Introduction
Liver cancer is the third main cause of
cancer-associated mortalities worldwide, with 782,500 new diagnosed
cases and 745,500 deaths estimated each year (1). Hepatitis B virus (HBV) and hepatitis C
virus (HCV) are the most critical known risk factors of liver
cancer (1). Although surgery and
chemotherapy have improved the survival time of patients with liver
cancer, a considerable number of patients still undergo recurrence
due to the resistance of cancer cells to chemotherapeutic drugs
(2). However, the chemoresistance
mechanisms of liver cancer remain unknown. Therefore, the
identification of drugs that increase sensitivity to liver cancer
chemotherapy is essential for the development of effective
therapies, which will be beneficial for patients.
Atorvastatin, a competitive inhibitor of β-hydroxy
β-methylglutaryl-CoA reductase, exerts beneficial effects on
circulating lipid levels and is used for the treatment and
prevention of coronary heart disease and stroke (3–5).
Additionally, atorvastatin has been proposed as an anticancer drug
candidate, since previous studies have demonstrated that
atorvastatin exerts antiproliferative, pro-apoptotic and
immunoregulatory effects (6–9). However, the underlying mechanisms of
atorvastatin-induced sensitization to chemotherapy in liver cancer
has not been elucidated.
The present study investigated the synergistic
effect of atorvastatin on cisplatin chemosensitivity and its
associated molecular mechanisms. Additionally, the role of the
Yes1-associated transcriptional regulator (YAP1) in liver cancer
cells was evaluated. Furthermore, cell survival and apoptosis in
liver cancer cell lines were analyzed using MTT assay and flow
cytometry, respectively.
Materials and methods
Cell culture
The human liver cancer HepG2 and Huh-7 cell lines
were purchased from the American Type Culture Collection. All cells
were grown in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (HyClone; Cytiva), 2 mM L-glutamine
(Gibco; Thermo Fisher Scientific, Inc.), 1% penicillin (100 U/ml)
and streptomycin (100 µg/ml; Gibco; Thermo Fisher Scientific,
Inc.). Cells were incubated at 37°C with 5% CO2 in a
humidified incubator and passaged at ≥80% confluence using trypsin
(Gibco; Thermo Fisher Scientific, Inc.).
Drug treatment
Firstly, HepG2 and Huh-7 cells were incubated with
different concentrations of atorvastatin (0, 10, 20, 30, 40, 50,
60, 70, 80, 90 and 100 µM; Selleck Chemicals) at 37°C for 24 h.
Secondly, 0, 10 and 100 µM atorvastatin combined with different
concentrations of cisplatin (Selleck Chemicals) or paclitaxel
(Selleck Chemicals) were incubated with HepG2 and Huh-7 cells for
24 h at 37°C. Since HpG2 and Huh-7 cells had different
sensitivities to cisplatin, the concentrations of cisplatin
incubated with HepG2 or Huh-7 cells were 0, 0.25, 0.5, 1 and 10
µg/ml, and 0, 1, 5, 10 and 20 µg/ml, respectively, while the
concentrations of paclitaxel incubated with HepG2 or Huh-7 cells
were 0, 100, 500, 800 and 1,000 µM. Finally, 4 µg/ml cisplatin
alone, 40 µM atorvastatin alone and 4 µg/ml cisplatin plus 40 µM
atorvastatin were incubated with HepG2 cells for 24 h at 37°C,
while 5 µg/ml cisplatin alone, 100 µM atorvastatin alone and 5
µg/ml cisplatin plus 100 µM atorvastatin were incubated with Huh-7
cells for 24 h at 37°C. Untreated cells were used as the control
check (CK). After treatment, the cell viability assay was
performed. Additionally, after treatment of Huh-7 cells with 5
µg/ml cisplatin alone, 100 µM atorvastatin alone, 300 µM paclitaxel
alone, 5 µg/ml cisplatin plus 100 µM atorvastatin and 5 µg/ml
cisplatin plus 300 µM paclitaxel for 24 h at 37°C, the flow
cytometric analysis of apoptosis was performed.
Cell viability assay
Cell viability of HepG2 and Huh-7 was tested in
vitro using MTT assays. A total of 1×104 cells were
seeded in 96-well plates. Following treatment, cells were incubated
with MTT solution (Sigma-Aldrich; Merck KGaA) in PBS for 3 h at
37°C according to the manufacturer's protocol. The purple formazan
was solubilized using DMSO. The absorbance was read on a microplate
reader at a wavelength of 490 nm (Molecular Devices, LLC). The
combination index (CI) values between 100 µM atorvastatin and
cisplatin in treating HepG2 and Huh-7 cells were calculated using
the following formula: Cell viability of cisplatin + atorvastatin
group / (cell viability of cisplatin group × cell viability of
atorvastatin group). The cut-off of CI value to determine whether a
synergistic effect was observed was 1.
Flow cytometric analysis of
apoptosis
Apoptosis was assessed using FITC-labeled Annexin-V
(BD Biosciences) and propidium iodide (PI; Sigma-Aldrich; Merck
KGaA) via flow cytometry. Briefly, following treatment for 24 h,
Huh-7 cells were collected and stained with 500 µl solution
containing Annexin V-FITC in the dark at room temperature for 30
min. This was followed by addition of PI for 5 min in the dark at
room temperature. Flow cytometry (FACSCanto; Becton, Dickinson and
Company) was used to detect fluorescent signals in the cells. Both
early and late apoptotic cells were calculated using FlowJo 7.6
(FlowJo LLC).
