Introduction
APRIN (also known as AS3 or PDS5B) is a
cohesin-associated protein and is involved in the regulation of
crucial cellular responses, such as chromatid cohesion, homologous
recombination, DNA repair and genomic integrity (1,2).
APRIN-deficient mice die shortly after birth and exhibit congenital
anomalies such as heart defects, short limbs and fusion of the
ribs, which underscores the essential function of the protein
(3).
Moreover, APRIN has been investigated as a putative
tumor suppressor. APRIN was initially studied as an
androgen-induced proliferative shutoff protein that inhibits the
proliferation of prostate cells that are androgen-dependent
(4,5). APRIN gene is located on chromosome 13,
where loss of heterozygosity is commonly detected in tumors
(6). Allelic imbalance of the
intragenic APRIN microsatellite repeat marker, D13S171, is
associated with invasive ductal breast carcinoma (7), lung carcinoma (8), prostate cancer (9) and esophageal carcinoma (10), suggesting APRIN as a putative tumor
suppressor.
While anomalies in APRIN gene expression lead to
increased cell proliferation, unfavorable diagnosis, and metastases
in various cancer types (6), there
is limited knowledge on the cellular mechanism of APRIN in these
cellular responses. Of particular note are the reports of decreased
expression of APRIN in tumors (2,11–13). Low
APRIN expression has been reported in tissue samples of breast
tumor and is associated with high histological grade estrogen
receptor-negative disease (2,11).
Furthermore, low expression levels of APRIN were observed in
gastric and colorectal cancer, as well as in pancreatic cancer
(12,13).
Investigation of APRIN in cellular responses
revealed distinct molecular mechanisms. The overexpression of APRIN
in pancreatic cancer cells resulted in the inhibition of cell
proliferation and invasion, whereas its downregulation led to
enhanced proliferation and cell motility via attenuation of Ptch2
expression; suggesting that the APRIN/Ptch2 axis regulates the
cellular responses of pancreatic cancer (13). APRIN associates with BRCA2 and
modulates DNA damage responses as well as homologous recombination
with implication in chemotherapy (2).
The present study investigated whether cancer cells
might employ their unique cellular regulators to exert cellular
responses upon variation in APRIN expression. The present findings
demonstrate that APRIN downregulation enhances cancer cell
proliferation via a novel IL-6/STAT3/cyclin D axis.
Materials and methods
Cell lines and treatments
A lung cancer cell line NCI-H460, an osteosarcoma
cell line U2OS and a prostate cancer cell line LNCaP were obtained
from American Type Culture Collection. Cell lines that stably
downregulate APRIN were generated by transducing the cell lines
with lentiviral particles (with 5×105 infectious units
of virus) that contain either control or APRIN shRNA (Santa Cruz
Biotechnology, Inc.; cat. no. SC-108080 or SC-61984-v,
respectively), as specified in the instruction manual. The viral
particles are provided as a ready-to-use product without the need
for cell packaging processes. Control shRNA lentiviral particles
encode a scrambled shRNA sequence that will not lead to the
specific degradation of any known mRNA. Briefly, 5×104
cells were incubated in a 12-well plate for 24 h and replenished
with 5 µg/ml polybrene-containing media. Cells were infected with
5×105 infectious units of virus. Viral
particle-transduced cells were selected and maintained in
puromycin-containing media. APRIN knockdown was confirmed by
western blot analysis. The whole procedure to establish the stable
cell lines took 30–45 days depending on the cell lines used. After
lentiviral particle transduction, it took 2–3 weeks to select
puromycin-resistant cells and additional 2–3 weeks to expand the
antibiotic-resistant cells for experiments. The cell lines were
very effective in establishing and maintaining APRIN
downregulation.
NCI-H460 and LNCaP cells were cultured in RPMI-1640
media, whereas U2OS cells were grown in DMEM, supplemented with 10%
fetal bovine serum (all from Welgene, Inc.), 100 U/ml penicillin
and 100 µg/ml streptomycin. Cell cultures were incubated at 37°C in
a humidified atmosphere of 5% CO2.
Cell proliferation assay
Cell proliferation was measured by MTT assay,
following the manufacturer's instruction (Thermo Fisher Scientific,
Inc.). Absorbance was measured at 570 nm by using microplate reader
Model 680 (Bio-Rad Laboratories, Inc.). In order to count the
number of cells directly, cells were seeded on 60-mm culture dish
at a density of 2×104 cells per dish, and incubated for
the indicated time period. Cells were washed with
phosphate-buffered saline (PBS), and collected following trypsin
treatment. Cells were counted by using Adam automated cell counter
(Nano-Tek).
Cell migration assay
Cell migration assay was performed by following the
manufacturer's instruction (Corning; Thermo Fisher Scientific,
Inc.), with some modification. Briefly, cells were seeded on the
upper layer of a 24-well Transwell plate (8.0-µm pore size;
Corning; Thermo Fisher Scientific, Inc.) at a density of
1×104 cells/well with serum-free media, whereas the
lower compartment was filled with RPMI-1640 culture media with 0.1%
serum. After 16 h in the cell culture incubator, cells that
migrated through the pores were visualized by staining at room
temperature for 2 h with 0.5% crystal violet solution in 20%
methanol. Stained cells were counted by microscopic observation
using an INFINITY2 light optical microscope (Lumenera Corporation)
at ×40 magnification and recorded as migrated cell population.
Wound healing assays were conducted following the
culture of cells up to 80% confluence. Cells were scratched with a
pipette tip and incubated with fresh RPMI-1640 medium supplemented
with 0.1% fetal bovine serum. Wound healing was observed under a
light optical microscope at ×10 magnification (INFINITY2; Lumenera
Corporation). Wound closure was expressed as the remaining area
uncovered by the cells. The scratched area at the 0-h time-point
was set to 1 (n=5). Wound area was analyzed with captured images
using the wound healing size tool of ImageJ v1.52S software
(National Institutes of Health).
