Introduction
Sezary syndrome is a rare type of non-Hodgkin
lymphoma, a common form of cutaneous T-cell lymphomas (1). Although the pathophysiology of Sezary
syndrome is not completely understood, it has been reported that a
RAS gene mutation is associated with its poor prognosis
(2). HUT78 is a Sezary syndrome cell
line, containing the neuroblastoma rat sarcoma oncogene
(NRAS)Q61K mutation and an amplified RAS/RAF/MEK
signaling pathway (2). This mutation
sensitizes the HUT78 cells toward suppression of the RAS/RAF/MEK
signaling pathway by NRAS knockdown as well as treatment with MEK
inhibitors (2). Phosphoinositide
3-kinase (PI3K)/Akt signaling is the second best validated NRAS
downstream (3), and melanoma cell
lines harboring NRASQ61K mutation are sensitive to the
combination of MEK/ERK and PI3K/Akt signaling inhibitors (4). However, HUT78 cells are refractory to
the PI3K inhibitors, maybe because of the existence of PTEN
(5), suggesting PI3K-independent
suppression of Akt activity is necessary.
Kinase inhibitors are indispensable in the current
therapy field for diseases with protein hyperphosphorylation,
especially cancer. However, there are various problems associated
with these inhibitors, such as drug resistance and limited targets.
Recently, reactivation of protein phosphatase 2A (PP2A) has
attracted a lot of attention as a different angle to block protein
hyperphosphorylation (6). PP2A is a
highly conserved eukaryotic serine/threonine protein phosphatase
that regulates a wide range of intracellular signal transduction
pathways, including MEK/ERK and PI3K/Akt (7–9). In
T-cell acute lymphoblastic leukemia (T-ALL) and chronic lymphocytic
leukemia, PP2A activity is low, and its reactivation exerts
antitumor effects (10,11). Perphenazine (PPZ) is a classically
used phenothiazine in neuroleptic-type anti-psychotic medications
(12). Previous research, including
our own, showed that PPZ exerts antitumor effects in T-ALL cells
through PP2A activation (10,13).
Because Akt is a substrate of PP2A, PP2A activation is expected to
directly inhibit Akt activity and induce apoptosis in cancer cells
that are resistant to PI3K inhibitors. However, it is unknown
whether PP2A reactivation by PPZ also exerts antitumor effects on
HUT78.
In this study, we revealed that PPZ exerts antitumor
effects on HUT78 by dephosphorylating Akt in a PP2A-dependent
manner. Interestingly, PPZ also dephosphorylated ERK1/2; however,
PP2A was not involved in this process, and an ERK1/2 inhibitor did
not suppress cell growth. In HUT78 cells, MEK inhibition led to
dephosphorylation of Akt, resulting in antitumor effects. Our
findings suggest that Akt inhibition by PP2A re-activation may be
an effective strategy for the treatment of NRAS-mutated
Sezary syndrome.
Materials and methods
Reagent and antibodies
The reagents used in this study were as follows: PPZ
and FR180204 (Sigma-Aldrich), Akt inhibitor VIII (Santa Cruz
Biotechnologies.), BCI (Merck Millipore), and U0126 (LC
Laboratories.). The antibodies used in this study were as follows:
Anti-p97/VCP, anti-B-Raf, anti-N-Myc downstream-regulated gene 1
(NDRG1) (GeneTex), anti-cleaved caspase 3 Asp175,
anti-pThr202/Tyr204 ERK1/2, anti-pThr308 Akt, anti-pSer473 Akt,
anti-pSer241 PDK1, anti-PDK1, anti-pThr346 (NDRG1),
anti-pSer217/221 MEK1/2, anti-pSer445 B-Raf, anti-pSer338 C-Raf,
anti-ERK1/2, and anti-Akt (Cell Signaling), anti-C-Raf (BD
Biosciences).
Cell culture
Human T cell acute lymphoblastic leukemia cell lines
(CCRF-CEM, Jurkat, TALL1) and the human Sezary syndrome cell line
(HUT78) were obtained from RIKEN BRC. The canine T cell acute
lymphoblastic leukemia cell line (UL-1) was kindly provided by Dr
Hajime Tsujimoto. MycoAlert Mycoplasma Detection kit (Lonza) was
used to test mycoplasma contamination. The cells were cultured in
RPMI1640 (Sigma-Aldrich) containing 10% fetal bovine serum (FBS,
Nichirei Biosciences) and antibiotic/antimycotic (Nacalai
Tesque).
Cell viability assay
Cells (4.0×104) were seeded on 96-well
plates, and drugs were added to the medium after 24 h. Following
three-day culture, cell viability assays were performed using Cell
Counting Kit-8 (CCK8, Dojindo) as previously described (13).
Flow cytometry
HUT78 cells were treated with PPZ (20 µM) for 12 h.
Apoptotic cells were stained with Annexin V-FITC Apoptosis
Detection Kit (Bio Vision) as per the manufacture's instructions.
Annexin V- and propidium iodide-positive cells were detected by
flow cytometer (BD Accuri; BD Biosciences) as previously described
(13).
Immunoblotting
Immunoblotting was performed as previously described
(13). Briefly, cell lysis buffer
containing 50 mM Tris-HCl (pH 8.0), 5 mM ethylenediaminetetraacetic
acid (EDTA) (pH 8.0), 5 mM ethylene glycol tetraacetic acid (EGTA),
1% Triton X-100, 1 mM Na3VO4, 20 mM sodium
pyrophosphate and Roche's complete protease inhibitor cocktail. The
protein concentrations were measured by DC protein assay kit
(Bio-Rad Laboratories, Inc.), and were separated by SDS-PAGE, then
transferred onto ClearTrans Nitrocellulose Membrane (Wako).