Western blotting
Huh-7 cells were lysed in RIPA lysis buffer
(Sigma-Aldrich; Merck KGaA), and protein concentration was
quantified using a BCA Protein Assay kit (Pierce; Thermo Fisher
Scientific, Inc.). Following protein separation (20 µg/lane) via 12
or 15% SDS-PAGE, proteins were transferred onto polyvinylidene
difluoride membranes. Subsequently, membranes were blocked in 5%
skimmed milk for 90 min at 37°C and incubated with primary
antibodies against caspase 3, caspase 9, poly-(ADP
ribose)-polymerase (PARP), YAP1 and β-actin overnight at 4°C.
Subsequently, membranes were incubated with an HRP-conjugated
secondary antibody for 2 h at 37°C.
The primary antibodies used for immunoblotting
included anti-caspase3 (1:1,000; cat. no. 9662; Cell Signaling
Technology, Inc.), anti-caspase9 (1:1,000; cat. no. 9508; Cell
Signaling Technology, Inc.), anti-PARP (1:1,000; cat. no. 9532;
Cell Signaling Technology, Inc.), YAP1 (1:1,000; cat. no. 14074;
Cell Signaling Technology, Inc.) and anti-β-actin (1:5,000; cat.
no. A5316; Sigma-Aldrich; Merck KGaA). The secondary antibodies
were HRP-conjugated anti-rabbit (1:3,000; cat. no. 7074; Cell
Signaling Technology, Inc.) and anti-mouse IgG antibodies (1:3,000;
cat. no. 7076; Cell Signaling Technology, Inc.). Protein bands were
detected using an ECL chemiluminescence reaction kit (EMD
Millipore).
Quantification of western blotting data, which was
performed using ImageJ 2.0 (National Institutes of Health) was
calculated as follows: i) Quantification of each protein density in
triplicate; ii) quantification of β-actin density in triplicate;
iii) dividing each protein density by the β-actin density to obtain
the relative band density in triplicate; and iv) setting each
replicate of relative density in the CK group as the control (as
one), and the relative density in other groups was calculated based
on the control.
Plasmid construction and
transfection
The human YAP1 coding sequence was synthesized and
subcloned into pcDNA3.1 (Addgene, Inc.). The integrity of the
respective plasmid constructs was confirmed by DNA sequencing (data
not shown). After 2×105 Huh-7 cells were seeded in
6-well plates overnight at 37°C, cells were transfected with 0.8 µg
plasmid using Lipofectamine® 2000 (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Additionally, control pcDNA3.1 was synthesized and served as a
negative control. Following incubation for 24 and 48 h at 37°C, the
overexpression efficiency of the plasmid was determined using
western blotting, as aforementioned. At 24 h after transfection,
Huh-7 cells transfected with the empty vector pcDNA3.1 were treated
with 5 µg/ml cisplatin alone or 5 µg/ml cisplatin plus 100 µM
atorvastatin and Huh-7 cells transfected with the pcDNA3.1-YAP1
were treated with 5 µg/ml cisplatin alone or 5 µg/ml cisplatin plus
100 µM atorvastatin for another 24 h at 37°C. Subsequently, the
flow cytometric analysis of apoptosis was performed.
Statistical analysis
Data are presented as the mean ± SD from ≥3 separate
experiments. All statistical analyses were performed using GraphPad