To carry out soft agar clonogenic
assay, trypsinized cells were mixed with 1.5% (at 55°C) agar
solution medium and then incubated in a 37°C incubator for 3–4
weeks
The number of colonies that were >200 µm in
diameter were counted using a light optical microscope at ×100
magnification (INFINITY2; Lumenera Corporation).
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted with TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) by following the
manufacturer's instruction. Total RNA (100 ng) was
reverse-transcribed using Superscript II (Invitrogen; Thermo Fisher
Scientific, Inc.), according to manufacturer's instructions. The
expression of mRNA was determined in triplicate by using SYBR
master mix kit (MBioTech) with a CFX96 system (Bio-Rad
Laboratories, Inc.). The thermocycling conditions consisted of an
initial denaturation step at 95°C for 10 min, followed by 40 cycles
of annealing at 60°C for 30 sec and extension at 72°C for 15 sec.
Relative mRNA expression levels were normalized to an endogenous
control GAPDH expression in the corresponding samples. Relative
quantification of gene expression was calculated using the
2−ΔΔCq method (14) using
the CFX manager software v2.1 (Bio-Rad Laboratories, Inc.). Primers
were purchased from Sigma-Aldrich (Merck KGaA). Primers used were
as follows: Cyclin D1 forward, 5′-GAACAAACAGATCATCCGCAAACA-3′;
cyclin D1 reverse, 5′-TGCTCCTGGCAGGCCCGGAGGCAG-3′; IL-6 forward,
5′-GTAGCCGCCCCACACAGA-3′; IL-6 reverse,
5′-CATGTCTCCTTTCTCAGGGCTG-3′; GAPDH forward,
5′-ATGACATCAAGAAGGTGGTG-3′; GAPDH reverse,
5′-CATACCAGGAAATGAGCTTG-3′.
Preparation of cell extracts and
western blot analysis
Western blot analysis was performed as previously
reported (15) with some variations
in the preparation of the cell extract and the antibodies used.
Cells were lysed in lysis buffer (Invitrogen), containing a
protease inhibitor and phosphatase inhibitor cocktails
(Sigma-Aldrich; Merck KGaA), on ice for 30 min. The following
primary antibodies (all diluted 1:1,000) were used for: APRIN (cat.
no. ab70299; Abcam), STAT3 (cat. no. 9139), pSTAT3 (cat. no. 9145),
cyclin D1 (cat. no. 2978) and cyclin D3 (cat. no. 2936) (Cell
Signaling Technology, Inc.), cyclin A (cat. no. sc-751), cyclin B1
(cat. no. sc-594), cyclin E (cat. no. sc-248) and β-actin (cat. no.
sc-81178) (Santa Cruz Biotechnology, Inc.). Peroxidase-conjugated
secondary antibodies (both 1:3,000; cat. no. A90-116P for
anti-mouse; cat. no. A120-101P for anti-rabbit) were purchased from
Bethyl Laboratories, Inc. The experiment was repeated at least
three times.
Cytokine array assay and enzyme-linked
immunosorbent assay (ELISA)
Control or APRIN-knockdown cells were seeded on
60-mm dishes at a density of 1×105 cells/dish. After
incubation for 24 h, supernatants from the cell cultures were
harvested and analyzed by using Human Cytokine Array Panel A (cat.
no. ARY005B; R&D Systems, Inc.) following the manufacturer's
instructions. The levels of multiple cytokines were simultaneously
detected in a sample. The levels of secreted cytokines were
normalized by the cell numbers and the resulting image data was
analyzed by ImageJ v1.52S program (National Institutes of Health;
http://imagej.nih.gov/ij/download.html).
For ELISA assay, IL-6 ELISA kit (cat. no. D6050) was
purchased from R&D Systems, Inc. Supernatant (100 µl) from the
cell cultures was applied to the ELISA kit and processed according
to manufacturer's instructions. The secreted IL-6 level was
normalized by the cell numbers.
Immunofluorescence analysis
A total of 2×104 cells/well were seeded
and cultured on cover slips in a 12-well plate for 24 h. Cells were
fixed with 4 % paraformaldehyde for 20 min at room temperature, and
permeabilized for 2 min at room temperature with 0.1 % Triton X-100
solution. Specimens were blocked with 2% BSA for 1 h at room
temperature. Immunostaining was performed with phospho-STAT3
(pSTAT3) primary antibody (1:200; cat. no. 9145; Cell Signaling
Technology, Inc.) and with Alexa 488-labeled anti-rabbit IgG
secondary antibody (1:500; cat. no. A-11034; Invitrogen; Thermo
Fisher Scientific, Inc.). Immunofluorescence images were acquired
using Axio Imager M2 microscope (Carl Zeiss AG) at ×400
magnification.
Chromatin immunoprecipitation (ChIP)
assay
ChIP assay was performed by using EZ-ChIP kit (cat.
no. 17-371, with Taq DNA polymerase included) from EMD Millipore,
following the manufacturer's instruction. Briefly, cells were
treated with 1/10 volume of 10% formaldehyde for 10 min at 37°C to
cross-link proteins to DNA. Soluble chromatin was subjected to
immunoprecipitation with anti-STAT3 antibody (Cell Signaling
Technology; cat. no. 9139). Amplification of the cyclin D1 promoter
sequence by PCR was carried out by using the following PCR primers:
5′-CGACCAAAGAGACAGAAC-3′ and 5′-TTAACCGGGAGAAACA-3′. The PCR
products were resolved on a 1.5% agarose gel and stained with
ethidium bromide. Thermocycling conditions were as follows: an
initial denaturation step at 94°C for 3 min, followed by 32 cycles
of denaturation at 94°C for 20 sec, annealing at 59°C for 30 sec
and extension at 72°C for 30 sec, and a final extension step at
72°C for 2 min.
