Introduction
Chronic obstructive pulmonary disease (COPD) is one
of the leading causes of death worldwide, and its prevalence and
mortality may increase further in the coming decades due to smoking
and exposure to noxious agents (1).
COPD is characterized by partially irreversible airflow obstruction
due to inflammation of the airways and destruction of lung
parenchyma (2). Tobacco smoke is the
main cause of COPD development and it can induce the release of
inflammatory cytokines, such as IL-1β, IL-6 and TNFα (3–5).
Currently, there is no effective and specific treatment for COPD.
Therefore, it is important to improve our understand of the
pathogenic mechanisms at the molecular level to develop new
therapeutic approaches.
Long non-coding (lnc)RNAs are a class of endogenous
cellular RNAs (>200 nucleotides) that lack protein-coding
capacity and serve regulatory roles in various physiopathological
processes (6). In previous years,
lncRNAs have been identified to have essential roles in various
types of disease, including COPD (7,8). For
example, lncRNA taurine upregulated 1 promotes airway remodeling by
inhibiting the miR-145-5p/dual specificity protein phosphatase 6
axis in CSE-induced COPD (9).
Moreover, another previous study reported that lncRNA MIR155 host
gene regulated M1/M2 macrophage polarization in the progression of
COPD (10). Maternally expressed
gene (MEG)3, located on human chromosome 14q32.3, is a maternally
imprinted gene and plays important roles in numerous diseases, such
as glioma, gastric cancer and Alzheimer's disease (11,12). In
addition, a previous report demonstrated that MEG3 is overexpressed
in COPD tissues (13). However, the
precise molecular mechanisms underlying MEG3 function in COPD
progression and development remains poorly understood.
Growing evidence has shown that lncRNAs can function
as competing endogenous (ce)RNAs to modulate gene expression
through sponging miRNA (14,15). miRNAs are small non-coding RNAs,
which negatively regulate gene expression and modulate diverse
physiological processes, such as differentiation, proliferation,
apoptosis and metabolism (16). An
increasing number of studies have shown that dysregulation of
miRNAs is associated with multiple pathological conditions,
including COPD (17,18). In particular, low expression of
miR-181a-2-3p has been identified in the serum of patients with
COPD (19). Nevertheless, to the
best of our knowledge, there is no evidence to support the
interaction between MEG3 and miR-181a-2-3p in COPD. Therefore, the
function of miR-181a-2-3p in the pathogenesis of COPD needs to be
investigated.
The present study used cigarette smoke extract
(CSE)-treated 16HBE cells as an in vitro model of COPD to
investigate the biological functions of MEG3 and miR-181a-2-3p in
COPD progression. In addition, the molecular mechanisms of MEG3 and
miR-181a-2-3p in CSE-stimulated 16HBE cells were investigated. The
results of the present study may provide a promising avenue for
treatment of COPD.
Materials and methods
Serum collection and RNA
isolation
Blood samples were obtained from 55 patients (median
age, 58; age range: 38–81 years; male: 36, females: 19) with COPD
and 47 healthy control individuals (median age: 53, age range:
36–73 years, male: 28, females; 19). These participants (no other
diseases) did not receive chemotherapy, radiotherapy or other
therapy prior to blood collection. All blood samples were obtained
from Changning County Hospital of Traditional Chinese Medicine
(Yibin, China). The participants provided written informed consent
and the study was approved by The Ethics Committee of Changning
County Hospital of Traditional Chinese Medicine (Yibin, China).
Blood samples from antecubital vein were collected
with a special tube containing separation gel and clot activator,
and then centrifuged at 1,700 × g for 10 min at room temperature.
The supernatant was transferred to a new tube and centrifuged at
850 × g for 30 min at 4°C to discard the cell debris. Next, the
final supernatant was transferred to labeled EP tubes and stored at
−80°C until RNA extraction. The miRNeasy Serum/Plasma kit (Qiagen
GmbH) was used to isolate total RNA from the serum.
Cell culture and transfection
Normal human bronchial epithelial cells (16HBE) were
purchased from BeNa Culture Collection and maintained in DMEM
(Hyclone; Cyvita) containing 10% (Gibco; Thermo Fisher Scientific,
Inc.) in a humidified atmosphere with 5% CO2 at 37°C.
For CSE treatment, 16HBE cells were exposed to various
concentrations (0, 1, 2 and 4%) of CSE for different times (0, 12,
24 and 48 h).
The small interfering (si)RNA against MEG3 (si-MEG3)
and siRNA scrambled control (si-NC; non-specific scrambled siRNA),
MEG3-overexpression vector and empty vector (vector), miR-181a-2-3p
mimic (miR-181a-2-3p) and its negative control (miR-NC),
miR-181a-2-3p inhibitor (anti-miR-181a-2-3p) and its negative
control (anti-miR-NC) were provided by Shanghai GenePharma Co.,
Ltd. The sequences were as follows: si-MEG3
(5′-GGGCTTCTGGAATGAGCAT-3′); si-NC (5′-UUCUCCGAACGUGUCACGUTT-3′);
miR-181a-2-3p mimic (5′-ACCACUGACCGUUGACUGUACC-3′); miR-NC
(5′-ACUCUAUCUGCACGCUGACUU-3′); miR-181a-2-3p inhibitor
(5′-GGUACAGUCAACGGUCAGUGGU-3′); anti-miR NC
(5′-CAGUACUUUUGUGUAGUACAA-3′). 16HBE cells were seeded into seeded
in six-well plates (5×105 cells/well) and then
transfected with oligonucleotide (50 nM miRNA mimic/inhibitor and
20 nM siRNA) or plasmid (2 µg) using the Lipofectamine®
3000 reagent (Invitrogen; Thermo Fisher Scientific, Inc.) when cell
confluence reached 60–70%. The cells were collected for subsequent
experimentation following 24 h of transfection at 37°C.
Non-transfected group was used as control group.
Preparation of CSE
The preparation of CSE was performed as previously
described (20). Briefly, the smoke
from 10 cigarettes (China Tobacco Hunan Industrial Co, Ltd.) was
bubbled through 25 ml of phosphate-buffered saline (PBS). The
suspension was adjusted to pH 7.2–7.4 and filtered using a
cellulose membrane (0.22 µm) to remove the bacteria. This solution
was regarded as 100% CSE and diluted with PBS to obtain
concentrations of 0, 1, 2 and 4%.
Cell viability assay
A Cell Counting Kit (CCK)-8 (Sangon Biotech Co.,
Ltd.) was utilized to evaluate cell viability. In brief, 16HBE
cells (100 µl) were seeded in 96-well plates overnight. After
treatment or/and transfection, CCK-8 (10 µl) reagent was added to
per well and then incubated for another 3 h. Lastly, optical
density (OD) value was examined under a microplate reader (Bio-Rad
Laboratories, Inc.) at 450 nm.
Apoptosis assay
An apoptosis assay was conducted using an Annexin
V-FITC/PI apoptosis detection kit (Sangon Biotech Co., Ltd.) to
determine the apoptosis. After treatment or/and transfection, 16HBE
cells were collected, centrifuged at 1,000 × g for 5 min 4°C,
washed three times with PBS and resuspended in binding buffer (300
µl). Subsequently, cells were double-stained with Annexin V-FITC
and PI for 20 min in the dark at room temperature. Lastly, the
apoptotic rate was then analyzed using flow cytometry (Guava
easyCyte HT; Luminex Corporation). The data were analyzed using
GuavaSoft 3.2 software (Luminex Corporation).