Prism 5.0 (GraphPad Software, Inc.) and SPSS 13.0 (SPSS, Inc.)
software packages. Statistical significance was determined using a
two-sided unpaired Student's t-test or one-way ANOVA followed by
Dunnett's or Tukey's multiple comparison test as appropriate.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Cytotoxicity of atorvastatin alone or
in combination with cisplatin or paclitaxel on liver cancer
cells
To determine whether atorvastatin could inhibit
liver cancer cell viability, two liver cancer cell lines, as well
as cisplatin and paclitaxel, were used for experiments.
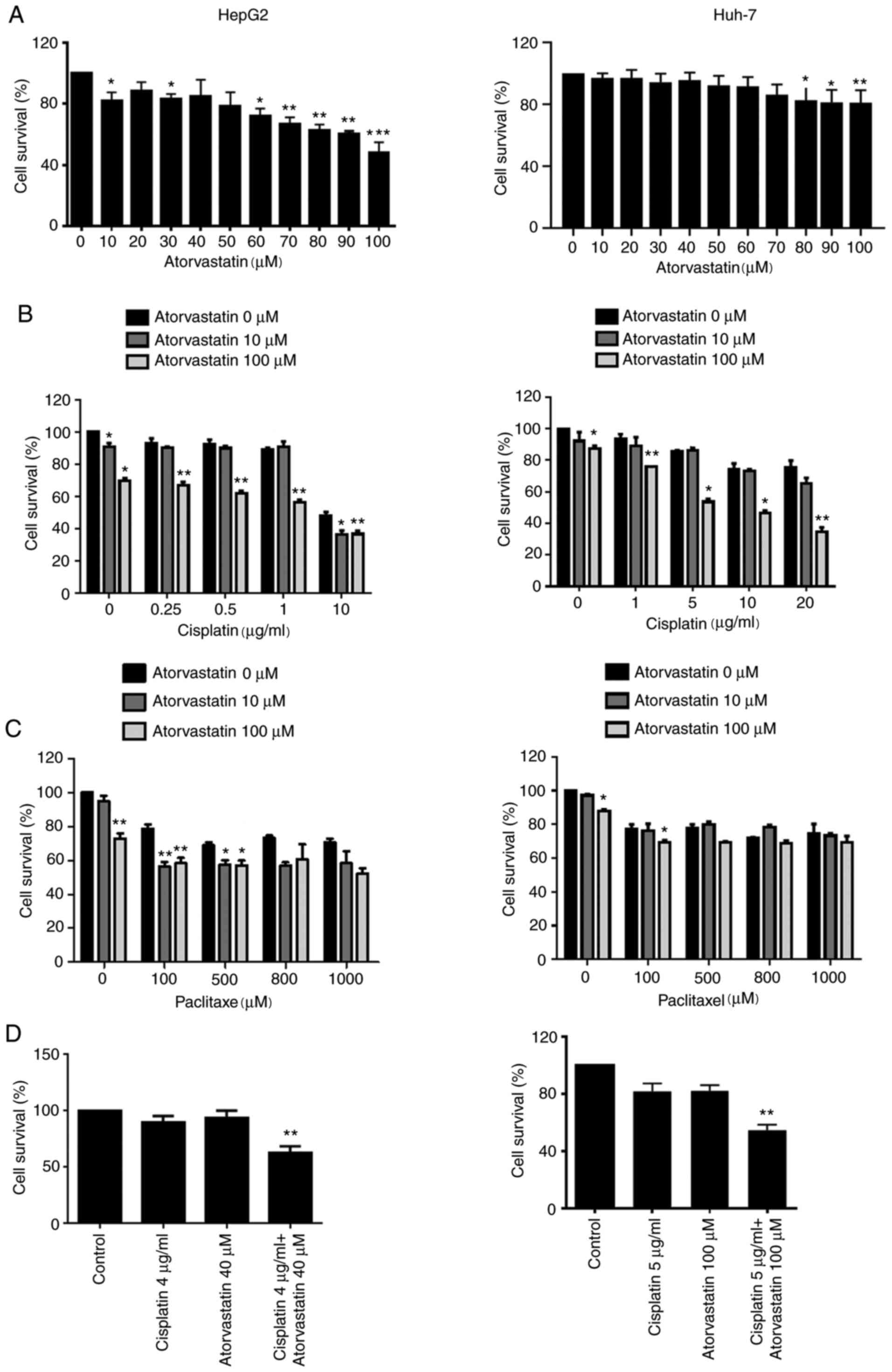
Cytotoxicity was evaluated using an MTT assay. The results revealed
that atorvastatin significantly suppressed HepG2 cell viability at
10, 30, 60, 70, 80, 90 and 100 µM, while Huh-7 cell viability was
only inhibited at 80, 90 and 100 µM (Fig. 1A). Since 10 µM was the lowest
concentration to inhibit cell viability and 100 µM was the highest
(Fig. 1A), these concentrations were
chosen for further experimentation. Additionally, the present study
examined whether combined treatment of atorvastatin with cisplatin
or paclitaxel exerted enhanced lethality on liver cancer cell
lines. As shown in Fig. 1B-D,
following co-treatment with the indicated concentrations of
atorvastatin and cisplatin or paclitaxel for 24 h, cells were
subjected to an MTT assay. The combination of atorvastatin and
cisplatin significantly inhibited cell viability in HepG2 and Huh-7
cells, but only slightly with paclitaxel. Using 100 µM atorvastatin
combined with 0–10 µg/ml or 0–20 µg/ml cisplatin significantly
inhibited HepG2 or Huh-7 cell viability compared with 0 µM
atorvastatin, respectively; additionally, 10 µM atorvastatin alone
and 10 µM atorvastatin combined with 10 µg/ml cisplatin
significantly inhibited HepG2 cell viability compared with 0 µM
atorvastatin (Fig. 1B). However,
only 100 µM paclitaxel combined with 100 µM atorvastatin or 100 µM
atorvastatin alone significantly inhibited cell viability in both
HepG2 and Huh-7 cells compared with 0 µM atorvastatin;
additionally, 10 µM atorvastatin with 100 or 500 µM paclitaxel
significantly inhibited HepG2 cell viability compared with 0 µM
atorvastatin (Fig. 1C). Further
experiments indicated that 4 µg cisplatin combined with 40 µM
atorvastatin significantly inhibited HepG2 cell viability and 5 µg
cisplatin combined with 100 µM atorvastatin significantly inhibited
Huh-2 cell viability compared with the control Fig. 1D). These results indicated that
atorvastatin may potentiate the chemosensitivity of liver cancer
cells to cisplatin. Furthermore, CI values were calculated based on
relative cell viability data, revealing that atorvastatin
synergized with 5–20 µg/ml cisplatin in killing Huh-7 cells (CI
values <1), but not HepG2 cells (CI values near or >1)
(Table I).
 | Table I.CI values between 100 µM atorvastatin
and cisplatin in treating liver cancer cells. |
Table I.
CI values between 100 µM atorvastatin
and cisplatin in treating liver cancer cells.
