Introduction
Breast cancer is the most common life-threatening
cancer in women globally, with an annual rate of new cases reaching
126/100,000 women and a death rate of ~30% (1). Although conventional treatment options,
including surgery, radiotherapy and chemotherapy, can successfully
cure patients or prolong patient survival in the majority of cases,
each of these therapies has limitations, such as the inability of
surgery to eliminate distant metastasis, the lack of durable
response to radiotherapy and drug resistance to chemotherapy
(2). Therefore, there is an urgent
need to develop new effective therapeutic approaches for breast
cancer treatment.
Oncolytic viruses can selectively infect, replicate
in, and kill cancer cells via induction of cancer cell lysis and
the host immune response in infected cancer cells, which results
from cancer antigen exposure in lysed cancer cells (3). Notably, oncolytic viruses do not harm
healthy cells. Oncolytic virus-based therapy has been regarded as a
potential novel therapeutic strategy for cancer treatment (4). Oncolytic viruses have been genetically
engineered to improve both the safety of treatment and selectivity
(5). Talimogene laherparepvec, a
genetically modified herpes simplex virus (HSV), has been approved
by the US Food and Drug Administration for clinical application in
advanced melanoma therapy (6).
Oncolytic HSV G47Δ is a third-generation replication-competent
HSV-1 vector derived from G207 with the deletion of the infected
cell protein 47 (ICP47) gene and both copies of the γ34.5 gene
(7). G47Δ has been used to treat
glioblastoma in clinical trials in Japan (7).
Temozolomide (TMZ) is an imidazotetrazine-derived
alkylating agent used as a first-line oral drug for the treatment
of malignant glioma because it is able to easily pass through the
blood-brain barrier due to its low molecular weight and
lipophilicity (8,9). In addition, TMZ has been used in
clinical trials for the treatment of advanced metastatic melanoma
(10,11). TMZ kills cancer cells via induction
of DNA alkylation and methylation damage in cancer cells (12). However, certain types of cancer cells
are able to repair TMZ-induced DNA damage, leading to resistance to
TMZ, while the genetically modified oncolytic HSV has a decreased
replication efficacy in cancer cells compared with naturally
occurring HSV, which both decreased the anti-cancer efficacy of
such therapies (13,14). Since the therapeutic targets of TMZ
and HSV are different, a strategy using a combination of TMZ and
HSV may notably enhance the efficacy of cancer therapy via a
complementary mechanism.
The present study investigated the combined role of
TMZ and G47Δ in regulating breast cancer cell behavior in
vitro and in vivo, and a preliminary mechanism was also
suggested. The results of the present study may provide valuable
insight into the development of novel therapeutic approaches to
treat breast cancer.
Materials and methods
Cell lines and culture
The human breast cancer cell lines SK-BR-3 and
MDA-MB-468, as well as the African green monkey kidney epithelial
cell line Vero, were gifts from Dr. Musheng Zeng (Sun Yat-sen
University Cancer Center, Guangzhou, China). These cells were grown
in Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% inactivated fetal calf
serum (FCS; Gibco; Thermo Fisher Scientific, Inc.), 100 IU/ml
penicillin, 100 µg/ml streptomycin and 2 mM L-glutamine. All cells
were maintained in a humidified incubator with 5% CO2 at
37°C.
Amplification of G47Δ
G47Δ was a gift from Dr. Samuel D. Rabkin (Harvard
Medical School, Boston, MA, USA) and diluted in 1% inactivated
FCS-containing PBS to infect Vero cells at a multiplicity of
infection (MOI) of 0.03, followed by incubation under standard
conditions (5% CO2 and 37°C) for 90 min. Viral inoculums
were then removed and replaced with 3% inactivated FCS-containing
DMEM, followed by incubation in 5% CO2 for 48–72 h at
34.5°C. Infected cells were collected when >90% of the cells
appeared round and refractile under a light microscope
(magnification, ×400) after calculation of 200 cells with two or
more fused nuclei vs. the total nuclei (fused and un-fused nuclei)
by two investigators and then resuspended in the virus buffer (20
mM Tris and 150 mM NaCl; pH 7.5). The resuspension solution was
subjected to three rapid freeze-thaw cycles for cell lysis and
virus release, followed by centrifugation at 500 × g and 4°C for 10
min. Next, the virus-containing supernatant was collected and
stored in multiple aliquots at −80°C until use. The viral titer was
determined via a plaque assay according to a previous studies
(15,16).
Cell viability assay
SK-BR-3 and MDA-MB-468 cells were seeded into 6-well
plates at a density of 3×105 cells/well and 37°C
incubated for 24 h. The cells were then treated with 2 µl DMSO
(mock), G47Δ (MOI, 0.01) and/or TMZ (6 mM for SK-BR-3 and 60 mM for
MDA-MB-468; Sigma-Aldrich; Merck KGaA) and cultured in 2 ml DMEM
containing 1% FCS at 37°C for 72 h. Next, 5 mg/ml MTT solution
(Sigma-Aldrich; Merck KGaA) was added to each well, and the cell
culture was incubated at 37°C for additional 4 h. The supernatant
was removed carefully, and DMSO was added to dissolve the blue
formazan crystals. The optical density was measured at 490 nm.
Chou-Talalay analysis of drug
synergy
Chou-Talalay analysis (17,18) was
performed to determine the combination index (CI) via assessment of
the cell growth inhibition in G47Δ and/or TMZ-treated tumor cells
using the following equation:
CI=(D)1/(Dx)1 +
(D)2/(Dx)2, where
(Dx)1 is the dose of agent 1 (e.g. G47Δ) required
to produce × percentage effect alone and (D)1 is
the dose of agent 1 required to produce the same × percentage
effect in combination with (D)2. Similarly,
(Dx)2 is the dose of agent 2 (e.g. TMZ) required
to produce × percentage effect alone and (D)2 is
the dose required to produce the same effect in combination with
(D)1. The denominators of the aforementioned CI
equation, (Dx)1 and (Dx)2, can
be determined by
Dx=Dm[fa/(1-fa)]1/m,
where Dm is the dose required for a 50% effect
(e.g. 50% inhibition of cell growth), fa is the
fraction affected by D (e.g. 0.5 if cell growth is inhibited
by 50%), and m is the coefficient of sigmoidicity of the
dose-effect curve. Different values of CI may be obtained to solve
the equation for different values of fa (e.g.
different degrees of inhibition of cell growth). CI<1 indicates
synergy, CI>1 indicates antagonism and CI=1 indicates an
additive effect. EnzFitter software, version 1.22 (Biosoft) was
used to determine the CI values.
Flow cytometry
A preliminary experiment was performed using SK-BR3
and MDA-MB468 cells after treated with a single drug for 48 h to
determine the IC50. Subsequently, these drug doses were
used to assess the effects on tumor apoptosis. In brief, the cells
were seeded into a 6-well plate at a density of 3×105
cells/well and cultured at 37°C for 24 h. The cells were then
treated with 2 µl DMSO (control), G47Δ (MOI, 0.01), and/or TMZ (6
µM for SK-BR-3 and 60 µM for MDA-MB-468) in DMEM containing 1% FCS
for 48 h or 72 h. For the cell cycle analysis, the cells were
cultured for 72 h and then collected, washed with PBS three times
and resuspended, followed by overnight fixation in 70% ethanol at
4°C. On the next day, the cells were incubated with RNase A at 37°C
for 1 h and then stained with 1 µg/ml propidium iodide (PI) in the
dark at 4°C for 30 min. Fluorescence of the stained cells was
detected for cell cycle analysis via the flow cytometer FACSCanto
II (BD Biosciences). The apoptosis assay was performed using an
Annexin V PE/7AAD kit (BD Biosciences) according to the
manufacturer's protocol. For apoptosis analysis, the cells were
cultured for 48 h and then collected and incubated with 1 µg/ml
FITC-Annexin V and PI in the dark at the room temperature for 10
min, and the fluorescent signals were analyzed using a FACSCanto II
flow cytometer (BD Biosciences).