Membranes were blocked with 0.5 or 3% skim milk, and treated with
primary antibodies in Tris-buffered saline (TBS) containing 0.05%
Tween-20 (TBS-T). After treating with secondary antibodies in
TBS-T, immunoreactive bands were detected using ECL Pro Western
Blotting Detection Reagent (PerkinElmer.) and visualized using a
LAS-3000 luminescent image analyzer (Fujifilm) or ImageQuant 800
(GE Healthcare). Valosin-containing protein (VCP) was used as a
loading control.
In vitro kinase assay
Active-MEK1 (50 ng) (SignalChem, M02-10G) and 200 ng
of unactive-ERK1 (SignalChem, M29-14U) were mixed into kinase
buffer (25 mM MOPS pH 7.2, 12.5 mM sodium glycerophosphate, 25 mM
MgCl2, 5 mM EGTA, 2 mM EDTA, and 0.25 mM DTT) with or
without PPZ (10 µM), and rotated for 1 h at 4°C. Thereafter, 25 mM
of ATP were added and incubated for 15 min at 30°C. The reaction
was stopped by adding sodium dodecyl sulfate (SDS) sample buffer
and samples were incubated for 5 min at 100°C. Total ERK1 and ERK1
phosphorylation were detected by immunoblotting.
Statistical analysis
The results are expressed as means ± standard
deviation. Student's t-tests were performed to compare two groups.
Comparison of three or more was performed using one-way analysis of
variance (ANOVA), followed by Fisher least significant difference
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
PPZ induces apoptosis in HUT78
cells
We investigated whether PPZ exerts antitumor effects
in Sezary syndrome cells as well as in T-ALL cells. In addition, we
also compared the sensitivity of the NRAS-mutated Sezary
syndrome cell line (HUT78) to PPZ with that of T-ALL cell lines
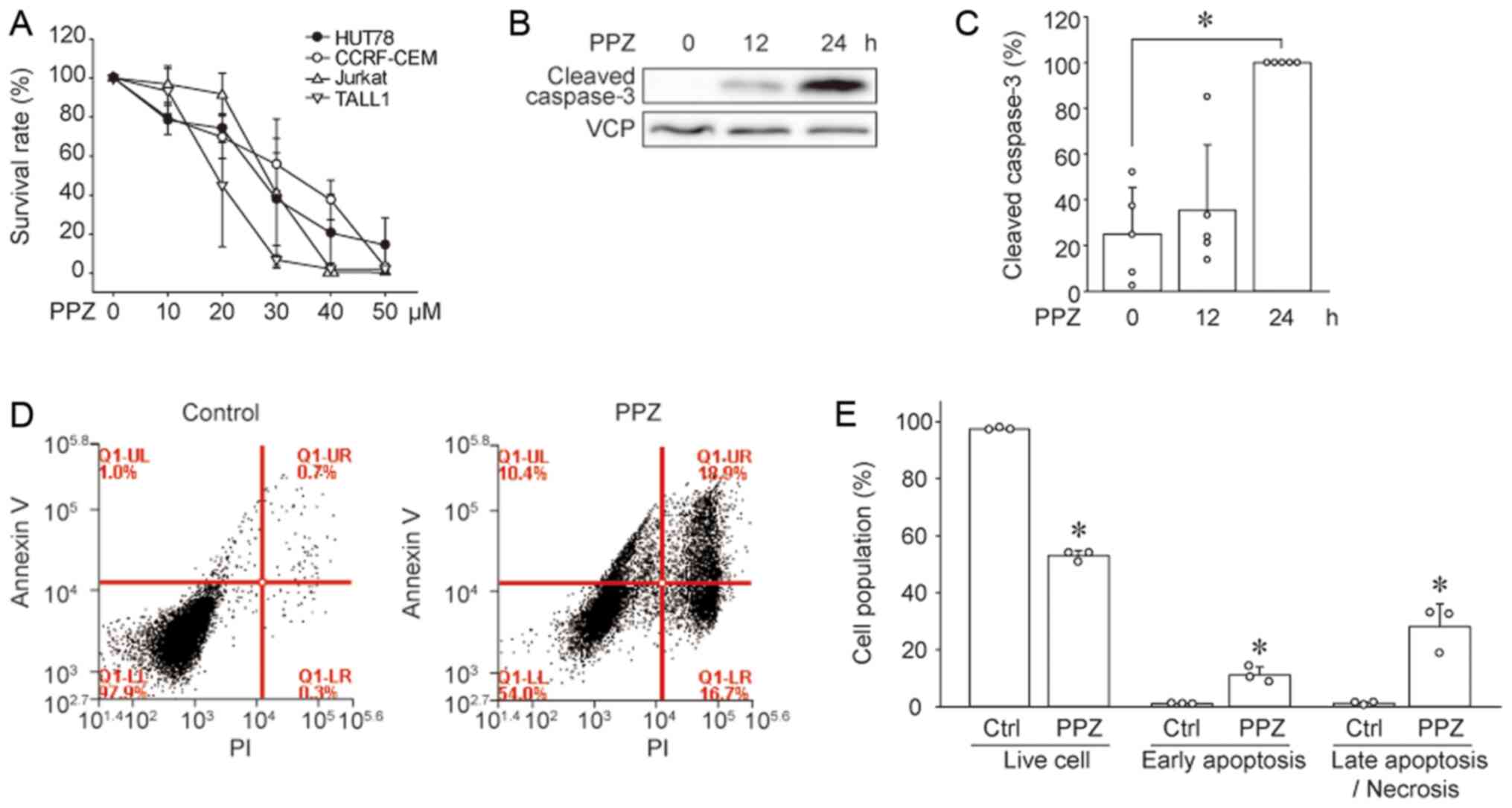
(CCRF-CEM, Jurkat, and TALL1). PPZ suppressed the viability of
HUT78 cells in a dose-dependent manner, and the same was observed
in T-ALL cell lines (Fig. 1A).
