 37°C for
24 h. Then, 0.01 µM Ev and 2 µM 16F16 were added to culture medium,
and the cells were cultured at 37°C for 72 h. After 3 washes with
phosphate buffered saline (PBS), the cells were lysed in 50 mM
Tris-HCl (pH 7.6)/0.5% SDS and sonicated for 20 min. The protein
concentration was quantified using Pierce 660 nm Protein Assay
Reagent (Thermo Fisher Scientific, Inc.), and protein samples were
used for western blot analysis.
Immunoprecipitation (IP) analysis
Cultured HuH-6 cells were plated in 100-mm dishes at
a density of 1.0×106 cells/dish and cultured at 37°C for
72 h. After washing with PBS, cells were lysed with IP Lysis Buffer
(cat. no. 87787; Thermo Fisher Scientific, Inc.) with protease
inhibitor cocktail (cat. no. P8340; dilution, 1:100; Sigma-Aldrich;
Merck KGaA), and incubated on ice for 10 min. The lysate was then
collected with a scraper and transferred to a 1.5-ml tube. The
lysate was centrifuged at 12,000 × g at 4°C for 5 min and the
supernatant was transferred to a new 1.5 ml tube. The protein
concentration was quantified using Pierce 660 nm Protein Assay
Reagent.
Immunoprecipitation was done in a solution
containing 500 µg protein, Protein A/G PLUS-Agarose (cat. no. 6200,
Santa Cruz Biotechnology, Inc.), and antibodies listed in Table I or isotype mouse IgG1 or rabbit IgG
(cat. nos. 5415 and 3900; dilution 1:100; both from Cell Signaling
Technology, Inc.) in 500 µl IP Lysis Buffer with protease inhibitor
cocktail at 4°C overnight. The mixture was applied to Sigma Prep
Spin Columns with Break-Away Tips (Sigma-Aldrich; Merck KGaA), and
the columns were washed with 500 µl IP Lysis Buffer 3 times. Then,
30 µl of Laemmli Sample Buffer (Bio-Rad Laboratories, Inc.) with
3-mercaptethanol was loaded into the columns, and the columns were
incubated at 95°C for 5 min. Protein samples were retrieved by
centrifugation at 100 × g for 3 min and the samples were used for
western blot analysis.
 | Table I.List of antibodies used in the
present study. |
Table I.
List of antibodies used in the
present study.
|
|
|
| Dilution |
|---|
|
|
|
|
|
|---|
| Antibody | Cat. no. | Company | WB | IP |
|---|
| PDIA3 | ab13506 | Abcam | 1:2,000 | 1:100 |
| TSC2 | 4308 | Cell Signaling
Technology, Inc. | 1:1,000 | – |
| mTOR | 2972 | Cell Signaling
Technology, Inc. | 1:1,000 | 1:100 |
| 4E-BP1 | 9644 | Cell Signaling
Technology, Inc. | 1:1,000 | 1:100 |
| p-4E-BP1
(Thr70) | 13396 | Cell Signaling
Technology, Inc. | 1:1,000 | – |
| S6K | 9202 | Cell Signaling
Technology, Inc. | 1:1,000 | 1:100 |
| p-S6K (Thr389) | 9234 | Cell Signaling
Technology, Inc. | 1:1,000 | – |
| β-actin | A5316 | Sigma-Aldrich;
Merck KGaA | 1:10,000 | – |
Western blot analysis
Protein samples were electrophoresed in 5–20%
polyacrylamide gel (e-PAGEL; ATTO Corp.) and transferred onto a
polyvinylidene difluoride membrane. After blocking with a mixture
of 5% skim milk and Tris-buffered saline/0.05% Tween-20 at room
temperature for 1 h, the membrane was incubated with antibodies
listed in Table I at 4°C overnight.
After washing with 25 mM Tris-HCl (pH 8.0)/150 mM NaCl/0.01% Triton
X, the membranes were incubated with horseradish
peroxidase-conjugated anti-mouse immunoglobulin antibody (True
Blot, cat. no. 18-8817-33; dilution, 1:10,000; Rockland Inc.),
anti-mouse immunoglobulin antibody (cat. no. A106PU; dilution,
1:10,000), or anti-rabbit immunoglobulin antibody (cat. no. A102PU;
dilution, 1:10,000, both from American Qualex Scientific Products,
Inc.) at room temperature for 1 h. The peroxidase activity was
detected as chemiluminescence using SuperSignal West Dura Extended
Duration Substrate (Thermo Fisher Scientific, Inc.). Positive bands
were quantified using Quantity One Software version 4.6.2 (Bio-Rad
Laboratories, Inc.). Protein expression and phosphorylation were
normalized to the expression of β-actin, and the levels in treated
cells were expressed as fold-change relative to untreated
cells.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Cells were plated on 60-mm dishes at
1.0×106 cells/well and cultured at 37°C for 24 h. Then,
0.01 µM Ev and/or 2 µM 16F16 were added to culture medium and the
cells were cultured at 37°C for 72 h. After a wash with PBS, total
RNA was extracted using TRIzol (Thermo Fisher Scientific, Inc.)
according to the recommended protocol. The concentration of total
RNA was measured using NanoDrop (Thermo Fisher Scientific,
Inc.).
Total RNA (200 ng) was treated with DNase I (Thermo
Fisher Scientific, Inc.) at room temperature for 15 min. cDNA was
reverse-transcribed from total RNA using a SuperScript VILO cDNA
Synthesis Kit (Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. Quantitative PCR was performed in a 20-µl
reaction mixture containing 1X TaqMan Fast Universal PCR Master Mix
(Thermo Fisher Scientific, Inc.), 1X TaqMan primers and probes and
reverse-transcribed cDNA. The TaqMan primers and probes were as
follows: vascular endothelial growth factor (VEGF) (Hs00900055_m1)
and 18S ribosome RNA (rRNA) (Hs03928990) (all from Thermo Fisher
Scientific, Inc.). The reaction was initiated with incubation at
95°C for 20 sec, followed by 40 cycles of incubation at 95°C for 1
sec and at 60°C for 20 sec. Alterations in fluorescence were
monitored using the Step One Plus Real-Time PCR System (Thermo
Fisher Scientific, Inc.). The expression level of VEGF was
standardized with that of 18S rRNA. The expression levels were
calculated by the 2−ΔΔCq method (28).
