Introduction
Inflammatory myofibroblastic tumor (IMT) is a rare
type of mesenchymal tumor with malignant potential that is
frequently observed in patients who are <16 years old, and is
rarely observed in adults (1). The
World Health Organization (WHO) 2020 definition of IMT suggests
that it is a distinctive, rarely metastasizing neoplasm composed of
myofibroblastic and fibroblastic spindle cells accompanied by an
inflammatory infiltrate of plasma cells, lymphocytes and/or
eosinophils (2). IMT exhibits a
predilection for the visceral soft tissues, most commonly observed
in the lungs, but can develop in any part of the body, including
the peritoneum, colon, liver, bladder, breast or nasal cavity
(3). IMT arising from somatic soft
tissues or extremities has been reported in isolated case reports
(4,5). IMTs are genetically heterogeneous
(2). Clonal rearrangement of the
anaplastic lymphoma kinase (ALK) gene on the short arm of
chromosome 2 at 2p23 with fusion of the 3 kinase region of the ALK
gene with various partners, including tropomyosin 3, tropomyosin 4,
clathrin heavy chain and RAN binding protein 2 (RANBP2), has been
identified in 50–70% of IMTs (2,3). Due to
the low incidence, particularly in adults, and lack of specificity
in clinical and imaging manifestations, IMTs are often misdiagnosed
(6). Epithelial inflammatory
myofibroblast sarcoma (EIMS), a rare malignant variant of IMT,
requires aggressive clinical measures, since patients with EIMS
often exhibit rapid local recurrence, and the disease is associated
with a high mortality rate (6). At
present, only isolated case reports of EIMS exist due to its rarity
(7–10).
The present study retrospectively analyzed the
clinical features, treatment and prognosis of 17 cases of
pathologically diagnosed IMT in patients ≥16 years old who were
diagnosed between 2010 and 2020, in order to assist clinicians in
improving the recognition and diagnosis of the disease.
Materials and methods
Study population
IMT tends to occur primarily in patients <16
years old, but rarely in those patients ≥16 years old (1). Therefore, a list of all IMT cases with
an age ≥16 years confirmed by pathology at The First Affiliated
Hospital of Nanjing Medical University (Nanjing, China) was
compiled. It was not possible to select younger patients, since the
Obstetrics, Gynecology and Paediatrics Departments are located at
the Women and Children's Branch Hospital (Nanjing, China).
Data were collected from 17 patients with IMT (age,
≥16 years) diagnosed between July 2010 and February 2020. The
diagnosis was confirmed by pathological analysis following surgery,
respiratory interventional techniques or needle biopsy. All
pathological sections were reassessed and the diagnosis was
confirmed by two or three pathologists. The present retrospective
analysis was approved by the Institutional Review Board of the
First Affiliated Hospital of Nanjing Medical University and written
informed consent was provided by subjects (adult patients) or
next-of-kin (patients <18 years old) for inclusion.
Thoracic or abdominal CT or MRI scans were performed
based on the location of the primary tumor in all patients and a
number of patients underwent a positron emission tomography
(PET-CT) scan or a form of endoscopy, including bronchoscopy,
gastroenterological endoscopy or cystoscopy, prior to surgery. The
present study retrospectively collected the main demographic
characteristics and clinical data, including sex, age, presenting
symptoms, blood examinations, location, size of tumors,
radiological features, endoscopic characteristics and surgical
methods, from the medical records. The patients were followed up
every 6 months until February 2020 through outpatient visits and/or
telephone contact.
Pathological review
The present study reviewed all pathological sections
to ensure they matched the criteria of the 5th edition of the WHO
classification of tumors of soft tissue and bone (2). Pathologists evaluated the pathological
sections for cell morphology, nuclear atypia, vascular invasion and
inflammatory components. Immunohistochemistry was performed on a
fully automated VENTANA Benchmark XT Stainer (Ventana Medical
Systems, Inc.; Roche Diagnostics). The specimens were fixed in 10%
neutral formaldehyde solution at room temperature for 24 h and
embedded into paraffin. The slides (3-µm-thick) were heated at 65°C
for 2 h. Deparaffinization, rehydration and antigen retrieval were
automatically completed. All the procedures were performed
according to the manufacturers protocol. The slides were incubated
overnight at 4°C with pre-diluted (ready-to-use) primary antibodies
(Fuzhou Maixin Biotech Co., Ltd.; Table
I) and then incubated with goat anti-mouse/rabbit IgG
HRP-conjugated polymer secondary antibodies (cat. no. KIT-5230;
Fuzhou Maixin Biotech Co., Ltd.) for 20–30 min at room temperature.
DAB+ chromogen (Fuzhou Maixin Biotech Co., Ltd.) was used to
produce dark brown color. Hematoxylin was used as a counter stain
for 5 min at room temperature. Sections were observed under a light
microscope (magnification, ×20). Multitumor tissue blocks
(including tissues of thyroid, lung, small intestine, liver,
tonsil, lymph nodes, prostate, fetal membranes, leiomyoma and lung
adenocarcinoma) were used as the positive and negative controls for
each immunohistochemical stain analyzed.
 | Table I.Primary antibodies used for
immunohistochemistry purchased from Fuzhou Maixin Biotech Co.,
Ltd. |
Table I.