Because PP2A reactivation has been reported to induce apoptosis in
cancer cells (10,14), we investigated whether PPZ also
induces apoptosis in HUT78 cells. Immunoblotting results revealed
that PPZ increased the expression of cleaved (active) caspase 3 in
HUT78 cells (Fig. 1B and C).
Moreover, flow cytometry analysis showed that PPZ treatment
resulted in an increased number of early-phase apoptotic cells,
shown as Annexin V-positive and propidium iodide (PI)-negative, as
well as Annexin V and PI positive late-phase apoptotic/necrotic
cells (Fig. 1D and E). These results
suggest that PPZ induces apoptosis in HUT78 cells.
PPZ induces dephosphorylation of PP2A
substrates
In T-ALL cells, PPZ induces dephosphorylation of
PP2A substrates, such as Akt and ERK1/2 (10). Immunoblotting results revealed that
PPZ also decreased phosphorylation levels of Akt and ERK1/2 in
HUT78 cells (Fig. 2A-D).
Phosphorylation at Thr308 and Ser473 in Akt is necessary for their
activation, and is regulated by PDK1 and mTORC2, respectively
(15,16). To reveal the effects of PPZ on
upstream kinases, Ser241 phosphorylation of PDK1, an index of PDK1
activity, and Thr346 phosphorylation of NDRG1, an index of mTORC2
activity, were analyzed by immunoblotting (15,17). PPZ
did not change PDK1 and NDRG1 phosphorylation levels (Fig. 2E and F), suggesting that PPZ induces
Akt dephosphorylation without affecting the upstream kinases. We
also investigated the effects of PPZ on upstream kinases of ERK1/2,
such as B-Raf, C-Raf, and MEK1/2 (18,19). PPZ
decreased phosphorylation level of MEK1/2, but it did not alter
B-Raf and C-Raf phosphorylation (Fig. 2G
and H). These data suggested that PPZ induces MEK/ERK
dephosphorylation without affecting the upstream kinases.
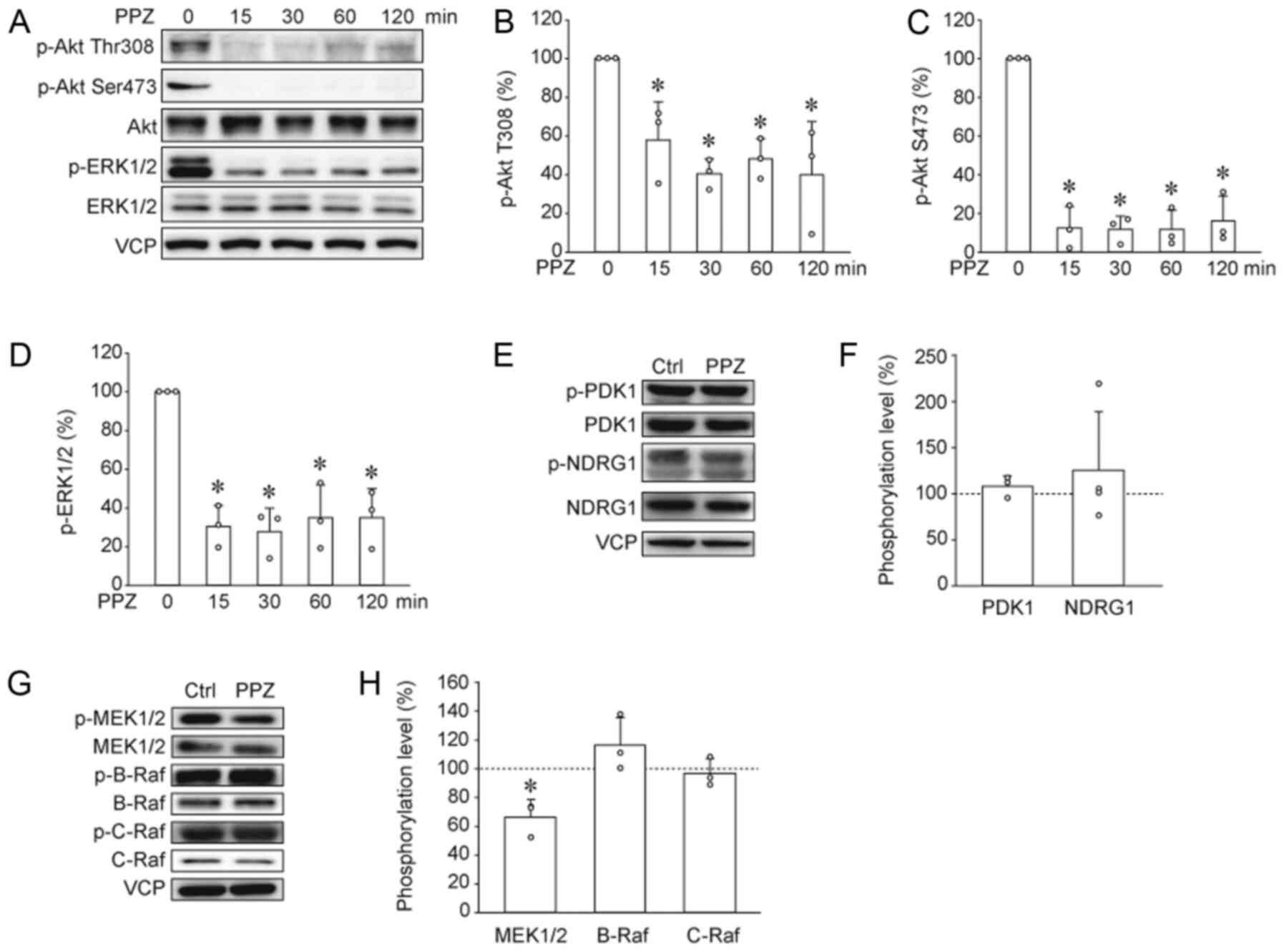 | Figure 2.PPZ induces dephosphorylation of
protein phosphatase 2A substrates. (A, E and G) Western blotting
and subsequent quantification was performed to analyze the effects
of PPZ on the phosphorylation of the following proteins: (B)
p-Thr308 Akt, (C) p-Ser473 Akt, (D) p-ERK1/2, (F) p-PDK1 and
p-NDRG1, (H) p-MEK1/2, p-B-Raf, and p-C-Raf. Data from three to
four independent experiments are presented. Ctrls were ethanol
treated. *P<0.05 vs. Ctrl. or PPZ 0 min. PPZ, perphenazine; p,
phosphorylated; NDRG1, N-Myc downstream-regulated gene 1; Ctrl,
control; VCP, valosin-containing protein. |
PPZ dephosphorylates Akt in
PP2A-dependent manner and dephosphorylates ERK1/2 with no
dependency on PP2A
Gutierrez et al reported that PPZ directly