Statistical analysis
All statistical analyses were performed using R
software. The half maximal-inhibitory concentration
(IC50) was calculated as the estimated value ± standard
error. All other data were expressed as mean ± standard deviation.
Comparison of data between 2 groups was performed using
Mann-Whitney U test. Three or more groups were conducted by
Kruskal-Wallis test followed by Dunn's post-hoc test. P<0.05 was
considered to indicate statistical significance.
Results
Expression of molecules in cultured
cells
Expression of TSC2 was not detected in Li-7 cells,
whereas TSC2 was expressed in HuH-6 cells (Fig. 1B). The expression of mTOR, S6 kinase
(S6K), 4E-binding protein 1 (4E-BP1), and PDIA3 was noted in both
HuH-6 and Li-7 cells, and their expression levels appeared
comparable between the 2 lines.
Viability of cultured cells treated
with everolimus and 16F16
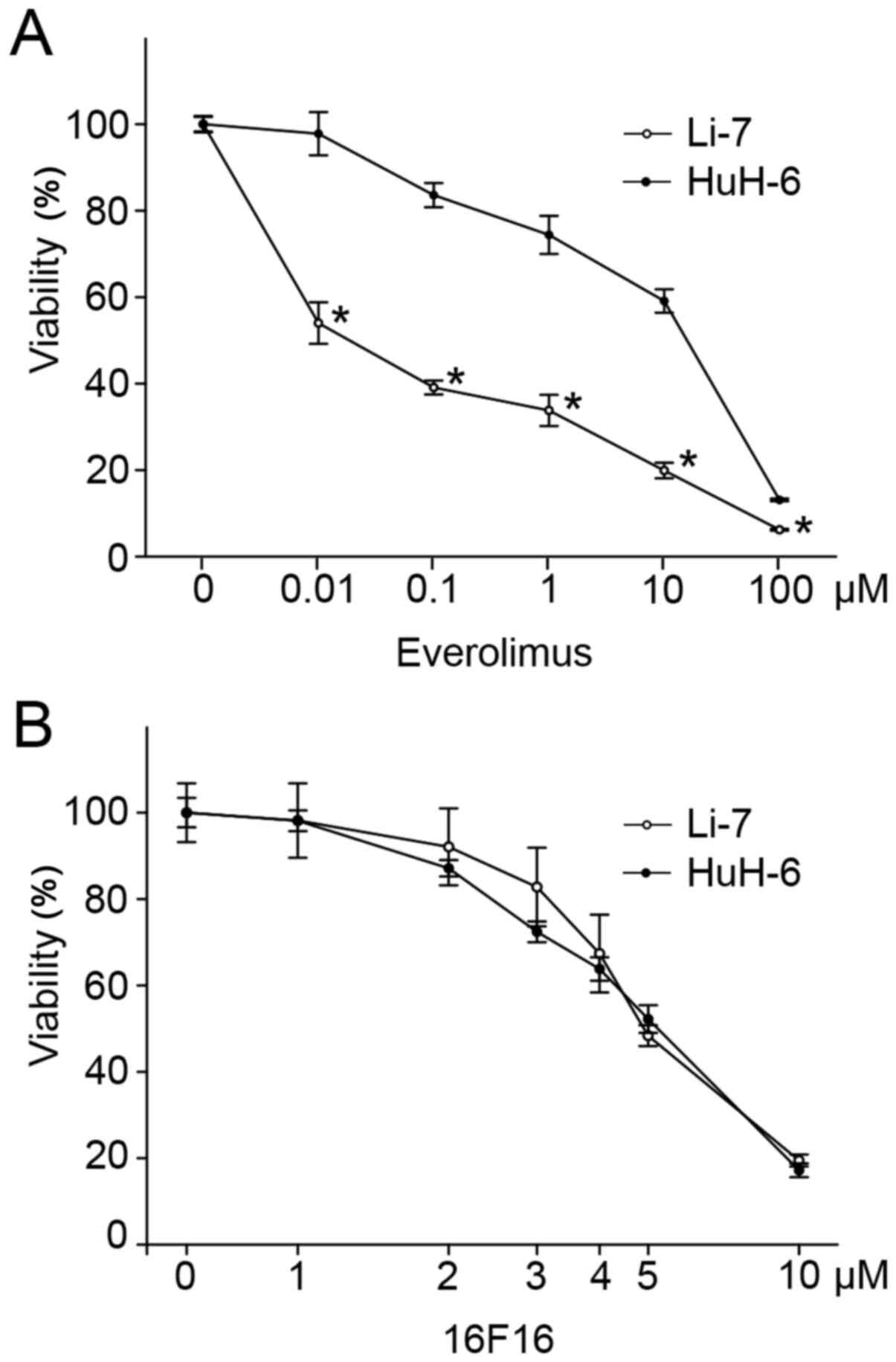
Li-7 cells were susceptible to Ev, and HuH-6 cells
were resistant to Ev (Fig. 2A).
Treatment with 0.01 µM Ev inhibited viability to 54.0±5.0% in Li-7
cells, but only to 97.8±5.0% in HuH-6 cells. The IC50 of
Ev was 0.02±0.01 µM in Li-7 cells and 9.26±4.44 µM in HuH-6 cells.
It was shown that high concentration of 16F16 reduces the viability
of culture liver cancer cells. Susceptibility to 16F16 was,
however, comparable between Li-7 and HuH-6 cells (Fig. 2B). The IC50 of 16F16 was
5.27±0.16 and 5.05±0.12 µM in Li-7 and HuH-6 cells,
respectively.
Susceptibility in Li-7 cells and resistance in HuH-6
cells to Ev was comparable with a previous report (18). The difference in the cell viability
by Ev between Li-7 and HuH-6 was evident at 0.01 µM. Although 16F16
reduced the cell viability at high concentration, the
susceptibility was same in Li-7 and HuH-6 cells. At low
concentration of 2 µM 16F16, the viability was suppressed only to
98.2±8.6% in Li-7 cells and 98.1±2.4% in HuH-6 cells. It was
considered that at this concentration, 16F16 did not show a
significant cytotoxic effect in either cell line. Depending on
these evidences, the subsequent experiments were performed under
treatment with 0.01 µM Ev alone, 2 µM 16F16 alone, and a
combination of 0.01 µM Ev and 2 µM 16F16.