Primary antibodies used for
immunohistochemistry purchased from Fuzhou Maixin Biotech Co.,
Ltd.
| Antibody | Catalogue.
number | Poly/monoclonal clone
number |
|---|
| ALK | MAB-0281 | 5A4 |
| SMA | Kit-0006 | 1A4 |
| Vim | MAB-0735 | MX034 |
| Des | MAB-0766 | MX046 |
| Ki-67 | MAB-0672 | MX006 |
| CKpan | Kit-0009 | AE1/AE3 |
| S-100 | Kit-0007 | 4C4.9 |
| CD21 | MAB-0339 | 2G9 |
| CD30 | MAB-0023 | Ber-H2 |
| CD117 | kit-0029 | YR145 |
| CD163 | MAB-0206 | 10D6 |
| CD68 | Kit-0026 | KP1 |
| HMB-45 | MAB-0098 | HMB45 |
| DOG1 | Kit-0035 | SP31 |
Immunohistochemical staining was performed on all
samples to confirm the pathological phenotype of IMT, and to
differentiate among diseases, such as inflammatory pseudotumours
(IPTs), sarcoma, cancer, gastrointestinal stromal tumor, dendritic
cell neoplasms or vascular tumors.
Fluorescence in situ hybridization
(FISH)
Dual color break-apart commercial probes of the ALK
kit (Abbott Pharmaceutical Co., Ltd.; cat. no. 06N38) were used for
FISH assays. Tissues were fixed in 10% neutral formaldehyde
solution for 24 h at room temperature, embedded into paraffin and
cut into 3-µm-thick slides. Subsequently, the slides were
deparaffinized in xylene for two times (15 min each), and
dehydrated in 95% ethanol (two times, 5 min each), 80% ethanol (2
min), 70% ethanol (2 min) and double-distilled H2O (2
min). The slides were then treated with protease K [0.1 mg/ml in 2X
SSC buffer (0.3 M NaCl, 0.03 M sodium citrate, pH 7.2)] for 10 min
at 37°C. The slides were washed with 2X SSC buffer for two times (5
min), post-fixed with 100% ethanol for 2 min and air-dried at room
temperature. For hybridization, 10 µl probe dissolved in
hybridization buffer (10% dextran sulfate, 2X SSC buffer, 50%
deionized formamide) was added to the tissues on slides, which were
placed in a Hybrite system (Abbott Pharmaceutical Co., Ltd.) for 5
min at 83°C and then 42°C for 4 h. After hybridization, the slides
were washed with 0.3% NP-40/0.4X SSC buffer for two times (2 min)
at room temperature to remove excess probes. Subsequently, the
slides were washed with 70% ethanol for 3 min and air-dried at room
temperature. Finally, 15 µl DAPI was added to slides for 5 min at
room temperature to stain the nucleus and mounted. The slides were
observed using a fluorescence microscope (DP70; Olympus
Corporation). For ALK rearrangement, the nuclei contained
broken-apart signals, and for no ALK rearrangement, the specimens
contained a set of immediately adjacent or fused orange/green
signals.
Statistical analysis
All statistical analyses were performed using the
software SPSS 25.0 (IBM Corp.). The data were presented as the mean
± SD. A Fishers exact test was used to analyze the data. P<0.05
was considered to indicate a statistically significant
difference.
Results
Patient characteristics
A total of 17 participants, including 12 male and 5
female patients, with a mean ± SD age of 34.76±13.79 years (range,
16–56 years) were included in the present study. The most common
locations of occurrence were the bronchus and lungs (9 cases,
52.94%), one of which was a patient with multiple sites of
involvement, including the mediastinum. The other sites of
involvement included colon and bladder (2 cases each; 11.76%), and
omentum majus, mesocolon, stomach and peritoneum (1 case each;
5.88%). Four cases had a history of smoking or alcoholism, and
another 4 cases had a history of trauma or surgery (data not
shown).
A total of 5 cases (29.41%) were discovered by
physical examination, regional pain in the lesions accounted for 4
cases (23.53%), and hemoptysis, melena or hematuria were the main
symptoms in 4 cases (23.53%), followed by tumor masses and fever (2
cases each; 11.76%). The time interval between symptom onset and
diagnosis was between 1 week and 8 years (data not shown). A
16-year-old patient with greater omentum IMT presented with fever,
anemia and thrombocytosis before surgery; however, the symptoms
gradually disappeared after surgery. The patient with EIMS (case
13) presented with 1 week of abdominal distention, and 3 days of
oliguria and diarrhea. The abdominal CT scan revealed abdominal and
pelvic effusion, a thickened peritoneum, and an unclear structure
in the mid-abdomen. Blood examination revealed that the white blood
cell and platelet counts were elevated.
Radiological features before
treatment
The most common radiological manifestations were
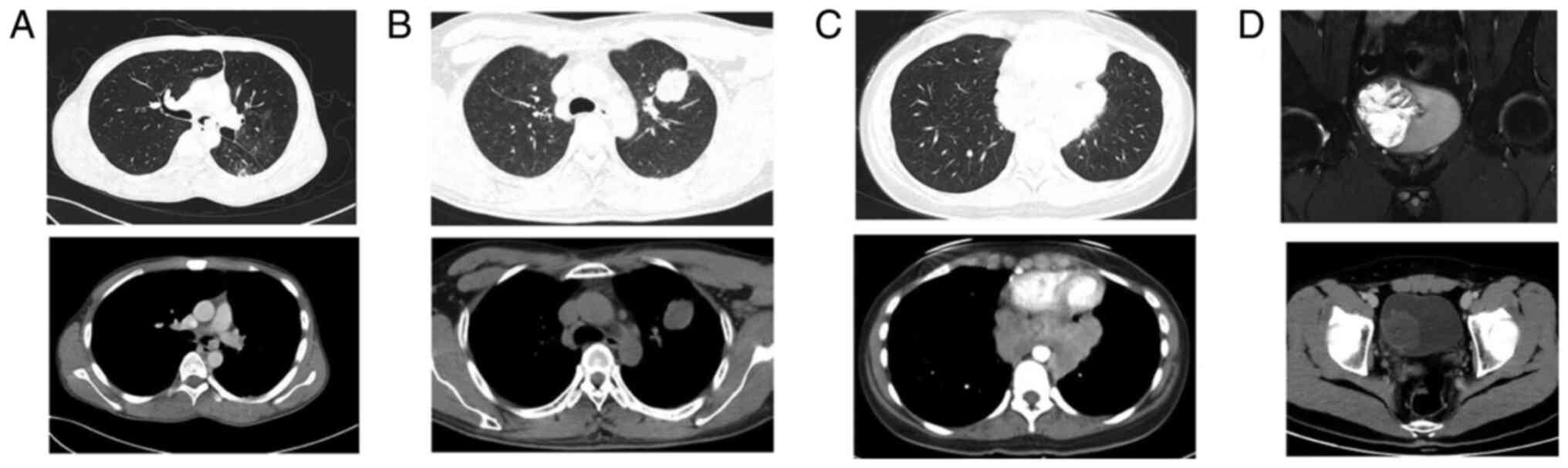
masses of different sizes (Fig. 1).