Reverse transcription-quantitative PCR
(RT-qPCR)
SK-BR-3 and MDA-MB-468 cells were treated as
aforementioned. Total RNA was isolated from cells using RNAiso Plus
(Takara Biotechnology Co., Ltd.), according to the manufacturer's
instructions. cDNA was synthesized via RT of RNA using PrimeScript
RT Enzyme Mix and SuperScript™ III First-Strand Synthesis SuperMix
(both Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. The qPCR products were amplified in the
7500 Real-Time PCR system (Applied Biosystems, Foster city, CA,
USA) using SYBR Premix Ex Taq (Invitrogen). The reaction conditions
were: 93°C For 2 min, then 93°C for 1 min, 55°C for 2 min, for a
total of 40 cycles. The primers used are presented in Table I. The relative RNA levels were
determined using the 2−ΔΔCq (19) method and was normalized to
β-actin.
 | Table I.Gene-specific primers for
quantitative PCR. |
Table I.
Gene-specific primers for
quantitative PCR.
| Gene | Sequence,
5′-3′ |
|---|
| ATM | Forward:
ACTGGCCTTAGCAAATGC |
|
| Reverse:
TTGCAGCCTCTGTTCGAT |
| ATR | Forward:
TGTCTGTACTCTTCACGGCATGTT |
|
| Reverse:
AAGAGGTCCACATGTCCGTGTT |
| H2AX | Forward:
CAGTGCTGGAGTACCTCAC |
|
| Reverse:
CTGGATGTTGGGCAGGAC |
| DNA-PKc | Forward:
CTGTGCAACTTCACTAAGTCCA |
|
| Reverse:
CAATCTGAGGACGAATTGCCTGADD34 |
| GADD34 | Forward:
GGAGGAAGAGAATCAAGCCA |
|
| Reverse:
TGGGGTCGGAGCCTGAAGAT |
| RRM1 | Forward:
TGGCCTTGTACCGATGCTG |
|
| Reverse:
GCTGCTCTTCCTTTCCTGTGTT |
| RRM2 | Forward:
GCGATTTAGCCAAGAAGTTCAGAT |
|
| Reverse:
CCCAGTCTGCCTTCTTCTTGA |
| β-actin | Forward:
TGGCACCCAGCACAATGAA |
|
| Reverse:
CTAAGTCATAGTCCGCCTAGAAGCA |
Western blotting
Cells were harvested and lysed in the ice-cold
radioimmunoprecipitation assay buffer containing 50 mM Tris-HCl,
150 mM NaCl, 1% Triton X-100, 0.5% SDS, 1 mM PMSF, 1 mM
Na3VO4, and 0.1% β-mercaptoethanol after
centrifugation at the top speed 8,000 × g for 10 min at 4°C. The
protein concentration was determined via the bicinchoninic acid
assay. Protein samples (20 µg each loading) were loaded and
separated by SDS-PAGE in 10% gels, and were then transferred onto
nitrocellulose membranes. The membranes were then blocked in 5%
bovine serum albumin (BSA; Sigma-Aldrich; Merck KGaA) in Tris-based
saline 0.05% Tween 20 (TBS-T) for 1 h at room temperature, followed
by overnight incubation with primary antibodies [histone H2AX
(H2AX; 1:1,000; cat. no. ab229914), γH2AX (1:1,000; cat. no.
ab243906), ATR (1:1,000; cat. no. ab2905), ATM (1:1,000; cat. no.
ab32420), DNA-dependent protein kinase, catalytic subunit (DNA-PKc;
1:1,000; cat. no. ab32566), growth arrest and DNA damage-inducible
protein GADD34 (GADD34; 1:500; cat. no. ab126075), or β-actin
(1:1,000; cat. no. ab8227); all from Abcam] at 4°C. Then, membranes
were washed three times with TBST and incubated with horseradish
peroxidase-conjugated goad anti-mouse IgG (H&L) (Cat. #ab6789,
Abcam) or rabbit anti-human IgG (H&L) (Cat. #ab6759, Abcam;
both at a dilution of 1:10,000) for 1 h at room temperature. After
three washes with TBST, the protein bands were detected using
enhanced chemiluminescence reagents (cat no. #35055; Pierce; Thermo
Fisher Scientific, Inc.). The western blot imagines were captured
and quantified by using a GBOX XT-16 chemiluminescent imager
(Syngene) after 20-min exposure of the membranes.
Animal experiments
All animal procedures were approved by the
Institutional Animal Care and Use Committee of The Third Affiliated
Hospital of Sun Yat-sen University (approval no. 11400700083061).
Female BALB/c nude mice with 4 weeks of age and 14–16 g body weight
were housed in clean cages and maintained in specific pathogen-free
‘barrier’ facility with the controlled temperature at 23°C, the
relative humidity of 40–70%, and a 12-h light/dark cycle with free
access to food and water. The mice were acclimated for 7 days
before the experiments. A total of 28 mice were randomly divided
into four groups (n=7/group): Mock, G47Δ, TMZ and G47Δ + TMZ. Mice
were intraperitoneally anesthetized with ketamine-xylazine,
followed by subcutaneous inoculation with 1×106 SK-BR-3
cells in the right hindlimb. Tumor growth was monitored daily, and
tumor size was measured every 4 days using a Vernier caliper. The
tumor volume was calculated as a × b2/2, where a is the
longest and b is the shortest tumor diameter. When the longest
tumor diameter reached ~5 mm, TMZ and/or G47Δ was administered to
the mice, except for those in the Mock group. For the TMZ group,
TMZ was intraperitoneally administered once a week at a dose of 50
mg/kg. For the G47Δ group, G47Δ was injected intratumorally once
every 3 days for a total of four times at a dose of
1×106 pfu/mouse. In the G47Δ plus TMZ group, TMZ and
G47Δ were administered in combination in the aforementioned manner.
The mice were sacrificed 60 days after inoculation or when the
longest tumor diameter reached 18 mm via CO2 and
cervical dislocation and all tumor xenografts were taken and
analyzed using different assays (see details in the corresponding
methods parts).
Statistical analysis
The data were expressed as the mean ± standard
deviation of the triplicated experiments and were statistically
analyzed using SPSS software (version 13.0; SPSS, Inc.).
Comparisons between two groups were conducted using the Student's
t-test and Pearson correlation analysis was used to assess
correlation. Comparisons of multiple groups was analyzed using the
one-way ANOVA followed by Bonferroni's correction. P<0.05 was
considered to indicate a statistically significant difference.
