Introduction
Lung cancer is the most common cancer worldwide
(1). In China in 2015, statistics
showed that the lung cancer had the highest rates of morbidity
(0.73 million new lung cancer cases) and mortality (0.6 million
lung cancer deaths) (2). In total,
~85% of lung cancer cases are non-small cell lung cancer (NSCLC).
Crizotinib, an inhibitor targeting the oncogenes hepatocyte growth
factor receptor (c-MET), ALK and ROS1, was approved in 2011 for use
due to its antitumor effects in patients with NSCLC displaying
abnormal c-MET pathway activation (3). c-MET is a hepatocyte growth factor
receptor-induced tyrosine kinase that, when aberrantly activated or
amplified, activates various signaling transduction cascades (for
example the PI3K/Akt/MAPK signaling pathways), promotes
tumorigenesis, metastasis, and drug resistance and leads to poor
prognosis (4,5).
Cellular metabolism is known to serve an important
role in tumor proliferation and migration. In the early 1960s,
Warburg showed that cancer cells are more likely to switch their
energy source from mitochondrial oxidative phosphorylation (in
normal cells) to glycolysis, and this switch has been named the
‘Warburg effect’ (6). Glycolysis is
inefficient in generating ATP but produces more intermediates that
promote tumor survival (7).
Metabolic alteration cells contribute to metastasis of some tumor
types (8,9), for example migratory/invasive breast
cancer cells specifically favor mitochondrial respiration, leading
to higher ATP levels compared with those found in the primary tumor
and circulating cells (10,11).
Crizotinib has been studied extensively regarding
its antiproliferative, antimetastatic and clinically beneficial
effects in lung cancer (12,13). However, more research is needed to
determine whether c-MET inhibition alters energy production in
NSCLC. Liu et al (14)
reported that c-Met induces cytochrome c release from mitochondria,
blocks c-Met-augmented loss of mitochondrial transmembrane
potential (Δψm) and inhibits apoptosis (14). A previous study showed that, in HepG2
cells, crizotinib does not affect mitochondrial respiration and has
a minimal effect on glycolysis (15). These conflicting findings highlight
the need to clarify the link between crizotinib and metabolism in
NSCLC.
The present study aimed to explore the role of
altered metabolic pattern following crizotinib treatment in A549
cells. To achieve this, the effects of crizotinib combined with
several metabolic inhibitors on energy production, Δψm, reactive
oxygen species (ROS), proliferation and migration in NSCLC were
analyzed. The level of apoptosis and autophagy were also analyzed.
These experiments aimed to assess the relationship between
crizotinib-induced metabolic change and the cancer cell death.
Materials and methods
Reagents
Crizotinib, rotenone and MG132 (Beyotime Institute
of Biotechnology) were dissolved in DMSO and stored at −80°C.
Before use, these reagents were diluted with RPMI-1640 culture
media, so that the final DMSO concentration was <0.1%. 2-DG
(Beyotime Institute of Biotechnology) was dissolved in pure water,
and diluted with culture media >100:1 before use. Light chain 3
I/II (LC3 I/II, cat. no. ab128025), and GAPDH (cat. no. ab9485)
antibodies were purchased from Abcam, phosphorylated c-MET
(p-c-MET; cat. no. 8198), c-MET (cat. no. 3077) and BAX (cat. no.
2772), BCL2 (cat. no. 4223), poly ADP-ribose polymerase (PARP; cat.
no. 9542) and tubulin (cat. no. 2146) antibodies were purchased
from Cell Signaling Technology, Inc.
Cell culture
The Clinical Research Center of Zhejiang Provincial
People's Hospital provided A549 cells which original purchased from
The Cell Bank of Type Culture Collection of the Chinese Academy of
Sciences. Cells were cultured in RPMI-1640 medium supplemented with
10% fetal bovine serum (FBS; Hyclone) and 1%
penicillin-streptomycin solution (Hyclone; Cyvita), and incubated
at 37°C with 5% CO2. Cells were seeded at 50,000
cells/well in 24-well or 6-well plates and cultured until 80–85%
confluent before experimental use.
Cell viability
After cells were treated with 0–100 µM crizotinib or
other inhibitors, including 10 nM rotenone, 10 mM 2-DG or 10 nM
MG132 for 24 h, 10 µl MTS solution (Promega Corporation) was added
to the supernatant. After 1 h of culture, an equivalent volume of
supernatant was transferred to 96-well plates to determine the
absorbance at 490 nm using an Absorbance Plate Reader (BioTek
Instruments, Inc.).
Metabolism analysis
After cells were treated for 24 h, 100 µl culture
supernatant was collected to determine the residual glucose
concentration using a glucose assay kit (Sigma-Aldrich; Merck KGaA)
and absorbance at 540 nm. Cells were washed with PBS, and 200 µl
ATP lysate was added to the collected homogenates for analysis as
previously described (16). Cellular
levels of ATP was determined by using an ATP Luminometric Assay kit
(Beyotime Institute of Biotechnology), lactate was tested by Lactic
Acid assay kit (Nanjing Jiancheng Bioengineering Institute), and
total protein was determined using the BCA Protein Assay kit
(Beyotime Institute of Biotechnology).
Mitochondrial membrane potential
After 24 h treatment with 1 µM crizotinib or other
inhibitors as aforementioned, cells were loaded with mitochondrial
membrane potential dye JC-1 (Beyotime Institute of Biotechnology)
for 20 min at 37°C. Cells were digested with trypsin and washed
with PBS in preparation for BD FACSCanto II flow cytometry (BD
Biosciences) according to manufacturer instructions. Red
fluorescence signified high mitochondrial membrane potential, and
green fluorescence presented low mitochondrial membrane potential.
Data were analyzed by using FlowJo software (version 10; FlowJo,
LLC).
ROS analysis
After 24 h treatment with 1 µM crizotinib or the
indicated inhibitors, cells were loaded with 2 mM ROS fluorescence
dye 2′,7′-dichlorodihydrofluorescein diacetate (Beyotime Institute
of Biotechnology) for 15 min at 37°C. Then cells were digested with
trypsin and resuspended with PBS for flow cytometry analysis as
aforementioned.
Cell proliferation
The ratio of cell proliferation was analyzed
following EdU incubation (Beyotime Institute of Biotechnology).
Briefly, after treatment with 1 µM crizotinib and various
inhibitors, 10 µM EdU solution was added to culture media and
incubated at 37°C with 5% CO2 for 2 h, then the cells
were fixed with 4% paraformaldehyde for 15 min and permeabilized
with 0.3% Triton X-100 for 15 min, both at room temperature. After
washed with PBS, the cells were stained by Click Additive Solution
and subjected to nuclear staining with Hoechst. Images were
captured using an inverted Nikon Eclipse Ti fluorescence microscope
(Nikon Corporation). After digested, suspended cells were used for
flow cytometer analysis as aforementioned and the EdU-positive cell
ratio was analyzed by using FlowJo software.
Cell migration
Cell migration assays were performed on 24-well
plates with Transwell inserts (BD Biosciences). In total,
2×104 cells were seeded into the upper chamber of the
Transwell plates without FBS in the RPMI-1640 culture media, while
lower chambers were RPMI-1640 with 10% FBS. In total, 1 µM
crizotinib and inhibitors were added to the media in both chambers.
After incubation for 18 h at 37°C with 5% CO2, cells in
the upper chamber were removed with a swab, while cells in the
lower chamber were fixed with 4% paraformaldehyde for 15 min at
room temperature, stained with crystal violet for 5 min at room
temperature, and manually counted in at least five random fields
under an inverted Nikon Eclipse Ti microscope.