Statistical analysis
Data were obtained by performing three independent
experiments and were presented as mean ± SEM. Data were analyzed
using GraphPad Prism 5 software (GraphPad Software, Inc.).
Differences between two groups were analyzed using the Student's
t-test. Multiple groups were analyzes using ANOVA followed by post
hoc test, such as Bonferroni (Figs. 1B,
C, F and 4C) or Tukey's test
(Fig. 4B). P<0.05 was considered
to indicate a statistically significant difference.
Results
Stable downregulation of endogenous
APRIN expression enhances cancer cell proliferation and
migration
In order to elucidate the role of APRIN in cancer
cell proliferation and migration, cancer cell lines that stably
downregulate endogenous APRIN expression were established by
transducing APRIN shRNA lentiviral particles. The APRIN shRNA
targeted and downregulated the expression of endogenous APRIN in a
human lung cancer cell line NCI-H460 and in an osteosarcoma cell
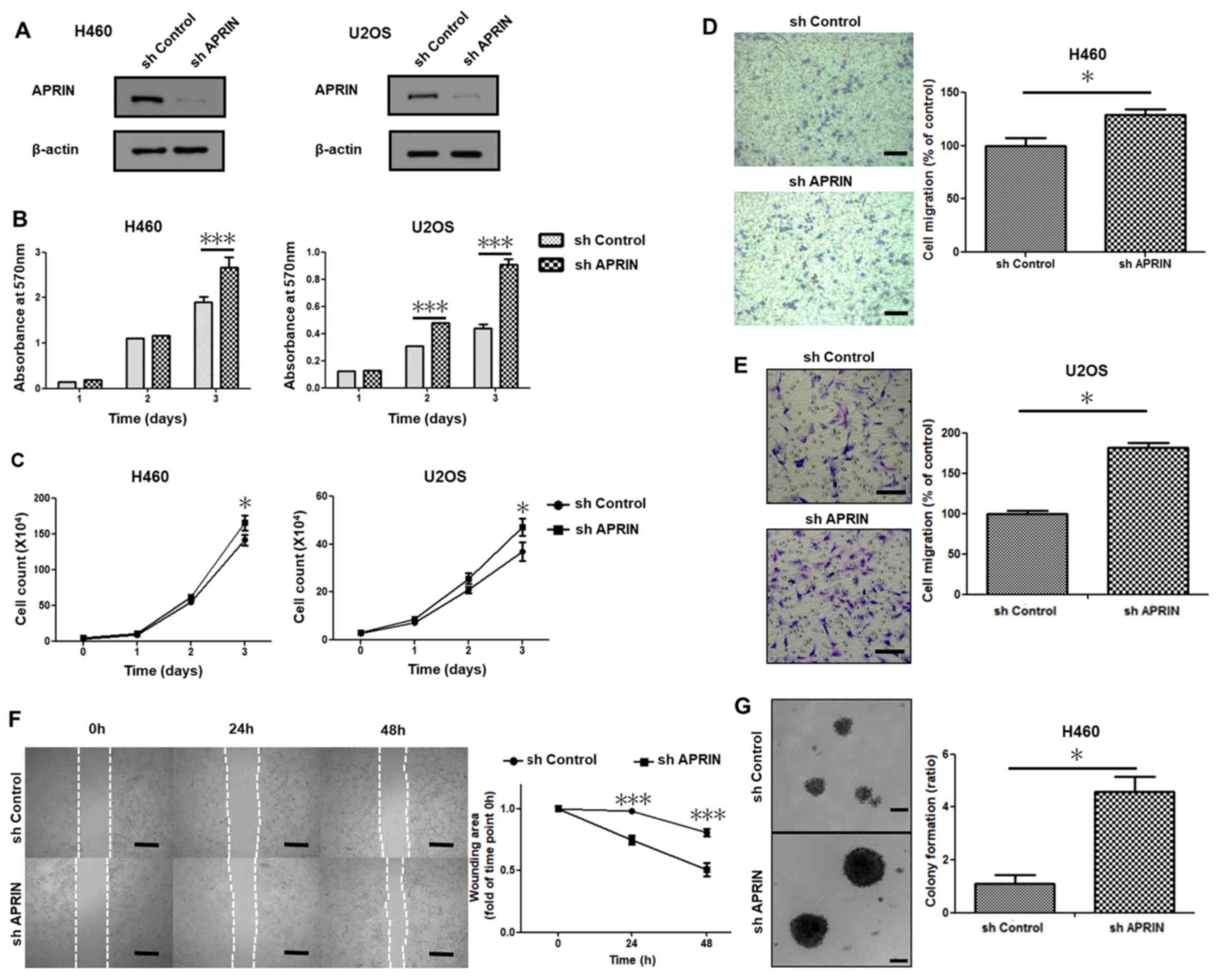
line U2OS, as shown by western blot analysis (Fig. 1A). MTT cell proliferation assay
revealed enhanced cell proliferation in APRIN-knockdown stable cell
lines compared with the control shRNA-transduced cell lines,
notably after 3 days (P<0.001; Fig.
1B). Cell count analysis also showed similar results
(P<0.05; Fig. 1C). These results
suggest that endogenous APRIN might inhibit cancer cell
proliferation.