activates PP2A and induces Akt and ERK1/2 dephosphorylation, and
this inhibitory effect was blocked by the PP2A inhibitor, okadaic
acid (OA), and by shRNA knockdown of specific PP2A subunits
(10). Thus, we tested whether OA
blocks the effects of PPZ in HUT78 cells (Fig. 3A). In HUT78 cells, OA induced Akt
phosphorylation, therefore, we need to adjust the exposure time. OA
blocked PPZ-induced Akt dephosphorylation; however, OA did not
block the dephosphorylation of ERK1/2. We previously reported that
PPZ dephosphorylated Akt and ERK1/2 in the canine T-ALL cell line
UL-1 (13). Similar to the effects
of OA on PPZ-induced dephosphorylations in HUT78 cells, OA blocked
PPZ-induced Akt dephosphorylation, but not ERK1/2 dephosphorylation
in UL-1 cells (Fig. 3B). These
results suggest that PPZ blocks Akt phosphorylation in a
PP2A-dependent manner, however, PP2A is not involved in PPZ-induced
ERK1/2 dephosphorylation. To clarify the molecular mechanism
associated with these effects of PPZ, we tested whether PPZ
directly inhibits MEK1/2 activity, using an in vitro kinase
assay (Fig. 3C and D). The
phosphorylation level of the MEK1/2 substrate, ERK1/2, was not
blocked by PPZ, suggesting PPZ may activate some phosphatase(s)
other than PP2A. Multiple phosphatases such as PP1, PP2B, PP2C, and
DUSP1/6, are involved in ERK1/2 dephosphorylation (20–23). We
analyzed the effects of the PP1, PP2B, PP2C, and DUSP1/6
inhibitors, tautomycin (Tau), cyclosporin A (CsA), sanguinarine,
and BCI, respectively, on PPZ-induced ERK1/2 dephosphorylation
(Fig. 3E). Our results show that
these phosphatase inhibitors did not block PPZ-induced ERK1/2
dephosphorylation. Therefore, we tested the pan-tyrosine
phosphatase inhibitor, Na3VO4, and observed
that Na3VO4 slightly, but significantly,
suppressed ERK1/2 dephosphorylation by PPZ (Fig. 3F and G), suggesting that a type of
tyrosine phosphatase or a dual specificity phosphatase is involved
in PPZ-induced ERK1/2 dephosphorylation.
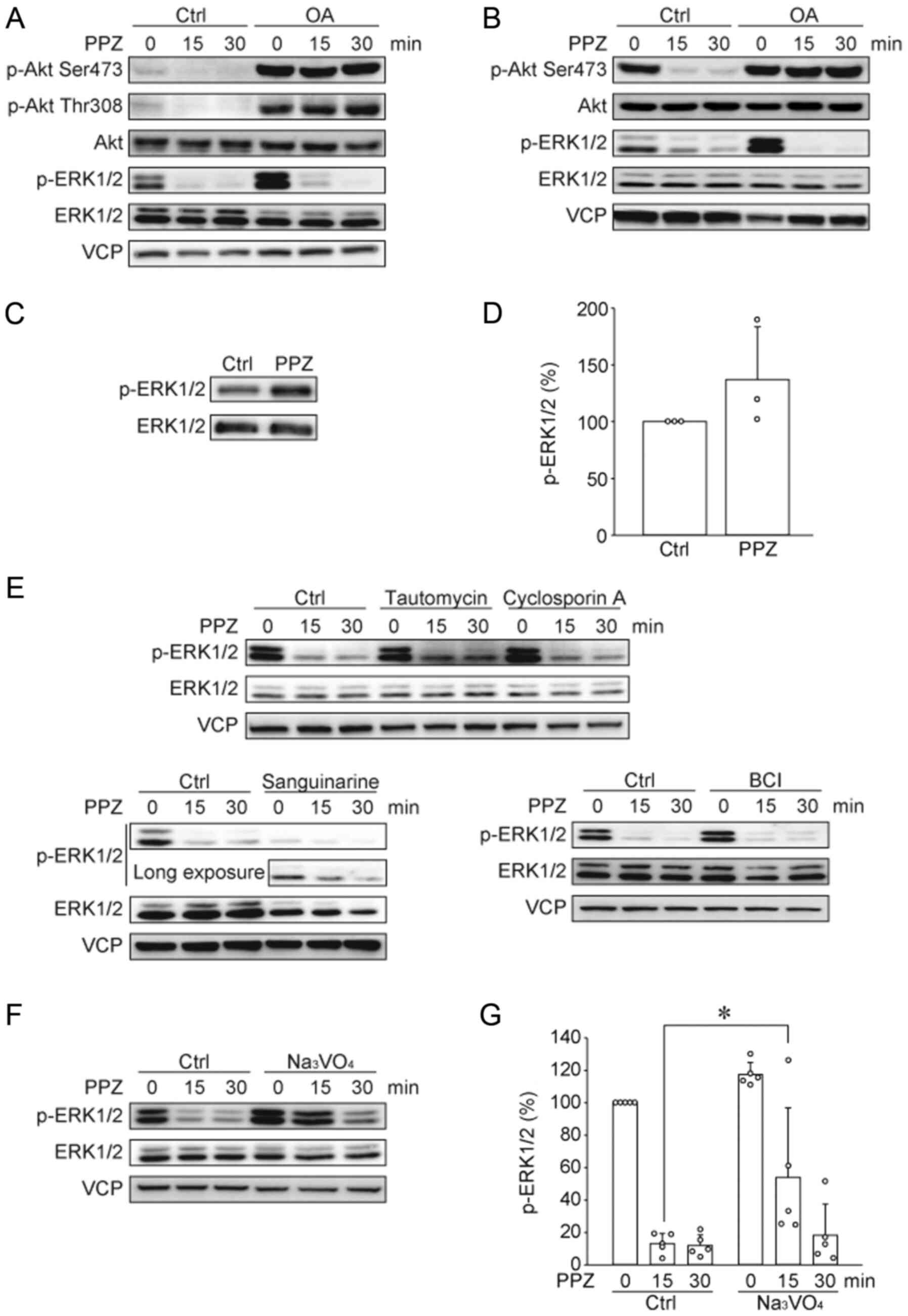 | Figure 3.PPZ dephosphorylates Akt in a
PP2A-dependent manner and dephosphorylates ERK1/2 with no
dependency on PP2A. The phosphorylation of Akt and ERK1/2 in (A)
HUT78 and (B) UL-1 cells were determined via western blotting.
Representative images from three independent experiments are
presented. The effect of PPZ (10 µM) on the activity of recombinant
MEK1/2 was analyzed by performing an in vitro kinase assay
with recombinant ERK1/2 as a substrate. Phosphorylation levels of
ERK1/2 were detected via (C) western blotting, with subsequent (D)