Effect of combination treatment with
everolimus and 16F16 on cultured cells
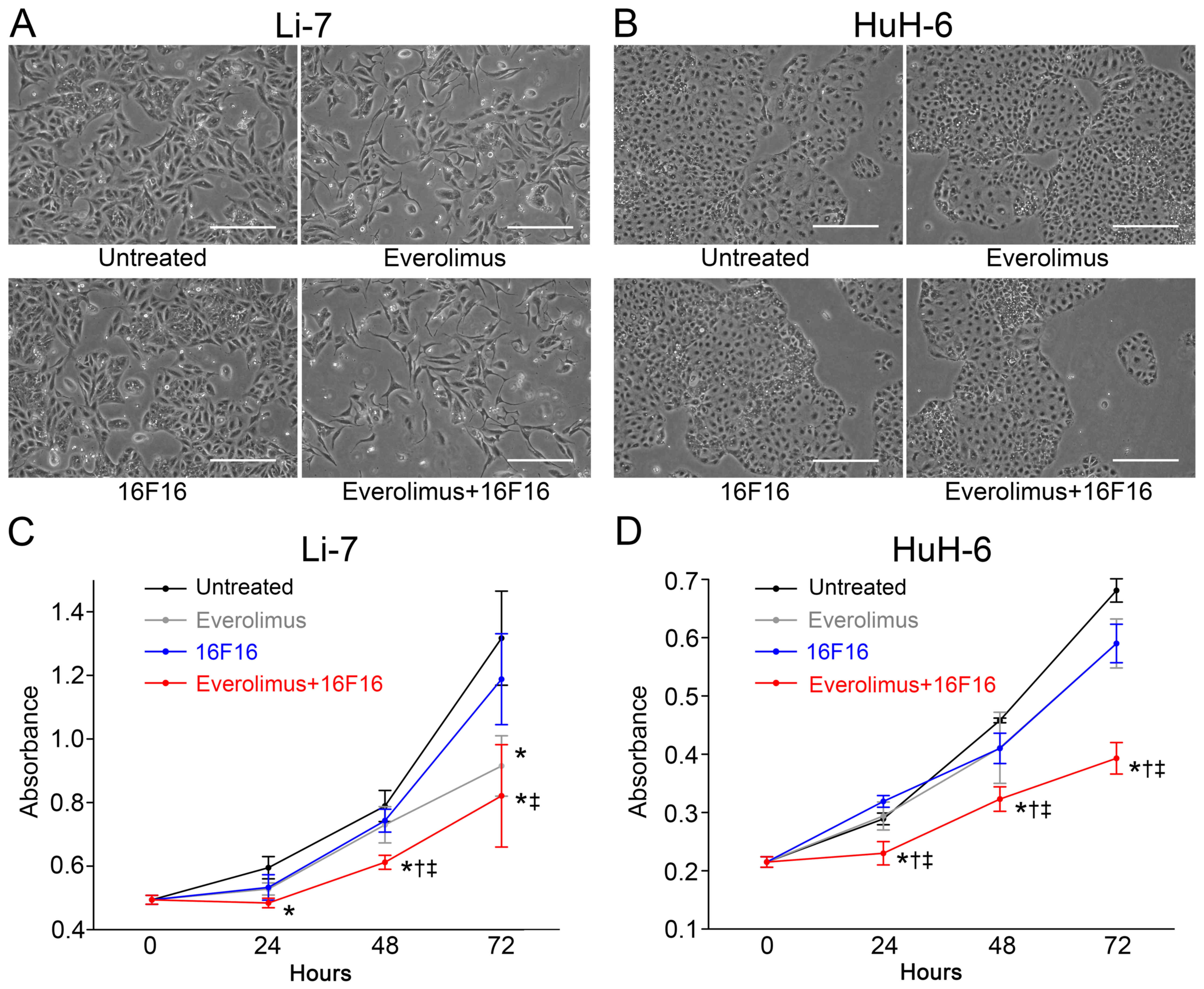
Li-7 cells treated with Ev alone appeared less
viable at 72 h compared with untreated Li-7 cells (Fig. 3A). 16F16 did not appear to affect
viability. Cultured cell treated with Ev and 16F16 appeared less
viable. Conversely, HuH-6 cells appeared viable at 72 h when
treated with Ev alone or 16F16 alone (Fig. 3B). Cultured cells treated with Ev and
16F16 in combination appeared to be less viable.
Proliferation of Li-7 cells was significantly
reduced to 69.5±7.2% by treatment with Ev alone compared with
untreated Li-7 cells at 72 h (Fig.
3C). Proliferation was reduced to 90.2±10.8% by treatment with
16F16 but was significantly suppressed to 62.3±12.2% by combination
treatment with Ev and 16F16. In HuH-6 cells, proliferation was
reduced to 86.7±6.1% by Ev alone and 86.6±4.8% by 16F16 alone,
whereas proliferation was significantly inhibited to 57.7±4.0% by
combination treatment with Ev and 16F16 (Fig. 3D).