| Cisplatin doses | CI values |
|---|
| HepG2 cells |
| 0.25
µg/ml | 1.034±0.028 |
| 0.5
µg/ml | 0.950±0.020 |
| 1
µg/ml | 0.978±0.103 |
| 10
µg/ml | 1.229±0.167 |
| Huh-7 cells |
| 1
µg/ml | 1.081±0.024 |
| 5
µg/ml | 0.834±0.028 |
| 10
µg/ml | 0.837±0.080 |
| 20
µg/ml | 0.613±0.081 |
Atorvastatin potentiates the
chemosensitivity of liver cancer cells by inducing apoptosis
Subsequently, whether the sensitization effect of
atorvastatin to cisplatin and paclitaxel involved the induction of
apoptosis was examined. Huh-7 cells were subjected to flow
cytometry analysis following treatment with 100 µM atorvastatin
alone or in combination with 5 µg/ml cisplatin or 300 µM
paclitaxel. The drug concentrations used for these experiments were
determined due to the following: i) 100 µM atorvastatin, but not 10
µM atorvastatin, significantly enhanced the effect of cisplatin in
suppressing relative Huh-7 cell viability (Fig. 1B) and 100 µM atorvastatin was
therefore chosen for subsequent experiments; ii) 5 µg/ml cisplatin
plus 100 µM atorvastatin achieved ~50% of Huh-7 cell inhibition
rate (Fig. 1B), therefore 5 µg/ml
cisplatin was chosen for subsequent experiments; and iii)
paclitaxel at various concentrations plus 100 µM atorvastatin did
not achieve 50% of Huh-7 cell inhibition rate (Fig. 1C), but 100 µM atorvastatin enhanced
the effect of paclitaxel on Huh-7 cell inhibition at 100 µM, but
not 500 µM paclitaxel, therefore 300 µM paclitaxel (the median
between 100 and 500) was chosen for subsequent experiments.
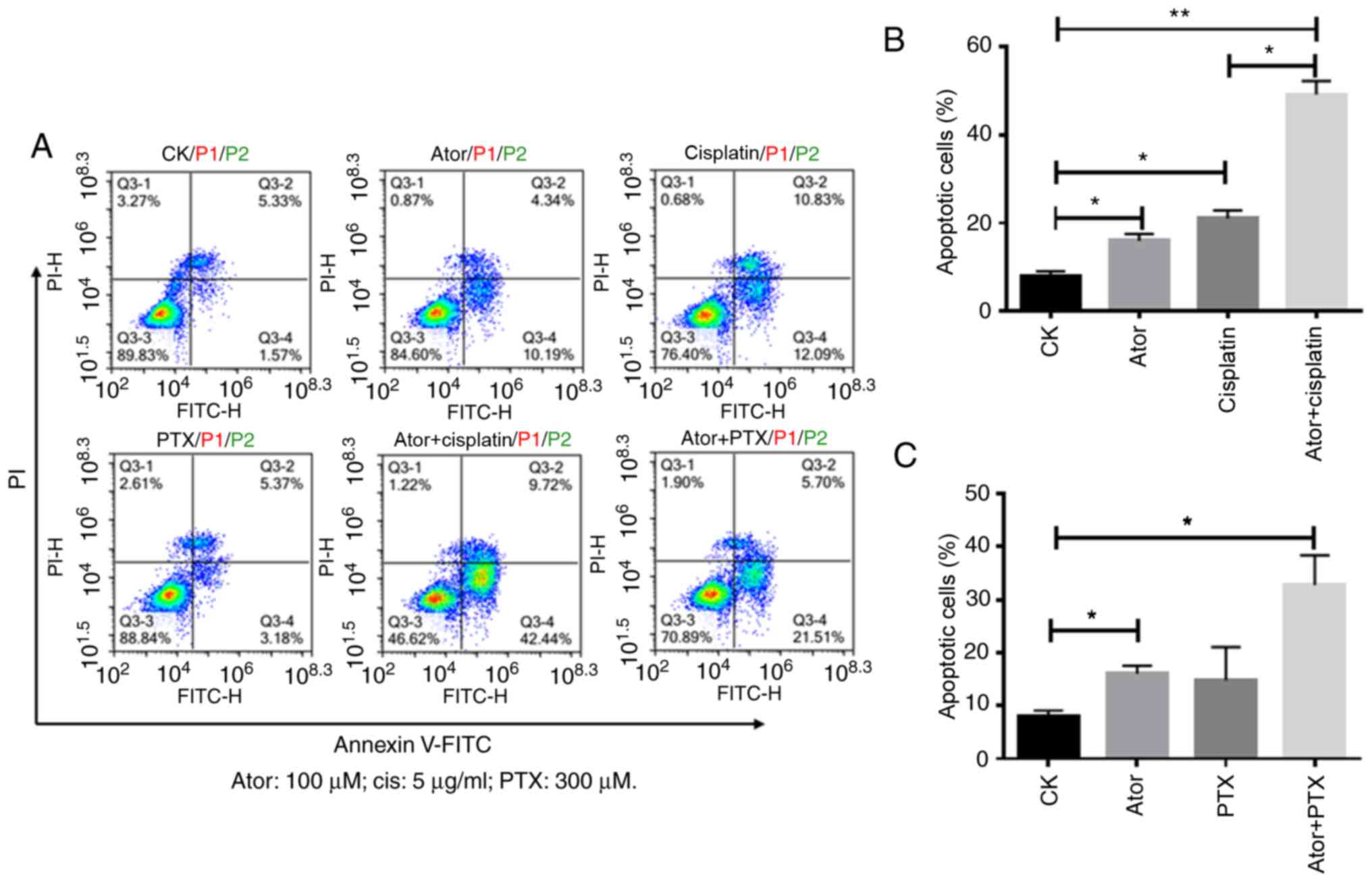
As shown in Fig. 2A and
B, atorvastatin significantly enhanced cisplatin-induced
apoptosis in Huh-7 cells. The percentage of Annexin-V+
cells increased from 16.37% (atorvastatin alone) and 23.12%
(cisplatin alone) to 54.62% (atorvastatin combined with cisplatin).
However, atorvastatin slightly enhanced paclitaxel-induced
apoptosis in Huh-7 cells. The percentage of Annexin-V+
cells increased from 16.37% (atorvastatin alone) and 14.35%
(paclitaxel alone) to 32.35% (atorvastatin combined with
paclitaxel) (Fig. 2A and C). The
present results suggested that atorvastatin significantly
potentiated cisplatin sensitivity in Huh-7 cells via inducing
apoptosis, while atorvastatin only slightly potentiated paclitaxel
sensitivity in Huh-7 cells.