Western blot assay
After treatment or/and transfection, 16HBE cells
were lysed in RIPA lysis buffer (Beyotime Institute of
Biotechnology) containing protease inhibitors to obtain total
protein. After quantification by using bicinchoninic acid protein
assay kit (Beyotime Institute of Biotechnology), an equal amount
(40 µg per lane) of protein was resolved by 10–12% SDS-PAGE and
then transferred onto PVDF membranes (0.2 µm; Beyotime Institute of
Biotechnology). Next, membranes were blocked using 5% non-fat milk
(Sangon Biotech Co., Ltd.) and then probed with specific primary
antibody against Bcl-2 (1:1,000; cat. no. ab196495), Bax (1:500;
cat. no. ab53154), cleaved caspase-3 (1:500; cat. no. ab49822) and
GAPDH (1:3,000, cat. no. ab37168) (all Abcam) for 12 h at 4°C.
Subsequently, all membranes were incubated in HRP-conjugated
anti-rabbit IgG (1:4,000; cat. no. D110058; Sangon Biotech Co.,
Ltd.). Finally, all protein bands were observed using the ECL
system (EMD Millipore). The protein levels were normalized by
GAPDH, and ImageJ software version 1.50d software (National
Institutes of Health) was used to evaluate the bands density.
ELISA assay
After treatment or/and transfection, the
concentrations of IL-1β, IL-6 and TNF-α were detected using the
following human ELISA kits: IL-1β (cat. no. E-EL-H0149c), IL-6
(cat. no. E-EL-H0102c) and TNF-α (cat. no. E-EL-H0109c (all
Elabscience, Inc.). The data were presented in terms of pg per
ml.
Lactate dehydrogenase (LDH) release
assay
After treatment or/and transfection, the level of
LDH released into cultured medium was measured using a LDH
Cytotoxicity Assay kit (Beyotime Institute of Biotechnology). The
results were presented as the percent of total LDH content.
RNA extraction and reverse
transcription quantitative (RT-q)PCR
Referring to instruction of manufacturers, total RNA
from 16HBE cells was isolated using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). For detecting MEG3
expression, the first strand of cDNA was synthesized using a Prime
Script RT reagent kit (Takara Bio, Inc.). For miR-181a-2-3p
expression detection, cDNA was synthesized using a TaqMan MicroRNA
Reverse Transcription kit (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The RT conditions were conducted as per the
manufacturer's protocols. Then, qPCR was performed using the
SYBR-Green Master Mix on 7500 Real-time PCR system (both Applied
Biosystems; Thermo Fisher Scientific, Inc.) with the following
thermocycling conditions: Pre-denaturation at 95°C for 15 sec,
followed by 40 cycles of denaturation at 95°C for 30 sec, annealing
at 60°C for 30 sec and extension at 72°C for 40 sec. The primers
used for RT-qPCR were listed as below: MEG3, Forward:
5′-CAGGATGGCAAAGGATGAAG-3′ and reverse:
5′-GCAGGTGAACACAAGCAAAGA-3′); miR-181a-2-3p, forward:
5′-GCGCGACCACTGACCGTTGAC3-3′ and reverse:
5′-ATCCAGTGCAGGGTCCGAGG-3′); GAPDH, forward:
5′-CGCTCTCTGCTCCTCCTGTTC-3′ and reverse:
5′-ATCCGTTGACTCCGACCTTCAC-3′); U6, forward:
5′-GCTTCGGCAGCACATATACTAAAAT-3′ and reverse:
5′-CGCTTCACGAATTTGCGTGTCAT-3′). The expression levels of MEG3 and
miR-181a-2-3p were calculated using the 2−ΔΔCq method
(21) and normalized to GAPDH and
U6, respectively.
Dual-luciferase reporter assay
Potential binding sites of MEG3 and miR-181a-2-3p
were predicted using DIANA tools (http://diana.imis.athena-innovation.gr/DianaTools/index.php).
The fragment of MEG3 containing the wild-type (WT) or mutant (MUT)
binding sites of miR-181a-2-3p was amplified and inserted into the
pmirGLO luciferase vector (Promega Corporation), namely WT-MEG3 or
MUT-MEG3. The miR-181a-2-3p or miR-NC was co-transfected with
WT-MEG3 or MUT-MEG3 into 16HBE cells using the Lipofectamine 3000
reagent for 48 h as aforementioned. Lastly, the luciferase activity
was determined by dual-luciferase reporter assay system (Promega
Corporation), followed by normalization to Renilla
luciferase activity.
RNA immunoprecipitation (RIP)
assay
To further verify the relationship between MEG3 and
miR-181a-2-3p, a Magna RIP kit (cat. no. 17-700; EMD Millipore) was
used for RIP assay. In brief, 16HBE cells were lysed in the RIP
lysis buffer, and then cell lysates were incubated in RIP buffer
containing magnetic beads (50 µl; cat. no. CS203178; EMD Millipore)
conjugated with anti-argonaute 2 (Anti-Ago2; cat. no. ab32381;
1:2,000; Abcam) or immunoglobulin G (Anti-IgG; cat. no. ab109489;
1:5,000, Abcam). Input and normal IgG were used as controls. After
that, immunoprecipitated RNAs were isolated by proteinase K (150
µl) at 55°C for 30 min to digest the protein. Lastly, purified RNAs
were determined using RTq-PCR as aforementioned.
Statistical analysis
Data are presented as the mean ± SD from at least
three independent experiments. The statistical differences between
two groups or multiple (>2) groups were assessed using unpaired
Student's t-test or one-way ANOVA followed by Tukey's post hoc
test. Statistical analyses were performed using Graph Prism 6.0
software (GraphPad Software Inc.). P<0.05 was considered to
indicate a statistically significant difference.
Results
CSE treatment represses cell viability
and promotes apoptosis, inflammation and cytotoxicity in 16HBE
cells
To explore the effect of CSE on COPD progression,
16HBE cells were exposed to CSE. The CCK-8 assay suggested that CSE
treatment decreased viability of 16HBE cells in a dose- and
time-dependent manner (Fig. 1A). The
apoptosis assay revealed that apoptosis was enhanced in 16HBE cells
exposed to CSE (Fig. 1B). Similarly,
CSE exposure increased the protein expression of Bax (pro-apoptotic
molecule) (22) and cleaved
caspase-3 (a key executor in apoptotic process) (23), but decreased the protein abundance of
Bcl-2 (anti-apoptotic molecule) (22) (Fig.
1C). Moreover, the levels of inflammatory cytokines, including
IL-1β, IL-6 and TNF-α, were examined using ELISA analysis in
CSE-induced 16HBE cells. The results reported that IL-1β, IL-6 and
TNF-α levels were increased in 16HBE cells after treatment of CSE
(Fig. 1D). LDH is a cytoplasmic
enzyme that is released when the plasma membrane is destroyed, and
can be measured in the supernatant as an indicator of cytotoxicity
(24). Results showed that LDH
release was increased in supernatants of 16HBE cells exposed to CSE
(Fig. 1E). Overall, these data
suggested that CSE might promote the progression of COPD.