Results
G47Δ and TMZ synergistically inhibit
breast cancer cell viability in vitro
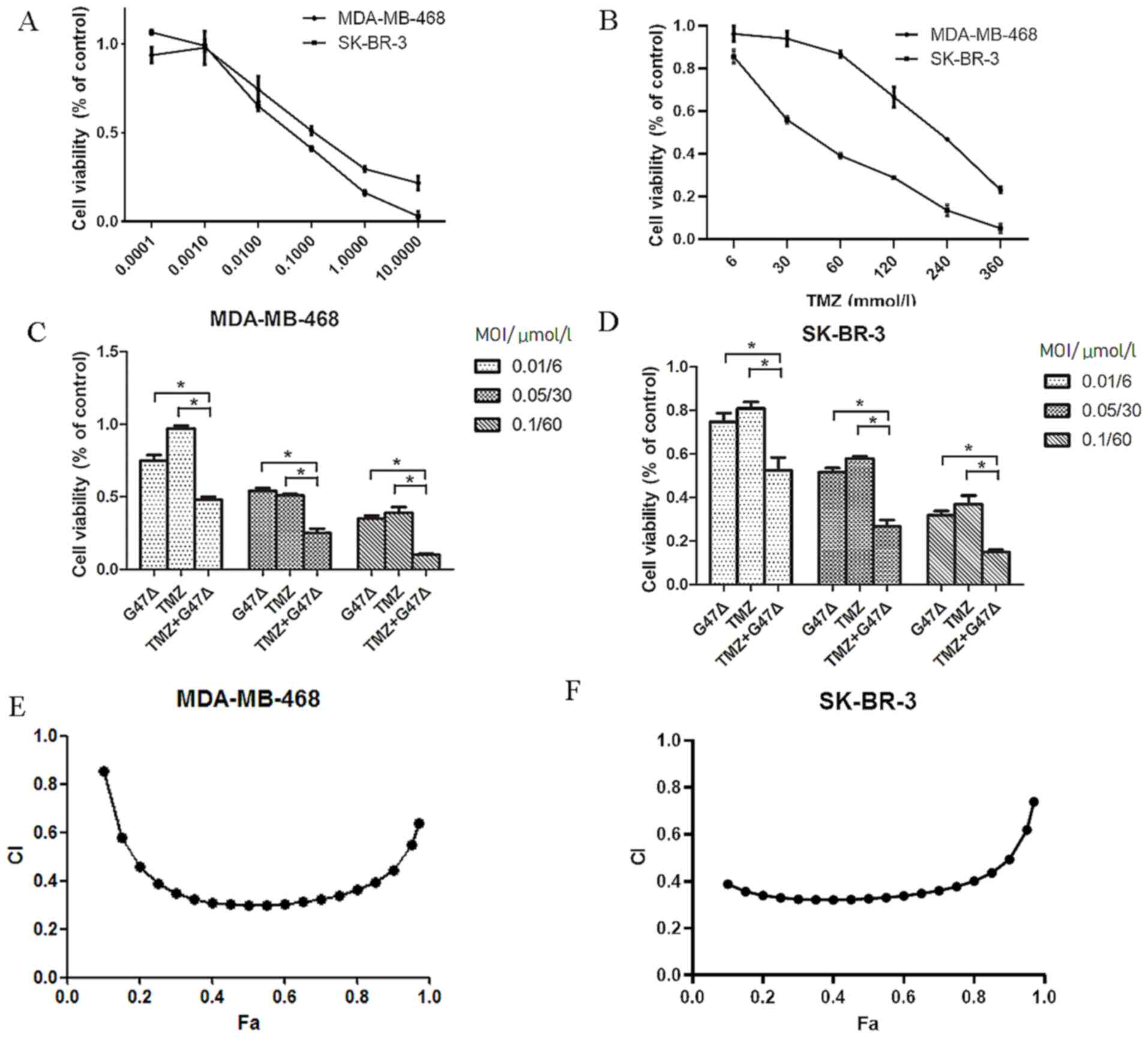
In order to investigate the combined effect of G47Δ
and TMZ on breast cancer cells, the individual effects of G47Δ and
TMZ on the viability of SK-BR-3 and MDA-MB-468 cells were assessed.
Treatment with G47Δ or TMZ alone inhibited viability of SK-BR-3 and
MDA-MB-468 cells in a dose-dependent manner, with median effective
doses (ED50) of 0.09 and 0.20 MOI for G47Δ, and 36.6 and
171.9 µM for TMZ in SK-BR-3 and MDA-MB-468 cells, respectively
(Fig. 1A and B). G47Δ and TMZ in
combination further suppressed breast cancer cell viability in a
dose-dependent manner, compared with G47Δ or TMZ alone (Fig. 1C and D), indicating a potential
synergy between G47Δ and TMZ in inhibiting cell viability. In order
to determine whether synergy existed between G47Δ and TMZ, the
Chou-Talalay analysis was performed for multiple G47Δ/TMZ ratios.
CI was 0.39–0.74 (CI, <0.9) in SK-BR-3 cells and 0.64–0.85 (CI,
<0.9) in MDA-MB-468 cells (Fig. 1E
and F). These results indicated that G47Δ and TMZ exhibited a
synergistic inhibitory effect on breast cancer cell growth.
G47Δ and TMZ synergistically induce
breast cancer cell cycle arrest in vitro
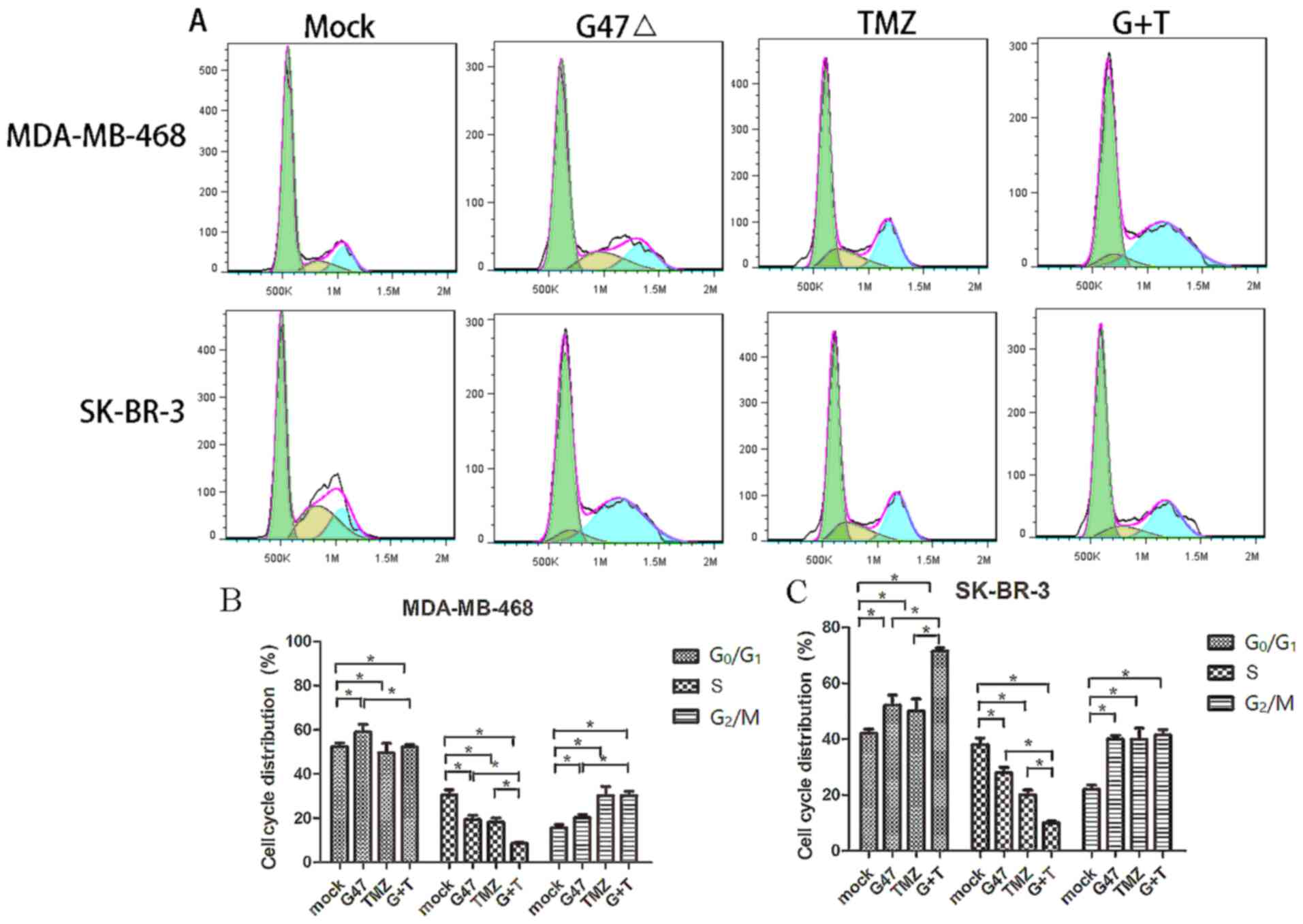
Cell cycle progression is associated with cell
division and growth (20). The
combined effect of G47Δ and TMZ on inhibition of breast cancer cell
cycle progression was investigated. G47Δ treatment alone induced
SK-BR-3 and MDA-MB-468 cell cycle arrest at the
G0/G1 phase of the cell cycle (52.1±2.1 and
58.9±3.6%, respectively), whereas TMZ alone induced SK-BR-3 and
MDA-MB-468 cell cycle arrest at the G2/M phase (39.9±3.7
and 30.2±4.1%, respectively) (Fig.