Apoptosis is involved in the
synergistic effect of atorvastatin on cisplatin sensitivity in
liver cancer cells
There are two fundamental pathways of apoptosis,
which are the extrinsic and intrinsic apoptosis pathways (10,11).
Cleavage of caspases and PARP are hallmarks of intrinsic and
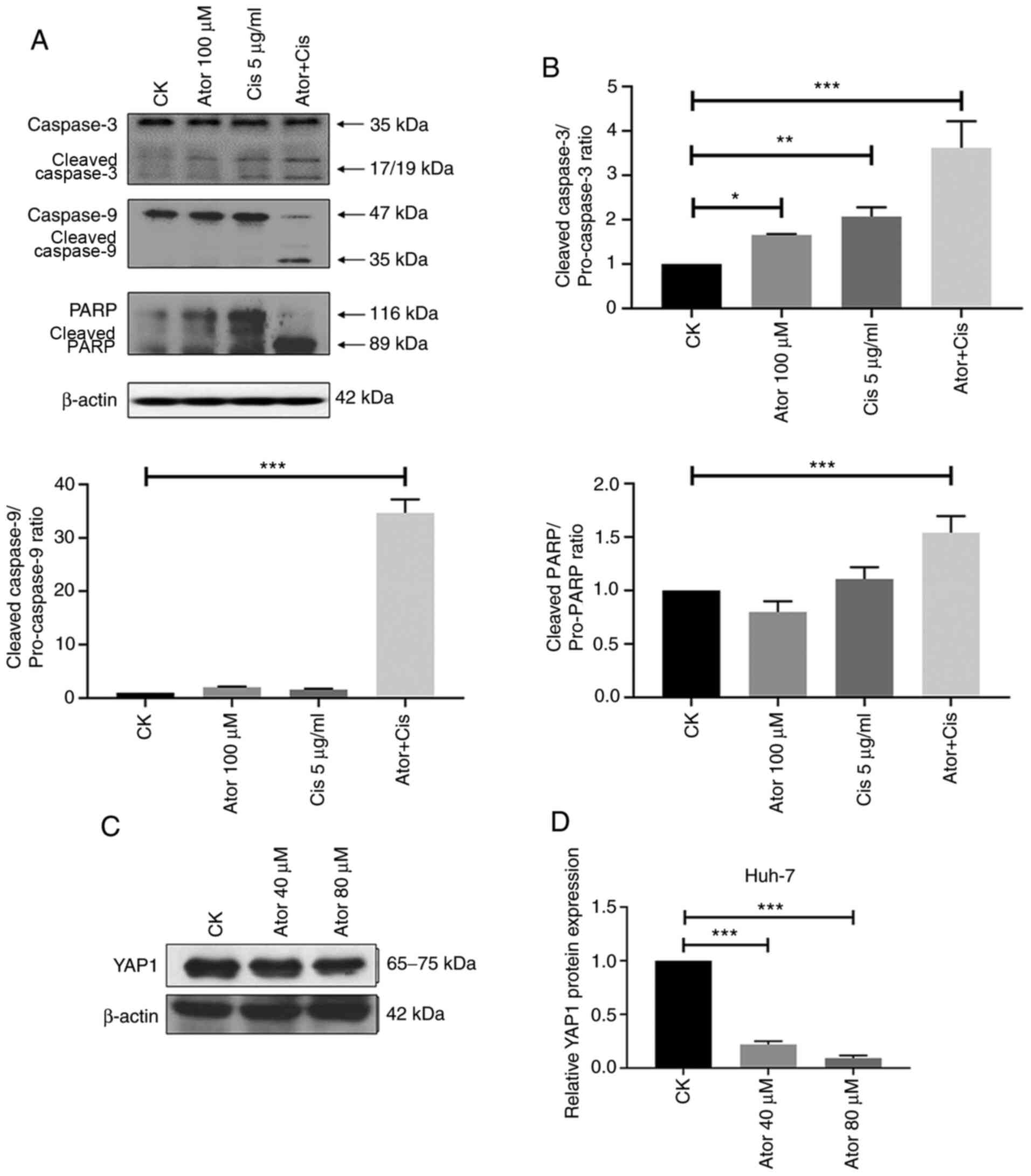
extrinsic apoptosis pathways activation (12). As shown in Fig. 3A and B, co-treatment with
atorvastatin and cisplatin in Huh-7 cells significantly increased
the cleavage of caspases 3 and 9, and PARP compared with CK.
Additionally, increasing evidence has demonstrated that increased
YAP1 expression is involved in liver cancer progression and
chemoresistance (13,14). To evaluate the effect of atorvastatin
treatment on the expression of YAP1 in liver cancer, the Huh-7
cells treated with atorvastatin. As shown in Fig. 3C and D, atorvastatin treatment
significantly inhibited YAP1 protein levels. The current
observations indicated that the intrinsic and extrinsic apoptotic
pathways and YAP1 may be involved in the synergistic effect of
atorvastatin on cisplatin sensitivity in liver cancer cells.