Migration of APRIN-knockdown cell lines was
investigated in Transwell migration assay. More APRIN-knockdown
cells (both NCI H460 or U2OS) migrated through the membrane
compared with the shRNA control cells (P=0.028 for H460 cells;
Fig. 1D). APRIN-depleted U2OS cells
showed more than 50% increase in migration compared with the
control (P<0.05; Fig. 1E). The
data from wound healing assay also showed similar results
(P<0.001; Fig. 1F). In addition,
larger colonies were observed in soft agar for APRIN-depleted
cells, suggesting higher malignancy upon APRIN depletion (Fig. 1G). These results suggest that
endogenous APRIN might inhibit cancer cell malignancy, in terms of
migration and anchorage independence.
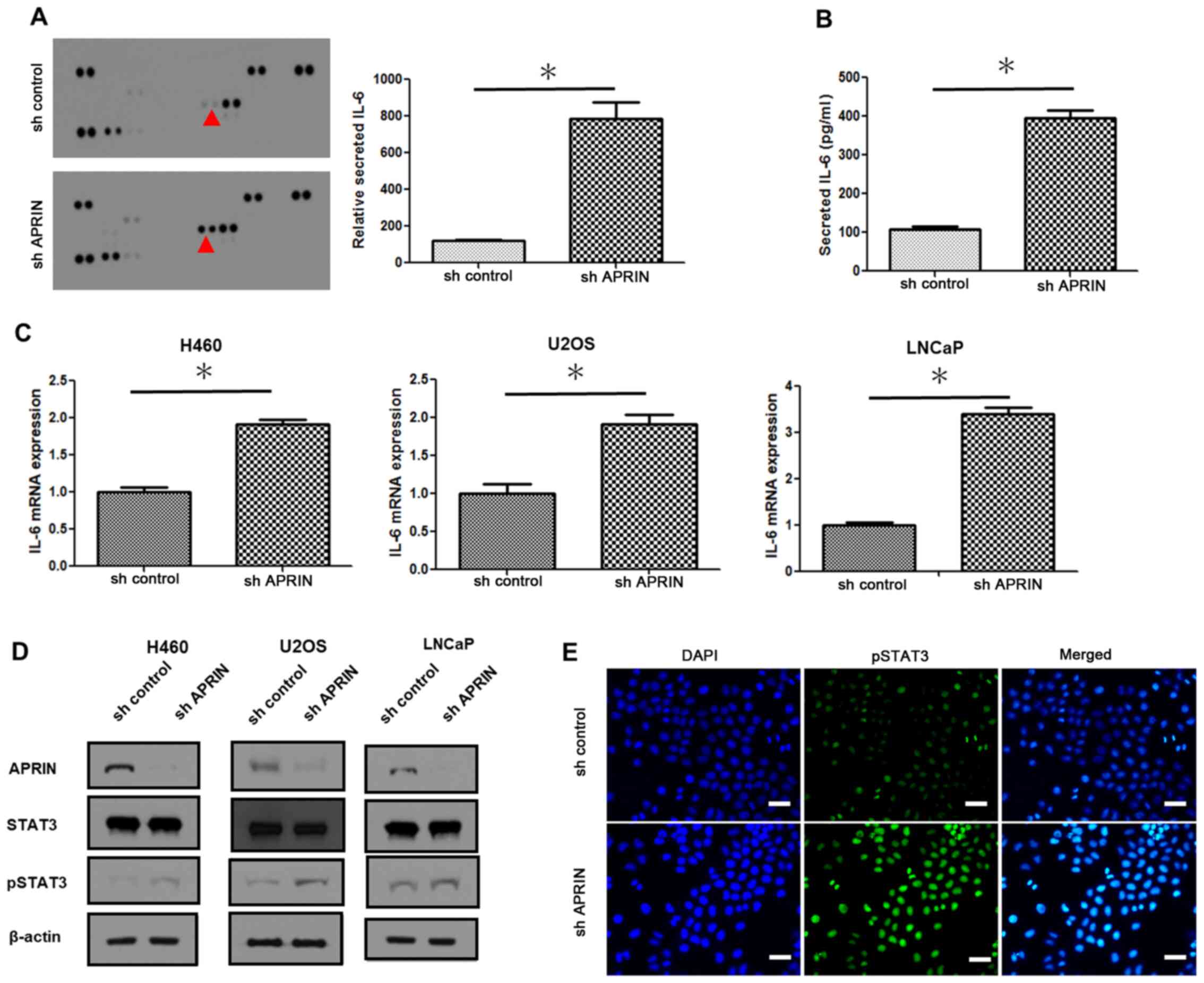
Downregulation of endogenous APRIN
increases IL-6 secretion and STAT3 activation
The mechanism underlying the effect of decreased
APRIN expression on the aforementioned cancer cell responses were
investigated. In order to identify such a mediator, cytokines that
exhibit differential expression between control and APRIN knockdown
cell lines were screened. Examination of cytokine array assay using
supernatants from the culture of NCI-H460 cells revealed a
prominent increase in IL-6 in APRIN-knockdown cell culture
(Fig. 2A). Measurement of the
intensity of the cytokine array data in Fig. 2A showed more than five-fold increase
in IL-6 in the APRIN-knockdown cell sample (P=0.0171; Fig. 2A graph). ELISA also confirmed the
data from the cytokine array; APRIN-knockdown NCI-H460 cell line
secreted significantly more IL-6 into the cell culture medium
compared with the control (P=0.005; Fig.
2B). RT-qPCR analysis of IL-6 mRNA expression showed
significant increase in APRIN-knockdown cells compared with the
control (P<0.0001 for H460; P=0.0025 for U2OS, and P<0.001
for LNCaP; Fig. 2C). APRIN knockdown
and control cells from NCI-H460, U2OS and LNCaP background showed
similar results (Fig. 2C).