Expression and phosphorylation of
molecules of the mTOR signaling pathway
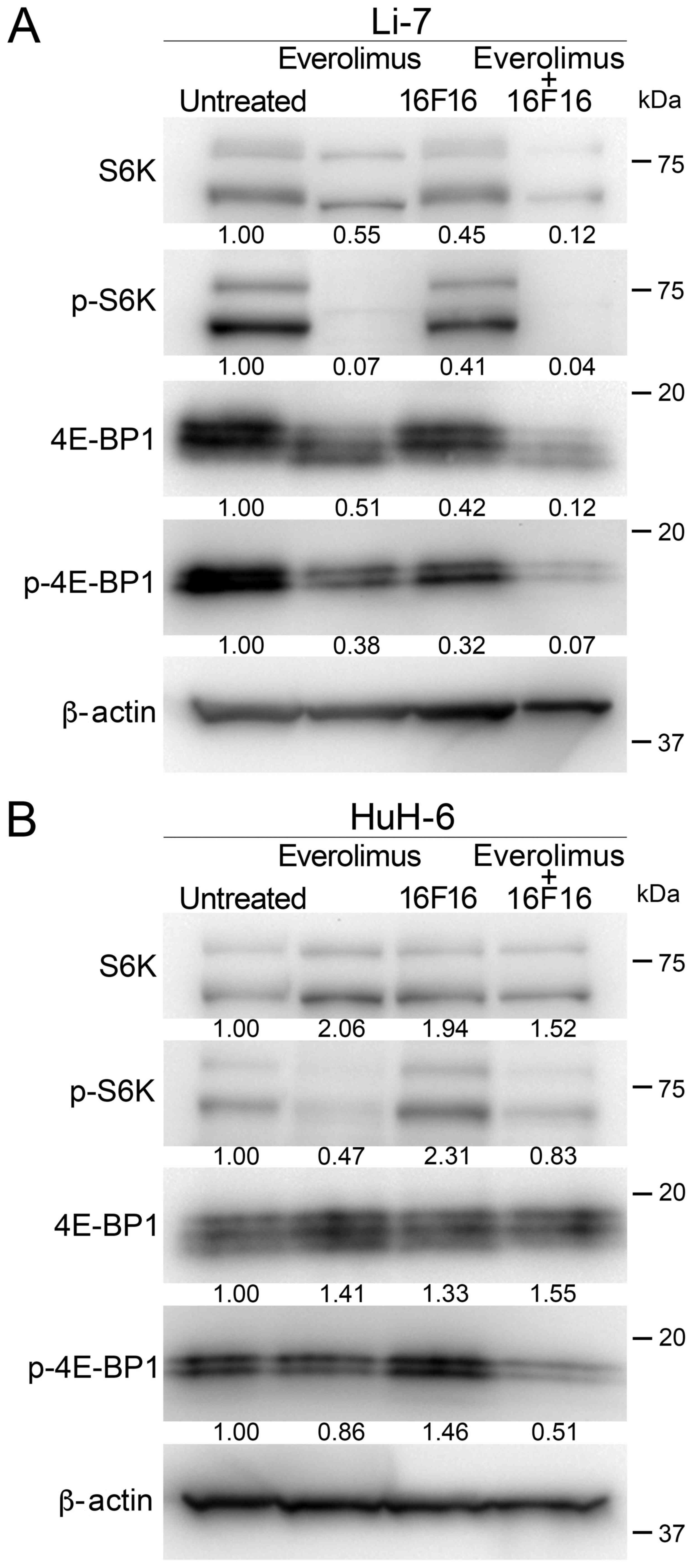
The expression and phosphorylation state of S6K and
4E-BP1, which are downstream molecules in mTORC1 signaling pathway,
were analyzed by western blot. In Li-7 cells, the expression of S6K
was slightly decreased by treatment with Ev or 16F16 and by the Ev
and 16F16 in combination, as compared with untreated cells
(Fig. 4A). p-S6K was reduced by
treatment with Ev. Treatment with 16F16 slightly reduced S6K
phosphorylation, and p-S6K was reduced by combination treatment
with Ev and 16F16. The expression of 4E-BP1 appeared to be reduced
in treated cells. p-4E-BP1 appeared to be slightly reduced in cells
treated with Ev alone and 16F16 alone. The phosphorylation of
4E-BP1 was reduced by combination treatment with Ev and 16F16
(Fig. 4A). In HuH-6 cells, the
expression of S6K was slightly elevated in treated cells. p-S6K was
reduced in cells treated with Ev alone. Phosphorylation was not
inhibited by treatment with 16F16, but it was reduced by the
combination treatment. Expression of 4E-BP1 was slightly elevated
in treated cells. Expression of p-4E-BP1 was comparable between
treatment with Ev alone and 16F16 alone, whereas phosphorylation
was reduced by the combination treatment (Fig. 4B).
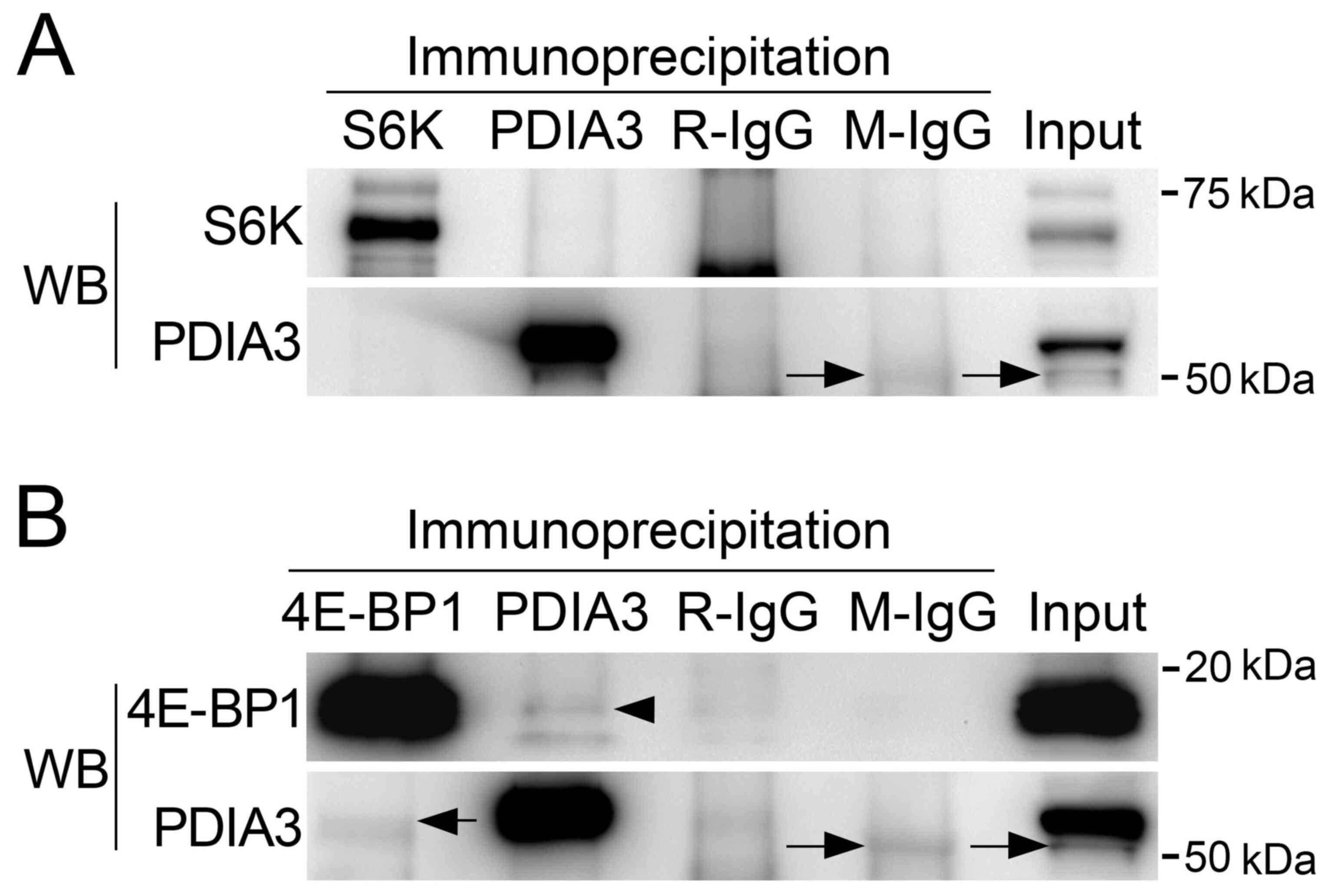
Immunoprecipitation analysis