Wound healing assay
A scratch wound assay was used to evaluate cell
migration. The cell monolayer was scratched with a 10-µl pipette
tip, and then washed three times with PBS. Afterwards cells were
treated for 18 h with 1 µM crizotinib and other inhibitors in
serum-free culture media. The images of scratch line were captured
at two fixed positions at the beginning and the end of the
experiment by Nikon light microscope at 100× magnification. These
images were analyzed by Adobe Photoshop (version CS6; Adobe) and
the wound recovery was calculated according to the reduction of
wound area.
Western blotting
A549 cells were homogenized using RIPA lysis buffer
(Beyotime Institute of Biotechnology) after exposure to 1 µM
crizotinib and the aforementioned inhibitors. Total protein of the
cell lysates were determined by using BCA Protein Assay kit as
aforementioned and then cell lysates were boiled for 15 min.
Equivalent amount of sample (50 µg protein per lane) were loaded
and separated by 10% SDS-PAGE, and then transferred to
polyvinylidene difluoride membranes (EMD Millipore). The membranes
were blocked with 5% skim milk powder for 2 h at room temperature
and then incubated with LC3 I/II, p-c-MET, c-MET, BAX, BCL2,
cleaved PARP, tubulin and GAPDH antibodies (all 1:1,000) overnight
at 4°C. Then the membranes were incubated with appropriate
HRP-conjugated goat anti-mouse (cat. no. HA1013) or goat anti-
rabbit (cat. no. HA1012) secondary antibodies (1:5,000; Hangzhou
HuaAn Biotechnology Co., Ltd.) for 1 h at room temperature.
Proteins were visualized using the ECL detection system (Beyotime
Institute of Biotechnology) and the Bio-Rad gel documentation
system (Bio-Rad Laboratories, Inc.). Protein bands was analyzed by
ImageJ software (version 1.49; National Institutes of Health).
Immunofluorescence
The level of LC3 was estimated by immunofluorescence
to analyze autophagy. A549 cells were seeded on glass coverslips
and cultured until 80% confluent. After undergoing treatment for 18
h with 1 µM crizotinib, the cells were fixed in 4% paraformaldehyde
for 15 min at room temperature and incubated with anti-LC3 I/II
antibody (1:200) at 4°C overnight. Then cells were incubated with
1:1,000 Alexa 488-conjugated antibody (cat. no. 4412; Cell
Signaling Technology) for 1 h at room temperature, after that DAPI
staining solution (Beyotime Institute of Biotechnology) was added
to cells for 5 min at room temperature to indicate nuclear
fluorescence. Fluorescence microscopy images were captured at ×400
magnification.
Statistical analysis
All experiments were repeated at least three times
and presented as mean ± SD (unless otherwise shown) and were
analyzed using one-way ANOVA followed by Tukey's post hoc test, or
Student's unpaired two-tailed t-test between two groups. GraphPad
Prism 6 software (GraphPad Software) was used for all analyses.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Crizotinib inhibits NSCLC cell
viability and ATP production
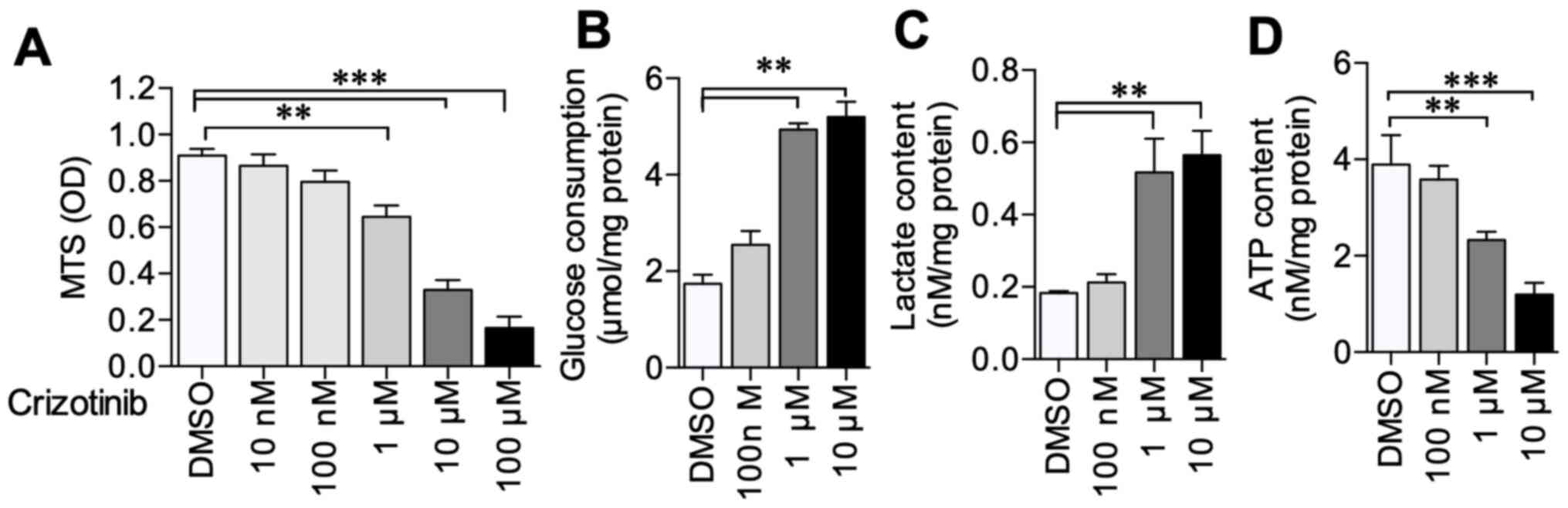
A549 cells were treated with 0–100 µM crizotinib to
evaluate crizotinib's effect on A549 cell viability and metabolism.
The results demonstrated that 1–100 µM crizotinib significantly
inhibited A549 cell viability compared with untreated cells
(Fig. 1A). Further experiments
demonstrated that 1 and 10 µM crizotinib treatment induced
increased glucose consumption (P<0.01) and lactate production
(P<0.01) compared with the DMSO control (Fig. 1B and C); however, ATP content
decreased (P<0.001) (Fig. 1D).
The experiments aforementioned were carried out on H1650 cell line
after 1 µM crizotinib treatment and observed the same results of
cell viability, glucose consumption, lactate and ATP production
(Fig. S1). These results suggested
that crizotinib inhibited NSCLC cell viability and alternated the
cell metabolism.