Increased activation of STAT3, a downstream mediator
of IL-6 receptor signaling (16),
was observed in APRIN-knockdown cell lines; demonstrated by STAT3
phosphorylation at tyrosine 705 residue (pSTAT3) (Fig. 2D). Western blot analysis show that
APRIN knockdown in NCI-H460, U2OS and LNCaP cells exhibited
increased levels of pSTAT3 (Fig.
2D).
Immunofluorescence analysis showed increased nuclear
localization of pSTAT3 in APRIN-knockdown cells (Fig. 2E). These results suggest that
downregulation of APRIN expression in cancer cells might induce
IL-6/STAT3-mediated cell responses. On the other hand, endogenous
APRIN may also function through modulation of IL-6/STAT3 signaling
pathway.
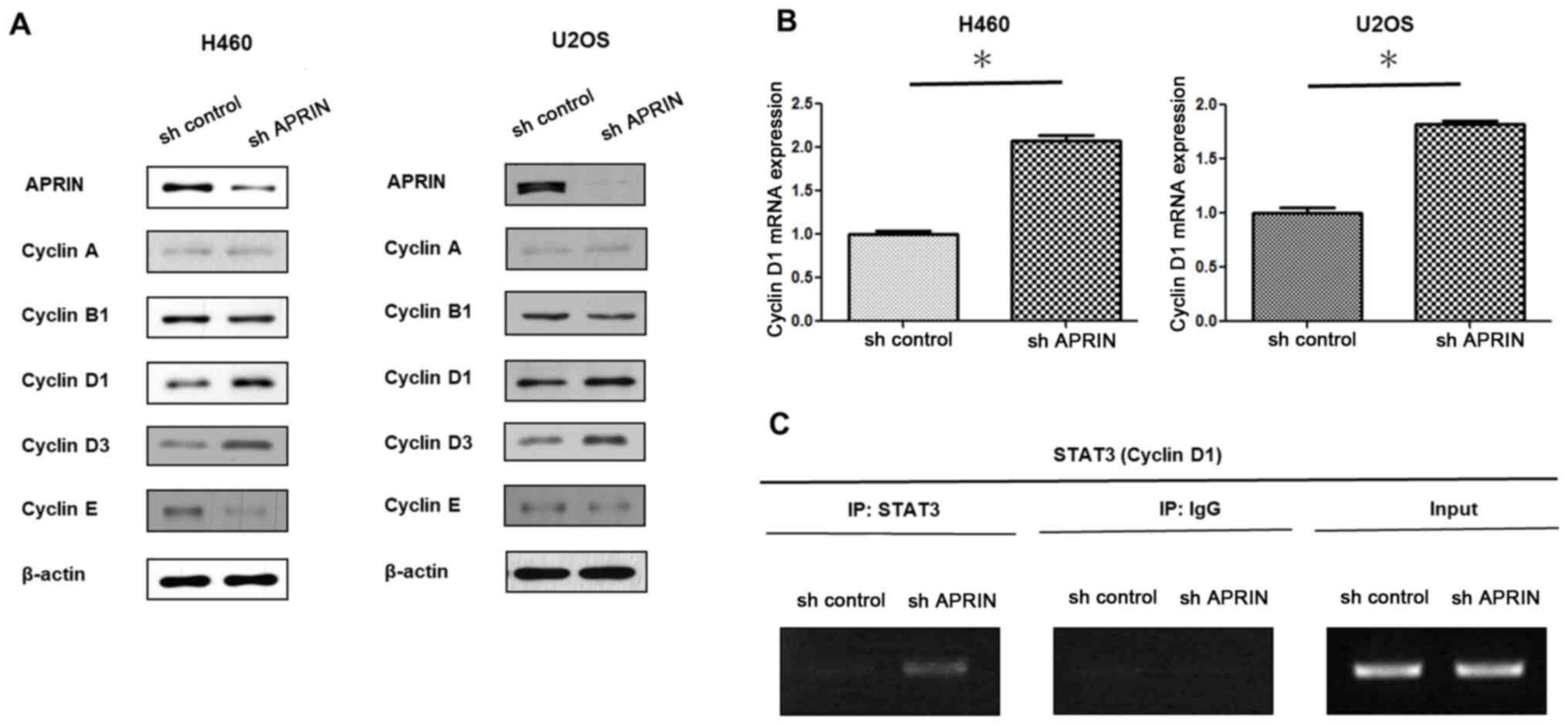
STAT3 upregulates cyclin D expression
in APRIN-knockdown cells
In order to test whether that IL-6/STAT3 signaling
regulates cellular responses such as proliferation in
APRIN-knockdown cells, the expression levels of cyclin family
proteins were determined. Western blot analysis showed notable
increase in cyclin D1 and D3 protein levels in APRIN-knockdown
cells (Fig. 3A). Both NCI-H460 lung
cancer cells and U2OS osteosarcoma cells showed similar results.
These data suggest that the APRIN-associated cell proliferation
responses might involve common regulatory components in different
cell lines. In accord with the protein expression, cyclin D1 mRNA
expression was also increased in APRIN knockdown cells as shown by
RT-qPCR results (P<0.0001 for H460 and P<0.0001 for U2OS
cells; Fig. 3B). As these results
suggest a transcriptional regulation for cyclin D1 expression,
whether STAT3 is involved in the regulation was examined. ChIP
assay showed significantly increased association of STAT3 with
cyclin D1 promoter sequence in APRIN-knockdown cells compared with
control cells (Fig. 3C). These
results suggest that STAT3 might be involved in the upregulation of
cyclin D1 in APRIN-knockdown cells. In other words, the results
suggest that endogenous APRIN might inhibit STAT3-regulated cyclin
D expression.