Whether PDIA3 forms a complex with S6K and 4E-BP1
was examined by immunoprecipitation using HuH-6 cell lysates. In
the sample immunoprecipitated with anti-PDIA3 antibody, no positive
band was observed in western blots with anti-S6K antibody (Fig. 5A). No positive band was detected in
western blotting with anti-PDIA3 antibody in the sample
immunoprecipitated with anti-S6K antibody (Fig. 5A). On the other hand, in the sample
immunoprecipitated with anti-PDIA3 antibody, a positive band was
detected in western blotting with anti-4E-BP1 antibody (Fig. 5B). Immunoprecipitated PDIA3 was
observed in western blotting with anti-PDIA3 antibody in the sample
immunoprecipitated with anti-4E-BP1 antibody (Fig. 5B). The immunoprecipitation analysis
demonstrated complex formation of PDIA3 with 4E-BP1 but not with
S6K.
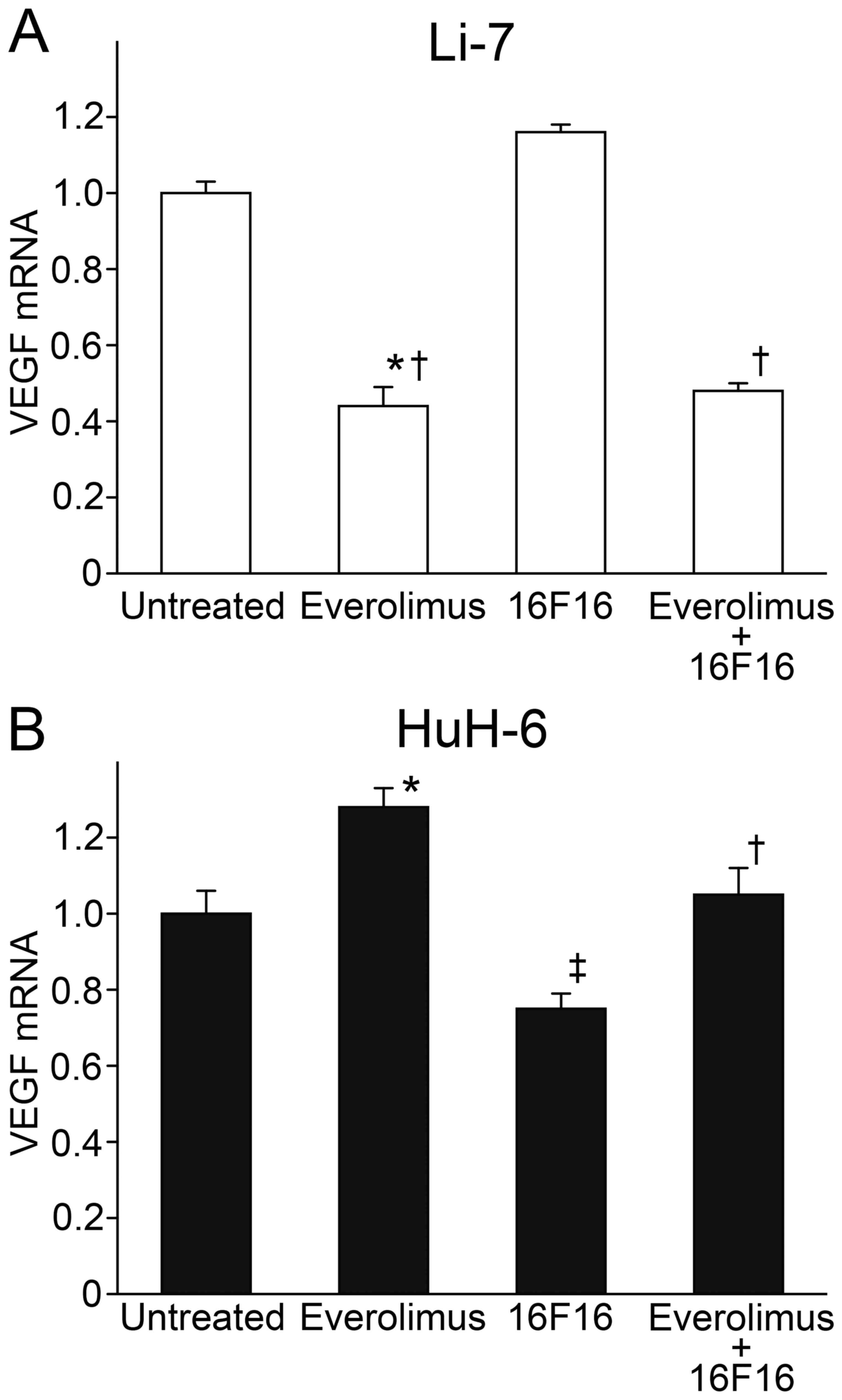
VEGF expression in cultured cells
mTORC1 signaling regulates the expression of VEGF,
which plays an important role in angiogenesis and the growth of
liver cancer. VEGF mRNA expression was examined in cultured cells
by RT-PCR. In Li-7 cells, the expression of VEGF mRNA was reduced
to 44.1±4.9 and 47.5±2.4% of untreated cells by treatment with Ev
alone and by combination treatment with Ev and 16F16, respectively
(Fig. 6A). In contrast, VEGF mRNA
expression was roughly comparable among HuH-6 cells, and there was
no apparent alteration by Ev or 16F16 (Fig. 6B).