2). By contrast, a combination of G47Δ and TMZ notably arrested
the cell cycle at the G0/G1 phase in SK-BR-3
cells (71.4±1.0 vs. G47Δ 52.1±2.1%; P=0.003) and at the
G2/M phase in MDA-MB-468 cells (41.5±2.0 vs. TMZ
30.2±4.1%; P=0.012). These data indicated that G47Δ and TMZ in
combination exhibited a more significant suppressive effect on
breast cancer cell cycle progression than G47Δ or TMZ alone,
suggesting a synergy between G47Δ and TMZ in the induction of
breast cancer cell cycle arrest.
G47Δ and TMZ synergistically promote
breast cancer cell apoptosis in vitro
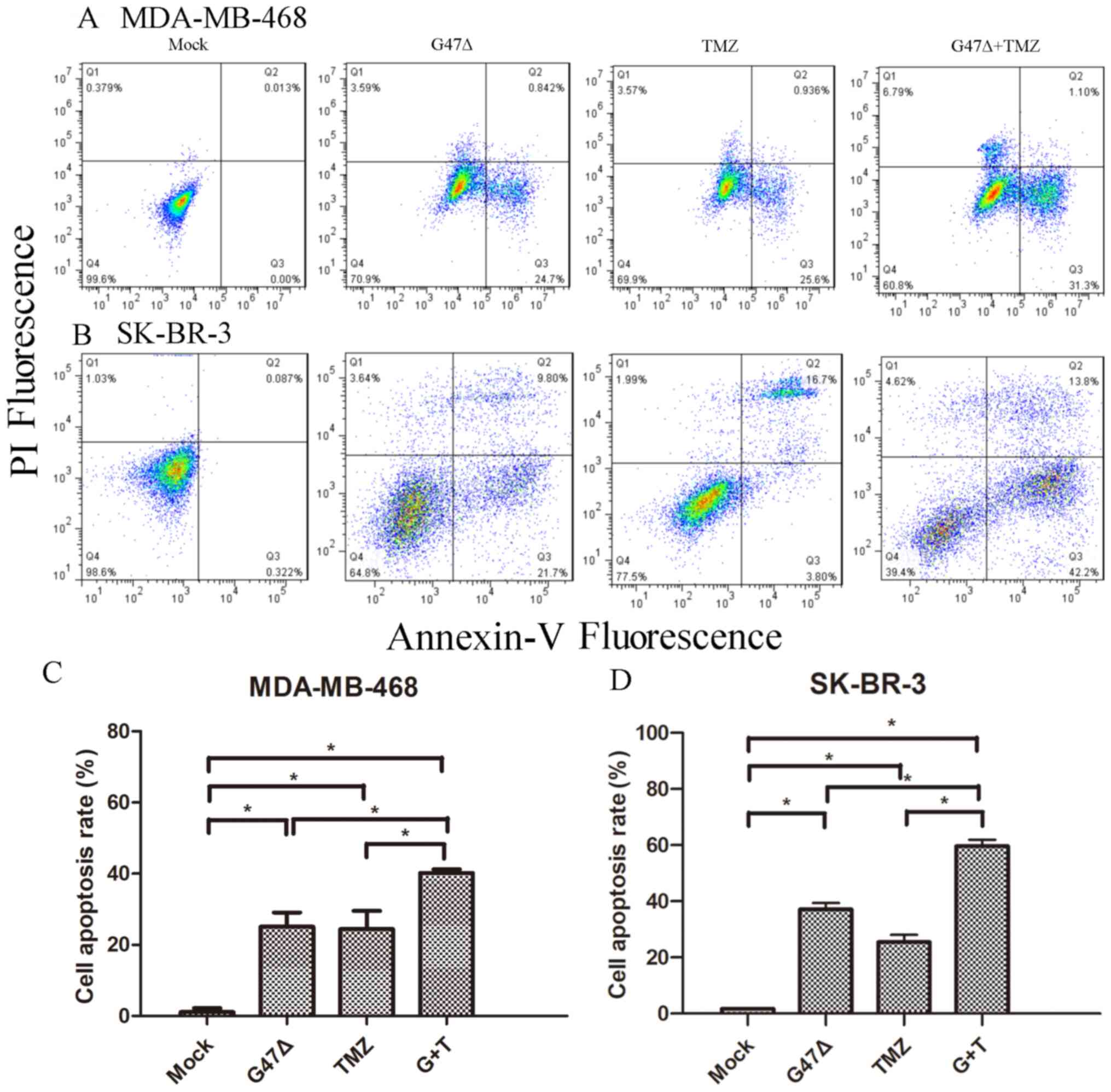
Cell apoptosis is a key process during tumor
development (21). Therefore, it was
investigated whether G47Δ and TMZ may exhibit a synergistic effect
on breast cancer cell apoptosis. G47Δ and TMZ individually markedly
induced SK-BR-3 cell apoptosis, compared with the mock control
group (37.10±2.30 and 25.50±2.60 vs. 1.70±0.26%, respectively)
(Fig. 3). Combined G47Δ and TMZ
significantly promoted SK-BR-3 cell apoptosis (59.60±2.25 vs.