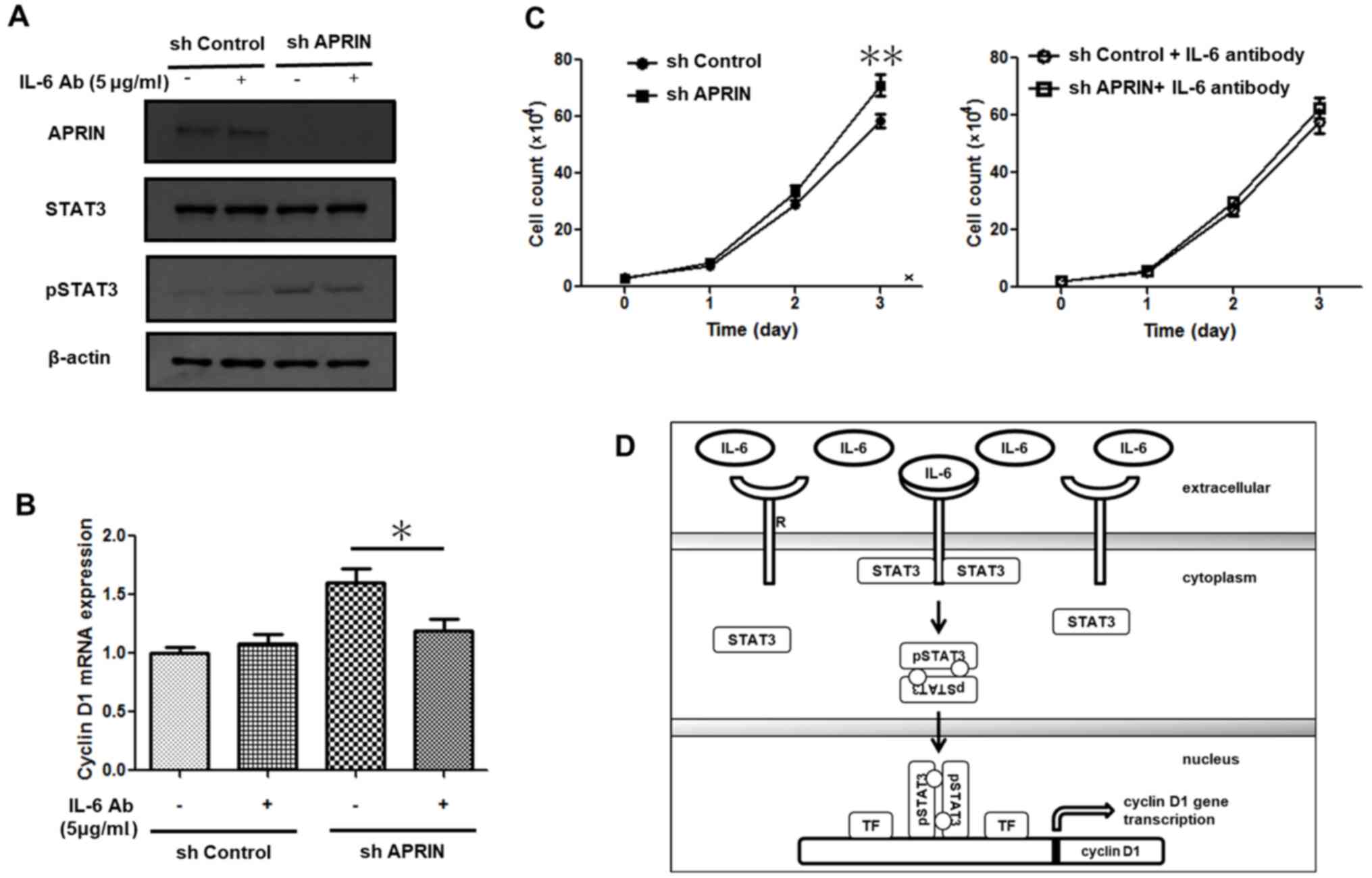
Treatment with an IL-6-neutralizing
antibody attenuates STAT3 activation, cyclin D1 mRNA expression and
proliferation in APRIN-knockdown cells
In order to demonstrate that IL-6 is responsible for
the downstream responses, such as increased STAT3 activation,
cyclin D1 expression and proliferation in APRIN-knockdown cells,
cells were treated with an IL-6-neutralizing antibody P620.
Treatment of the antibody decreased phosphorylated STAT3 (pSTAT3)
in APRIN-knockdown cells, as well as in control cells, whereas
STAT3 protein levels in the cells were constant (Fig. 4A). Western blot analysis showed a
slight increase in pSTAT3 in APRIN-knockdown cells compared with
that of control cells, despite the treatment with IL-6-neutralizing
antibody. This result may reflect that the amount of secreted IL-6
in APRIN-knockdown cell culture is significantly more than that of
control cells and that the antibody addition resulted in partial
neutralization of IL-6. These data suggest that APRIN
knockdown-induced IL-6 modulates STAT3 activation.
It was also observed that the neutralizing antibody
treatment significantly decreased the mRNA expression of cyclin D1
in APRIN-knockdown cells (P<0.05; Tukey's test; Fig. 4B). Moreover, increased proliferation
of APRIN-knockdown cells were attenuated by the antibody treatment
(P<0.01; Fig. 4C). Therefore,
APRIN may regulate cancer cell proliferation via IL-6/STAT3/cyclin
D1 pathway (Fig. 4D).
Discussion
Since APRIN has been studied as a growth inhibitory
gene with potential tumor suppressor functions, its association
with various cancer cells has been examined, including in prostate
(5), esophageal (17,18),
head and neck (19), and pancreatic
cancer cells (13). While the
precise mechanism for the negative regulation of cancer cell
proliferation by APRIN is still unclear, a few studies have
identified distinct regulators. For example, it has been shown that
overexpression of APRIN inhibits proliferation and promotes
apoptosis in P19 embryonal carcinoma cells (20). Another study suggested that APRIN
upregulates Ptch2 in pancreatic cancer (PC) cells and that this
APRIN/Ptch2 axis inhibits cell proliferation and invasion in PC
cells (13).