Discussion
This is the first study to show enhancement of the
antiproliferative effect of inhibitor for mTOR by inhibitor for
PDIA3 in liver cancer. Two cell lines of liver cancer, one
susceptible and one resistant to mTOR inhibitor, were used in the
present study. Combination treatment with Ev and 16F16 suppressed
the proliferation of cultured liver cancer cells, and the
suppression was associated with reduced phosphorylation of 4E-BP1,
which formed a complex with PDIA3. It is noteworthy that
enhancement was evident in a cultured liver cancer cell that was
resistant to mTOR inhibitor.
It is thought that the activation of mTOR is
associated with the pathogenesis and aggressiveness of liver
cancer. In vitro and animal studies have shown that mTOR
inhibitors could be effective for the treatment of liver cancer
(11,13,15,29).
However, clinical trials have failed to demonstrate significant
improvement in the prognosis in of patients with liver cancer
(17). To date, no mTOR inhibitors
have been approved for the treatment of liver cancer. This may be
due in part to the incomplete inhibition of mTOR and the activation
of other signaling pathways such as MAPK and PI3K by feedback
mechanism (30). Subpopulation
analysis of liver cancer treated with Ev suggested that liver
cancer with loss of TSC2 is susceptible to Ev, but liver cancer
with the expression of TSC2 is resistant to Ev. To overcome the
incomplete effect of mTOR inhibitors for liver cancer, combination
therapies have been utilized. Concomitant inhibition of mTOR and
MAPK or DNA replication was shown to possibly be effective for the
treatment of liver cancer (31–33).
The association of PDIA3 with mTOR was shown in a
previous study by Ramirez-Rangel and colleagues (23). It was demonstrated that knock-down of
PDIA3 by specific siRNA suppressed the proliferation, while
overexpression enhanced proliferation, and cell proliferation was
correlated with phosphorylation of S6K and 4E-BP1. It was also
shown that PDIA3 forms a complex with mTORC1 but not with mTORC2
(23). 16F16 may have an effect
similar to that of siRNA for PDIA3, since a high concentration of
16F16 reduced the cell viability and proliferation of cultured
liver cancer cells. However, enhancement of the antiproliferative
effect of Ev by 16F16 was achieved at a suboptimal concentration of
16F16, at which cellular viability was not affected. Although the
phosphorylation of S6K was inhibited by Ev alone, the
phosphorylation of 4E-BP1 was reduced by combination treatment with
Ev and 16F16. This may suggest that the mTOR-4E-BP1 signaling
pathway is more affected by the integrity of the complex supported
by PDIA3. This is accounted for by the formation of a complex of
PDIA3 with 4E-BP1 but not with S6K, as shown in the
immunoprecipitation assay.
There was a difference in the association of cell
proliferation with the phosphorylation of S6K and 4E-BP1 between
Li-7 and HuH-6 cells, which were susceptible and resistant to Ev,
respectively. Cell proliferation was inhibited by Ev alone in Li-7
cells, whereas proliferation was only inhibited by combination
treatment with Ev and 16F16 in HuH-6 cells. This may be explained
by the expression of TSC2, which is a molecule upstream of mTORC1
and inhibits mTOR function (18,34).
Thus, molecular analysis of TSC2 expression may be useful to
determine cases that can benefit most from combination treatment
with Ev and 16F16. However, there could be a difference in the
mechanism of pathogenesis between cultured liver cancer cell lines.
It may be also accounted for by the origin of the cultured cells;
Li-7 was derived from HCC, and Huh-6 was derived from
hepatoblastoma. It is plausible that the inhibition of
proliferation is a synergistic effect due to mTOR inhibition and an
unknown molecule involved in the pathogenesis by 16F16. The further
investigation on the pathogenesis of liver cancer is needed.
Angiogenesis plays an important role in the
progression of liver cancer (25,35). It
has been shown that VEGF released from liver cancer initiates
angiogenesis and vascular formation (35,36). In
the present study, VEGF expression was reduced by treatment with Ev
in Li-7 cells but was unchanged by treatment with Ev and or Ev and
16F16 in combination in HuH-6 cells. For the treatment of liver
cancer, which is resistant to Ev, the combination of Ev and 16F16
is effective for inhibiting proliferation but not sufficiently
effective for inhibiting angiogenesis. Anti-angiogenic therapy may
therefore be needed as an adjunct to combination treatment with Ev
and 16F16.
The present study was in vitro study using
cultured liver cells. There are a couple of issues that needs to be
addressed in the future study. 16F16 was used at the concentration,
at which the viability of cultured liver cancer cells was not
affected. However, the toxicity of 16F16 was not fully elucidated
in vivo. Further, the effect of mTOR inhibitor for human
liver cancer is controversial (17).
Thus, the effect of combination treatment of Ev and 16F16 needs to
be verified in animal model inoculated with cultured liver cancer
cells. In liver cancer, the recurrence soon after the resection of
the primary tumor is not infrequent. A novel therapy to prevent the
survival and induction of cancer stem cell is expected. It is
considered that mTORC1 signaling is involved in the development and
maintenance of stem cells (37).
Further study is needed to examine whether the combination
treatment of Ev and 16F16 suppress the development of cancer stem
cell of liver cancer.
It has been shown that the level of PDIA3 expression
is associated with the prognosis of liver cancer (21). Liver cancer with high PDIA3
expression follows an unfavorable course and is characterized by
high proliferation activity and a low frequency of cell death. The
expression of PDIA3 and its association with prognosis has been
reported for other carcinomas (21,22,38–41).
Furthermore, knock-down of PDIA3 may enhance the effect of
radiation therapy in laryngeal cancer (38) and breast cancer (42). The addition of a PDIA3 inhibitor may
be beneficial to enhance the effect of chemotherapy and radiation
therapy. PDIA3 may form a complex with other molecules that are
involved in the carcinogenesis and behavior of tumors. Targeting of
chaperone protein would be a novel strategy for enhancing the
therapeutic effects of small molecule inhibitors, radiation, and
chemotherapy.