37.10±2.30 and 25.50±2.60%; both P<0.05). Similarly, a
combination of G47Δ and TMZ further enhanced MDA-MB-468 cell
apoptosis (40.2±1.1 vs. 25.1±4.4 and 24.4±5.1%, respectively; both
P<0.05 vs. control group). Collectively, these data suggested
that G47Δ and TMZ synergistically decreased tumor cell
proliferation via induction of breast cancer cell apoptosis.
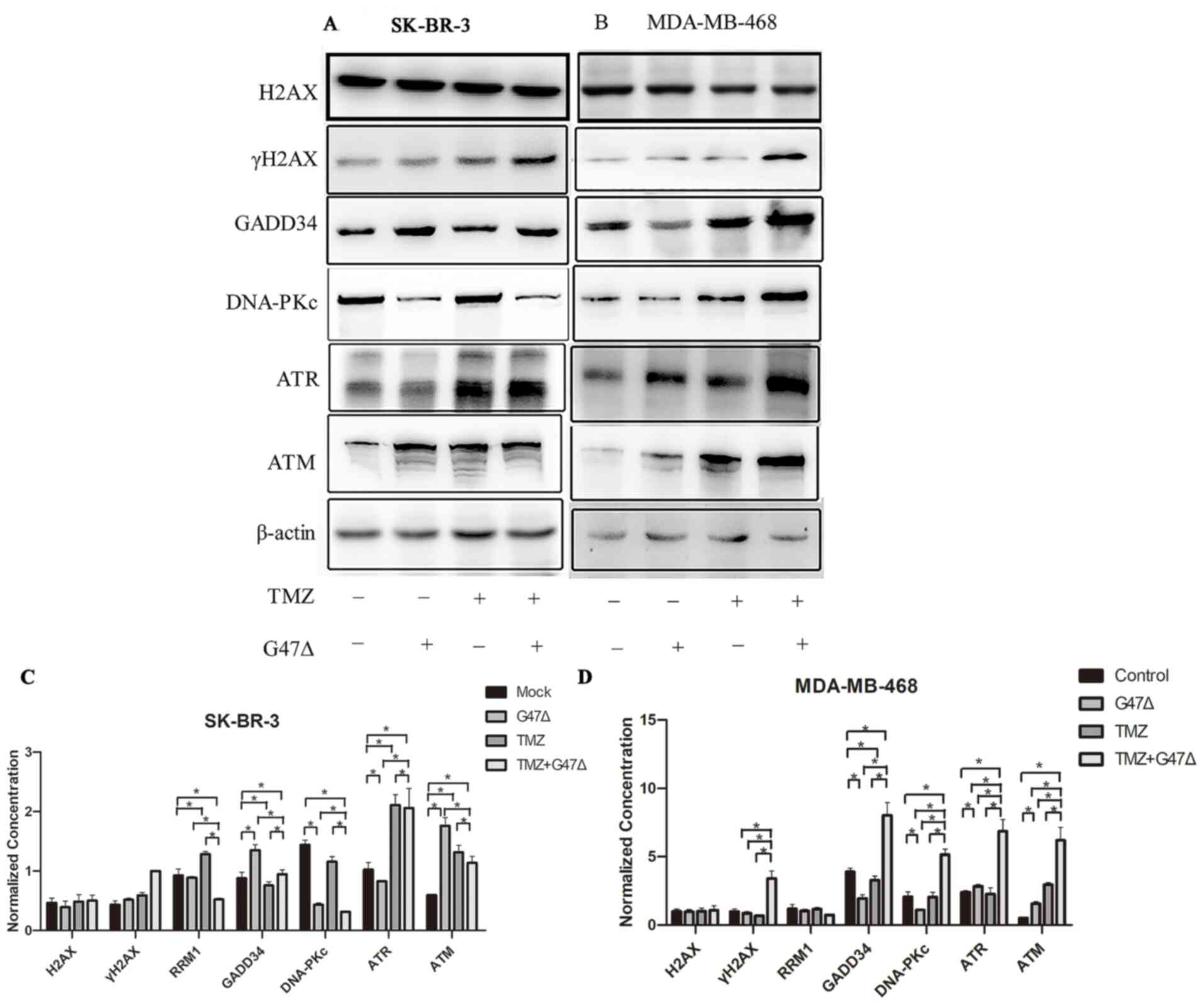
G47Δ and TMZ synergistically regulate
the expression levels of DNA damage-associated genes in breast
cancer cells
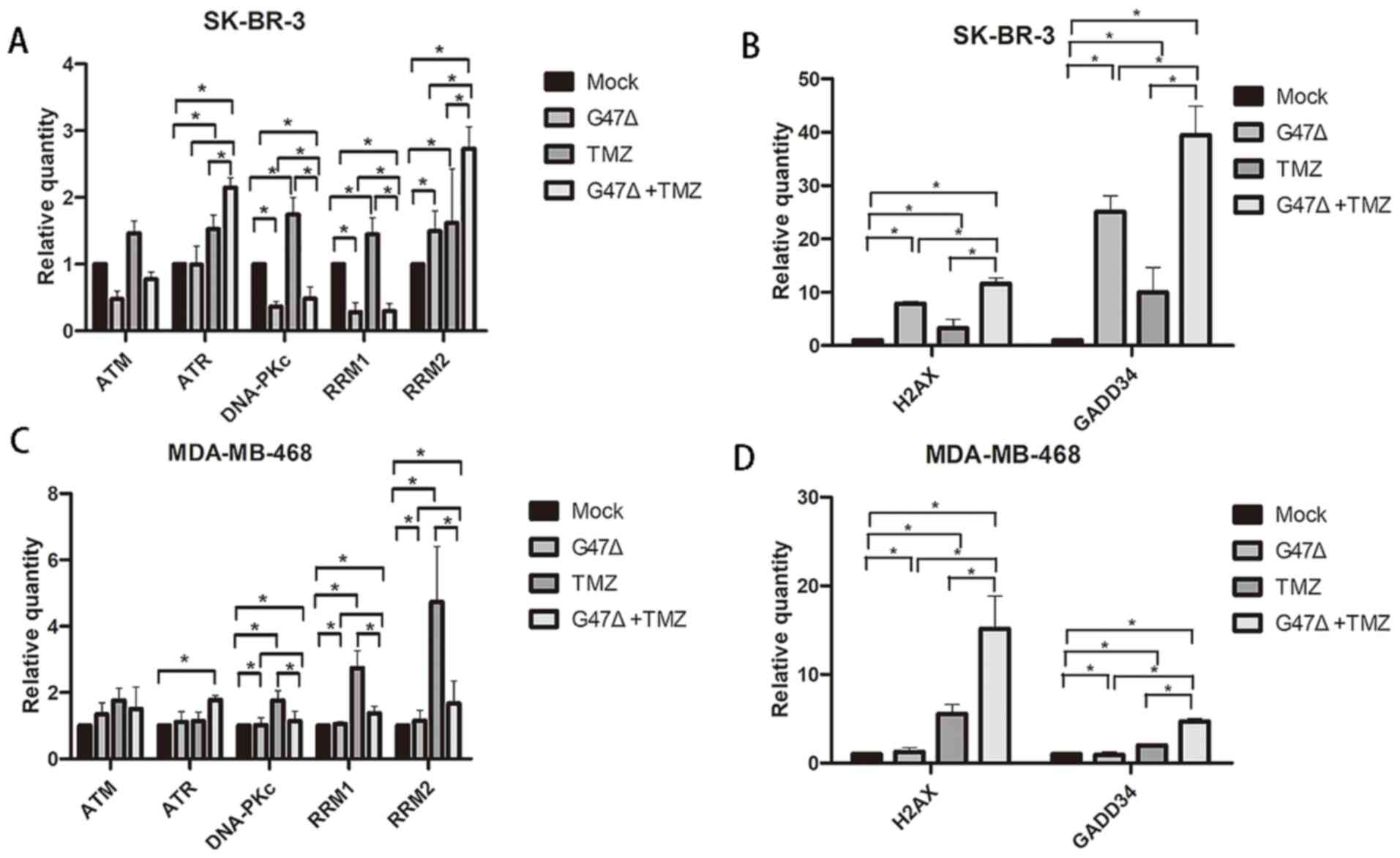
Cell cycle arrest and apoptosis are triggered by DNA
damage in cells (22). Therefore, it
was determined whether G47Δ and TMZ serve a synergistic role in
induction of DNA damage in breast cancer cells. The results showed
that G47Δ or TMZ alone induced expression of γH2AX protein but not
the H2AX mRNA level; γH2AX is a sensitive molecular marker of DNA
double-strand breaks (23), in
SK-BR-3 cells (Figs. 4 and 5). G47Δ and TMZ in combination further
augmented the individual effect of G47Δ or TMZ on the mRNA and
protein expression levels of H2AX and γH2AX, respectively. In
addition, G47Δ and TMZ in combination notably promoted G47Δ- or
TMZ-induced expression levels of GADD34, TM, DNA-PKc, RRM1, RRM2,
and ATR, which are key DNA damage response genes (24). Similar results were also observed in
MDA-MB-468 cells (Figs. 4 and
5). These findings indicated that
G47Δ and TMZ synergistically upregulated the expression levels of
DNA damage-associated genes, and may thus induce DNA damage and
trigger the DNA damage response, which in turn leads to breast
cancer cell cycle arrest and apoptosis (Fig. 3).