Crizotinib does not suppress
metabolism in 2-DG-treated cells
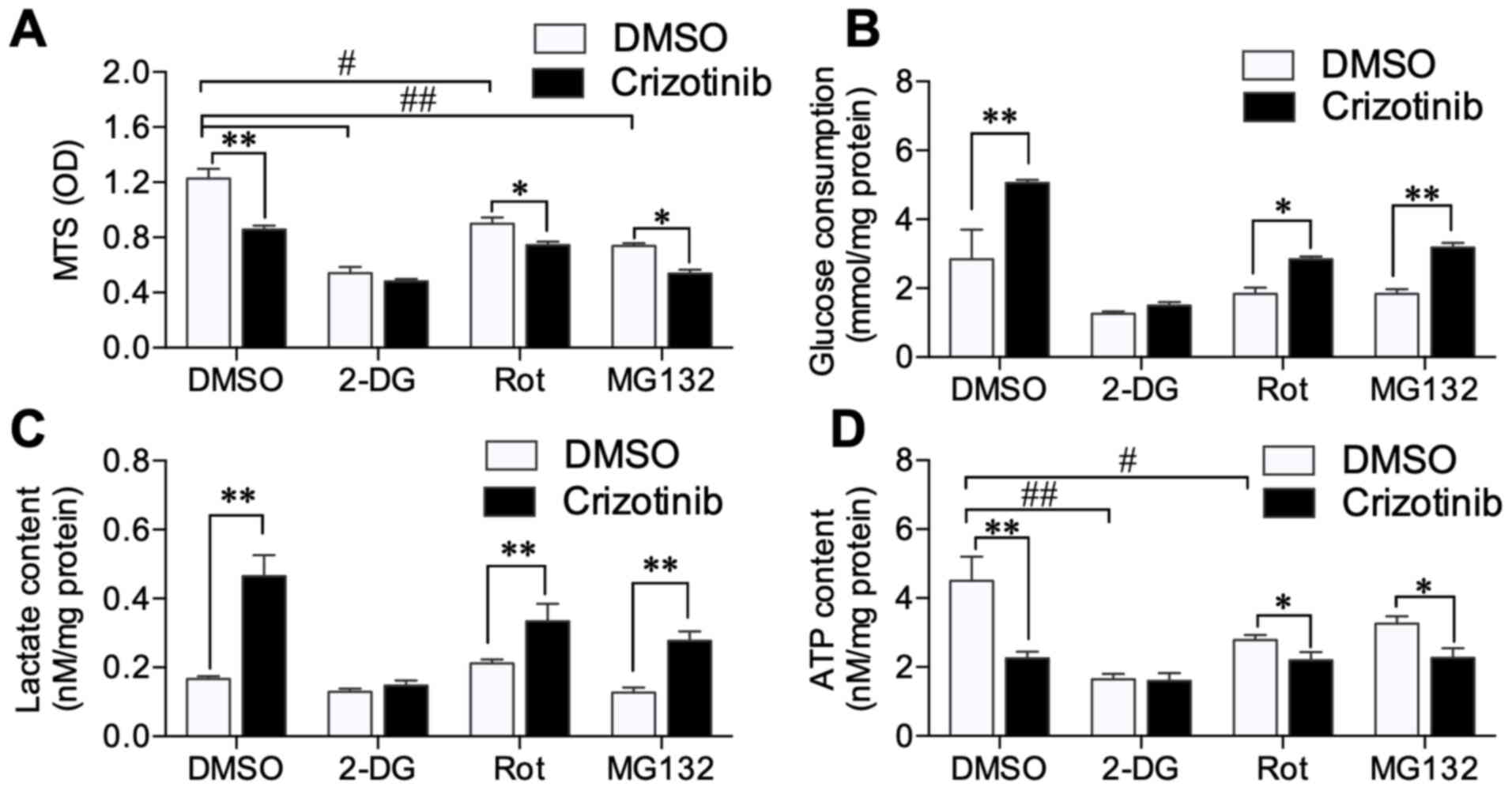
Overall, 10 nM rotenone (mitochondrial complex I
inhibitor) (17), 10 mM 2-DG
(glycolysis inhibitor) (18) or 10
nM MG132 (proteasome inhibitor) (19) were added to A549 cells, in the
presence or absence of crizotinib, to explore the mechanism by
which crizotinib interrupts energy metabolism. All inhibitors
rotenone, 2-DG and MG132 decreased A549 cell viability (Fig. 2A). Combined 1 µM crizotinib with
rotenone (P<0.05) or MG132 (P<0.05), further inhibited cell
viability; however, 1 µM crizotinib did not decrease cell viability
in conjunction with 2-DG-treatment (Fig.
2A). Crizotinib further increased glucose consumption and
lactate production in cells treated with rotenone (P<0.05 and
P<0.01) and MG132 (both P<0.01), but not with 2-DG (Fig. 2B and C). ATP production was decreased
in cells treated with 2-DG (P<0.01) and rotenone (P<0.05).
Crizotinib did not further inhibit ATP production mediated by 2-DG
treatment (Fig. 2D). These results
indicated that crizotinib enhanced A549 cell glucose consumption
and anaerobic respiration; however, still induced low ATP
levels.
Metabolic inhibitors interrupt
crizotinib-mediated inhibition of cell proliferation
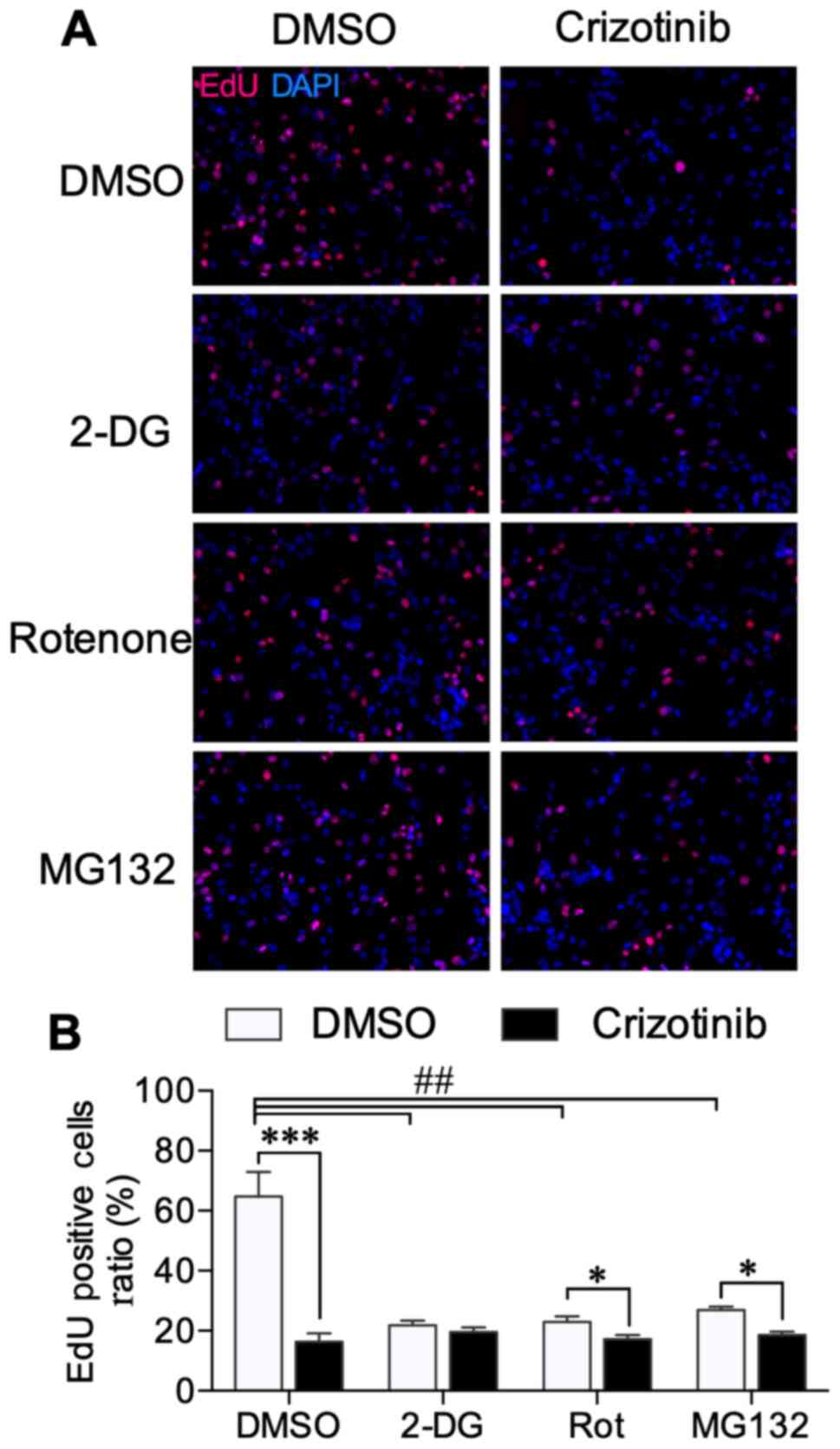
Cell proliferation was assessed using EdU
incorporation, and typical microscope images are shown in Fig. 3A. EdU-positive cell ratios were
detected using flow cytometry, and the typical histograms are shown
in Fig. S2. Similar to results
obtained with cell viability, EdU data showed that all 2-DG,
rotenone, and MG132 treatments inhibited the ratio of proliferating
cells (Fig. 3B). Crizotinib further
decreased the proliferation ratio in rotenone (P<0.05) and
MG132-treated cells (P<0.05), but not in 2-DG-treated cells
(Fig. 3B). These results further
verified the relationship between cell proliferation and
crizotinib-induced metabolic alternation.