The CT or MRI diagnoses before treatment were as follows: 10
patients were diagnosed with malignant tumors, 2 with lymphoma, 3
cases were considered benign lesions, including 2 cases of
granulomatous disease and 1 pulmonary sclerosing hemangioma. In
addition, 2 cases of peritoneal effusion were separately considered
to be stromal tumor and tuberculous peritonitis. Additionally, 3
patients who underwent PET-CT all exhibited
18F-fluorodeoxyglucose uptake, with a diagnosis of
malignant tumor with lymph node metastasis in 1 case and
granulomatous disease in 2 cases (data not shown).
Treatment and follow-up
Among the 17 patients, 1 patient was diagnosed with
EIMS by needle biopsy under the guidance of ultrasound and survived
only 3 weeks from symptom onset, and thus did not undergo surgery.
The remaining 16 patients underwent tumor resection by surgery or
respiratory interventional techniques. Furthermore, 2 cases
relapsed, and both of these patients underwent surgery again, and 1
of these patients was administered crizotinib following
reoperation, while the other received local interventional therapy
following recurrence due to negative ALK expression.
All patients were followed up until February 2020
through outpatient visits and/or phone calls to patients themselves
or family members. One patient was lost to follow-up, 1 patient
died, 2 local recurrence patients (case 9 and 16) were confirmed to
be alive, and the remaining 13 patients were cured. The follow-up
time of each patient was >1 year. The average follow-up time was
48.93 months (range, 18–114 months).
The clinical characteristics, primary locations,
tumor sizes, treatment and prognosis of the 17 patients are
summarized in Table II.
| Table II.Clinical characteristics of the 17
patients. |
Table II.
Clinical characteristics of the 17
patients.
| Case | Sex | Age at onset,
years | Symptoms | Location | Maximum size of
tumor, cm | Treatment | Follow-up,
months | Prognosis |
|---|
| 1. | Male | 45 | Physical
examination | LUL | 6.5 | Lobectomy | 47 | Cure |
| 2. | Male | 17 | Cough, sputum,
fever | LMB | 1.5 | Interventional
therapy by bronchoscope | 23 | Cure |
| 3 | Female | 29 | Chest pain | RUL | 2.0 | Wedge resection | 48 | Cure |
| 4 | Female | 33 | Physical
examination | RUL | 0.7 | Wedge resection | 31 | Cure |
| 5 | Male | 50 | Hemoptysis | RUL | 2.5 | Segmentectomy | 18 | Cure |
| 6 | Female | 53 | Chest pain | RML | 3.0 | Lobectomy | 90 | Cure |
| 7 | Male | 46 | Physical
examination | RML | 1.8 | Lobectomy | 28 | Cure |
| 8 | Male | 43 | Physical
examination | RML | 3.8 | Lobectomy | 26 | Cure |
| 9 | Female | 18 | Physical
examination Difficulty in swallowing | Mediastina
Mediastina, LLL, esophagus | 5.0 Mediastina,
4.5; LLL, 6.0; cardia, 3.5 | Mediastinal mass
resection Mediastinal and esophageal lesion resection and wedge
resection | 67 | Relapsed 15 months
after surgery, reoperation, relapsed again, local interventional
therapy |
| 10 | Female | 16 | Fever, anemia,
thrombocytosis | Omentum majus | 11.0 | Tumorectomy | 0.3 | Lost to
follow-up |
| 11 | Male | 22 | Abdominal mass | Mesocolon | 7.0 | Tumorectomy | 114 | Cure |
| 12 | Male | 23 | Abdominal mass | Colon
sigmoideum | 15.0 | Exploratory
laparotomy | 86 | Cure |
| 13 | Male | 29 | Abdominal
distension, diarrhea, oliguria | Peritoneum | Large mass | Tumor puncture | 0 | Death |
| 14 | Male | 56 | Intermittent
abdominal pain | Ileocecal
colon | 4.0 | Right
hemicolectomy | 47 | Cure |
| 15 | Male | 47 | Intermittent
hematuria | Bladder | 2.0 | Radical cystectomy
and ileum orthotopic neobladders | 20 | Cure |
| 16 | Male | 21 | Gross
hematuria | Bladder | 6.0 | Partial
cystectomy | 19 | Relapsed 2 months
after |
|
|
|
|
| Bladder | 1.5 | Transurethral
resection of bladder tumor |
| surgery,
reoperation, then administration of crizotinib |
| 17 | Male | 43 | Dull pain in upper
abdomen, black stool | Antrum | 4.5 | Distal subtotal
gastrectomy | 70 | Cure |
Clinical characteristics by age group
and location of the lesion
In total, 9 patients were <40 years old and had a
mean age of onset of 23.11 years. The patients who exhibited
recurrence and the patient with EIMS were all in this group. The
lesion locations were the lungs/mediastinum (4 cases), omentum
majus/mesocolon/peritoneum (3 cases), and colon and bladder (1 case
each). The other 8 patients were >40 years old, the mean age of
onset was 47.88 years, all patients underwent surgery and were
cured, and the lesion locations were the lungs (5 cases), and
stomach, bladder and colon (1 case each; Table III).
| Table III.Clinical characteristics based on age
and intra/extra-thoracic location. |
Table III.