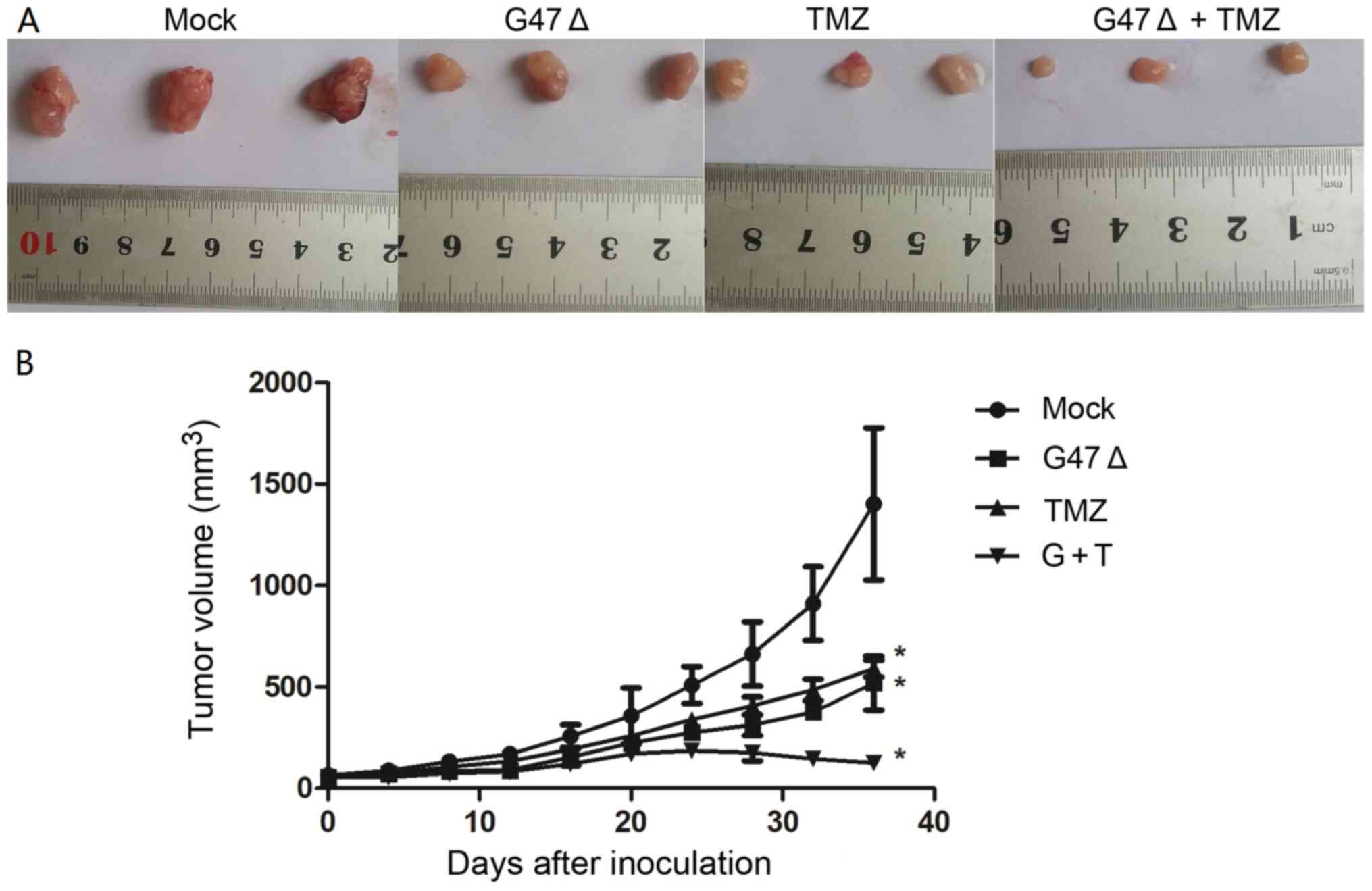
G47Δ and TMZ synergistically suppress
breast cancer cell-derived tumor growth in vivo
In order to further investigate the combined role of
G47Δ and TMZ in breast cancer cell tumorigenesis in vivo, a
breast cancer xenograft model was established by inoculating
SK-BR-3 cells into nude mice, which were then treated with G47Δ and
TMZ individually or in combination. As shown in Fig. 6, G47Δ or TMZ alone significantly
decreased the size of SK-BR-3 cell-derived tumor xenografts in a
time-dependent manner, compared with the mock group [519.0±133.3
(n=6) and 591.3±41.8 (n=7) vs. 1,402.3±375.3 mm3 (n=6)
at 36 days after inoculation, respectively; both P<0.05]. A
combination of G47Δ and TMZ further enhanced the inhibitory effect
of individual G47Δ or TMZ on tumor growth [125.0±7.6 (n=7) vs.
519.0±133.3 (n=6) and 591.3±41.8 mm3 (n=7) at 36 days
after inoculation, respectively; both P<0.05 vs. control group].
These data indicated that combined treatment with G47Δ and TMZ
decreased breast cancer cell-derived tumor growth more effectively
than treatment with G47Δ or TMZ alone, suggesting a synergy between
G47Δ and TMZ in suppressing breast cancer cell tumorigenesis in
vivo.