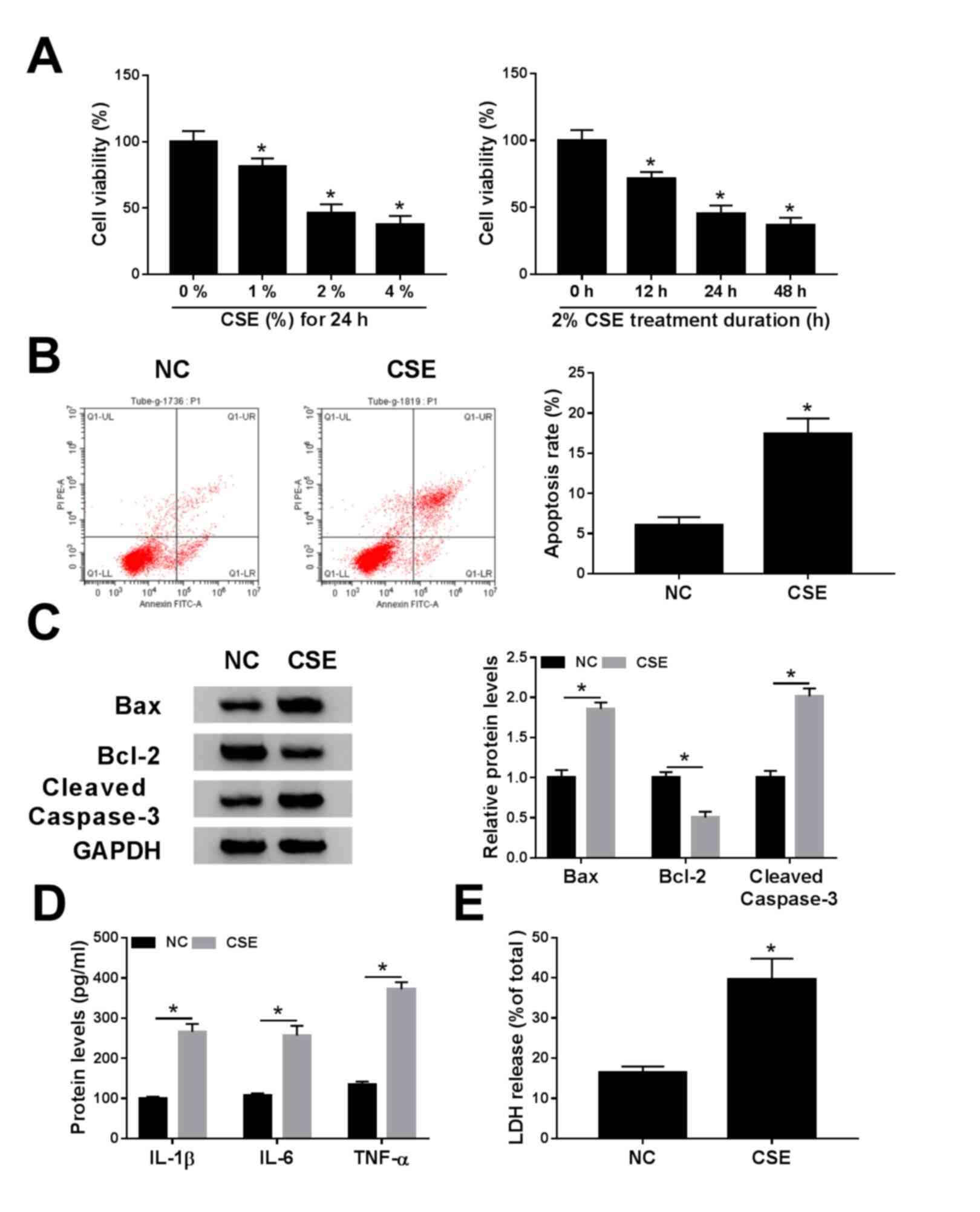 | Figure 1.Effects of CSE on cell viability,
apoptosis, inflammation and cytotoxicity in 16HBE cells. (A) Cell
Counting Kit-8 assay was employed to determine the cell viability
in 16HBE cells exposed to various concentrations (0, 1, 2 and 4%)
of CSE for 0, 12, 24 or 48 h. (B-E) 16HBE cells were exposed to CSE
(2% for 24 h). (B) Apoptosis rate was determined using flow
cytometry analysis. (C) Western blotting was conducted to measure
the protein levels of Bax, Bcl-2 and cleaved caspase-3. (D) Levels
of IL-1β, IL-6 and TNF-α were examined using ELISA assays. (E) LDH
release was measured by LDH release assay. *P<0.05 vs. 0% or NC.
CSE, cigarette smoke extract; NC, negative control. |
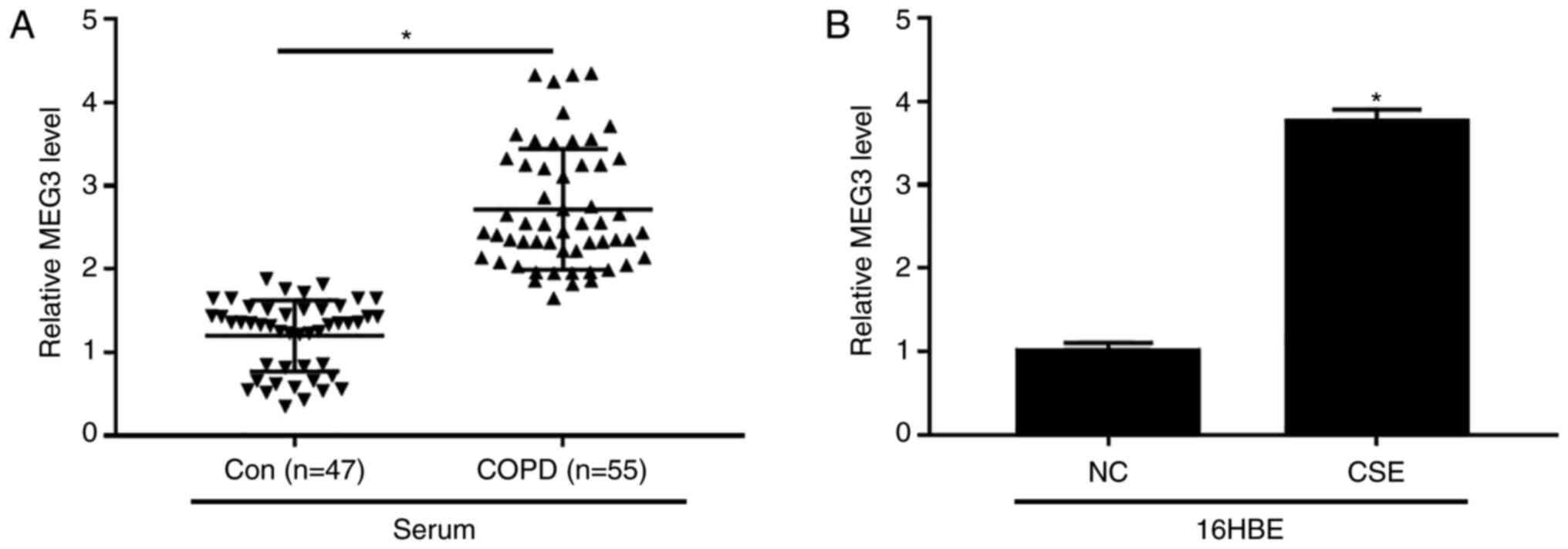
Increased MEG3 expression in serum of
patients with COPD and CSE-treated 16HBE cells
The expression of MEG3 in serum of patients with
COPD and CSE-exposed 16HBE cells was examined. The data
demonstrated that the level of MEG3 was evidently increased in
serum of patients with COPD compared with control group (Fig. 2A). Additionally, CSE exposure also
enhanced the expression of MEG3 in 16HBE cells (Fig. 2B). These results suggested that MEG3
might play an important role in COPD progression.