Acknowledgements
The authors would like to acknowledge the assistance
of Ms. Kiyoko Kawahara for cell culture, Mr. Takenori Fujii and Ms.
Yoko Kawamoto for help with western blotting, and Mr. Kiyoshi
Teduka for help with reverse transcription-quantitative PCR (all
from Department of Integrated Diagnostic Pathology, Nippon Medical
School, Tokyo, Japan).
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YK, RW and ZN designed the study and wrote the
manuscript draft. YK performed histological examinations. YK, HT
and RO conducted biochemical examinations and data analyses. RW and
SK performed statistical analyses. YK, KI, MK and NT conducted cell
culture experiments and prepared figures/tables. YK, RK and HY
performed RT-qPCR. HY and ZN supervised the experimental design and
manuscript writing. All authors agree to be accountable for all
aspects of the work in ensuring that questions related to the
accuracy or integrity of any part of the work are appropriately
investigated and resolved. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
4E-BP1
|
4E-binding protein 1
|
|
Ev
|
everolimus
|
|
HCC
|
hepatocellular carcinoma
|
|
mTOR
|
mechanistic target of rapamycin
|
|
mTORC1
|
mTOR complex 1
|
|
PBS
|
phosphate-buffered saline
|
|
PDIA3
|
protein disulfide isomerase A member
3
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
|
S6K
|
S6 kinase
|
|
TSC2
|
tuberous sclerosis complex 2
|
|
VEGF
|
vascular endothelial growth factor
|
References
|
1
|
Altekruse SF, McGlynn KA and Reichman ME:
Hepatocellular carcinoma incidence, mortality, and survival trends
in the United States from 1975 to 2005. J Clin Oncol. 27:1485–1491.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Eso Y and Marusawa H: Novel approaches for
molecular targeted therapy against hepatocellular carcinoma.
Hepatol Res. 48:597–607. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Forner A, Reig M and Bruix J:
Hepatocellular carcinoma. Lancet. 391:1301–1314. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Grandhi MS, Kim AK, Ronnekleiv-Kelly SM,
Kamel IR, Ghasebeh MA and Pawlik TM: Hepatocellular carcinoma: From
diagnosis to treatment. Surg Oncol. 25:74–85. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yoshida H, Taniai N, Yoshioka M, Hirakata
A, Kawano Y, Shimizu T, Ueda J, Takata H, Nakamura Y and Mamada Y:
Current status of laparoscopic hepatectomy. J Nippon Med Sch.
86:201–206. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Swamy SG, Kameshwar VH, Shubha PB, Looi
CY, Shanmugam MK, Arfuso F, Dharmarajan A, Sethi G, Shivananju NS
and Bishayee A: Targeting multiple oncogenic pathways for the
treatment of hepatocellular carcinoma. Target Oncol. 12:1–10. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Conciatori F, Ciuffreda L, Bazzichetto C,
Falcone I, Pilotto S, Bria E, Cognetti F and Milella M: mTOR
Cross-talk in cancer and potential for combination therapy. Cancers
(Basel). 10:232018. View Article : Google Scholar
|
|
8
|
Fasolo A and Sessa C: Targeting mTOR
pathways in human malignancies. Curr Pharm Des. 18:2766–2777. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Matter MS, Decaens T, Andersen JB and
Thorgeirsson SS: Targeting the mTOR pathway in hepatocellular
carcinoma: Current state and future trends. J Hepatol. 60:855–865.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tian T, Li X and Zhang J: mTOR Signaling
in cancer and mTOR inhibitors in solid tumor targeting therapy. Int
J Mol Sci. 20:7552019. View Article : Google Scholar
|
|
11
|
Villanueva A, Chiang DY, Newell P, Peix J,
Thung S, Alsinet C, Tovar V, Roayaie S, Minguez B, Sole M, et al:
Pivotal role of mTOR signaling in hepatocellular carcinoma.
Gastroenterology. 135:1972–1983, 1983.e1-11. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Buitrago-Molina LE, Pothiraju D, Lamlé J,
Marhenke S, Kossatz U, Breuhahn K, Manns MP, Malek N and Vogel A:
Rapamycin delays tumor development in murine livers by inhibiting
proliferation of hepatocytes with DNA damage. Hepatology.
50:500–509. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Engl T, Rutz J, Maxeiner S, Juengel E,
Roos F, Khoder W, Bechstein WO, Nelson K, Tsaur I, Haferkamp A, et
al: mTOR inhibition reduces growth and adhesion of hepatocellular
carcinoma cells in vitro. Mol Med Rep. 16:7064–7071. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sahin F, Kannangai R, Adegbola O, Wang J,
Su G and Torbenson M: mTOR and P70 S6 kinase expression in primary
liver neoplasms. Clin Cancer Res. 10:8421–8425. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Semela D, Piguet AC, Kolev M, Schmitter K,
Hlushchuk R, Djonov V, Stoupis C and Dufour JF: Vascular remodeling
and antitumoral effects of mTOR inhibition in a rat model of
hepatocellular carcinoma. J Hepatol. 46:840–848. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Thomas HE, Mercer CA, Carnevalli LS, Park
J, Andersen JB, Conner EA, Tanaka K, Matsutani T, Iwanami A, Aronow
BJ, et al: mTOR inhibitors synergize on regression, reversal of
gene expression, and autophagy in hepatocellular carcinoma. Sci
Transl Med. 4:139ra842012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhu AX, Kudo M, Assenat E, Cattan S, Kang
YK, Lim HY, Poon RT, Blanc JF, Vogel A, Chen CL, et al: Effect of
everolimus on survival in advanced hepatocellular carcinoma after
failure of sorafenib: The EVOLVE-1 randomized clinical trial. JAMA.
312:57–67. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huynh H, Hao HX, Chan SL, Chen D, Ong R,
Soo KC, Pochanard P, Yang D, Ruddy D, Liu M, et al: Loss of
tuberous sclerosis complex 2 (TSC2) is frequent in hepatocellular
carcinoma and predicts response to mTORC1 inhibitor everolimus. Mol
Cancer Ther. 14:1224–1235. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Turano C, Gaucci E, Grillo C and
Chichiarelli S: ERp57/GRP58: A protein with multiple functions.