quantification from three independent experiments. (E) HUT78 cells
were pre-incubated with tautomycin (1 µM), cyclosporin A (10 µM)
and sanguinarine (10 µM) for 4 h, as well as with BCI (10 µM) for 1
h. Then cells were treated with PPZ (20 µM) for 15 and 30 min. The
phosphorylation of ERK1/2 was detected by western blotting.
Representative images from two to three independent experiments are
presented. HUT78 cells, following pre-incubation with
Na3VO4 (300 µM) for 1 h, were treated with
PPZ (20 µM) for 15 and 30 min. (F) Representative images and (G)
quantitative data from three independent experiments were
presented. *P<0.05 as indicated. PPZ, perphenazine; PP2A,
protein phosphatase 2A; Ctrl, control; VCP, valosin-containing
protein; p, phosphorylated; OA, okadaic acid. |
Akt pathway is important for the
survival of HUT78 cells
To determine which signal transduction pathway is
important for the survival of HUT78 cells, Akt or ERK1/2, we
analyzed the effects of an Akt inhibitor (Akt inhibitor VIII) and
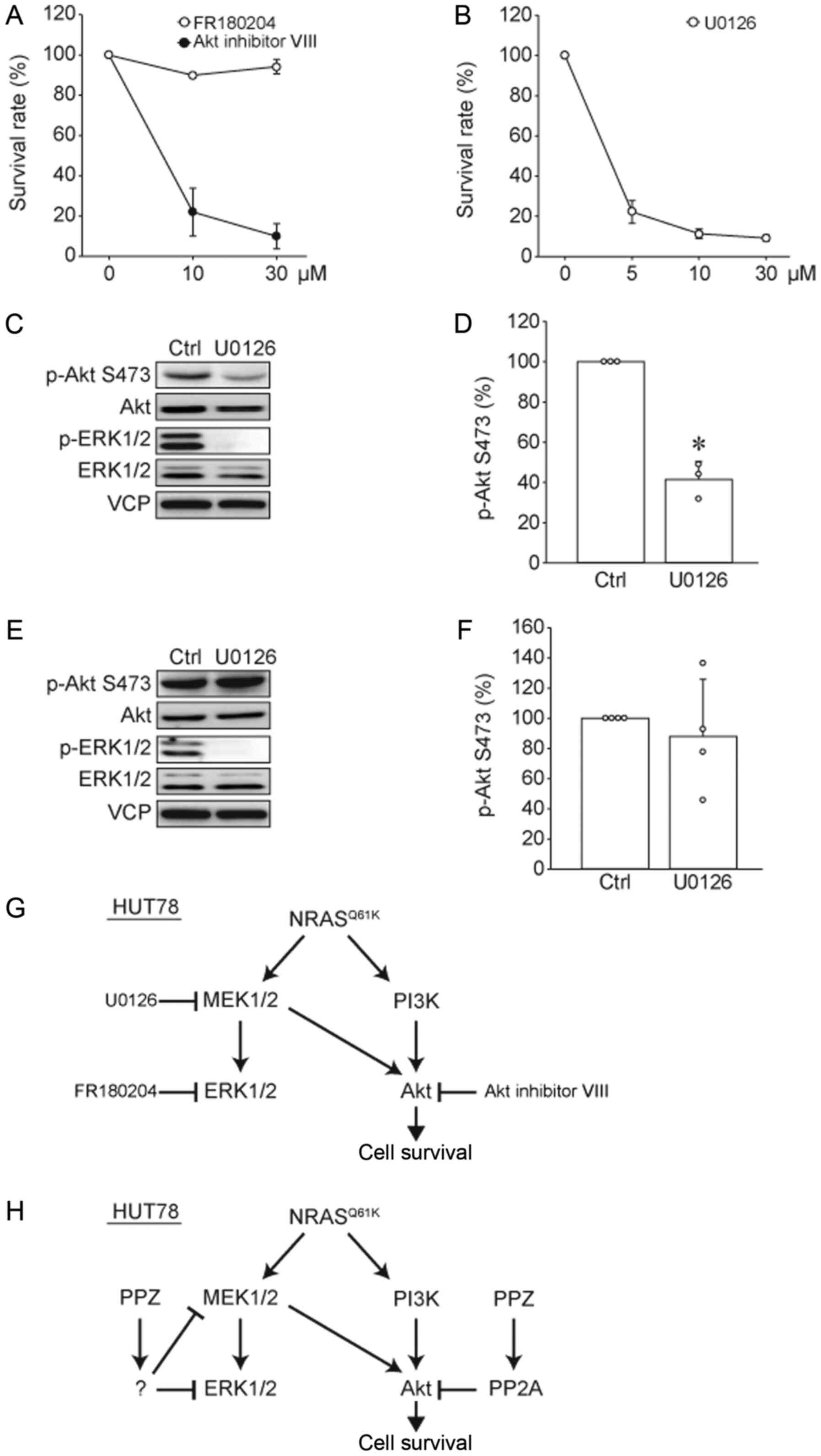
an ERK1/2 inhibitor (FR180204) on cell viability (Fig. 4A). Interestingly, the Akt inhibitor
induced cell death in HUT78 cells in a dose-dependent manner, while
the ERK1/2 inhibitor had no effect on cell viability. Furthermore,
the MEK1/2 inhibitor, U0126, showed antitumor effects on HUT78
cells (Fig. 4B). To clarify this
discrepancy, we studied the effects of this MEK1/2 inhibitor on the
phosphorylation levels of Akt and ERK1/2 (Fig. 4C and D). U0126 suppressed not only
ERK1/2 phosphorylation but also Akt phosphorylation, suggesting
that MEK1/2 activity positively regulates Akt phosphorylation in
HUT78 cells. We previously reported that UL-1 cells were sensitive
to Akt inhibitor VIII, but not to FR180204 or U0126 (13). Therefore, we examined the effects of
U0126 on Akt phosphorylation in UL-1 cells (Fig. 4E and F). We found that in UL-1 cells,
U0126 suppressed ERK1/2 phosphorylation, but did not affect Akt
phosphorylation. These results suggested that MEK inhibitor
suppressed the survival of HUT78 cells by inhibiting Akt activity
(Fig. 4G), and Akt inhibition is
important for the antitumor effects of PPZ.
Discussion
Recently, reactivation of PP2A has attracted a lot
of attention as a promising approach for cancer therapy. Small
molecule activators of PP2A (SMAPs) specifically activates the PP2A
B56α subunit (6) and exerts
anticancer effects on various cancers models such as lung cancer,
breast cancer, endometrial cancer, and pancreatic neuroendocrine