Clinical characteristics based on age
and intra/extra-thoracic location.
|
| Age | Lesion
location |
|---|
|
|
|
|
|---|
| Clinical
characteristics | ≤40 years, n | >40 years,
n | P-value | Intra-thoracic,
n | Extra-thoracic,
n | P-value |
|---|
| Sex |
|
| 0.29 |
|
| 0.29 |
|
Male | 5 | 7 |
| 5 | 7 |
|
|
Female | 4 | 1 |
| 4 | 1 |
|
| Lesion
location |
|
| 0.64 |
|
|
|
|
Intra-thoracic | 4 | 5 |
|
|
|
|
|
Extra-thoracic | 5 | 3 |
|
|
|
|
| Neoplasm
invasiveness |
|
| 0.64 |
|
| 0.35 |
| No | 4 | 5 |
| 6 | 3 |
|
|
Yes | 5 | 3 |
| 3 | 5 |
|
| Initial
treatment |
|
| >0.99 |
|
| 0.06 |
| MIS or
endoscopy | 5 | 6 |
| 8 | 3 |
|
| Routine
surgery | 3 | 2 |
| 1 | 4 |
|
|
Non-surgery | 1 | 0 |
| 0 | 1 |
|
| Lymph node
dissection |
|
| <0.01 |
|
| >0.99 |
|
Yes | 1 | 8 |
| 5 | 4 |
|
| No | 7 | 0 |
| 4 | 3 |
|
| NA | 1 | 0 |
| 0 | 1 |
|
| Relapse |
|
| 0.08 |
|
| 0.44 |
|
Yes | 2 | 0 |
| 1 | 1 |
|
| No | 5 | 8 |
| 8 | 5 |
|
| NA | 2 | 0 |
| 0 | 2 |
|
In total, 9 patients exhibited a primary tumor
located in the lungs/mediastinum (52.94%), and the remaining 8
patients exhibited tumors located in the abdomen. Cases of IMT in
the oral and maxillofacial region, breast or nervous system were
not observed in the present study.
Pathological characteristics
The resected tumor sizes ranged between 0.7 and 15