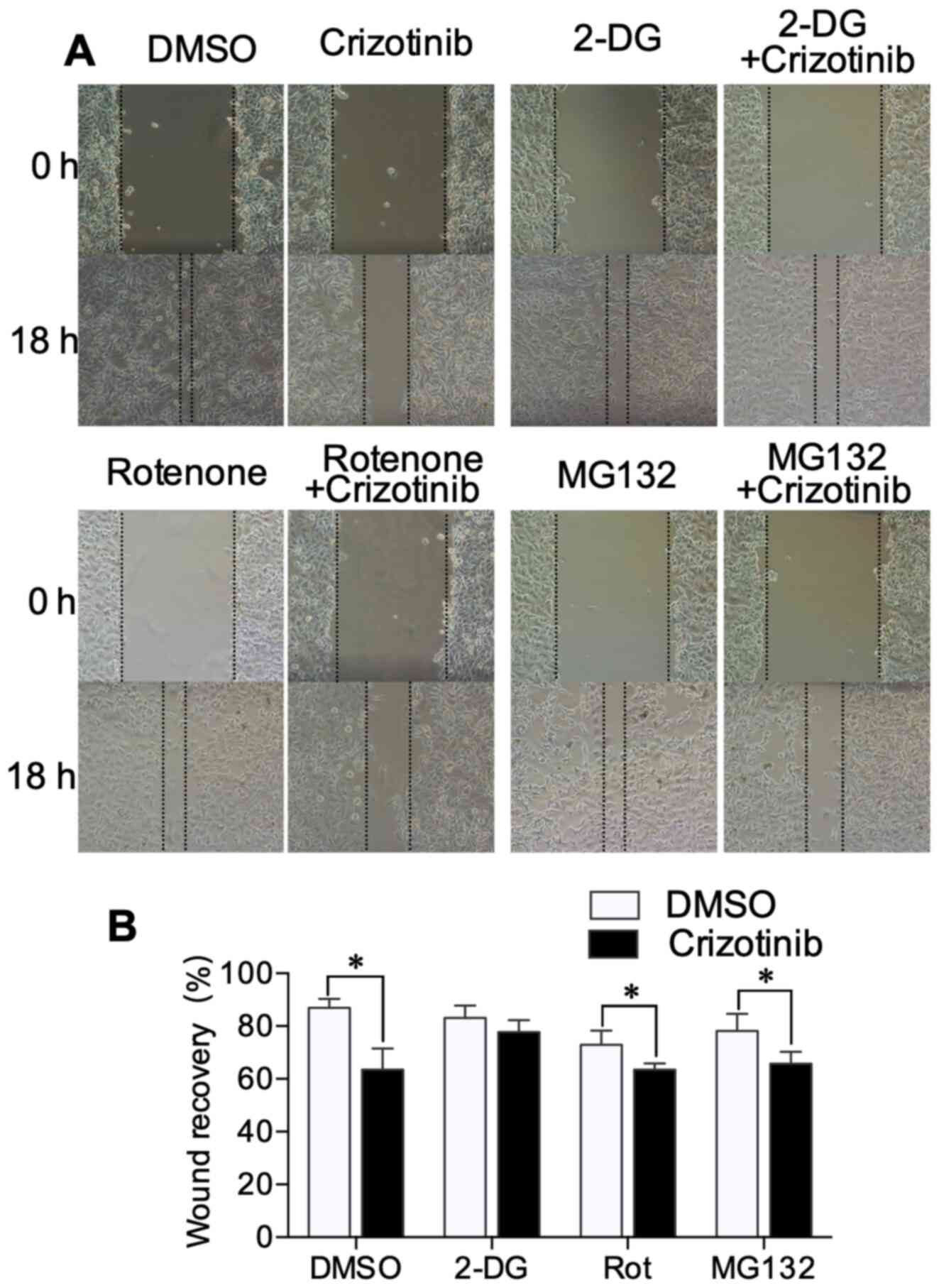
Metabolic inhibitors interrupt
crizotinib-mediated inhibition of cell migration
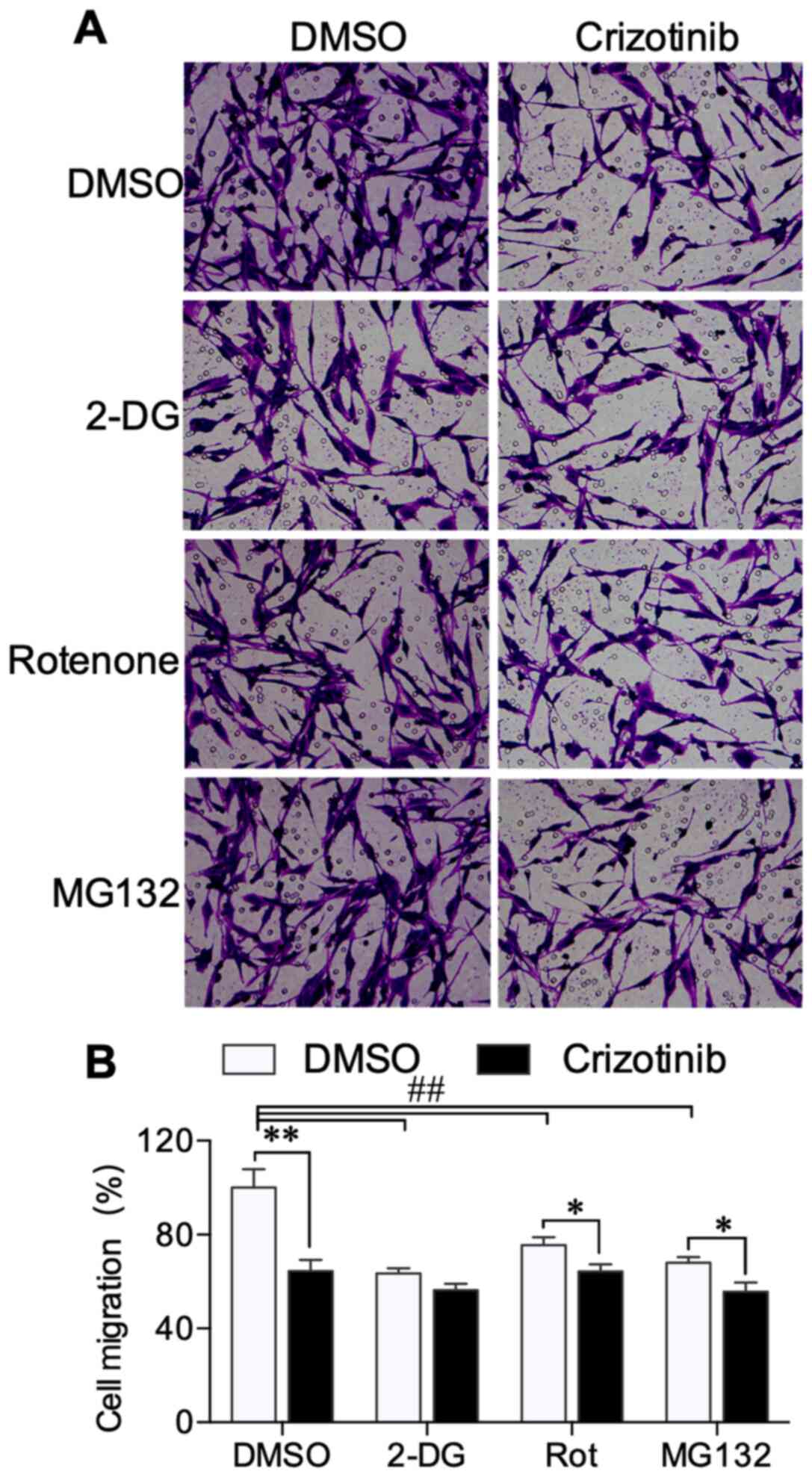
Transwell and wound healing assays were performed to
explore the effect of crizotinib on A549 cell migration. Transwell
experiments demonstrated that crizotinib inhibited A549 cell
migration across the membrane. 2-DG, rotenone and MG132 also
decreased cell migration (Fig. 4A).