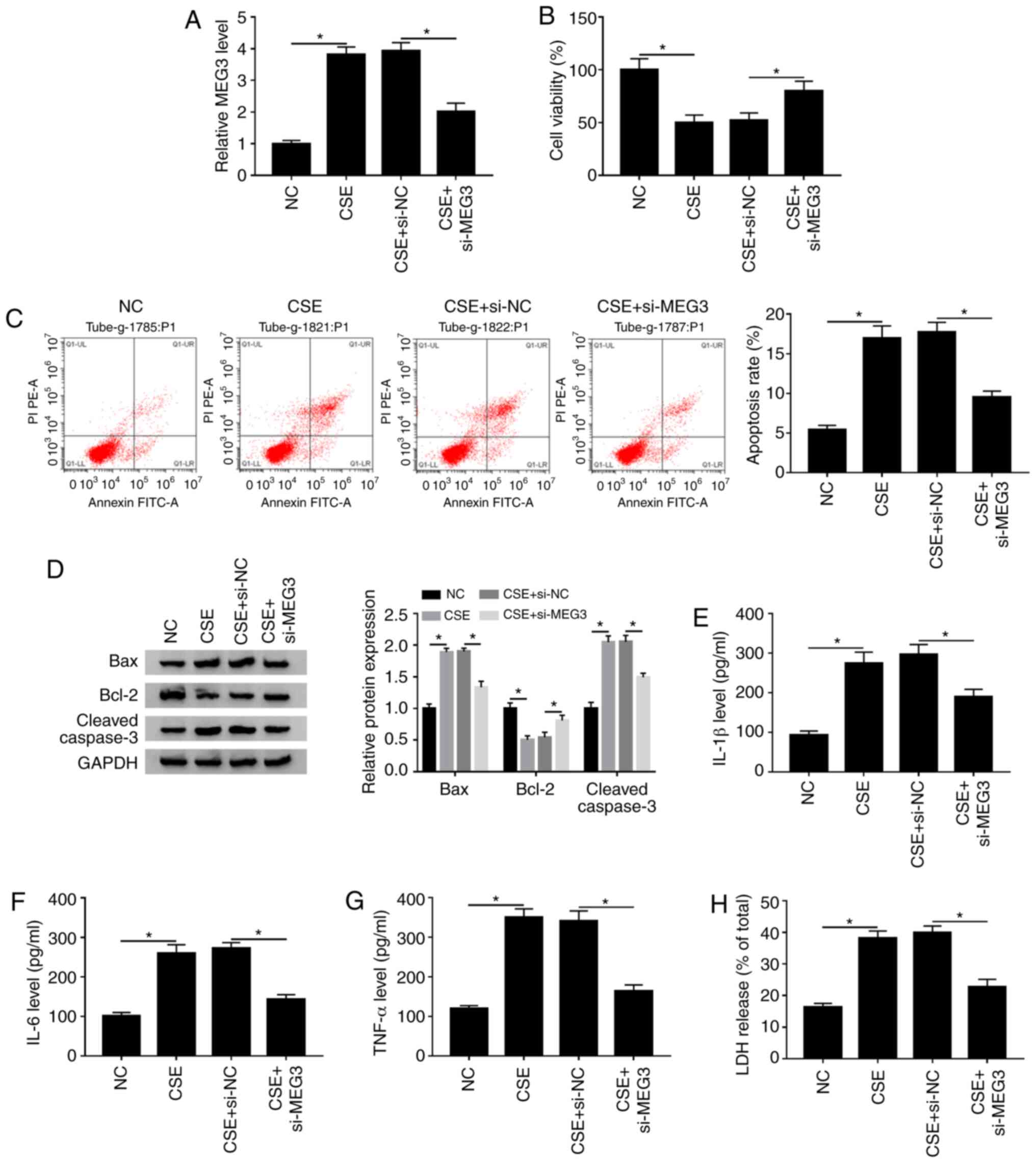
MEG3-knockdown attenuates CSE-induced
apoptosis, inflammation and cytotoxicity in 16HBE cells
To investigate whether MEG3 was involved in the
CSE-mediated the progression of COPD, si-NC or si-MEG3 was
transfected into CSE-treated 16HBE cells. It was reported that the
expression of MEG3 was decreased after transfection with si-MEG in
16HBE cells compared with the si-NC (Fig. S1A), suggesting that MEG3 was
successfully knocked down in 16HBE cells. The RT-qPCR assay showed
that knockdown of MEG3 obviously reversed increase of MEG3
expression caused by CSE in 16HBE cells (Fig. 3A). The CCK-8 assay indicated that
MEG3 interference markedly abrogated the inhibitory effect of CSE
treatment on the viability of 16HBE cells (Fig. 3B). Next, the impact of MEG3 on
apoptosis of CSE-treated 16HBE cells was further explored. As shown
in Fig. 3C and D, silencing MEG3
weakened the effect of CSE treatment on promotion of apoptotic
rate, Bax and cleaved caspase-3 expression decreased and the level
of Bcl-2 increased compared with the NC group. Moreover, knockdown
of MEG3 also abated the promoting effects of CSE treatment on
IL-1β, IL-6 and TNF-α levels as well as LDH release compared with
the NC (Fig. 3E-H). These findings
suggested that MEG3-downregulation overcame CSE-mediated decreased
cell viability and decreased CSE-induced apoptosis, inflammation
and cytotoxicity in 16HBE cells.
miR-181a-2-3p is a direct target of
MEG3
It is widely reported that lncRNAs can exert their
functions through binding with their downstream miRNAs (25). Thus, the predicted target miRNAs of
MEG3 were identified using DIANA tools. As presented in Fig. 4A, MEG3 contained potential binding
sites of miR-181a-2-3p. Moreover, the transfection efficiency of
miR-181a-2-3p and anti-miR-181a-2-3p was examined using RTq-PCR.
The results showed that transfection of miR-181a-2-3p increased the
expression of miR-181a-2-3p compared with miR-NC group, and
transfection of anti-miR-181a-2-3p decreased the expression of
miR-181a-2-3p (Fig. S1B),
indicating that miR-181a-2-3p and anti-miR-181a-2-3p were
successfully transfected into 16HBE cells. Subsequently, the
interaction between MEG3 and miR-181a-2-3p was validated by
dual-luciferase reporter and RIP assays. Results showed that
miR-181a-2-3p-overexpression significantly decreased the luciferase
activity of WT-MEG3 in16HBE cells, whereas miR-181a-2-3p
upregulation had no significant impact on the luciferase activity
of MUT-MEG3 (Fig. 4B). RIP analysis
demonstrated that the enrichment of MEG3 and miR-181a-2-3p was
greatly enhanced in Anti-Ago2 group compared with the Anti-IgG
group (Fig. 4C). Next, miR-181a-2-3p
expression in serum of patients with COPD and CSE-induced 16HBE
cells was measured. As exhibited in Fig.
4D and E, miR-181a-2-3p abundance was significantly decreased
in serum of patients with COPD and CSE-exposed 16HBE cells compared
with respective controls. In addition, the effect of MEG3 on
expression of miR-181a-2-3p in 16HBE cells was further explored.
The transfection efficiency of MEG3 was assessed using RTq-PCR. As
a result, MEG3 expression was significantly increased in 16HBE
cells transfected with MEG3 compared with vector group, suggesting
MEG3 had been successfully transfected into 16HBE cells (Fig. 4F). Furthermore, it was reported that
knockdown of MEG3 enhanced miR-181a-2-3p abundance, whereas
MEG3-overexpression decreased the levels of miR-181a-2-3p (Fig. 4G). Taken together, these data
demonstrated that miR-181a-2-3p could directly bind to MEG3 and was
negatively modulated by MEG3.