cm in the largest dimension, with a mean size of 4.59 cm. Gross
examination revealed that the tumors exhibited soft to firm
variegated appearance with grey white areas, and parts of the mass
presenting a polypoid-like appearance protruding into the bronchus,
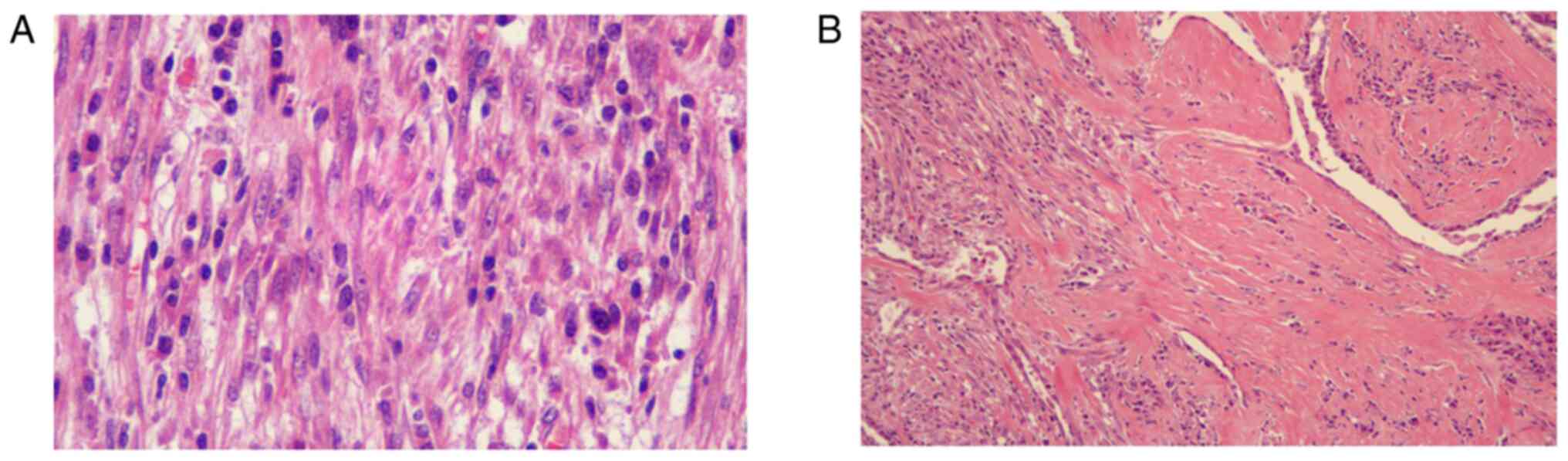
bladder or colon (data not shown). Microscopically, the plump
spindle myofibroblasts and fibroblast-like tumor cells were
arranged loosely or densely with a fascicular or storiform
architecture in a loose edematous or collagenous stroma, with a
prominent presence of acute and chronic inflammatory cell
infiltration, consisting primarily of plasma cells and lymphocytes
(Fig. 2). According to
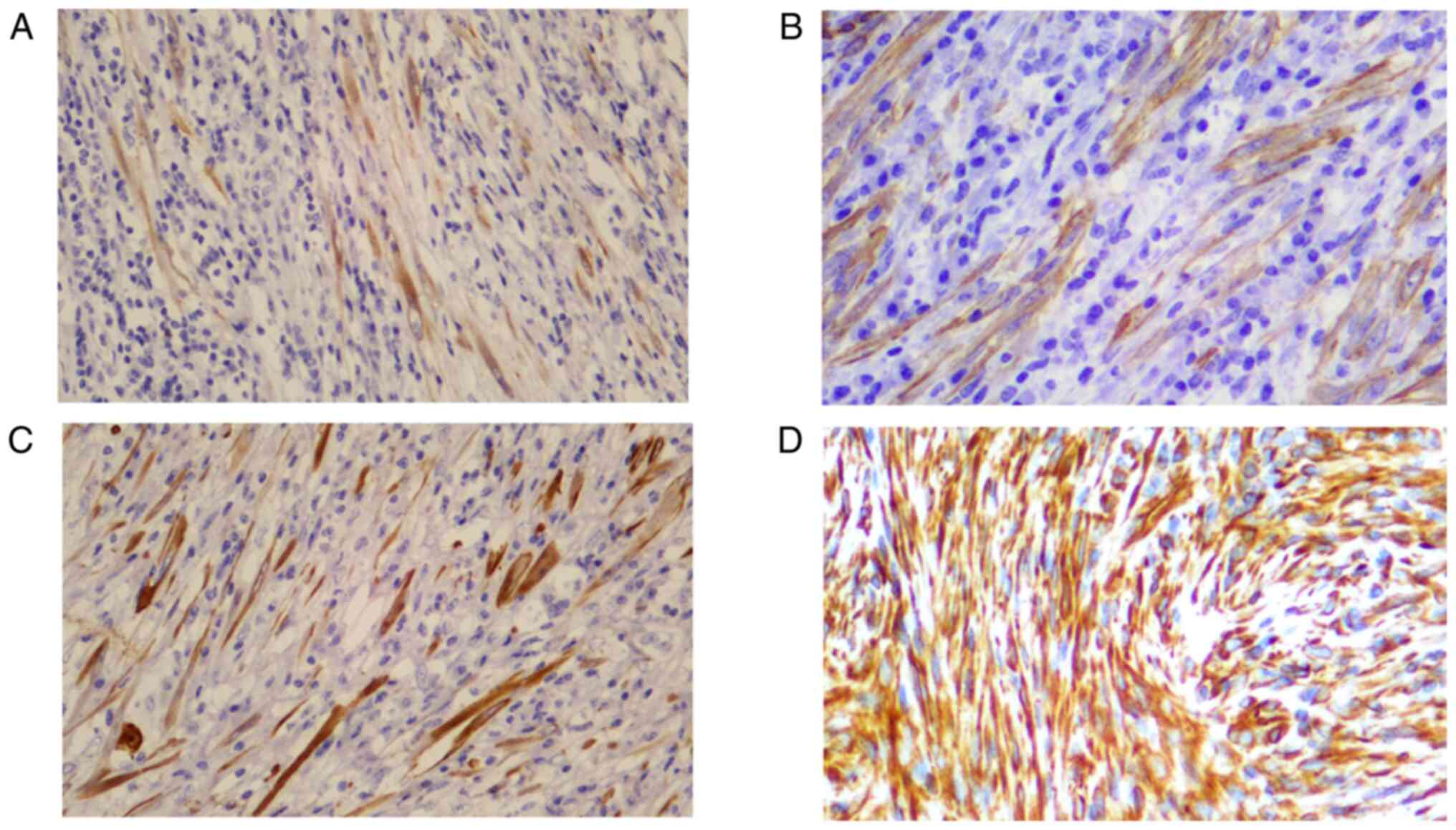
immunohistochemistry (Table IV),
the positive rate of ALKp80 expression (Fig. 3A) was the highest (13/17), followed
by SMA (12/17; Fig. 3B), CKp (6/17;
Fig. 3C), Vim (5/17; Fig. 3D) and Des (4/17). Identification of
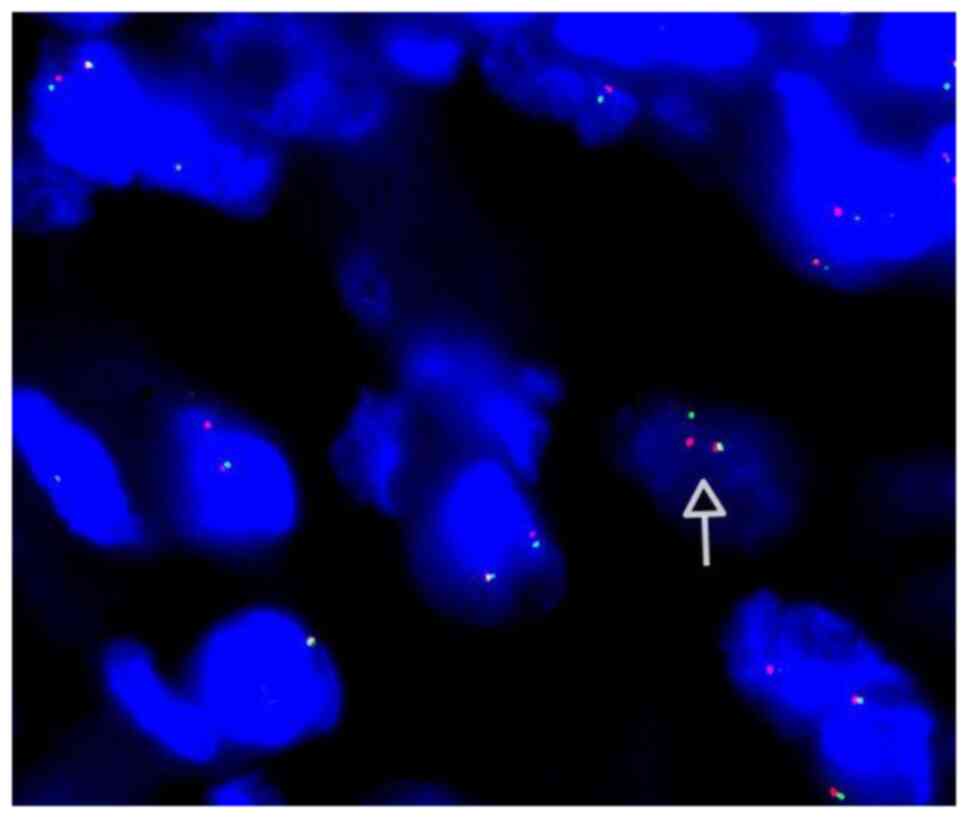
ALK gene rearrangement by FISH was performed in 5 patients, and
only 2 demonstrated evidence of ALK rearrangement (Fig. 4). In the patient with EIMS, the Ki-67
index was 10%, and in the two relapsed patients, the Ki-67 index
was 5%+ and 3% (data not shown); only in 2 patients, the Ki-67
index was 10%+ (Table IV).