Atorvastatin enhances the effect of
cisplatin on treating liver cancer cells via regulating YAP1
expression
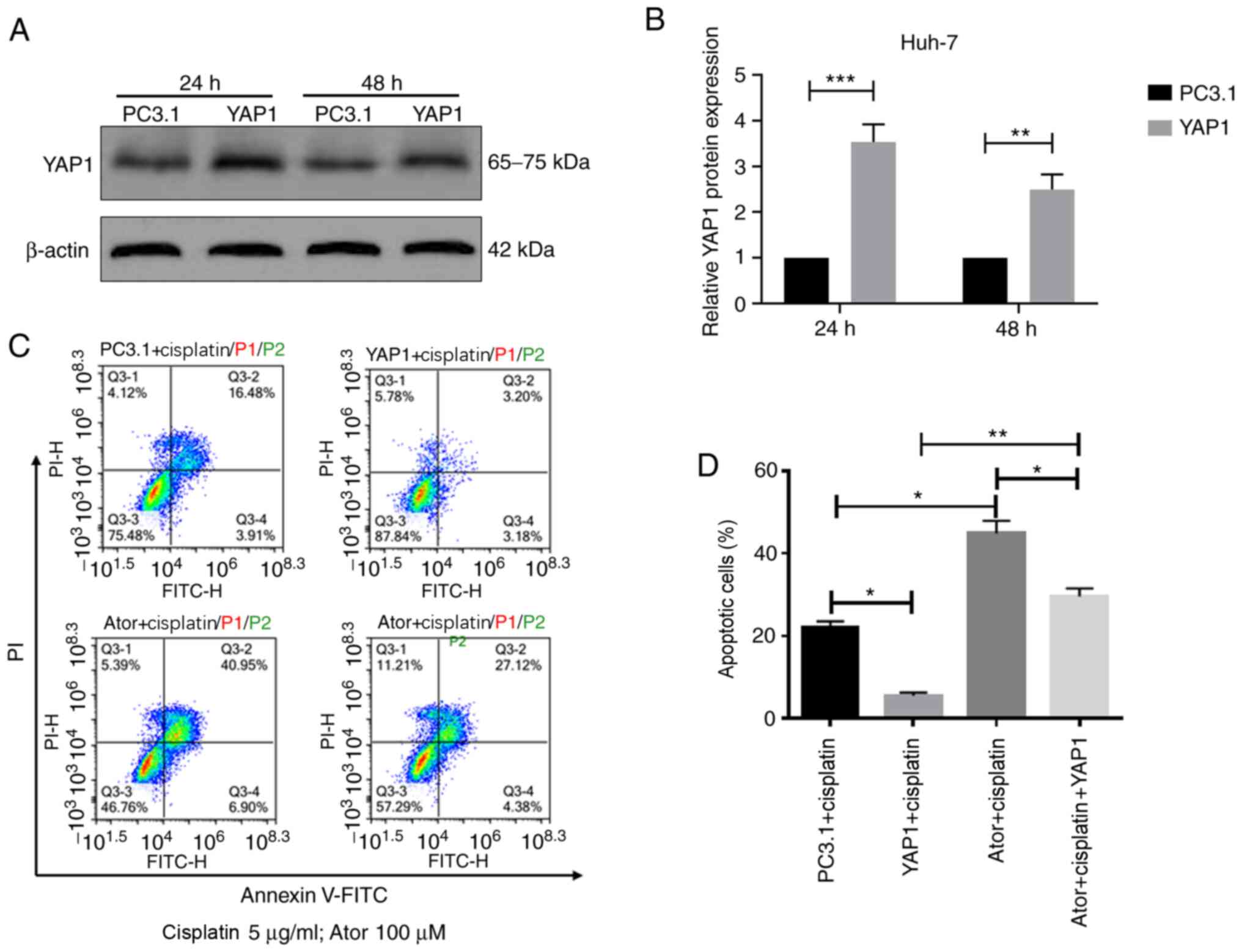
To further confirm whether atorvastatin enhanced
cisplatin chemosensitivity via YAP1, Huh-7 cells were transfected
with a YAP1 overexpression plasmid or an empty vector pcDNA3.1 The
transfection efficiency was verified using western blotting,
revealing that YAP1 levels were significantly increased in Huh-7
cells transfected with YAP1 expression plasmid after both 24 and 48
h of transfection (Fig. 4A and
B).
Huh-7 cells were transfected with empty vector
pcDNA3.1 or pcDNA3.1-YAP1 plasmid and incubated for 24 h.
Subsequently, cells were treated with 5 µg/ml cisplatin and 100 µM
atorvastatin for another 24 h. Following treatment, flow cytometry
was performed to determine the apoptotic cell percentage by
co-staining with Annexin V-FITC and PI (Fig. 4C). YAP1 overexpression significantly
attenuated the apoptosis mediated by the combination of
atorvastatin and cisplatin (Fig.
4D). Overall, the present results indicated that atorvastatin
may sensitize liver cancer cells to cisplatin, at least partially
via inhibiting YAP1.
Discussion
To date, increasing evidence has associated the YAP1
oncogene to tumorigenesis of several types of cancer, including
pancreatic ductal adenocarcinoma, lung cancer, colon cancer,
prostate cancer and liver cancer (15–19).
YAP1 is the downstream effector of the Hippo signaling pathway, and
in cooperation with the TEA domain transcription factor 1,
increased YAP1 expression stimulates a number of target genes
responsible for cell viability and apoptosis (20,21).
Several studies have demonstrated that increased YAP1 expression is
associated with elevated drug resistance in numerous cancer cells,
such as neuroblastoma, esophageal cancer and colorectal cancer
cells (22–25). The present study investigated the
mechanism of the synergistic effects of YAP1 with cisplatin.
Firstly, the present study revealed that atorvastatin inhibited
liver cancer cell viability in a dose-dependent manner. Secondly,
the present study demonstrated that sub-cytotoxic levels of
atorvastatin sensitized HepG2 and Huh-7 cells to different
concentrations of cisplatin and paclitaxel using an MTT assay.