When combined with rotenone (P<0.05) and MG132 (P<0.05), but
not 2-DG treatment, crizotinib further suppressed A549 cell
migration (Fig. 4B). The wound
healing assay showed that 2-DG, rotenone and MG132 treatment did
not alter the wound recovery ratio. Crizotinib treatment
significantly reduced the wound recovery compared with the
DMSO-treated group (P<0.01). For cells treated with 2-DG in the
Transwell experiment, crizotinib did not further narrow the width
of the scratch wound (Fig. 5A and
B). These results suggested the crizotinib-mediated inhibition
of cell migration may be associated with altered metabolism.
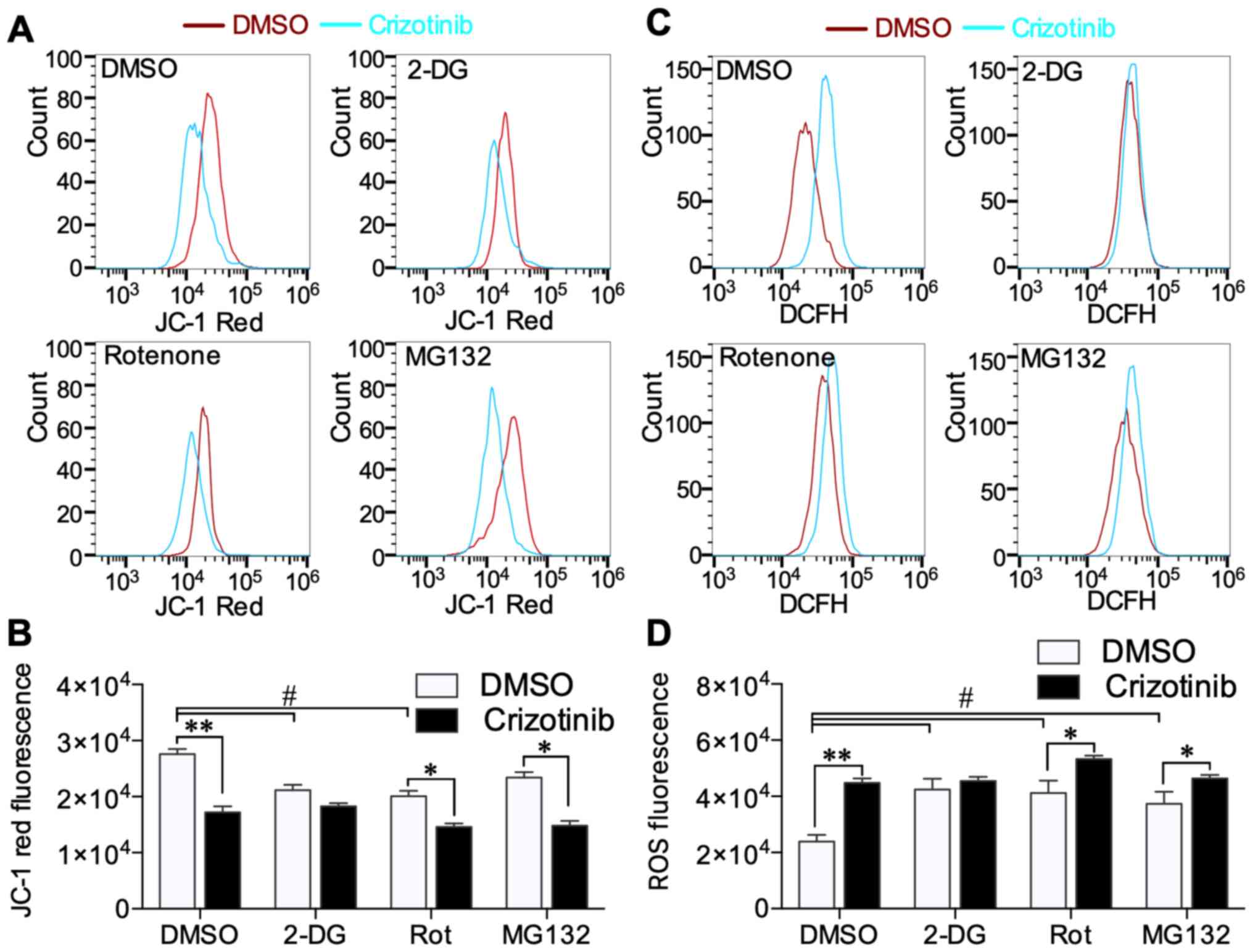
Crizotinib decreases the Δψm and
increases ROS content
It was determined that crizotinib induced A549 cell
Δψm and ROS content using flow cytometry. Crizotinib substantially
decreased high Δψm level (JC-1 red fluorescence) compared with the
DMSO-treated group (P<0.01) (Fig.
6A). 2-DG and rotenone decreased high Δψm level compared with
cells only treated with DMSO, while crizotinib treatment further
decreased Δψm in rotenone- (P<0.05) and MG132-treated cells
(P<0.05) (Fig. 6B). Low Δψm level
signal (JC-1 green fluorescence) was not significantly different
between any of the groups (Fig.
S3). Crizotinib induced significantly high ROS level
(P<0.01), while 2-DG, rotenone and MG132 alone increased ROS
level in A549 cells compared with the DMSO only treated group
(P<0.05) (Fig. 6C). Only in
2-DG-treated cells did crizotinib not further increase ROS content
(Fig. 6D). These results showed that
crizotinib inhibited mitochondrial respiration and increased ROS,
and these crizotinib effects were limited by 2-DG-mediated
inhibition of glycolysis.