| Table IV.Pathological characteristics based on
age and intra/extra-thoracic location |
Table IV.
Pathological characteristics based on
age and intra/extra-thoracic location
|
| Age | Lesion
location |
|---|
|
|
|
|
|---|
| Pathological
characteristics | ≤40 years, n | >40 years,
n | P-value | Intra-thoracic,
n | Extra-thoracic,
n | P-value |
|---|
| Maximum tumor
diameter, cm |
|
| 0.13 |
|
| 0.05 |
| ≤5 | 4 | 7 |
| 8 | 3 |
|
|
>5 | 5 | 1 |
| 1 | 5 |
|
| ALK rearrangements
by immunohistochemistry |
|
| >0.99 |
|
| 0.58 |
|
Positive | 7 | 6 |
| 6 | 7 |
|
|
Negative | 2 | 2 |
| 3 | 1 |
|
| SMA staining |
|
| 0.13 |
|
| >0.99 |
|
Positive | 8 | 4 |
| 6 | 6 |
|
|
Negative | 1 | 4 |
| 3 | 2 |
|
| CKp staining |
|
| >0.99 |
|
| <0.01 |
|
Positive | 3 | 3 |
| 0 | 6 |
|
|
Negative | 6 | 5 |
| 9 | 2 |
|
| Vim staining |
|
| >0.99 |
|
| 0.13 |
|
Positive | 3 | 2 |
| 1 | 4 |
|
|
Negative | 6 | 6 |
| 8 | 4 |
|
| Des staining |
|
| >0.99 |
|
| 0.29 |
|
Positive | 2 | 2 |
| 1 | 3 |
|
|
Negative | 7 | 6 |
| 8 | 5 |
|
| Ki-67 index |
|
| 0.21 |
|
| >0.99 |
|
≤10% | 9 | 6 |
| 8 | 7 |
|
|
>10% | 0 | 2 |
| 1 | 1 |
|
Discussion
IMTs are rare mesenchymal neoplasms which exhibit
low to intermediate malignant potential, and are composed of
spindle shaped myofibroblasts often accompanied by the presence of
inflammatory cells (3,11). IMTs are referred to by several terms,
including IPT, histiocytoma, fibrous histiocytoma, xanthoma,
xanthofibroma, xantogranuloma and plasma cell granuloma (1). IPT has been the most widely used term,
and IMT has previously been considered to be a type of IPT. At
present, some clinicians still use the terms ITP and IMT
interchangeably. IPT is an inflammatory reactive mass, which is
currently considered to be an IgG4-related disease (12). Although they exhibit certain
similarities with regards to morphology, IMT and IPT are different
diseases. The therapeutic principles are also different. IMT is
treated using surgical excision, whereas IPT can be managed more
conservatively (3). Generally, the
proliferation and nuclear atypia of spindle cells in IMT are more
marked, and ALK-1 staining is positive in some patients (13). Lymphoplasmacytic infiltrate,
storiform-fibrosis and obliterative phlebitis (14) are more prominent in IgG4-related IPT,
whereas ALK expression is negative (12,13).
Next generation sequencing-based RNA fusion assays have been
suggested to be a primary method, which may be used to provide a
more accurate diagnosis than immunohistochemical platforms and
FISH-based assays of ALK (14).
IMT affects patients of a broad age range, but
generally presents in patients <16 years old (1). The most common anatomical location of
IMT is the lung; however, theoretically, any site may be involved,
including the mesentery, retroperitoneum, mediastinum, somatic soft
tissues, larynx, uterus, bone and central nervous system (3). In the present study, the primary
locations were the lungs and the intra-abdominal regions, 1 case
presented with a tumor in the mediastinum, and cases in other
organs were not detected.
To the best of our knowledge, the etiology and
pathogenesis of IMT is unknown. Trauma, surgery, inflammation and
infection may contribute to the development of IMT (15). Epstein-Barr virus (16) and human herpesvirus-8 viral infection
(3) are also considered to be
associated with IMT; however, additional evidence is required, and
a causal relationship has not been established. In the present
study, 4 patients had a history of trauma or surgery, but the time
span between trauma or surgery and IMT onset was 2–10 years, and
the location of trauma or surgery was not the same as that of IMT.