Subsequently, the synergistic effect of atorvastatin on cisplatin
or paclitaxel sensitivity was analyzed, revealing that this
mechanism involved apoptosis induction in Huh-7 cells subjected to
flow cytometry analysis following treatment with atorvastatin alone
or in combination with cisplatin or paclitaxel. The present results
suggested that atorvastatin may regulate the intrinsic and
extrinsic apoptotic pathways to increase cell sensitivity to
cisplatin and paclitaxel. In addition, western blotting was
performed to evaluate the protein expression levels of cleaved
caspase 3 and 9, and PARP, which were all upregulated in Huh-7
cells co-treated with atorvastatin and cisplatin compared with
CK.
Finally, the YAP1 protein was further investigated,
since increased YAP1 expression is best known as a regulator of
cell viability, survival and chemoresistance (26–29). The
present study demonstrated that YAP1 levels were decreased by
atorvastatin treatment in Huh-7 cells. Furthermore, transfecting
Huh-7 cells with pcDNA3.1-YAP1 expression plasmid significantly
reversed the apoptosis mediated by the combination of atorvastatin
with cisplatin. Therefore, the current data revealed that
atorvastatin may potentiate the chemosensitivity of liver cancer
cells to cisplatin by regulating YAP1 expression.
Despite the findings of the present study, there are
still some limitations. First, the combined effect of atorvastatin
plus cisplatin or paclitaxel on cell apoptosis, and protein
expression levels, such as YAP1, were detected in a single cell
line; therefore, further validation in multiple cell lines is
required in future studies. Second, the deeper molecular mechanism
of atorvastatin plus cisplatin treatment via YAP1 requires further
exploration. Third, due to lack of funding, in vivo
validation was not performed in the present study and should
therefore be performed in future studies.
In conclusion, the current results demonstrated that
elevated levels of YAP1 in liver cancer may serve a role in cancer
cell chemoresistance. Although other downstream target genes may
also be involved in regulating apoptosis following atorvastatin
treatment, the present data illustrated that atorvastatin may
potentiate chemosensitivity to cisplatin in liver cancer cells by
regulating YAP1, which may serve a role as an apoptosis suppressor.
Therefore, the results of the present study indicated that
atorvastatin plus cisplatin therapy may be a potential strategy for
the treatment of chemoresistant liver cancer.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Medical Science
and Technology Planning Project of Zhejiang Province (grant nos.
2018RC023 and 2020KY064).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GS designed the experiments. LG, JZ, HZ and ZZ
performed the experiments. LG and JZ analyzed the data. LG wrote
the manuscript. All authors read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patients consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar
|
|
2
|
Singh MP, Cho HJ, Kim JT, Baek KE, Lee HG
and Kang SC: Morin Hydrate Reverses Cisplatin Resistance by
Impairing PARP1/HMGB1-Dependent Autophagy in Hepatocellular
Carcinoma. Cancers (Basel). 11:9862019. View Article : Google Scholar
|
|
3
|
Stone NJ, Robinson JG, Lichtenstein AH,
Bairey Merz CN, Blum CB, Eckel RH, Goldberg AC, Gordon D, Levy D,
Lloyd-Jones DM, et al American College of Cardiology/American Heart
Association Task Force on Practice Guidelines, : 2013 ACC/AHA
guideline on the treatment of blood cholesterol to reduce
atherosclerotic cardiovascular risk in adults: A report of the
American College of Cardiology/American Heart Association Task
Force on Practice Guidelines. J Am Coll Cardiol. 63:2889–B2934.
2014. View Article : Google Scholar
|
|
4
|
Amarenco P, Bogousslavsky J, Callahan A
III, Goldstein LB, Hennerici M, Rudolph AE, Sillesen H, Simunovic
L, Szarek M, Welch KM, et al Stroke Prevention by Aggressive
Reduction in Cholesterol Levels (SPARCL) Investigators, : High-dose
atorvastatin after stroke or transient ischemic attack. N Engl J
Med. 355:549–559. 2006. View Article : Google Scholar
|
|
5
|
Zhang L, Lv H, Zhang Q, Wang D, Kang X,
Zhang G and Li X: Association of SLCO1B1 and ABCB1 Genetic Variants
with Atorvastatin-induced Myopathy in Patients with Acute Ischemic
Stroke. Curr Pharm Des. 25:1663–1670. 2019. View Article : Google Scholar
|
|
6
|
Wu J, Wong WW, Khosravi F, Minden MD and
Penn LZ: Blocking the Raf/MEK/ERK pathway sensitizes acute
myelogenous leukemia cells to lovastatin-induced apoptosis. Cancer
Res. 64:6461–6468. 2004. View Article : Google Scholar
|
|
7
|
Sun HY and Singh N: Antimicrobial and
immunomodulatory attributes of statins: Relevance in solid-organ
transplant recipients. Clin Infect Dis. 48:745–755. 2009.
View Article : Google Scholar
|
|
8
|
Osmak M: Statins and cancer: Current and
future prospects. Cancer Lett. 324:1–12. 2012. View Article : Google Scholar
|
|
9
|
Kong Y, Cao XN, Zhang XH, Shi MM, Lai YY,
Wang Y, Xu LP, Chang YJ and Huang XJ: Atorvastatin enhances bone
marrow endothelial cell function in corticosteroid-resistant immune
thrombocytopenia patients. Blood. 131:1219–1233. 2018. View Article : Google Scholar
|
|
10
|
Green DR: Apoptotic pathways: Paper wraps
stone blunts scissors. Cell. 102:1–4. 2000. View Article : Google Scholar
|
|
11
|
Wang X: The expanding role of mitochondria
in apoptosis. Genes Dev. 15:2922–2933. 2001.
|
|
12
|
Nuñez G, Benedict MA, Hu Y and Inohara N:
Caspases: The proteases of the apoptotic pathway. Oncogene.