tumor (24–27). SMAPs were engineered from
phenothiazine parent compounds supporting the activation effects of
PPZ on PP2A.
Gutierrez et al reported that PPZ activates
PP2A to dephosphorylate Akt and ERK1/2 in human T-ALL cell line
KOPT-K1 (10). These reports were
based on results from experiments using OA as well as shRNA targets
in PP2A. Similarly, in the present study, we observed that OA
blocked the effects of PPZ on Akt dephosphorylation; however, OA
did not block PPZ-induced ERK1/2 dephosphorylation in HUT78 and
UL-1 cells. One possible explanation for this discrepancy is that
Gutierrez et al (10) used 1
µM of OA to block ERK1/2 dephosphorylation; 1 µM of OA is known to
inhibit not only the activity of PP2A, but also that of PP1, and a
lower dose, such as 100 nM, needs to be used to specifically
inhibit PP2A (28). Moreover, in
their results from studies using shRNA, Gutierrez et al
(10) showed that PP2A knockdown
effectively inhibited PPZ-induced Akt dephosphorylation, but only
slightly suppressed ERK1/2 dephosphorylation. Overall, these data
suggest that although PPZ dephosphorylates Akt in a PP2A-dependent
manner, PP2A is not a major phosphatase in PPZ-induced ERK1/2
dephosphorylation (Fig. 4H). It has
been reported that PP2A activity on ERK1/2 is regulated by the
immediate early response 3 protein (IER3 or IEX-1) (9). IER3 enhances the phosphorylation of
PP2A regulatory subunit by ERK1/2 leading to suppression of PP2A
activity (9). Because RAS
oncogene mutation induces IER3 expression (29), and HUT78 cells express NRAS
mutation (2), an increase in IER3
protein levels may be one of the processes that could explain the
lack of involvement of PP2A in PPZ-induced ERK1/2
dephosphorylation.
Which phosphatase is responsible for PPZ-induced
ERK1/2 dephosphorylation? The results of our in vitro kinase
assay suggest that PPZ does not directly inhibit MEK1/2 kinase
activity. Inhibitors of PP1, PP2B, PP2C, and DUSP1/6 activity did
not block PPZ-induced ERK1/2 dephosphorylation; however, the
pan-tyrosine phosphatase inhibitor, Na3Vo4,
partially inhibited ERK1/2 dephosphorylation, suggesting that a
kind of tyrosine or dual specificity phosphatase(s) may be involved
in PPZ-induced ERK1/2 dephosphorylation. The dual specificity
phosphatase 4, DUSP4, dephosphorylates ERK1/2 (30). We were not able to further
investigate this point owing to the unavailability of a specific
DUSP4 inhibitor. A genetic screening assay using genome editing or
si/shRNA will need to be conducted in future, to identify the
responsible phosphatase(s).