Additionally, 4 patients had a history of smoking, but the IMT
locations were not all in bronchus or lung. However, in the patient
with a history of alcoholism, the lesion was located in the gastric
antrum. Therefore, whether these were associated with the
occurrence of IMT was unclear. In addition, 2 patients were
assessed for Epstein-Barr encoded RNA and both were negative,
suggesting that IMT was not associated with Epstein-Barr virus
infection.
Patients usually present with a mass or nonspecific
symptoms, which vary according to the location, size and growth
pattern of the tumor; additionally, the radiological features are
also non-specific (3). In the
present study, the diagnoses based on imaging prior to surgery were
mostly cancer, stromal tumor or lymphoma. Out of 6 patients, some
underwent gastroenterological endoscopy, while others received
bronchoscopy or cystoscopy prior to surgery; however, all had
negative results. After surgery, immunohistochemistry analysis was
required to confirm the pathological diagnosis. A 16-year-old
patient with IMT presented with fever, anemia and thrombocytosis
before surgery, the large tumor (11×8×8 cm in the greater omentum)
required an increased nutrient supply, which resulted in anemia,
while the inflammatory stimulation of the tumor caused fever and
thrombocytosis. All these parameters gradually returned to normal
following surgery. The histological characteristics of IMT include
variable spindle cell proliferation in a myxoid to collagenous
stroma, with prominent inflammatory infiltrate composed primarily
of plasma cells and lymphocytes, with the occasional presence of
neutrophils and eosinophils (3).
Immunohistochemistry is performed primarily to confirm the
immunophenotype of myofibroblasts and to exclude other diseases.
ALK rearrangement is relatively common in IMT, while it is negative
in leiomyosarcoma, sarcomatoid carcinoma, embryonal
rhabdomyosarcoma or reactive myofibroblastic proliferations
(17). The present study revealed a
76% positive rate of ALK staining, in line with previous literature
showing that ALK-1 positive staining is observed in 60–89% of cases
(18). ALK-positive patients appear
to have a favorable prognosis (11,15). For
patients with ALK-positive IMT with unresectable tumors or with
advanced stage IMT, ALK inhibition is an effective therapy
(19). Watanabe et al
(20) reported on a patient with
ALK-negative primary pulmonary IMT who achieved remission when
treated with macrolide drugs-clarithromycin. The Ki-67 index in the
patient with EIMS was 10%, and in the two patients with relapse it
was 5%+ and 3%, respectively, which was not higher than that of the
other patients. A high Ki-67 index may be a risk factor for tumor
progression, but was not associated with relapse.
EIMS is a rare variant of IMT, which is clinically
distinct from conventional cases in that it follows an aggressive
clinical course and has a high mortality rate (6). EIMS predominantly originates
intraabdominally, with most cases arising in the omentum and
mesentery, and adult males appear to be more susceptible to EIMS
(7). There are some common features
observed in patients with EIMS, including round-to-epithelioid
tumor cells, abundant myxoid stroma with inflammatory infiltrates
composed primarily of neutrophils, immuno-positivity for nuclear
membrane ALK staining, RANBP2-ALK gene fusion and Des positive
staining (7,8). In IMTs, inflammatory infiltrates are
composed primarily of plasma cells and lymphocytes (3), immuno-positive staining for ALK
expression is predominantly observed in the cytoplasm and membrane
(9), and SMA expression is more
commonly observed than Des (3). Lee
et al (9) reported a novel
ribosome binding protein 1 (RRBP1)-ALK fusion gene in EIMS, and, to
the best of our knowledge, RRBP1-ALK and RANBP2-ALK are the only
recurrent oncogenic mechanisms identified to date in EIMS. Although
the prognosis of patients with EIMS is poor, there are still a few
cases of successful remission with a sustained response to the ALK
inhibitor crizotinib (10).
Furthermore, programmed cell death 1 ligand 1 expression has been
detected in patients with EIMS (7).
Therefore, whether immune checkpoint blockade may serve as a novel
therapeutic approach for treatment of EIMS should be studied
further. In the present study, one patient was diagnosed with
peritoneum EIMS, although ALK fusion gene detection was positive,
the patient still succumbed to the disease, as the disease was
present at an advanced stage at symptom onset and progressed
rapidly thereafter.
During the collection of clinical data and
rechecking of the pathological data, it was identified that IMTs
were occasionally over-diagnosed. Under most circumstances,
myofibroblast hyperplasia was diagnosed as IMT. Therefore, one
should be wary of diagnosing a middle-aged or elderly patient with
IMT. The present study was limited by the small number of cases,
and ALK gene rearrangement assessment by FISH was only performed in
some patients. A lack of younger cases was another limitation of
the present study. Due to this selection bias, the positive rate of
ALK-positive staining may be different from that in patients of all
ages with IMT. Additional clinical data are required to identify
prognostic features which could be used to guide treatment and
predict the outcome.
In conclusion, IMT is a low to intermediate-grade
tumor which exhibits low potential for malignancy, and has a
tendency for local recurrence and a small risk of distant
metastasis. The clinical manifestations and imaging findings are
usually non-specific. Once the histopathological diagnosis is
confirmed, radical surgery is preferred and routine follow-up is
necessary.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors contributions
WS conceived the study, collected and analysed the
patient data, and was a major contributor in writing the
manuscript. YZ designed the study and performed the histological
and immunohistochemistry examination. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
The present retrospective analysis was approved by
the Institutional Review Board of the First Affiliated Hospital of
Nanjing Medical University and written informed consent was
provided by the subjects (adult patients)/next-of-kin (patients
<18 years old) for inclusion.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Oztuna F, Pehlivanlar M, Abul Y, Tekinbas
C, Ozoran Y and Ozlu T: Adult inflammatory myofibroblastic tumor of
the trachea: Case report and literature review. Respir Care.