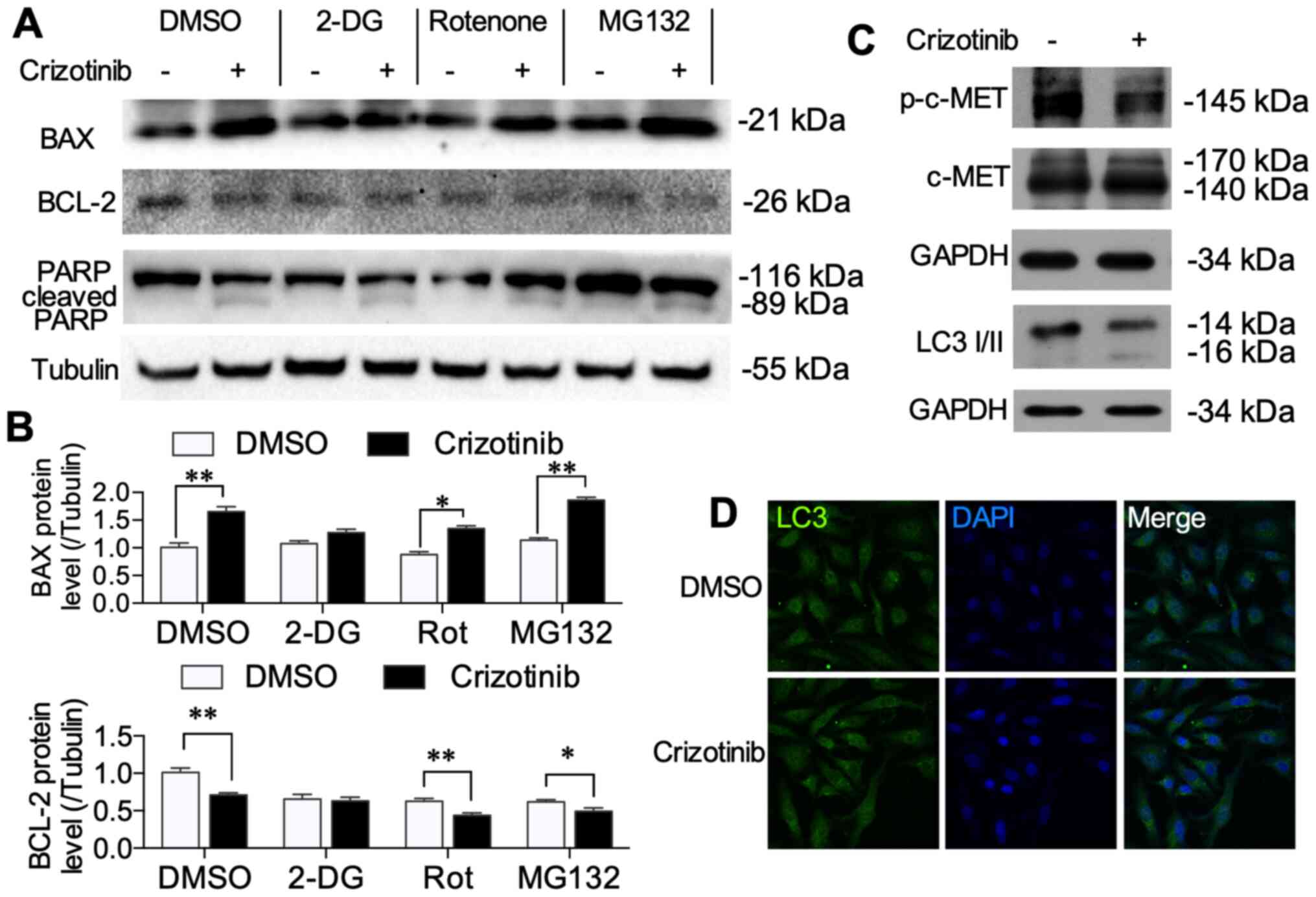
Crizotinib treatment induces
mitochondrial-related apoptosis
It was determined that crizotinib and inhibitors
induced apoptosis using western blotting. Crizotinib increased BAX
(P<0.01) and inhibited BCL-2 protein expression (P<0.01)
(Fig. 7A). All inhibitors (2-DG,
rotenone and MG132) alone did not change the protein levels of BAX
or BCL-2. However, treatment with 2-DG did not affect
crizotinib-induced BAX and BCL-2 alternations (Fig. 7B). Crizotinib enhanced cleaved PARP
protein level, which is an indispensable signal in apoptosis
cascade reaction (20), while the
inhibitors did not change the crizotinib-induced cleaved PARP level
(Fig. 7A). LC3 I/II protein levels
were also evaluated using western blot and immunofluorescence to
determine whether crizotinib induced autophagy. In A549 cells, 1 µM
crizotinib decreased p-c-MET levels (Fig. 6C). Crizotinib neither increased LC3
I/II protein levels (Fig. 7C), nor
induced autolysosome aggregation (Fig.
7D). These results suggested that crizotinib activated the
mitochondrial pathway of apoptosis, which is associated with
crizotinib-induced changes in metabolic pattern.
Discussion
In the present study, crizotinib treatment inhibited
A549 cell proliferation, migration, ATP production and Δψm, while
crizotinib induced high glucose consumption, lactate and ROS
content and activated mitochondrial-related apoptosis signals.
However crizotinib treatment did not further suppress proliferation
and migration ability after 2-DG-mediated inhibition of glycolysis.
These data indicated that crizotinib inhibits mitochondrial
respiration and, to compensate, enhances anaerobic respiration, but
still failed to maintain sufficient ATP content. Crizotinib-induced
insufficient ATP supply may play an important role in its antitumor
effects.
Energy production is important for all cancer cell
activity (9). Glucose metabolism
produces a supply of the energy source ATP and numerous
intermediaries necessary for cell proliferation and migration.
Cancer cells usually display altered metabolic pattern to survive.
Enhanced glycolysis or anaerobic respiration in cancer cells
metabolizes more glucose compared with normal cells, which mainly
depended on aerobic respiration (21), and accumulating lactate produces an
acidic microenvironment (7,22). Thus, therapeutic targeting of
glycolysis is an important and effective antitumor strategy
(23,24). However, a growing number of studies
have shown that some cancer cells do not show enhanced glycolysis,
but rather still rely on oxidative phosphorylation (25,26).
Previously, the complexity of metabolic reprogramming was
demonstrated by differences in glucose metabolism in primary,
circulating and metastatic tumor cells (9,10).
It was reported that inhibition of c-MET enhances
oridonin-mediated reduction of Δψm, resulting in the apoptosis of
A549 cells (14). Therefore, it was
hypothesized that crizotinib might also alter metabolism in NSCLC
cells. In the present study, 1–100 µM crizotinib was shown to
inhibit A549 cell viability, and 1 µM crizotinib was shown to
inhibit ATP production and reduce Δψm; however, this treatment also
stimulated glucose consumption and lactate production. A previous
study suggested that there is a close link between attenuated
mitochondrial bioenergetic function and enhanced glycolysis in A549
cells (27). The current results
indicated that crizotinib suppressed mitochondrial oxidative
phosphorylation and may compensate via enhanced glycolysis and
anaerobic respiration in A549 cells. However, this compensation
still failed to maintain ATP levels. It was also observed this
phenomenon in another NSCLC cell line, H1650 cells, which showed
similar results after treatment with 1 µM crizotinib (Fig. S1). However, in head and neck
squamous cell carcinoma, crizotinib caused a notable decrease in
glycolytic capacity and inhibited lactate production (28). In hepatic cancer HepG2 cells,
crizotinib did not affect mitochondrial oxidation and only weakly
affected glycolysis (15). These
findings suggest the crizotinib may exert different effects in
different types of cancer depending on different metabolic pattern.