Cell Mol Biol Lett. 16:539–563. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hettinghouse A, Liu R and Liu CJ:
Multifunctional molecule ERp57: From cancer to neurodegenerative
diseases. Pharmacol Ther. 181:34–48. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Takata H, Kudo M, Yamamoto T, Ueda J,
Ishino K, Peng WX, Wada R, Taniai N, Yoshida H, Uchida E, et al:
Increased expression of PDIA3 and its association with cancer cell
proliferation and poor prognosis in hepatocellular carcinoma. Oncol
Lett. 12:4896–4904. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kondo R, Ishino K, Wada R, Takata H, Peng
WX, Kudo M, Kure S, Kaneya Y, Taniai N, Yoshida H, et al:
Downregulation of protein disulfide isomerase A3 expression
inhibits cell proliferation and induces apoptosis through STAT3
signaling in hepatocellular carcinoma. Int J Oncol. 54:1409–1421.
2019.PubMed/NCBI
|
|
23
|
Ramírez-Rangel I, Bracho-Valdés I,
Vázquez-Macías A, Carretero-Ortega J, Reyes-Cruz G and
Vázquez-Prado J: Regulation of mTORC1 complex assembly and
signaling by GRp58/ERp57. Mol Cell Biol. 31:1657–1671. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hoffstrom BG, Kaplan A, Letso R, Schmid
RS, Turmel GJ, Lo DC and Stockwell BR: Inhibitors of protein
disulfide isomerase suppress apoptosis induced by misfolded
proteins. Nat Chem Biol. 6:900–906. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang ZF and Poon RT: Vascular changes in
hepatocellular carcinoma. Anat Rec (Hoboken). 291:721–734. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hirohashi S, Shimosato Y, Kameya T, Koide
T, Mukojima T, Taguchi Y and Kageyama K: Production of
alpha-fetoprotein and normal serum proteins by xenotransplanted
human hepatomas in relation to their growth and morphology. Cancer
Res. 39:1819–1828. 1979.PubMed/NCBI
|
|
27
|
Doi I: Establishment of a cell line and
its clonal sublines from a patient with hepatoblastoma. Gan.
67:1–10. 1976.PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huynh H, Chow KH, Soo KC, Toh HC, Choo SP,
Foo KF, Poon D, Ngo VC and Tran E: RAD001 (everolimus) inhibits
tumour growth in xenograft models of human hepatocellular
carcinoma. J Cell Mol Med. 13:1371–1380. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Carracedo A, Ma L, Teruya-Feldstein J,
Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma
SC, et al: Inhibition of mTORC1 leads to MAPK pathway activation
through a PI3K-dependent feedback loop in human cancer. J Clin
Invest. 118:3065–3074. 2008.PubMed/NCBI
|
|
31
|
Newell P, Toffanin S, Villanueva A, Chiang
DY, Minguez B, Cabellos L, Savic R, Hoshida Y, Lim KH,
Melgar-Lesmes P, et al: Ras pathway activation in hepatocellular
carcinoma and anti-tumoral effect of combined sorafenib and
rapamycin in vivo. J Hepatol. 51:725–733. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Piguet AC, Saar B, Hlushchuk R, St-Pierre
MV, McSheehy PM, Radojevic V, Afthinos M, Terracciano L, Djonov V
and Dufour JF: Everolimus augments the effects of sorafenib in a
syngeneic orthotopic model of hepatocellular carcinoma. Mol Cancer
Ther. 10:1007–1017. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang Z, Zhou J, Fan J, Qiu SJ, Yu Y, Huang
XW and Tang ZY: Effect of rapamycin alone and in combination with
sorafenib in an orthotopic model of human hepatocellular carcinoma.
Clin Cancer Res. 14:5124–5130. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ferrin G, Guerrero M, Amado V,
Rodriguez-Peralvarez M and De la Mata M: Activation of mTOR
signaling pathway in hepatocellular carcinoma. Int J Mol Sci.
21:12662020. View Article : Google Scholar
|
|
35
|
Liu K, Zhang X, Xu W, Chen J, Yu J, Gamble
JR and McCaughan GW: Targeting the vasculature in hepatocellular
carcinoma treatment: Starving versus normalizing blood supply. Clin
Transl Gastroenterol. 8:e982017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Morse MA, Sun W, Kim R, He AR, Abada PB,
Mynderse M and Finn RS: The role of angiogenesis in hepatocellular
carcinoma. Clin Cancer Res. 25:912–920. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Meng D, Frank AR and Jewell JL: mTOR
signaling in stem and progenitor cells. Development.
145:dev1525952018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Choe MH, Min JW, Jeon HB, Cho DH, Oh JS,
Lee HG, Hwang SG, An S, Han YH and Kim JS: ERp57 modulates STAT3
activity in radioresistant laryngeal cancer cells and serves as a
prognostic marker for laryngeal cancer. Oncotarget. 6:2654–2666.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chung H, Cho H, Perry C, Song J, Ylaya K,
Lee H and Kim JH: Downregulation of ERp57 expression is associated
with poor prognosis in early-stage cervical cancer. Biomarkers.
18:573–579. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shimoda T, Wada R, Kure S, Ishino K, Kudo
M, Ohashi R, Fujita I, Uchida E, Yoshida H and Naito Z: Expression
of protein disulfide isomerase A3 and its clinicopathological
association in gastric cancer. Oncol Rep. 41:2265–2272.
2019.PubMed/NCBI
|
|
41
|
Zou H, Wen C, Peng Z, Shao YΥ, Hu L, Li S,
Li C and Zhou HH: P4HB and PDIA3 are associated with tumor
progression and therapeutic outcome of diffuse gliomas. Oncol Rep.
39:501–510. 2018.PubMed/NCBI
|
|
42
|
Hussmann M, Janke K, Kranz P, Neumann F,
Mersch E, Baumann M, Goepelt K, Brockmeier U and Metzen E:
Depletion of the thiol oxidoreductase ERp57 in tumor cells inhibits
proliferation and increases sensitivity to ionizing radiation and
chemotherapeutics. Oncotarget. 6:39247–39261. 2015. View Article : Google Scholar : PubMed/NCBI
|