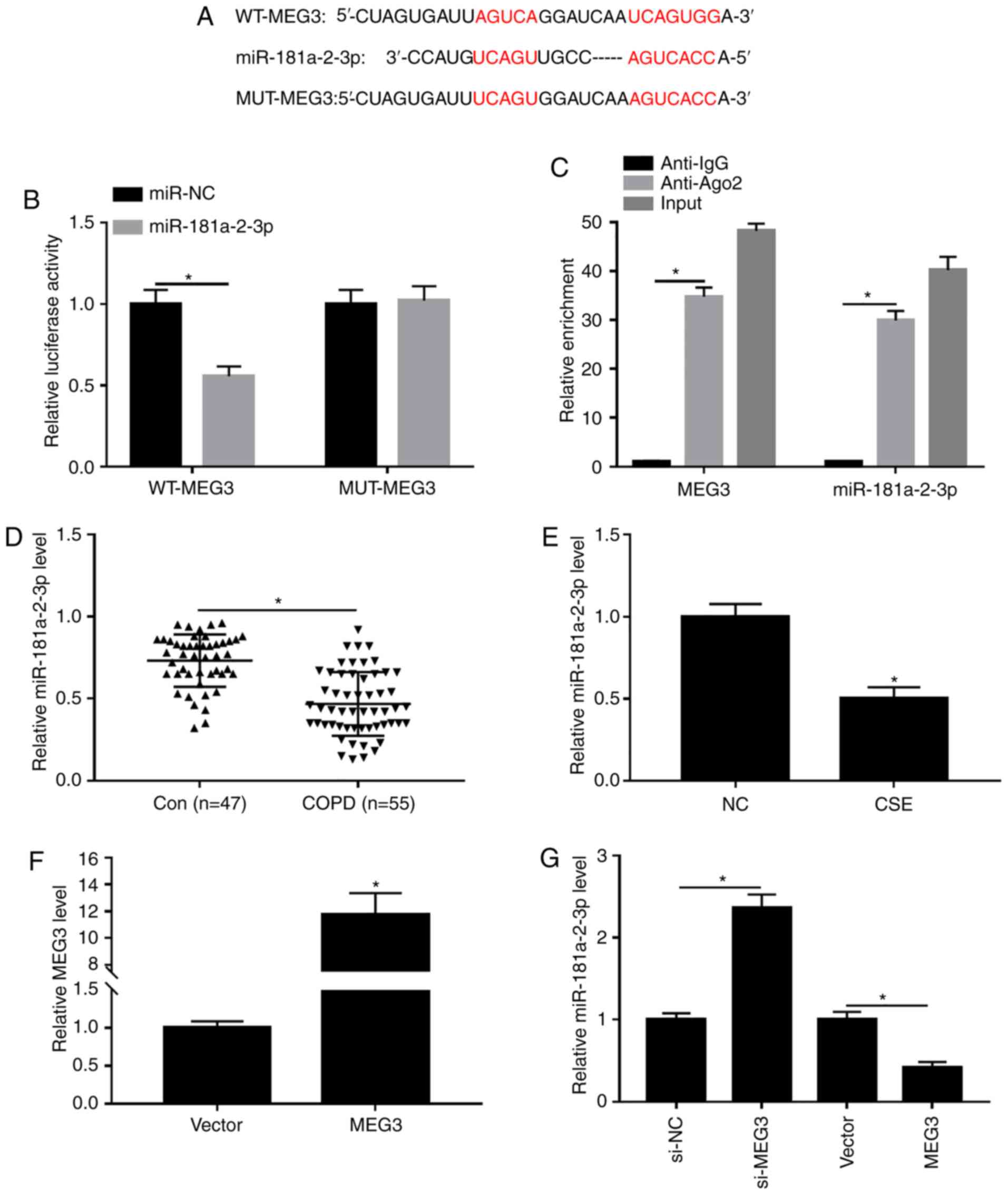 | Figure 4.Interaction between miR-181a-2-3p and
MEG3. (A) Putative binding sites between miR-181a-2-3p and MEG3
were predicted using DIANA tools. (B) Effect of
miR-181a-2-3p-overexpression on luciferase activities of WT-MEG3
and MUT-MEG3 was evaluated using a dual-luciferase luciferase
reporter assay. (C) Enrichment of MEG3 or miR-181a-2-3p was
measured using an RNA immunoprecipitation assay in 16HBE cells
incubated with Anti-Ago2 or Anti-IgG. (D) Relative abundance of
miR-181a-2-3p was detected using RT-qPCR in serum of patients with
COPD or healthy controls. (E) Relative miR-181a-2-3p expression was
analyzed using RT-qPCR in16HBE cells and 16HBE cells treated with
CSE (2%, 24 h). (F) Expression of MEG3 was assessed by RT-qPCR
assay in 16HBE cells transfected with vector or MEG3. (G) RT-qPCR
was carried out to evaluate the level of miR-181a-2-3p in 16HBE
cells transfected with si-NC, si-MEG3, vector or MEG3. *P<0.05
vs. respective control. MEG3, maternally expressed gene 3; CSE,
cigarette smoke extract; NC, negative control; si, small
interfering; WT, wild-type; MUT, mutant; RT-q, reverse
transcription-quantitative; COPD, chronic obstructive pulmonary
disease; vector, MEG3-overexpression vector negative control. |
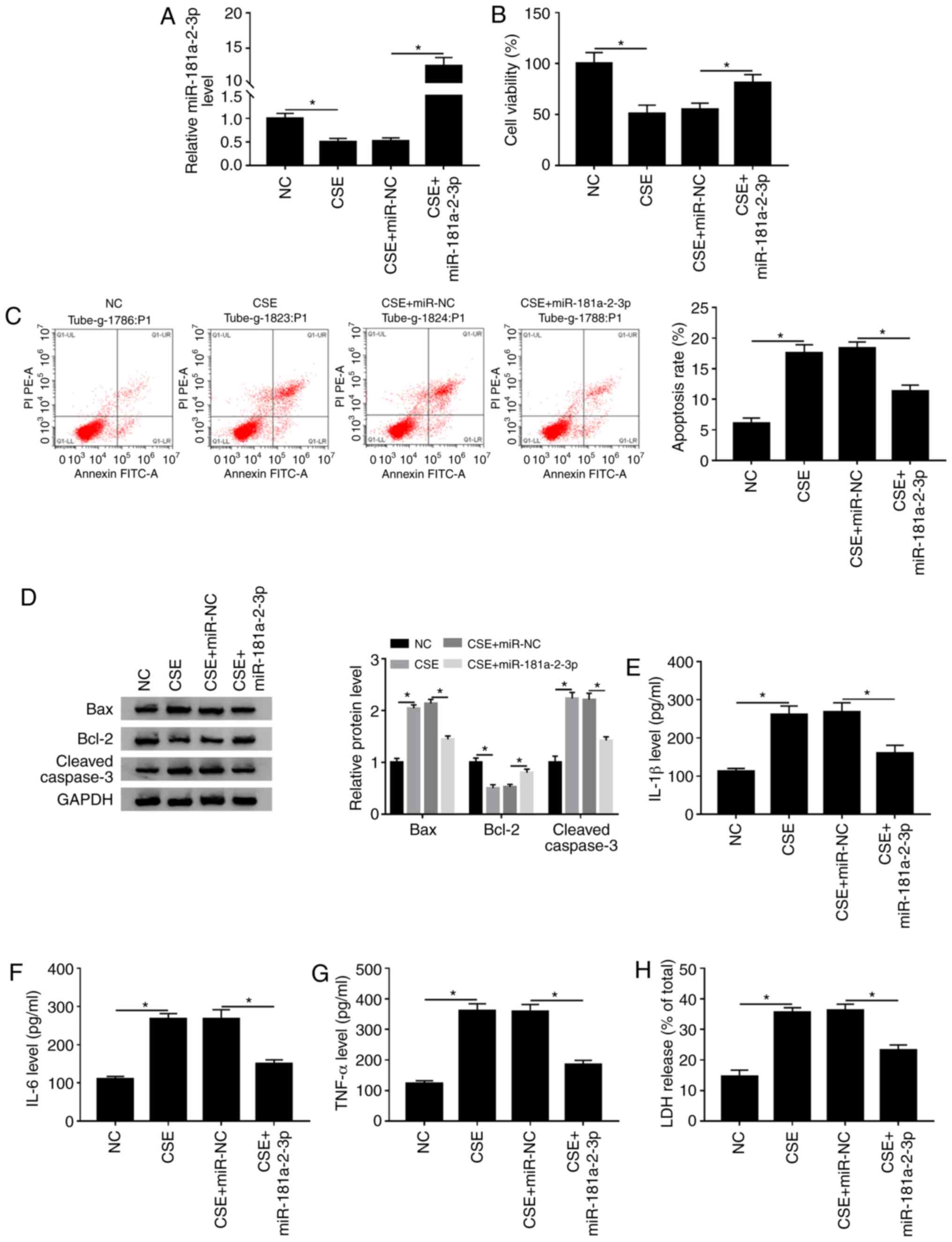
miR-181a-2-3p inhibits CSE-induced
apoptosis, inflammation and cytotoxicity in 16HBE cells
To determine the biological function of
miR-181a-2-3p in CSE-treated 16HBE cells, the overexpression
plasmid of miR-181a-2-3p was constructed. The data demonstrated
that treatment with CSE led to a significant decrease in
miR-181a-2-3p level compared with the NC, while this effect was
abated by addition of miR-181a-2-3p (Fig. 5A). Subsequently, it was examined
whether miR-181a-2-3p upregulation could play biological roles in
cell viability, apoptosis, inflammation and cytotoxicity in
CSE-exposed 16HBE cells. It was reported that miR-181a-2-3p
upregulation reversed the effect of CSE on the inhibition of cell
viability in 16HBE cells (Fig. 5B).