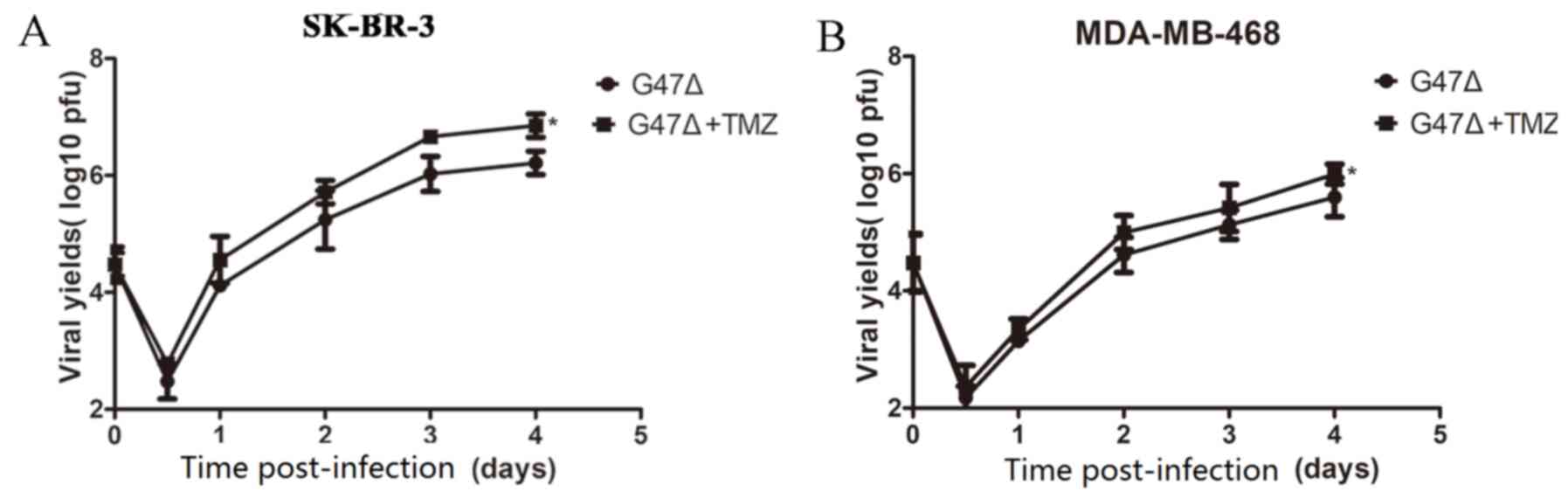
TMZ accelerates G47Δ replication in
vitro
The mechanism underlying the synergistic inhibitory
effect of G47Δ and TMZ on breast cancer cell tumorigenesis was
further investigated. The yield of G47Δ increased in a
time-dependent manner in SK-BR-3 and MDA-MB-468 cells treated with
G47Δ and TMZ together, compared with that in cells treated with
G47Δ alone (Fig. 7). These findings
indicated that a synergy between G47Δ and TMZ in inhibiting breast
cancer cell tumorigenesis may be at least partially due to
acceleration of G47Δ replication by TMZ.
Discussion
Genetically modified replication-competent oncolytic
HVS strains have been used as oncolytic virus therapy in which
cancer cells are killed via a direct oncolytic effect of the virus
and induction of host immunity (25). However, due to the highly attenuated
replication ability of these genetically altered viruses, including
HSV G47Δ, oncolytic virus therapy is commonly used in combination
with chemotherapy or radiotherapy to improve the efficiency of
cancer treatment.
In the present study, the combination of G47Δ and
TMZ induced stronger cytotoxicity than G47Δ or TMZ alone in breast
cancer cells. This synergy is likely due to the distinct mechanisms
of G47Δ and TMZ in killing breast cancer cells. For example, TMZ
induces cancer cell DNA damage/repair, as evidenced by TMZ-induced
upregulation of the DNA damage response genes ATR and GADD34. By
contrast, G47Δ replicates in and lyses cancer cells. A previous
study demonstrated that TMZ-induced DNA repair notably enhanced
HSV-mediated oncolysis in primary brain tumor cells via promotion
of HSV replication (26). Therefore,
TMZ treatment may induce the sensitivity of breast cancer cells to
G47Δ infection, resulting in accelerated G47Δ replication and
augmented cancer cell lysis. The present study confirmed that TMZ
may promote G47Δ replication in breast cancer cells in
vitro.
The present study also demonstrated that G47Δ and
TMZ in combination synergistically induced breast cancer cell
apoptosis. A previous study demonstrated that HSV can induce robust
apoptosis of TMZ-resistant glioma cells both in vitro and
in vivo, indicating synergy between G47Δ and TMZ (27). DNA damage promotes cell apoptosis if
such DNA damage is not repaired (22,28,29). In
TMZ-resistant breast cancer, repairing TMZ-induced DNA damage
promotes G47Δ replication in breast cancer cells. These cells are
lysed, triggering the host immune response and resulting in
cytokine-induced cancer cell apoptosis (30). In addition, the synergistic role of
G47Δ and TMZ in DNA damage may interfere with DNA replication in
breast cancer cells, further affecting cancer cell division. The
present study demonstrated that G47Δ and TMZ exhibited a
synergistic effect on induction of breast cancer cell cycle arrest.
The cell cycle was arrested at different phases in SK-BR-3 and
MDA-MB-468 cells, which is likely due to the different genetic
background between these two cell lines (31).
The synergy between G47Δ and TMZ in regulation of
breast cancer cell behavior was further verified in a nude mouse
xenograft model. The in vivo synergy in inhibition of breast
cancer cell-derived tumor xenograft growth may be due to the
effects of treatments on breast cancer cell viability, apoptosis,
cycle arrest and DNA damage/repair. The present study may provide
valuable information for the potential clinical application of G47Δ
and TMZ in breast cancer treatment.
However, the present study has certain limitations.
For example, although the combined effect of G47Δ and TMZ on breast
cancer cell behaviors (due to acceleration of G47Δ replication by
TMZ) was demonstrated, the underlying mechanism by which TMZ
promoted G47Δ replication was not investigated in detail. Moreover,
it was not assessed whether G47Δ may serve a role in increasing the
sensitivity of breast cancer cells to TMZ. The present study also
did not identify the optimal dose combination of G47Δ and TMZ to
suppress breast cancer cell tumorigenesis in vitro and in
vivo. Finally, clinical or preclinical data were not available
to assess the therapeutic value of G47Δ and TMZ in combination for
breast cancer development and progression, although previous
studies have demonstrated anti-breast cancer activity in
vitro (32–34). Further investigation is required to
elucidate these points. In conclusion, the present study
demonstrated that the combined administration of G47Δ and TMZ
effectively suppressed breast cancer cell-derived tumor growth
in vivo, compared with the administration of G47Δ or TMZ
alone. Synergy between G47Δ and TMZ was at least partially mediated
via TMZ-induced acceleration of G47Δ replication, and such a
synergy in breast cancer cells in vitro and in vivo
provides novel insight into the future development of a therapeutic
strategy against breast cancer.
Acknowledgements
Not applicable.
Funding
The present study was funded in part by a grant from
the Xinjiang Medical University Research and Innovation Fund
Project (grant no. XYDCX201677).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JF and RL designed the research. JF performed the
main experiments and drafted the paper. HJ participated in the
construction of the animal models. LC analyzed the data. BM
participated the study design, interpreted the data and prepared
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal procedures were approved by the
Institutional Animal Care and Use Committee of The Third Affiliated
Hospital of Sun Yat-sen University (approval no.