The primary lung cancer A549 cell line depends partly on
mitochondrial oxidative phosphorylation for ATP production
(29,30) and A549 cells consume much more oxygen
compared with metastatic tumor cells, such as H1299 and H460
(31).
Additional experiments aimed to explore the details
of crizotinib treatment on cell metabolism. A549 cells were
pretreated with 2-DG, rotenone and MG132 to block specific
pathways. 2-DG competes with glucose and can be transported into
cells, but cannot be used as a substrate in glycolysis (18). It has been reported that 2-DG
decreases the migration of triple-negative breast cancer cells,
supporting a link between metabolic dysfunction and decreased
migration (32). A previous study
demonstrated that 2-DG attenuates ATP production, and also inhibits
mitochondrial bioenergetics (33).
2-DG combined with other antitumor drugs, such as metformin,
propranolol or docetaxel, effectively prevents cell proliferation
and induces apoptosis in prostate cancer cells and other solid
tumors, for example NSCLC and adenoid-cystic carcinoma (34,35). The
present study observed that 2-DG inhibited ATP production,
proliferation and migration of A549 cells. However, crizotinib in
2-DG-pretreated cells did not further inhibit ATP production, Δψm
or ROS level. These data indicated that crizotinib enhanced cell
glycolysis, which can be blocked by 2-DG pretreatment, and
consequently induced notably lower glucose consumption and ATP
production. These results demonstrated that different metabolic
patterns in cancer cells may cause variable responses to crizotinib
therapy.
Rotenone is a specific inhibitor of the
mitochondrial respiratory chain complex I (17). Rotenone decreases breast cancer
cells, pancreatic cancer cells and lung cancer cell survival in low
glucose conditions in vitro (17). The present study showed that rotenone
alone notably suppressed ATP production, Δψm and cell
proliferation. Combined with rotenone, treatment with crizotinib
further decreased cell proliferation and migration, which suggested
that crizotinib affects not only mitochondrial oxidation but also
other signaling pathways.
Low Δψm induces cytochrome c release from the
mitochondria and also initiates apoptosis (36). The BCL family of proteins serve a
role in regulation of mitochondrial membrane permeability and the
mitochondrial apoptotic pathway (37). The present study determined the
pro-apoptotic BAX and antiapoptotic BCL-2 protein levels (37). The results showed that crizotinib
induced high BAX and low BCL-2 protein expression, while in
2-DG-pretreated cells, crizotinib did not further change the BAX
and BCL-2 protein levels, which was consistent with the results in
the metabolism and Δψm experiments. It has been reported that
crizotinib induces apoptosis in H2228 lung cancer cells (38). The present study detected cleaved
PARP to analyze A549 cell apoptosis. The data showed that
crizotinib induced high cleaved PARP levels in A549 cells with or
without metabolic inhibitors, which suggested that crizotinib
induced apoptosis.
Reportedly, 4–8 µM crizotinib treatment induces
autophagy in lung cancer cell lines, including SPC-A1 and H827
(39). There is evidence that
autophagy-induced protein degeneration can fuel cellular
metabolism, and autophagy inhibition impairs the proliferation of
tumor cells (40). However, in the
present study, LC3 I/II protein was not significantly increased or
aggregated following treatment with l µM crizotinib. It was also
demonstrated that the ubiquitin-proteasome inhibitor MG132 was
ineffective in blocking the effect of crizotinib on ATP production,
cell proliferation and migration. These findings indicated that low
concentrations of crizotinib may not interrupt protein degradation
via autophagy or the ubiquitin-proteasome pathway. The present
study indicated that the alternation of metabolic pattern serves
important roles in the antitumor effect of crizotinib, which may
contribute to different therapy outcomes in a clinical setting.
However, in the present study, the molecular mechanisms
underpinning crizotinib-induced metabolic alternation were not
resolved, therefore in the future more molecular experiments are
needed to investigate these.
In conclusion, the present study reported that
crizotinib treatment inhibited proliferation, migration and
cellular ATP production and induced mitochondrial apoptosis in A549
cells. Further experiments with metabolic inhibitors clarified that
crizotinib shifted metabolic pattern and suppressed ATP supply,
leading to low mitochondrial function and compensatory high
anaerobic respiration. These alternations of metabolic pattern and
insufficient ATP supply may play important roles in anti-tumor
effect of crizotinib.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from The
National Natural Science Foundation of China (grant no. 81600595),
The Natural Science Foundation of Zhejiang Province (grant nos.
LY17H160063, LQ16H070003 and LQ17H160017) and The Medicine and
Health Research Foundation of Zhejiang Province (grant nos.
2017KY196 and 2020KY012).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributors
SY and KH designed this study. SY, HZ, YC and KL
performed the experiments. SY, SJ and HK performed data analyses.
SY and HK wrote the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
NSCLC
|
non-small cell lung cancer
|
|
Δψm
|
mitochondrial transmembrane
potential
|
References
|
1
|
Puxty K, Grant CH, McLoone P, Sloan B,
Quasim T, Hulse K and Morrison DS: Factors associated with
intensive care admission in patients with lung cancer: A
population-based observational study of 26,731 patients. BMC Pulm
Med. 20:362020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Watermann I, Schmitt B, Stellmacher F,
Müller J, Gaber R, Kugler C, Reinmuth N, Huber RM, Thomas M, Zabel
P, et al: Improved diagnostics targeting c-MET in non-small cell
lung cancer: Expression, amplification and activation? Diagn
Pathol. 10:1302015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Szturz P, Raymond E, Abitbol C, Albert S,
de Gramont A and Faivre S: Understanding c-MET signalling in
squamous cell carcinoma of the head & neck. Crit Rev Oncol
Hematol. 111:39–51. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Moro-Sibilot D, Cozic N, Pérol M, Mazières
J, Otto J, Souquet PJ, Bahleda R, Wislez M, Zalcman G, De Guibert
S, et al: Crizotinib in c-MET- or ROS1-positive NSCLC: Results of
the AcSe phase II trial. Ann Oncol. 30:1985–1991. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Warburg O: On respiratory impairment in
cancer cells. Science. 124:269–270. 1956.PubMed/NCBI
|
|
7
|
Zhao Y, Butler EB and Tan M: Targeting
cellular metabolism to improve cancer therapeutics. Cell Death Dis.
4:e5322013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pan H, Wang BH, Li ZB, Gong XG, Qin Y,
Jiang Y and Han WL: Mitochondrial superoxide anions induced by
exogenous oxidative stress determine tumor cell fate: An individual
cell-based study. J Zhejiang Univ Sci B. 20:310–321. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang D and Kim J: Mitochondrial retrograde
signalling and metabolic alterations in the tumour
microenvironment. Cells. 8:2752019. View Article : Google Scholar
|
|
10
|
LeBleu VS, O'Connell JT, Gonzalez Herrera
KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A,
Domingos Chinen LT, Rocha RM, et al: PGC-1α mediates mitochondrial
biogenesis and oxidative phosphorylation in cancer cells to promote
metastasis. Nat Cell Biol. 16:992–1003, 1-15. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dupuy F, Tabaries S, Andrzejewski S, Dong
Z, Blagih J, Annis MG, Omeroglu A, Gao D, Leung S, Amir E, et al:
PDK1-dependent metabolic reprogramming dictates metastatic
potential in breast cancer. Cell Metab. 22:577–589. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Camidge DR, Kim HR, Ahn MJ, Yang JC, Han
JY, Lee JS, Hochmair MJ, Li JY, Chang GC, Lee KH, et al: Brigatinib
versus crizotinib in ALK-positive non-small-cell lung cancer. N
Engl J Med. 379:2027–2039. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu YL, Yang JC, Kim DW, Lu S, Zhou J, Seto
T, Yang JJ, Yamamoto N, Ahn MJ, Takahashi T, et al: Phase II study
of crizotinib in East Asian patients with ROS1-positive advanced
non-small-cell lung cancer. J Clin Oncol. 36:1405–1411. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu Y, Liu JH, Chai K, Tashiro S, Onodera
S and Ikejima T: Inhibition of c-Met promoted apoptosis, autophagy
and loss of the mitochondrial transmembrane potential in
oridonin-induced A549 lung cancer cells. J Pharm Pharmacol.