Moreover, it was shown that miR-181a-2-3p upregulation decreased
CSE-induced apoptosis (Fig. 5C). In
addition, western blotting demonstrated that
miR-181a-2-3p-overexpression decreased CSE-mediated promotion of
Bax and cleaved caspase-3 expression and increased Bcl-2 expression
(Fig. 5D). Furthermore,
miR-181a-2-3p-overexpression also abated the promotive effects of
CSE on inflammatory cytokine (IL-1β, IL-6 and TNF-α) levels and LDH
release (Fig. 5E-H). Collectively,
these findings demonstrated that miR-181a-2-3p repressed
CSE-induced apoptosis, inflammation and cytotoxicity in16HBE
cells.
miR-181a-2-3p-knockdown partly abates
the inhibitory effect of MEG3 interference on apoptosis,
inflammation and cytotoxicity in CSE-treated 16HBE cells
Based on the aforementioned findings, it was
speculated that MEG3-knockdown inhibited CSE- induced apoptosis,
inflammation and cytotoxicity via regulating miR-181a-2-3p. To
validate this hypothesis, rescue experiments were performed in
CSE-exposed 16HBE cells. The data showed that transfection of
si-MEG3 reversed the inhibitory effect of CSE on miR-181a-2-3p
expression, whereas co-transfection of anti-miR-181a-2-3p again
weakened the promotive effect of MEG3-knockdown on miR-181a-2-3p
level (Fig. 6A). Moreover,
miR-181a-2-3p interference inhibited the promotion of cell
viability mediated si-MEG3 in CSE-treated 16HBE cells (Fig. 6B). Furthermore, miR-181a-2-3p
interference effectively abated the antiapoptotic effect induced by
silencing MEG3 in CSE-treated 16HBE cells (Fig. 6C). In addition, miR-181a-2-3p
downregulation reversed the impact of si-MEG3 on inhibition of Bax
and cleaved caspase-3 protein expression, and promotion of Bcl-2
protein level in CSE-treated 16HBE cells (Fig. 6D). Besides, the suppressive effects
of MEG3-knockdown on inflammatory cytokine (IL-1β, IL-6 and TNF-α)
levels and LDH release were also abolished by
miR-181a-2-3p-downregulation in CSE-exposed 16HBE cells (Fig. 6E-H). To sum up, these data indicated
that MEG3-knockdown inhibited CSE-mediated apoptosis, inflammation
and cytotoxicity by upregulating miR-181a-2-3p.
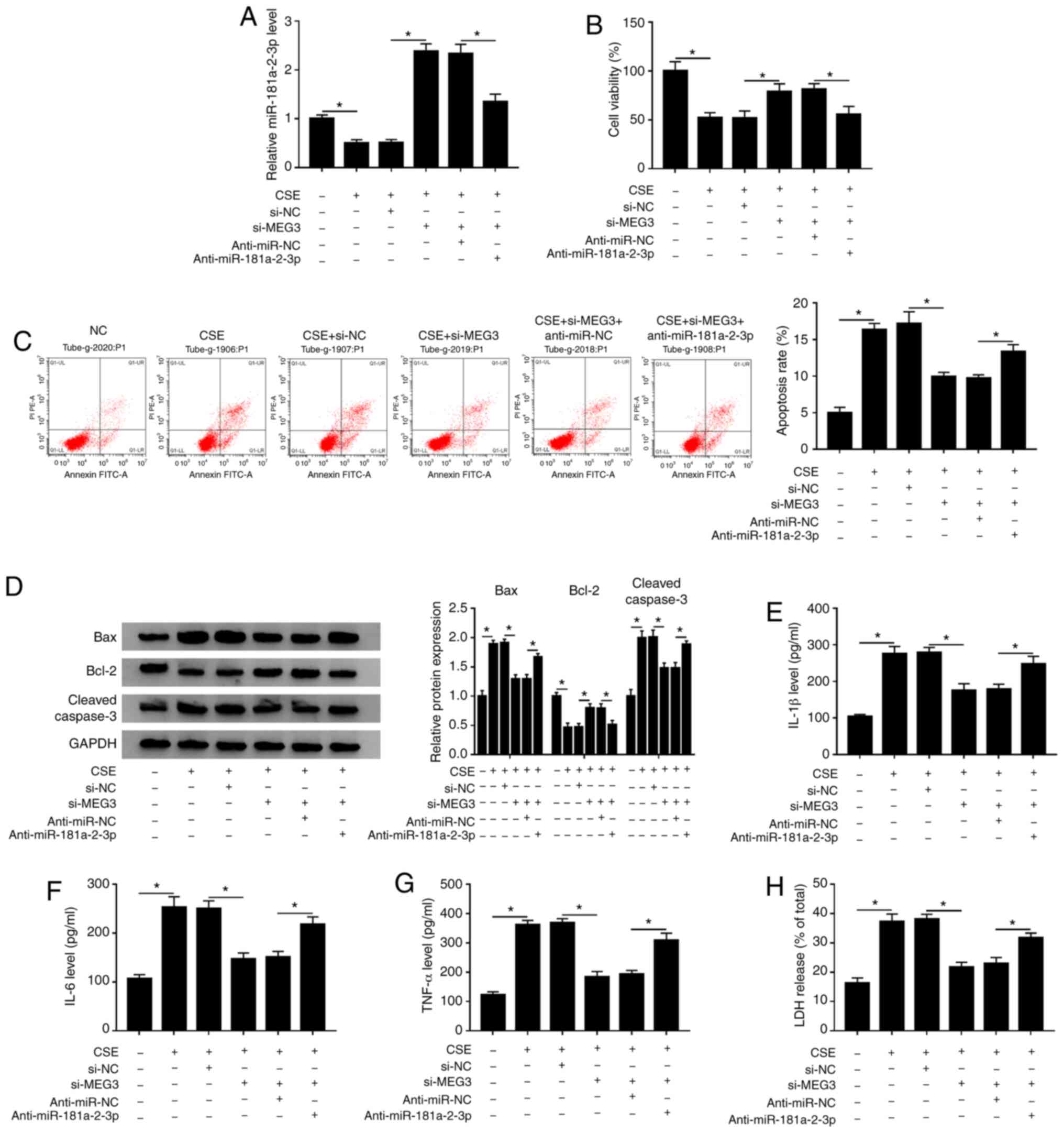 | Figure 6.miR-181a-2-3p downregulation reverses
the suppressive effects of MEG3-knockdown on apoptosis,
inflammation and cytotoxicity in CSE-treated 16HBE cells. 16HBE
cells were transfected with si-NC, si-MEG3, si-MEG3 + anti-miR-NC,
or si-MEG3 + anti-miR-181a-2-3p before treatment of CSE. (A)
Relative miR-181a-2-3p abundance was analyzed using RT-qPCR. (B)
Cell Counting Kit-8 analysis was applied to assess the cell
viability. (C) Flow cytometry analysis was performed to measure the
apoptotic rate. (D) Western blotting was conducted to examine the
protein levels of Bax, Bcl-2 and cleaved caspase-3. Levels of (E)
IL-1β, (F) IL-6 and (G) TNF-α were analyzed using ELISA kits. (H)
LDH release assay was used to evaluate cytotoxicity. *P<0.05 vs.