17:3237–3245. 1998. View Article : Google Scholar
|
|
13
|
Wang J, Li H, Xia C, Yang X, Dai B, Tao K
and Dou K: Downregulation of CENPK suppresses hepatocellular
carcinoma malignant progression through regulating YAP1.
OncoTargets Ther. 12:869–882. 2019. View Article : Google Scholar
|
|
14
|
Chen M, Wu L, Tu J, Zhao Z, Fan X, Mao J,
Weng Q, Wu X, Huang L, Xu M, et al: miR-590-5p suppresses
hepatocellular carcinoma chemoresistance by targeting YAP1
expression. EBioMedicine. 35:142–154. 2018. View Article : Google Scholar
|
|
15
|
Camargo FD, Gokhale S, Johnnidis JB, Fu D,
Bell GW, Jaenisch R and Brummelkamp TR: YAP1 increases organ size
and expands undifferentiated progenitor cells. Curr Biol.
17:2054–2060. 2007. View Article : Google Scholar
|
|
16
|
Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim
J, Xie J, Ikenoue T, Yu J, Li L, et al: Inactivation of YAP
oncoprotein by the Hippo pathway is involved in cell contact
inhibition and tissue growth control. Genes Dev. 21:2747–2761.
2007. View Article : Google Scholar
|
|
17
|
Harvey KF, Zhang X and Thomas DM: The
Hippo pathway and human cancer. Nat Rev Cancer. 13:246–257. 2013.
View Article : Google Scholar
|
|
18
|
Janse van Rensburg HJ, Azad T, Ling M, Hao
Y, Snetsinger B, Khanal P, Minassian LM, Graham CH, Rauh MJ and
Yang X: The Hippo Pathway Component TAZ Promotes Immune Evasion in
Human Cancer through PD-L1. Cancer Res. 78:1457–1470. 2018.
View Article : Google Scholar
|
|
19
|
Wei H, Wang F, Wang Y, Li T, Xiu P, Zhong
J, Sun X and Li J: Verteporfin suppresses cell survival,
angiogenesis and vasculogenic mimicry of pancreatic ductal
adenocarcinoma via disrupting the YAP-TEAD complex. Cancer Sci.
108:478–487. 2017. View Article : Google Scholar
|
|
20
|
Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu
J, Lin JD, Wang CY, Chinnaiyan AM, et al: TEAD mediates
YAP-dependent gene induction and growth control. Genes Dev.
22:1962–1971. 2008. View Article : Google Scholar
|
|
21
|
Zhao B, Li L, Lei Q and Guan KL: The
Hippo-YAP pathway in organ size control and tumorigenesis: An
updated version. Genes Dev. 24:862–874. 2010. View Article : Google Scholar
|
|
22
|
Keren-Paz A, Emmanuel R and Samuels Y: YAP
and the drug resistance highway. Nat Genet. 47:193–194. 2015.
View Article : Google Scholar
|
|
23
|
Coggins GE, Farrel A, Rathi KS, Hayes CM,
Scolaro L, Rokita JL and Maris JM: YAP1 Mediates Resistance to
MEK1/2 Inhibition in Neuroblastomas with Hyperactivated RAS
Signaling. Cancer Res. 79:6204–6214. 2019. View Article : Google Scholar
|
|
24
|
Li F, Xu Y, Liu B, Singh PK, Zhao W, Jin
J, Han G, Scott AW, Dong X, Huo L, et al: YAP1-Mediated CDK6
Activation Confers Radiation Resistance in Esophageal Cancer -
Rationale for the Combination of YAP1 and CDK4/6 Inhibitors in
Esophageal Cancer. Clin Cancer Res. 25:2264–2277. 2019. View Article : Google Scholar
|
|
25
|
Lee KW, Lee SS, Kim SB, Sohn BH, Lee HS,
Jang HJ, Park YY, Kopetz S, Kim SS, Oh SC, et al: Significant
association of oncogene YAP1 with poor prognosis and cetuximab
resistance in colorectal cancer patients. Clin Cancer Res.
21:357–364. 2015. View Article : Google Scholar
|
|
26
|
Vazquez-Marin J, Gutierrez-Triana JA,
Almuedo-Castillo M, Buono L, Gomez-Skarmeta JL and Mateo JL:
Wittbrodt JandMartinez-Morales JR: yap1b, a divergent Yap/Taz
family member, cooperates with yap1 in survival and morphogenesis
via common transcriptional targets. Development. 146:dev1732862019.
View Article : Google Scholar
|
|
27
|
Song Y, Sun Y, Lei Y, Yang K and Tang R:
YAP1 promotes multidrug resistance of small cell lung cancer by
CD74-related signaling pathways. Cancer Med. 9:259–268. 2020.
View Article : Google Scholar
|
|
28
|
Shi J, Li F, Yao X, Mou T, Xu Z, Han Z,
Chen S, Li W, Yu J, Qi X, et al: The HER4-YAP1 axis promotes
trastuzumab resistance in HER2-positive gastric cancer by inducing
epithelial and mesenchymal transition. Oncogene. 37:3022–3038.
2018. View Article : Google Scholar
|
|
29
|
Errico A: Targeted therapies: Hippo
effector YAP1 inhibition - towards a new therapeutic option to
overcome drug resistance. Nat Rev Clin Oncol. 12:1902015.
View Article : Google Scholar
|