65:1622–1642. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mingard C, Paech F, Bouitbir J and
Krahenbuhl S: Mechanisms of toxicity associated with six tyrosine
kinase inhibitors in human hepatocyte cell lines. J Appl Toxicol.
38:418–431. 2018. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hao K, Kong FP, Gao YQ, Tang JW, Chen J,
Evans AM, Lightman SL, Chen XQ and Du JZ: Inactivation of
corticotropin-releasing hormone-induced insulinotropic role by
high-altitude hypoxia. Diabetes. 64:785–795. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Palorini R, Simonetto T, Cirulli C and
Chiaradonna F: Mitochondrial complex I inhibitors and forced
oxidative phosphorylation synergize in inducing cancer cell death.
Int J Cell Biol. 2013:2438762013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhu Z, Jiang W, McGinley JN and Thompson
HJ: 2-Deoxyglucose as an energy restriction mimetic agent: Effects
on mammary carcinogenesis and on mammary tumor cell growth in
vitro. Cancer Res. 65:7023–7030. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harhouri K, Navarro C, Depetris D, Mattei
MG, Nissan X, Cau P, De Sandre-Giovannoli A and Lévy N:
MG132-induced progerin clearance is mediated by autophagy
activation and splicing regulation. EMBO Mol Med. 9:1294–1313.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu X, Kang J, Wang H and Huang T:
Mitochondrial ROS contribute to oridonin-induced HepG2 apoptosis
through PARP activation. Oncol Lett. 15:2881–2888. 2018.PubMed/NCBI
|
|
21
|
Kim JH, Nam B, Choi YJ, Kim SY, Lee JE,
Sung KJ, Kim WS, Choi CM, Chang EJ, Koh JS, et al: Enhanced
glycolysis supports cell survival in EGFR-mutant lung
adenocarcinoma by inhibiting autophagy-mediated EGFR degradation.
Cancer Res. 78:4482–4496. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Anemone A, Consolino L, Arena F, Capozza M
and Longo DL: Imaging tumor acidosis: A survey of the available
techniques for mapping in vivo tumor pH. Cancer Metastasis Rev.
38:25–49. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Martinez-Outschoorn UE, Peiris-Pagés M,
Pestell RG, Sotgia F and Lisanti MP: Cancer metabolism: A
therapeutic perspective. Nat Rev Clin Oncol. 14:1132017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Avagliano A, Ruocco MR, Aliotta F, Belviso
I, Accurso A, Masone S, Montagnani S and Arcucci A: Mitochondrial
flexibility of breast cancers: A growth advantage and a therapeutic
opportunity. Cells. 8:4012019. View Article : Google Scholar
|
|
25
|
Weinberg F, Hamanaka R, Wheaton WW,
Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger
GR and Chandel NS: Mitochondrial metabolism and ROS generation are
essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA.
107:8788–8793. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vyas S, Zaganjor E and Haigis MC:
Mitochondria and cancer. Cell. 166:555–566. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu M, Neilson A, Swift AL, Moran R,
Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J,
et al: Multiparameter metabolic analysis reveals a close link
between attenuated mitochondrial bioenergetic function and enhanced
glycolysis dependency in human tumor cells. Am J Physiol Cell
Physiol. 292:C125–C136. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kumar D, New J, Vishwakarma V, Joshi R,
Enders J, Lin F, Dasari S, Gutierrez WR, Leef G, Ponnurangam S, et
al: Cancer-associated fibroblasts drive glycolysis in a targetable
signaling loop implicated in head and neck squamous cell carcinoma
progression. Cancer Res. 78:3769–3782. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hensley CT, Faubert B, Yuan Q, Lev-Cohain
N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, et al:
Metabolic heterogeneity in human lung tumors. Cell. 164:681–694.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Faubert B, Li KY, Cai L, Hensley CT, Kim
J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al:
Lactate metabolism in human lung tumors. Cell. 171:358–371.e9.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cruz-Bermúdez A, Vicente-Blanco RJ,
Laza-Briviesca R, García-Grande A, Laine-Menéndez S, Gutiérrez L,
Calvo V, Romero A, Martín-Acosta P, García JM and Provencio M:
PGC-1alpha levels correlate with survival in patients with stage
III NSCLC and may define a new biomarker to metabolism-targeted
therapy. Sci Rep. 7:166612017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
O'Neill S, Porter RK, McNamee N, Martinez
VG and O'Driscoll L: 2-Deoxy-D-Glucose inhibits aggressive
triple-negative breast cancer cells by targeting glycolysis and the
cancer stem cell phenotype. Sci Rep. 9:37882019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Maximchik P, Abdrakhmanov A, Inozemtseva
E, Tyurin-Kuzmin PA, Zhivotovsky B and Gogvadze V:
2-Deoxy-D-glucose has distinct and cell line-specific effects on
the survival of different cancer cells upon antitumor drug
treatment. FEBS J. 285:4590–4601. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Raez LE, Papadopoulos K, Ricart AD,
Chiorean EG, Dipaola RS, Stein MN, Rocha Lima CM, Schlesselman JJ,
Tolba K, Langmuir VK, et al: A phase I dose-escalation trial of
2-deoxy-D-glucose alone or combined with docetaxel in patients with
advanced solid tumors. Cancer Chemother Pharmacol. 71:523–530.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Brohée L, Peulen O, Nusgens B, Castronovo
V, Thiry M, Colige AC and Deroanne CF: Propranolol sensitizes
prostate cancer cells to glucose metabolism inhibition and prevents
cancer progression. Sci Rep. 8:70502018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guerra-Castellano A, Díaz-Quintana A,
Pérez-Mejías G, Elena-Real CA, González-Arzola K, García-Mauriño
SM, De la Rosa MA and Díaz-Moreno I: Oxidative stress is tightly
regulated by cytochrome c phosphorylation and respirasome factors
in mitochondria. Proc Natl Acad Sci USA. 115:7955–7960. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Peña-Blanco A and García-Sáez AJ: Bax, Bak
and beyond-mitochondrial performance in apoptosis. FEBS J.
285:416–431. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lu H, Wu S, Chen H, Huang Y, Qiu G, Liu L
and Li Y: Crizotinib induces apoptosis of lung cancer cells through
JAK-STAT pathway. Oncol Lett. 16:5992–5996. 2018.PubMed/NCBI
|
|
39
|
You L, Shou J, Deng D, Jiang L, Jing Z,
Yao J, Li H, Xie J, Wang Z, Pan Q, et al: Crizotinib induces
autophagy through inhibition of the STAT3 pathway in multiple lung
cancer cell lines. Oncotarget. 6:40268–40282. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kimmelman AC and White E: Autophagy and
tumor metabolism. Cell Metab. 25:1037–1043. 2017. View Article : Google Scholar : PubMed/NCBI
|