respective control. MEG3, maternally expressed gene 3; CSE,
cigarette smoke extract; NC, negative control; si, small
interfering; RT-q, reverse transcription-quantitative; COPD,
chronic obstructive pulmonary disease. |
Discussion
COPD is a progressive lung disease that is primarily
caused by cigarette smoke-induced chronic inflammation (26). Increasing evidence has suggested that
lncRNAs are commonly dysregulated in a range of diseases and serve
as critical regulators of pathological processes, such as
differentiation, immunity and inflammation (27,28).
Therefore, research on lncRNA may help improve the diagnosis and
treatment of COPD.
As a relatively well-studied lncRNA, MEG3 has been
confirmed to widely participate in the progression of multiple
human diseases. For instance, Lu et al (29) demonstrated that MEG3 represses
non-small cell lung cancer cell proliferation and promotes
apoptosis through increased p53 expression. Qiu et al
(30) highlighted that
MEG3-downregulation aggravated retinal vessel dysfunction in
vivo through activating PI3K/Akt signaling pathway. Besides,
several studies have demonstrated that the expression of MEG3 is
enhanced in tissues of patients with COPD and might participate in
the development of the disease (13,31,32).
Consistent with these results, the present study reported that MEG3
expression was increased in serum of patients with COPD and
CSE-stimulated 16HBE cells. Furthermore, it was observed that
si-MEG3 reversed CSE-induced apoptosis, inflammation and
cytotoxicity. These results combined with previous studies
demonstrated that MEG3 serves a role in the progression of
COPD.
Several studies have suggested that lncRNAs exert
their functions via interacting with miRNA (33,34). For
example, lncRNA X-inactive specific transcript promotes glioma
tumorigenicity and angiogenesis through serving as a molecular
sponge of miR-29 (35). lncRNA
metallothionein 1J, pseudogene functions as a ceRNA to regulate
F-box/WD repeat-containing protein 7 in gastric cancer by
competitively binding to miR-92a-3p (36). MEG3 is also involved in a variety of
diseases by serving as a sponge for miRNA. For example,
MEG3-knockdown inhibits osteosarcoma cell viability, migration and
invasion and promotes apoptosis through sponging miR-127 (37). Moreover, MEG3 suppresses the
proliferation of chronic myeloid leukemia cells by acting as a
sponge of miRNA-21 (38).
Understanding the exact molecular mechanism underlying the
biological effects of lncRNAs may contribute to the development of
lncRNA-directed diagnosis and therapy for COPD. Using DIANA tools,
the present study observed that MEG3 contained binding sites for
miR-181a-2-3p. Subsequently, the prediction was confirmed using
luciferase reporter and RIP assays. miR-181a-2-3p, a member of
miR-181 family, has is abnormally expressed in numerous diseases,
such as cervical cancer (39), child
acute lymphoblastic leukemia (40)
and gastric cancer (41). Some
studies have shown that miR-181a serves as a novel marker for the
inflammatory response (42,43). Besides, Kim et al (19) showed that miR-181a-2-3p expression is
decreased in lung tissues and serum of patients with COPD, and its
knockdown promotes inflammatory responses in cadmium-treated
bronchial epithelial cells. The present study reported that
miR-181a-2-3p abundance was decreased in the serum of patients with
COPD and CSE-stimulated 16HBE cells. Some recent studies have shown
that hemolysis, which occurs during blood collection or sample
processing, can have significant impact on the levels of certain
miRNAs in plasma and serum (44–46).
However, hemolysis miRNA controls were not considered in the plasma
analysis. In future studies, hemolysis miRNA controls (miR-23a and
−451a) should be included in the plasma analysis. The function
experiments demonstrated that overexpression of miR-181a-2-3p
blocked the pro-apoptosis, pro-inflammation and pro-cytotoxicity
effects induced by CSE in 16HBE cells, suggesting that
miR-181a-2-3p might relieve these CSE-induced effects in COPD.
Rescue experiments were performed to determine whether the function
of MEG3 was regulated by miR-181a-2-3p. As expected, miR-181a-2-3p
interference reversed the inhibitory effects of MEG3-knockdown on
apoptosis, inflammation and cytotoxicity in CSE-stimulated 16HBE
cells. As miRNAs exert their biological functions through
modulating their downstream targets (47), future research should continue to
investigate the downstream targets of miR-181a-2-3p to understand
the underlying mechanism of its function in COPD.
In conclusion, the present study demonstrated that
MEG3-knockdown inhibited CSE-induced apoptosis, inflammation and
cytotoxicity in 16HBE cells by upregulating miR-181a-2-3p. The
study reported a novel molecular mechanism of COPD progression and
might help us to improve our understanding the pathogenesis of
COPD. Furthermore, understanding this mechanism might accelerate
the development of lncRNA-targeted diagnostic and therapeutic
agents for COPD.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
SF and YR conceived and designed the study. SF, WZ
and HZ performed the experiments. CW performed the statistical
analysis and interpreted the data. YR drafted the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by The Ethics Committee of
Changning County Hospital of Traditional Chinese Medicine (Yibin,
China). Written informed consent was obtained from all
participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pauwels RA and Rabe KF: Burden and
clinical features of chronic obstructive pulmonary disease (COPD).
Lancet. 364:613–620. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nussbaumer-Ochsner Y and Rabe KF: Systemic
manifestations of COPD. Chest. 139:165–173. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tanni SE, Pelegrino NR, Angeleli AY,
Correa C and Godoy I: Smoking and tumor necrosis factor-alpha
mediated systemic inflammation in COPD patients. J Inflamm (Lond).
7:292010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rusznak C, Mills PR, Devalia JL, Sapsford
RJ, Davies RJ and Lozewicz S: Effect of cigarette smoke on the
permeability and IL-1 β and sICAM-1 release from cultured human
bronchial epithelial cells of never-smokers, smokers, and patients
with chronic obstructive pulmonary disease. Am J Respir Cell Mol
Biol. 23:530–536. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu H, Yang S, Wu X, Zhao J, Zhao J, Ning
Q, Xu Y and Xie J: Interleukin-33/ST2 signaling promotes production
of interleukin-6 and interleukin-8 in systemic inflammation in
cigarette smoke-induced chronic obstructive pulmonary disease mice.
Biochem Biophys Res Commun. 450:110–116. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wilusz JE, Hongjae S and Spector DL: Long
noncoding RNAs: Functional surprises from the RNA world. Genes Dev.
23:1494–1504. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wapinski O and Chang HY: Long noncoding
RNAs and human disease. Trends Cell Biol. 21:354–361. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lalevée S and Feil R: Long noncoding RNAs
in human disease: Emerging mechanisms and therapeutic strategies.