58:e72–e76. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yamamoto H: WHO Classification of Tumours
Editorial Board. Fibroblastic/myofibroblastic Tumors. WHO
Classification of Tumors of Soft Tissue and Bone. 5th edition.
International Agency for Research on Cancer; Lyon, France: pp.
109–112. 2020
|
|
3
|
Gleason BC and Hornick JL: Inflammatory
myofibroblastic tumours: Where are we now? J Clin Pathol.
61:428–437. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ding R, Li X, Zhu XM, Song QX, Fan QH,
Zhang ZH and Gong QX: Inflammatory myofibroblastic tumor arising
from soft tissues of extremities harboring a novel CLIP2-ALK
fusion. Pathol Int. 70:798–803. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu H, Lin J, Yang P, Shen H and Yang H:
Whether inflammatory myofibroblastic tumor of the thigh relapses
after surgical excision? Int J Clin Exp Med. 8:11584–11588.
2015.PubMed/NCBI
|
|
6
|
Mariño-Enríquez A, Wang WL, Roy A,
Lopez-Terrada D, Lazar AJ, Fletcher CD, Coffin CM and Hornick JL:
Epithelioid inflammatory myofibroblastic sarcoma: An aggressive
intra-abdominal variant of inflammatory myofibroblastic tumor with
nuclear membrane or perinuclear ALK. Am J Surg Pathol. 35:135–144.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Du X, Gao Y, Zhao H, Li B, Xue W and Wang
D: Clinicopathological analysis of epithelioid inflammatory
myofibroblastic sarcoma. Oncol Lett. 15:9317–9326. 2018.PubMed/NCBI
|
|
8
|
Sarmiento DE, Clevenger JA, Masters GA,
Bauer TL and Nam BT: Epithelioid inflammatory myofibroblastic
sarcoma: A case report. J Thorac Dis. 7:E513–E516. 2015.PubMed/NCBI
|
|
9
|
Lee JC, Li CF, Huang HY, Zhu MJ,
Mariño-Enríquez A, Lee CT, Ou WB, Hornick JL and Fletcher JA: ALK
oncoproteins in atypical inflammatory myofibroblastic tumours:
Novel RRBP1-ALK fusions in epithelioid inflammatory myofibroblastic
sarcoma. J Pathol. 241:316–323. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu Q, Kan Y, Zhao Y, He H and Kong L:
Epithelioid inflammatory myofibroblastic sarcoma treated with ALK
inhibitor: A case report and review of literature. Int J Clin Exp
Pathol. 8:15328–15332. 2015.PubMed/NCBI
|
|
11
|
Shukla PS and Mittal K: Inflammatory
myofibroblastic tumor in female genital tract. Arch Pathol Lab Med.
143:122–129. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chougule A, Bal A, Das A, Agarwal R, Singh
N and Rao KL: A comparative study of inflammatory myofibroblastic
tumors and tumefactive IgG4-related inflammatory lesions: The
relevance of IgG4 plasma cells. Appl Immunohistochem Mol Morphol.
24:721–728. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhu L, Li J, Liu C, Ding W, Lin F, Guo C
and Liu L: Pulmonary inflammatory myofibroblastic tumor versus
IgG4-related inflammatory pseudotumor: Differential diagnosis based
on a case series. J Thorac Dis. 9:598–609. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Taylor MS, Chougule A, MacLeay AR, Kurzawa
P, Chebib I, Le L and Deshpande V: Morphologic overlap between
inflammatory myofibroblastic tumor and IgG4-related disease:
Lessons from next-generation sequencing. Am J Surg Pathol.
43:314–324. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pecoraro Y, Diso D, Anile M, Russo E,
Patella M and Venuta F: Primary inflammatory myofibroblastic tumor
of the trachea. Respirol Case Rep. 2:147–149. 2014.PubMed/NCBI
|
|
16
|
Wang S, Chen L, Cao Z, Mao X, Zhang L and
Wang B: Inflammatory myofibroblastic tumor of the lumbar spinal
canal: A Case Report With Literature Review. Medicine (Baltimore).
96:e64882017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sukov WR, Cheville JC, Carlson AW, Shearer
BM, Piatigorsky EJ, Grogg KL, Sebo TJ, Sinnwell JP and Ketterling
RP: Utility of ALK-1 protein expression and ALK rearrangements in
distinguishing inflammatory myofibroblastic tumor from malignant
spindle cell lesions of the urinary bladder. Mod Pathol.
20:592–603. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cook JR, Dehner LP, Collins MH, Ma Z,
Morris SW, Coffin CM and Hill DA: Anaplastic lymphoma kinase (ALK)
expression in the inflammatory myofibroblastic tumor: A comparative
immunohistochemical study. Am J Surg Pathol. 25:1364–1371. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mossé YP, Voss SD, Lim MS, Rolland D,
Minard CG, Fox E, Adamson P, Wilner K, Blaney SM and Weigel BJ:
Targeting ALK with crizotinib in pediatric anaplastic large cell
lymphoma and inflammatory myofibroblastic tumor: A children's
oncology group study. J Clin Oncol. 35:3215–3221. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Watanabe H, Uruma T, Tazaki G, Tajiri T,
Kikuchi R, Itoh M, Aoshiba K and Nakamura H: Remission of
ALK-negative primary pulmonary inflammatory myofibroblastic tumor
on treatment with clarithromycin: A case report and review of the
literature. Oncol Lett. 11:1757–1761. 2016. View Article : Google Scholar : PubMed/NCBI
|