The anti-proliferative role of APRIN in cancer cells
was examined in the present study. Our current findings demonstrate
that APRIN downregulation enhanced cancer cell proliferation via a
novel IL-6/STAT3/cyclin D axis. APRIN depletion also increased cell
migration and anchorage-independent growth (Fig. 1D-G). While significant differences in
the expression of typical EMT markers, such as E/N-cadherin, snail
or slug (data not shown) could not be observed, wound healing assay
and Transwell migration assay clearly showed enhanced cell
migration in APRIN-depleted cells (Fig.
1D-F). Investigation of the unidentified factors which are
responsible for the cell migration might provide insights into the
APRIN-associated cellular responses. Notably, it would be
interesting to screen IL-6/STAT3-regulated factors that are
involved in cell migration.
Stable downregulation of APRIN expression in a lung
cancer cell line NCI-H460 resulted in prominent increase in IL-6
(Fig. 2). Since the cytokine levels
were measured by using culture media supernatant, these findings
demonstrated that IL-6 is secreted from the cell line and is
responsible for the downstream cellular responses. Indeed, one of
the downstream regulators, STAT3 was activated in the
APRIN-downregulated cell line (Fig.
2D). Treatment of the cell culture with IL-6-neutralizing
antibody attenuated STAT3 activation and its downstream target gene
cyclin D1 expression (Fig. 4). These
findings demonstrate that prominent paracrine production of IL-6 is
responsible for the enhanced activation of STAT3 in
APRIN-downregulated cells.
It was reported that many lung cancer cell lines
exhibit variable levels of activated STAT3 (pSTAT3) (21). Identification of the regulatory
factors that are responsible for STAT3 activation might have
implications for the development of targeted therapy in cancer
(22). Depletion of STAT3 itself in
the cells affected cell viability limiting further investigation.
However, the present results showed that blockade of IL-6 secretion
attenuates the enhanced growth of the APRIN-depleted lung cancer
cell line (Fig. 4C), demonstrating
potential therapeutic benefit of IL-6 inhibition.
While IL-6 is upregulated in lung cancer patient and
is associated with decreased cancer survival (23–25), its
association with APRIN is first shown in the present study. The
mechanism by which APRIN downregulation leads to IL-6 upregulation
in lung cancer cells is unknown. APRIN depletion might cause
pleiotropic effects in cellular responses including activities of
transcription factors. Transcriptional mediators of IL-6 gene such
as NF-κB, AP-1 and CREB were reported (26–28).
Interestingly, the involvement of STAT3 with NF-κB has been
suggested in IL-6 gene induction (29). Thus, sequence of events may be
envisioned where APRIN depletion activates a multitude of
transcription factors and some of these in turn induce IL-6
expression. Increased IL-6 may activate downstream transcription
factors including STAT3, which might reciprocally amplify IL-6
production. The resulting augmented activity of IL-6 and STAT3
might lead to enhanced expression of cyclin D1 (Fig. 4D). Further examination should reveal
how APRIN depletion regulates the IL-6/STAT3/cyclin D axis.
A study suggests that loss of APRIN expression could
sensitize breast cancer cells to DNA damaging agents (2). Characterizing the responses of various
cancer cells with aberrant APRIN expression to diverse therapeutic
agents, may provide crucial data to develop therapeutic approaches
for APRIN/IL-6/STAT3-associated cancer.
Acknowledgements
Not applicable.
Funding
This study was supported by a grant of the Korea
Institute of Radiological and Medical Sciences (grant no.
50531-2020) funded by Ministry of Science and ICT, Republic of
Korea.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
SB and SK designed and supervised the experiments.
MS and MK conducted the experiments. SB, SK and MS wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Xiong B and Gerton JL: Regulators of the
cohesin network. Annu Rev Biochem. 79:131–153. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brough R, Bajrami I, Vatcheva R, Natrajan
R, Reis-Filho JS, Lord CJ and Ashworth A: APRIN is a cell cycle
specific BRCA2-interacting protein required for genome integrity
and a predictor of outcome after chemotherapy in breast cancer.
EMBO J. 31:1160–1176. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang B, Jain S, Song H, Fu M, Heuckeroth
RO, Erlich JM, Jay PY and Milbrandt J: Mice lacking sister
chromatid cohesion protein PDS5B exhibit developmental
abnormalities reminiscent of Cornelia de Lange syndrome.
Development. 134:3191–3201. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Geck P, Szelei J, Jimenez J, Lin TM,
Sonnenschein C and Soto AM: Expression of novel genes linked to the
androgen-induced, proliferative shutoff in prostate cancer cells. J
Steroid Biochem Mol Biol. 63:211–218. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Geck P, Maffini MV, Szelei J, Sonnenschein
C and Soto AM: Androgen-induced proliferative quiescence in
prostate cancer cells: The role of AS3 as its mediator. Proc Natl
Acad Sci USA. 97:10185–10190. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Geck P, Sonnenschein C and Soto AM: The
D13S171 marker, misannotated to BRCA2, links the AS3 gene to
various cancers. Am J Hum Genet. 69:461–463. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Beckmann MW, Picard F, An HX, van Roeyen
CR, Dominik SI, Mosny DS, Schnürch HG, Bender HG and Niederacher D:
Clinical impact of detection of loss of heterozygosity of BRCA1 and
BRCA2 markers in sporadic breast cancer. Br J Cancer. 73:1220–1226.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gorgoulis VG, Kotsinas A, Zacharatos P,
Mariatos G, Liloglou T, Tsoli E, Kokotas S, Fassoulas C, Field JK
and Kittas C: Association of allelic imbalance at locus D13S171
(BRCA2) and p53 alterations with tumor kinetics and chromosomal
instability (aneuploidy) in nonsmall cell lung carcinoma. Cancer.