Epigenomics. 7:877–879. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gu W, Yuan Y, Wang L, Yang H, Li S, Tang Z
and Li Q: Long non-coding RNA TUG1 promotes airway remodelling by
suppressing the miR-145-5p/DUSP6 axis in cigarette smoke-induced
COPD. J Cell Mol Med. 23:7200–7209. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li N, Liu Y and Cai J: LncRNA MIR155HG
regulates M1/M2 macrophage polarization in chronic obstructive
pulmonary disease. Biomed Pharmacother. 117:1090152019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li J, Bian EB, He XJ, Ma CC, Zong G, Wang
HL and Zhao B: Epigenetic repression of long non-coding RNA MEG3
mediated by DNMT1 represses the p53 pathway in gliomas. Int J
Oncol. 48:723–733. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Peng W, Si S, Zhang Q, Li C, Zhao F, Wang
F, Yu J and Ma R: Long non-coding RNA MEG3 functions as a competing
endogenous RNA to regulate gastric cancer progression. J Exp Clin
Cancer Res. 34:792015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song B, Ye L, Wu S and Jing Z: Long
non-coding RNA MEG3 regulates CSE-induced apoptosis and
inflammation via regulating miR-218 in 16HBE cells. Biochem Biophys
Res Commun. 521:368–374. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta Stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kumar MS, Armenteros-Monterroso E, East P,
Chakravorty P, Matthews N, Winslow MM and Downward J: HMGA2
functions as a competing endogenous RNA to promote lung cancer
progression. Nature. 505:212–217. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ardekani AM and Naeini MM: The role of
microRNAs in human diseases. Avicenna J Med Biotechnol. 2:161–179.
2010.PubMed/NCBI
|
|
17
|
Hobbs BD and Tantisira KG: MicroRNAs in
COPD: Small molecules with big potential. Eur Respir J.
53:19005152019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Diao X, Zhou J, Wang S and Ma X:
Upregulation of miR-132 contributes to the pathophysiology of COPD
via targeting SOCS5. Exp Mol Pathol. 105:285–292. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim J, Kim DY, Heo HR, Choi SS, Hong SH
and Kim WJ: Role of miRNA-181a-2-3p in cadmium-induced inflammatory
responses of human bronchial epithelial cells. J Thorac Dis.
11:3055–3069. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Richter A, O'Donnell RA, Powell RM,
Sanders MW, Holgate ST, Djukanovic R and Davies DE: Autocrine
ligands for the epidermal growth factor receptor mediate
interleukin-8 release from bronchial epithelial cells in response
to cigarette smoke. Am J Respir Cell Mol Biol. 27:85–90. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Danpure C: Lactate dehydrogenase and cell
injury. Cell Biochem Funct. 2:144–148. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Edlich F: BCL-2 proteins and apoptosis:
Recent insights and unknowns. Biochem Biophys Res Commun.
500:26–34. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Porter AG and Jänicke RU: Emerging roles
of caspase-3 in apoptosis. Cell Death Differ. 6:99–104. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jin B, Jin H, Wu HB, Xu JJ and Li B: Long
non-coding RNA SNHG15 promotes CDK14 expression via miR-486 to
accelerate non-small cell lung cancer cells progression and
metastasis. J Cell Physiol. 233:7164–7172. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rabe KF, Hurd S, Anzueto A, Barnes PJ and
Zielinski J: Global strategy for the diagnosis, management, and
prevention of chronic obstructive pulmonary disease: GOLD executive
summary. Am J Respir Crit Care Med. 176:532–555. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Manel E: Non-coding RNAs in human disease.
Nat Rev Genet. 12:861–874. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yan B, Wang ZH, Liu JY, Tao ZF, Li XM and
Qin J: Long noncoding RNAs: Versatile players in biologcial
processes and human disorders. Epigenomics. 6:375–379. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lu KH, Li W, Liu XH, Sun M, Zhang ML, Wu
WQ, Xie WP and Hou YY: Long non-coding RNA MEG3 inhibits NSCLC
cells proliferation and induces apoptosis by affecting p53
expression. BMC Cancer. 13:4612013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qiu GZ, Tian W, Fu HT, Li CP and Liu B:
Long noncoding RNA-MEG3 is involved in diabetes mellitus-related
microvascular dysfunction. Biochem Biophys Res Commun. 471:135–141.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tang W, Shen Z, Guo J and Sun S: Screening
of long non-coding RNA and TUG1 inhibits proliferation with TGF-β
induction in patients with COPD. Int J Chron Obstruct Pulmon Dis.
11:2951–2964. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li X, Zheng M, Pu J, Zhou Y, Hong W, Fu X,
Peng Y, Zhou W, Pan H, Li B and Ran P: Identification of abnormally
expressed lncRNAs induced by PM2.5 in human bronchial epithelial
cells. Biosci Rep. 38:BSR201715772018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Militello G, Weirick T, John D, Döring C,
Dimmeler S and Uchida S: Screening and validation of lncRNAs and
circRNAs as miRNA sponges. Brief Bioinform. 18:780–788.
2017.PubMed/NCBI
|
|
34
|
Momen-Heravi F and Bala S: Emerging role
of non-coding RNA in oral cancer. Cell Signal. 42:134–143. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cheng Z, Li Z, Ma K, Li X, Tian N, Duan J,
Xiao X and Wang Y: Long non-coding RNA XIST promotes glioma
tumorigenicity and angiogenesis by acting as a molecular sponge of
miR-429. J Cancer. 8:4106–4116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang G, Li S, Lu J, Ge Y, Wang Q, Ma G,
Zhao Q, Wu D, Gong W, Du M, et al: LncRNA MT1JP functions as a
ceRNA in regulating FBXW7 through competitively binding to
miR-92a-3p in gastric cancer. Mol Cancer. 17:872018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang Y and Kong D: Knockdown of lncRNA
MEG3 inhibits viability, migration, and invasion and promotes
apoptosis by sponging miR-127 in osteosarcoma cell. J Cell Biochem.
119:669–679. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li Z, Yang L, Liu X, Nie Z and Luo J: Long
noncoding RNA MEG3 inhibits proliferation of chronic myeloid
leukemia cells by sponging microRNA21. Biomed Pharmacother.
104:181–192. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ravindresh C: let-7i-5p, miR-181a-2-3p and
EGF/PI3K/SOX2 axis coordinate to maintain cancer stem cell
population in cervical cancer. Sci Rep. 8:78402018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nabhan M, Louka ML, Khairy E, Tash F,
Ali-Labib R and El-Habashy S: MicroRNA-181a and its target Smad 7
as potential biomarkers for tracking child acute lymphoblastic
leukemia. Gene. 628:253–258. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gorur A, Balci Fidanci S, Dogruer Unal N,
Ayaz L, Akbayir S, Yildirim Yaroglu H, Dirlik M, Serin MS and Tamer
L: Determination of plasma microRNA for early detection of gastric
cancer. Mol Biol Rep. 40:2091–2096. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xie W, Li Z, Li M, Xu N and Zhang Y:
miR-181a and inflammation: miRNA homeostasis response to
inflammatory stimuli in vivo. Biochem Biophys Res Commun.
430:647–652. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Weidong X, Mengnan L, Naihan X, Qing L,
Nunu H, Jie H, Yaou Z and Tobias E: miR-181a regulates inflammation
responses in monocytes and macrophages. PLoS One. 8:e586392013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kirschner MB, Edelman JJ, Kao SC, Vallely
MP, van Zandwijk N and Reid G: The impact of hemolysis on cell-free
microRNA biomarkers. Front Genet. 4:942013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Myklebust MP, Rosenlund B, Gjengstø P,
Bercea BS, Karlsdottir Á, Brydøy M and Dahl O: Quantitative PCR
measurement of miR-371a-3p and miR-372-p is influenced by
hemolysis. Front Genet. 10:4632019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kirschner MB, Kao SC, Edelman JJ,
Armstrong NJ, Vallely MP, van Zandwijk N and Reid G: Haemolysis
during sample preparation alters microRNA content of plasma. PLoS
One. 6:e241452011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 13:215–233. 2009. View Article : Google Scholar
|