11400700083061).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
TMZ
|
temozolomide
|
|
HSV
|
herpes simplex virus
|
|
MOI
|
multiplicity of infection
|
|
CI
|
combination index
|
References
|
1
|
Shah R, Rosso K and Nathanson SD:
Pathogenesis, prevention, diagnosis and treatment of breast cancer.
World J Clin Oncol. 5:283–298. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tinoco G, Warsch S, Gluck S, Avancha K and
Montero AJ: Treating breast cancer in the 21st century: Emerging
biological therapies. J Cancer. 4:117–132. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lou E: Oncolytic herpes viruses as a
potential mechanism for cancer therapy. Acta Oncol. 42:660–671.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chiocca EA and Rabkin SD: Oncolytic
viruses and their application to cancer immunotherapy. Cancer
Immunol Res. 2:295–300. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Burke J, Nieva J, Borad MJ and Breitbach
CJ: Oncolytic viruses: Perspectives on clinical development. Curr
Opin Virol. 13:55–60. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rehman H, Silk AW, Kane MP and Kaufman HL:
Into the clinic: Talimogene laherparepvec (T-VEC), a first-in-class
intratumoral oncolytic viral therapy. J Immunother Cancer.
4:532016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fukuhara H, Ino Y and Todo T: Oncolytic
virus therapy: A new era of cancer treatment at dawn. Cancer Sci.
107:1373–1379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mannas JP, Lightner DD, Defrates SR,
Pittman T and Villano JL: Long-term treatment with temozolomide in
malignant glioma. J Clin Neurosci. 21:121–123. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sengupta S, Marrinan J, Frishman C and
Sampath P: Impact of temozolomide on immune response during
malignant glioma chemotherapy. Clin Dev Immunol. 2012:8310902012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bafaloukos D, Tsoutsos D, Kalofonos H,
Chalkidou S, Panagiotou P, Linardou E, Briassoulis E, Efstathiou E,
Polyzos A, Fountzilas G, et al: Temozolomide and cisplatin versus
temozolomide in patients with advanced melanoma: A randomized phase
II study of the hellenic cooperative oncology group. Ann Oncol.
16:950–957. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dronca RS, Allred JB, Perez DG, Nevala WK,
Lieser EA, Thompson M, Maples WJ, Creagan ET, Pockaj BA, Kaur JS,
et al: Phase II study of temozolomide (TMZ) and everolimus (RAD001)
therapy for metastatic melanoma: A north central cancer treatment
group study, N0675. Am J Clin Oncol. 37:369–376. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang J, Stevens MF and Bradshaw TD:
Temozolomide: Mechanisms of action, repair and resistance. Curr Mol
Pharmacol. 5:102–114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kondo N, Takahashi A, Mori E, Noda T,
Zdzienicka MZ, Thompson LH, Helleday T, Suzuki M, Kinashi Y,
Masunaga S, et al: FANCD1/BRCA2 plays predominant role in the
repair of DNA damage induced by ACNU or TMZ. PLoS One.
6:e196592011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fu X, Tao L, Wang PY, Cripe TP and Zhang
X: Comparison of infectivity and spread between HSV-1 and HSV-2
based oncolytic viruses on tumor cells with different receptor
expression profiles. Oncotarget. 9:21348–21358. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Baer A and Kehn-Hall K: Viral
concentration determination through plaque assays: Using
traditional and novel overlay systems. J Vis Exp.
e520652014.PubMed/NCBI
|
|
16
|
Han C and Yang C: Viral plaque analysis on
a wide field-of-view, time-lapse, on-chip imaging platform.
Analyst. 139:3727–3734. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chou TC: The median-effect principle and
the combination index for quantitation of synergism and antagonism.
Chou TC and Rideout DC: Synergism and antagonism in chemotherapy.
Academic Press; San Diego: pp. 61–102. 1991
|
|
18
|
Khafif A, Schantz SP, Chou TC, Edelstein D
and Sacks PG: Quantitation of chemopreventive synergism between
(−)-epigallocatechin-3-gallate and curcumin in normal, premalignant
and malignant human oral epithelial cells. Carcinogenesis.
19:419–424. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vallin J and Grantham J: The role of the
molecular chaperone CCT in protein folding and mediation of
cytoskeleton-associated processes: Implications for cancer cell
biology. Cell Stress Chaperones. 24:17–27. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Morata G and Ballesteros-Arias L: Cell
competition, apoptosis and tumour development. Int J Dev Biol.
59:79–86. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Roos WP and Kaina B: DNA damage-induced
cell death by apoptosis. Trends Mol Med. 12:440–450. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mah LJ, El-Osta A and Karagiannis TC:
gammaH2AX: A sensitive molecular marker of DNA damage and repair.
Leukemia. 24:679–686. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Thomas SE, Malzer E, Ordóñez A, Dalton LE,
van't Wout EF, Liniker E, Crowther DC, Lomas DA and Marciniak SJ:
p53 and translation attenuation regulate distinct cell cycle
checkpoints during endoplasmic reticulum (ER) stress. J Biol Chem.
288:7606–7617. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Watanabe D: Medical application of herpes
simplex virus. J Dermatol Sci. 57:75–82. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Aghi M, Rabkin S and Martuza RL: Effect of
chemotherapy-induced DNA repair on oncolytic herpes simplex viral
replication. J Natl Cancer Inst. 98:38–50. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jahan N, Lee JM, Shah K and Wakimoto H:
Therapeutic targeting of chemoresistant and recurrent glioblastoma
stem cells with a proapoptotic variant of oncolytic herpes simplex
virus. Int J Cancer. 141:1671–1681. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Norbury CJ and Zhivotovsky B: DNA
damage-induced apoptosis. Oncogene. 23:2797–2808. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Borges HL, Linden R and Wang JY: DNA
damage-induced cell death: Lessons from the central nervous system.
Cell Res. 18:17–26. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Prestwich RJ, Errington F, Diaz RM, Pandha
HS, Harrington KJ, Melcher AA and Vile RG: The case of oncolytic
viruses versus the immune system: Waiting on the judgment of
Solomon. Hum Gene Ther. 20:1119–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Holliday DL and Speirs V: Choosing the
right cell line for breast cancer research. Breast Cancer Res.
13:2152011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Melisko ME, Assefa M, Hwang J, DeLuca A,
Park JW and Rugo HS: Phase II study of irinotecan and temozolomide
in breast cancer patients with progressing central nervous system
disease. Breast Cancer Res Treat. 177:401–408. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bobustuc GC, Kassam AB, Rovin RA, Jeudy S,
Smith JS, Isley B, Singh M, Paranjpe A, Srivenugopal KS and Konduri
SD: MGMT inhibition in ER positive breast cancer leads to CDC2,
TOP2A, AURKB, CDC20, KIF20A, Cyclin A2, Cyclin B2, Cyclin D1, ERα
and Survivin inhibition and enhances response to temozolomide.
Oncotarget. 9:29727–29742. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Garza-Morales R, Gonzalez-Ramos R, Chiba
A, Montes de Oca-Luna R, McNally LR, McMasters KM and
Gomez-Gutierrez JG: Temozolomide enhances triple-negative breast
cancer virotherapy in vitro. Cancers (Basel). 10:1442018.
View Article : Google Scholar
|