89:1933–1945. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Edwards SM, Dunsmuir WD, Gillett CE,
Lakhani SR, Corbishley C, Young M, Kirby RS, Dearnaley DP, Dowe A,
Ardern-Jones A, et al CRC/BPG UK Familial Prostate Cancer Study
Collaborators, : Immunohistochemical expression of BRCA2 protein
and allelic loss at the BRCA2 locus in prostate cancer. Int J
Cancer. 78:1–7. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Harada H, Tanaka H, Shimada Y, Shinoda M,
Imamura M and Ishizaki K: Lymph node metastasis is associated with
allelic loss on chromosome 13q12-13 in esophageal squamous cell
carcinoma. Cancer Res. 59:3724–3729. 1999.PubMed/NCBI
|
|
11
|
Chen W, Salto-Tellez M, Palanisamy N,
Ganesan K, Hou Q, Tan LK, Sii LH, Ito K, Tan B, Wu J, et al:
Targets of genome copy number reduction in primary breast cancers
identified by integrative genomics. Genes Chromosomes Cancer.
46:288–301. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim MS, An CH, Yoo NJ and Lee SH:
Frameshift mutations of chromosome cohesion-related genes SGOL1 and
PDS5B in gastric and colorectal cancers with high microsatellite
instability. Hum Pathol. 44:2234–2240. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ma J, Cui Y, Cao T, Xu H, Shi Y, Xia J,
Tao Y and Wang ZP: PDS5B regulates cell proliferation and motility
via upregulation of Ptch2 in pancreatic cancer cells. Cancer Lett.
460:65–74. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta DeltaC(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chung HJ, Yoon SI, Shin SH, Koh YA, Lee
SJ, Lee YS and Bae S: p53-mediated enhancement of radiosensitivity
by selenophosphate synthetase 1 overexpression. J Cell Physiol.
209:131–141. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Levy DE and Lee CK: What does Stat3 do? J
Clin Invest. 109:1143–1148. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Harada H, Uchida N, Shimada Y, Kumimoto H,
Shinoda M, Imamura M and Ishizaki K: Polymorphism and allelic loss
at the AS3 locus on 13q12-13 in esophageal squamous cell carcinoma.
Int J Oncol. 18:1003–1007. 2001.PubMed/NCBI
|
|
18
|
Zhang Y, Huang X, Qi J, Yan C, Xu X, Han Y
and Wang M: Correlation of genomic and expression alterations of
AS3 with esophageal squamous cell carcinoma. J Genet Genomics.
35:267–271. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Reis EM, Ojopi EP, Alberto FL, Rahal P,
Tsukumo F, Mancini UM, Guimarães GS, Thompson GM, Camacho C,
Miracca E, et al Head and Neck Annotation Consortium, : Large-scale
transcriptome analyses reveal new genetic marker candidates of
head, neck, and thyroid cancer. Cancer Res. 65:1693–1699. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou X, Kong X, Xu W and Chen J:
Overexpression of APRIN inhibits differentiation and proliferation
and promotes apoptosis in P19 embryonal carcinoma cells. Mol Biol
Rep. 40:491–495. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gao SP, Mark KG, Leslie K, Pao W, Motoi N,
Gerald WL, Travis WD, Bornmann W, Veach D, Clarkson B, et al:
Mutations in the EGFR kinase domain mediate STAT3 activation via
IL-6 production in human lung adenocarcinomas. J Clin Invest.
117:3846–3856. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu H and Jove R: The STATs of cancer--new
molecular targets come of age. Nat Rev Cancer. 4:97–105. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chang CH, Hsiao CF, Yeh YM, Chang GC, Tsai
YH, Chen YM, Huang MS, Chen HL, Li YJ, Yang PC, et al: Circulating
interleukin-6 level is a prognostic marker for survival in advanced
nonsmall cell lung cancer patients treated with chemotherapy. Int J
Cancer. 132:1977–1985. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pine SR, Mechanic LE, Enewold L,
Chaturvedi AK, Katki HA, Zheng YL, Bowman ED, Engels EA, Caporaso
NE and Harris CC: Increased levels of circulating interleukin 6,
interleukin 8, C-reactive protein, and risk of lung cancer. J Natl
Cancer Inst. 103:1112–1122. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Songür N, Kuru B, Kalkan F, Özdilekcan C,
Çakmak H and Hizel N: Serum interleukin-6 levels correlate with
malnutrition and survival in patients with advanced non-small cell
lung cancer. Tumori. 90:196–200. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xiao W, Hodge DR, Wang L, Yang X, Zhang X
and Farrar WL: Co-operative functions between nuclear factors
NFkappaB and CCAT/enhancer-binding protein-β (C/EBP-β) regulate the
IL-6 promoter in autocrine human prostate cancer cells. Prostate.
61:354–370. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sehgal PB: Regulation of IL6 gene
expression. Res Immunol. 143:724–734. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Squarize CH, Castilho RM, Sriuranpong V,
Pinto DS Jr and Gutkind JS: Molecular cross-talk between the
NFkappaB and STAT3 signaling pathways in head and neck squamous
cell carcinoma. Neoplasia. 8:733–746. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang J, Liao X, Agarwal MK, Barnes L,
Auron PE and Stark GR: Unphosphorylated STAT3 accumulates in
response to IL-6 and activates transcription by binding to
NFkappaB. Genes Dev. 21:1396–1408. 2007. View Article : Google Scholar : PubMed/NCBI
|