We found that pharmacological inhibition of Akt, but
not of ERK1/2, suppressed HUT78 cell growth. These results suggest
that PP2A-dependent Akt dephosphorylation plays an important role
in the antitumor effects of PPZ. Interestingly, a MEK inhibitor
effectively suppressed HUT78 cell growth, consistent with a
previous report which showed that three types of MEK inhibitors
induced apoptosis in HUT78 cells (2). That study reported that HUT78 cells
harbored the NRASQ61K mutation that leads to enhanced
MEK/ERK signaling (2). Based on our
results using an ERK1/2 inhibitor, ERK1/2 activation by
NRASQ61K mutation is not involved in cell survival in
HUT78 cell lines. We revealed that a MEK inhibitor inhibited Akt
phosphorylation in HUT78 cells; however, in UL-1 cells that are
resistant to MEK inhibitor, pharmacological inhibition of MEK did
not affect Akt phosphorylation. These results suggest that
NRASQ61K mutation leads to Akt phosphorylation through
MEK1/2 activation, and that Akt activity plays an important role in
the survival of HUT78 cells.
As for the HUT78 cells, Akt inhibitor is enough to
kill cells and PP2A activator may be not necessary to use. PP2A
activators suppress multiple signals not only Akt but also such as
c-myc and E2F (31,32). Therefore, the effect of the PP2A
activator is not limited to Akt inhibition, and the usage of Akt
inhibitors and PP2A activators may be different from drug-resistant
point of view. It was reported that long-term treatment with Akt
inhibitor (MK-2206) induces drug resistant in pancreatic
neuroendocrine tumors (PNETs). On the other hand, PNETs did not
become resistant to PP2A activator even after long-term treatment
(25). Overall, our data suggests
that PPZ-induced Akt dephosphorylation by PP2A activation may be an
attractive antitumor strategy for NRAS-mutated Sezary
syndrome.
Acknowledgements
Not applicable.
Funding
This study was partially supported by Japan Society
for the Promotion of Science (grant nos. 17H03915, 18J13124 and
18K05994).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
ST and TO conceived and designed the present study,
analyzed and interpreted the data, and wrote/revised the
manuscript. ST acquired the most of the data. NK acquired the data
for apoptosis and interpreted the data. TM acquired the FACS data.
KS provided facilities, materials and suggestions, and interpreted
the data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CsA
|
cyclosporin A
|
|
NRAS
|
neuroblastoma rat sarcoma oncogene
|
|
OA
|
okadaic acid
|
|
PI
|
propidium iodide
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
PP2A
|
protein phosphatase 2A
|
|
PPZ
|
perphenazine
|
|
SDS
|
sodium dodecyl sulfate
|
|
T-ALL
|
T-cell acute lymphoblastic
leukemia
|
|
Tau
|
tautomycin
|
|
VCP
|
valosin-containing protein
|
References
|
1
|
Foss FM and Girardi M: Mycosis fungoides
and sezary syndrome. Hematol Oncol Clin North Am. 31:297–315. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kiessling MK, Oberholzer PA, Mondal C,
Karpova MB, Zipser MC, Lin WM, Girardi M, Macconaill LE, Kehoe SM,
Hatton C, et al: High-throughput mutation profiling of CTCL samples
reveals KRAS and NRAS mutations sensitizing tumors toward
inhibition of the RAS/RAF/MEK signaling cascade. Blood.
117:2433–2440. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vu HL and Aplin AE: Targeting mutant NRAS
signaling pathways in melanoma. Pharmacol Res. 107:111–116. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Petit V, Raymond J, Alberti C, Pouteaux M,
Gallagher SJ, Nguyen MQ, Aplin AE, Delmas V and Larue L: C57BL/6
congenic mouse NRASQ61K melanoma cell lines are highly
sensitive to the combination of Mek and Akt inhibitors in vitro and
in vivo. Pigment Cell Melanoma Res. 32:829–841. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Uddin S, Hussain A, Al-Hussein K,
Platanias LC and Bhatia KG: Inhibition of phosphatidylinositol
3′-kinase induces preferentially killing of PTEN-null T leukemias
through AKT pathway. Biochem Biophys Res Commun. 320:932–938. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Leonard D, Huang W, Izadmehr S, O'Connor
CM, Wiredja DD, Wang Z, Zaware N, Chen Y, Schlatzer DM, Kiselar J,
et al: Selective PP2A enhancement through biased heterotrimer
stabilization. Cell. 181:688–701.e16. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wlodarchak N and Xing Y: PP2A as a master
regulator of the cell cycle. Crit Rev Biochem Mol Biol. 51:162–184.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kuo YC, Huang KY, Yang CH, Yang YS, Lee WY
and Chiang CW: Regulation of phosphorylation of Thr-308 of Akt,
cell proliferation, and survival by the B55alpha regulatory subunit
targeting of the protein phosphatase 2A holoenzyme to Akt. J Biol
Chem. 283:1882–1892. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Letourneux C, Rocher G and Porteu F:
B56-containing PP2A dephosphorylate ERK and their activity is
controlled by the early gene IEX-1 and ERK. EMBO J. 25:727–738.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gutierrez A, Pan L, Groen RW, Baleydier F,
Kentsis A, Marineau J, Grebliunaite R, Kozakewich E, Reed C,
Pflumio F, et al: Phenothiazines induce PP2A-mediated apoptosis in
T cell acute lymphoblastic leukemia. J Clin Invest. 124:644–655.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zonta F, Pagano MA, Trentin L, Tibaldi E,
Frezzato F, Trimarco V, Facco M, Zagotto G, Pavan V, Ribaudo G, et
al: Lyn sustains oncogenic signaling in chronic lymphocytic
leukemia by strengthening SET-mediated inhibition of PP2A. Blood.
125:3747–3755. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hartung B, Sampson S and Leucht S:
Perphenazine for schizophrenia. Cochrane Database Syst Rev.
2015:CD0034432015.
|
|
13
|
Tsuji S, Yabe R, Usui T, Mizuno T, Ohama T
and Sato K: Anti-tumor effects of perphenazine on canine lymphoma.
J Vet Med Sci. 78:1293–1298. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ruvolo PP, Deng X, Ito T, Carr BK and May
WS: Ceramide induces Bcl2 dephosphorylation via a mechanism
involving mitochondrial PP2A. J Biol Chem. 274:20296–20300. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Alessi DR, James SR, Downes CP, Holmes AB,
Gaffney PR, Reese CB and Cohen P: Characterization of a
3-phosphoinositide-dependent protein kinase which phosphorylates
and activates protein kinase Balpha. Curr Biol. 7:261–269. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sarbassov DD, Guertin DA, Ali SM and
Sabatini DM: Phosphorylation and regulation of Akt/PKB by the
Rictor-mTOR complex. Science. 307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
García-Martínez JM and Alessi DR: mTOR
complex 2 (mTORC2) controls hydrophobic motif phosphorylation and
activation of serum- and glucocorticoid-induced protein kinase 1
(SGK1). Biochem J. 416:375–385. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Alessi DR, Saito Y, Campbell DG, Cohen P,
Sithanandam G, Rapp U, Ashworth A, Marshall CJ and Cowley S:
Identification of the sites in MAP kinase kinase-1 phosphorylated
by p74raf-1. EMBO J. 13:1610–1619. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou B, Wang ZX, Zhao Y, Brautigan DL and
Zhang ZY: The specificity of extracellular signal-regulated kinase
2 dephosphorylation by protein phosphatases. J Biol Chem.
277:31818–31825. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu XD, Liu YN, Zhang ZY, Ma ZA, Suo ZW and
Yang X: Spinophilin-targeted protein phosphatase-1 alleviated
inflammatory pain by negative control of MEK/ERK signaling in
spinal cord dorsal horn of rats. J Neurosci. 35:13989–14001. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Y, Lin DH, Wang ZJ, Jin Y, Yang B
and Wang WH: K restriction inhibits protein phosphatase 2B (PP2B)
and suppression of PP2B decreases ROMK channel activity in the CCD.
Am J Physiol Cell Physiol. 294:C765–C773. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Saidak Z, Giacobbi AS, Louandre C, Sauzay
C, Mammeri Y and Galmiche A: Mathematical modelling unveils the
essential role of cellular phosphatases in the inhibition of
RAF-MEK-ERK signalling by sorafenib in hepatocellular carcinoma
cells. Cancer Lett. 392:1–8. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Risom T, Wang X, Liang J, Zhang X, Pelz C,
Campbell LG, Eng J, Chin K, Farrington C, Narla G, et al:
Deregulating MYC in a model of HER2+ breast cancer mimics human
intertumoral heterogeneity. J Clin Invest. 130:231–246. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Umesalma S, Kaemmer CA, Kohlmeyer JL,
Letney B, Schab AM, Reilly JA, Sheehy RM, Hagen J, Tiwari N, Zhan
F, et al: RABL6A inhibits tumor-suppressive PP2A/AKT signaling to
drive pancreatic neuroendocrine tumor growth. J Clin Invest.
130:1641–1653. 2019. View Article : Google Scholar
|
|
26
|
Taylor SE, O'Connor CM, Wang Z, Shen G,
Song H, Leonard D, Sangodkar J, LaVasseur C, Avril S, Waggoner S,
et al: The highly recurrent PP2A Aα-subunit mutation P179R alters
protein structure and impairs PP2A enzyme function to promote
endometrial tumorigenesis. Cancer Res. 79:4242–4257. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tohmé R, Izadmehr S, Gandhe S, Tabaro G,
Vallabhaneni S, Thomas A, Vasireddi N, Dhawan NS, Ma'ayan A, Sharma
N, et al: Direct activation of PP2A for the treatment of tyrosine
kinase inhibitor-resistant lung adenocarcinoma. JCI Insight.
4:e1256932019. View Article : Google Scholar
|
|
28
|
Favre B, Turowski P and Hemmings BA:
Differential inhibition and posttranslational modification of
protein phosphatase 1 and 2A in MCF7 cells treated with
calyculin-A, okadaic acid, and tautomycin. J Biol Chem.
272:13856–13863. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Garcia MN, Grasso D, Lopez-Millan MB,
Hamidi T, Loncle C, Tomasini R, Lomberk G, Porteu F, Urrutia R and
Iovanna JL: IER3 supports KRASG12D-dependent pancreatic cancer
development by sustaining ERK1/2 phosphorylation. J Clin Invest.
124:4709–4722. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chu Y, Solski PA, Khosravi-Far R, Der CJ
and Kelly K: The mitogen-activated protein kinase phosphatases
PAC1, MKP-1, and MKP-2 have unique substrate specificities and
reduced activity in vivo toward the ERK2 sevenmaker mutation. J
Biol Chem. 271:6497–6501. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Farrington CC, Yuan E, Mazhar S, Izadmehr
S, Hurst L, Allen-Petersen BL, Janghorban M, Chung E, Wolczanski G,
Galsky M, et al: Protein phosphatase 2A activation as a therapeutic
strategy for managing MYC-driven cancers. J Biol Chem. 295:757–770.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Enjoji S, Yabe R, Tsuji S, Yoshimura K,
Kawasaki H, Sakurai M, Sakai Y, Takenouchi H, Yoshino S, Hazama S,
et al: Stemness is enhanced in gastric cancer by a SET/PP2A/E2F1
axis. Mol Cancer Res. 16:554–563. 2018. View Article : Google Scholar : PubMed/NCBI
|