Introduction
Hereditary multiple osteochondromas (HMO) is
characterized by multiple cartilage-capped bony projections
(exostoses) that usually arise from the metaphysis of long bones
(1). It is also known as hereditary
multiple exostoses, multiple cartilaginous exostoses,
osteochondromas and diaphyseal aclasis (2). The prevalence of HMO is ~1:50,000, and
the male: Female ratio is 1.5:1 (3,4).
Although HMO can be asymptomatic and diagnosed incidentally, it can
disrupt bone growth and cause short stature, unequal limb lengths
and joint deformities with significant morbidity (5,6). The
most serious complication of HMO is the malignant transformation
into chondrosarcoma, occurring in 0.5–5% of the patients (6). Therefore, clinical and radiological
follow up is crucial for the management of patients with HMO.
However, there is currently no standard follow-up protocol for HMO.
Genetic analysis of EXT genes to identify patients with HMO
at higher risk of developing severe disease or malignant
transformation may contribute to the future management of such
patients.
HMO is an autosomal dominant inherited disease with
a penetrance of 100% (5). Genetic
analysis of HMOs in different populations identified two main
causative genes, namely exostosis 1 (EXT1) and exostosis 2
(EXT2) (5,7,8).
Mutations in the EXT1 and EXT2 genes account for
>90% of all HMO cases (7,9). EXT1 and EXT2 both encode
for a glycosyltransferase required for heparan sulfate (HS)
synthesis and polymerization as HS proteoglycans (HSPGs) (8). The role of EXT1 and EXT2 proteins in HS
synthesis involves the formation of heterocomplex of both proteins
(1). HSPGs play a key role in the
regulation of different signaling pathways involved in chondrocyte
proliferation and differentiation in the growth plate (8).
The present study investigated 16 Jordanian index
cases from nine different unrelated families with confirmed
diagnosis of HMO. The different clinical characteristics in
addition to the mutational spectrum of the EXT genes and the
expression of their corresponding proteins were evaluated in this
group of patients.
Materials and methods
Patients with HMO
The present study was conducted at several
Orthopedic Surgery clinics over a 2-year period between January
2018 and December 2019. A total of 42 individuals were included in
this study, among which 16 were diagnosed with HMO. Of the
patients, 12 were men and 4 were women who had a mean age of 13.9
years (age range, 6–27 years). HMO diagnosis was confirmed by
either histopathological or radiological examinations. These
patients were evaluated clinically, and their available
radiological examinations were reviewed. Disease severity was
determined according to the severity score described by Mordenti
et al (10) (Table I). Blood samples were collected from
all participants. Paraffin-embedded tissues of patients with HMO
who underwent surgical excision were available from the archives of
Pathology Department. Informed consent was obtained from all
individuals who participated in this study. The study protocol was
approved by the Human Research Ethics Committee.
 | Table I.HMO severity score described by
Mordenti et al (10). |
Table I.
HMO severity score described by
Mordenti et al (10).
| Class | Subclass |
|---|
| I: No
deformities-no functional limitations |
|
| IA | ≤5 sites with
exostosis |
| IB | >5 sites with
exostosis |
| II: Deformities-no
functional limitations |
|
| IA | ≤5 sites with
deformities |
| IB | >5 sites with
deformities |
| III:
Deformities-functional limitations |
|
|
IIIA | 1 site with
functional limitations |
|
IIIB | >1 site with
functional limitations |
Molecular analysis
Genomic DNA from the patients and their available
family members was extracted from peripheral blood samples using a
Gentra Puregene kit (Qiagen GmbH) following the manufacturers
instructions. The quality and concentration of the DNA was
determined by NanoDrop 2000 V7.3.1 (Thermo Fisher Scientific,
Inc.). All exons and exon-flanking intron sequences of EXT1
(NM_000127) and EXT2 (NM_000401) genes were amplified by PCR
(polymerase chain reaction). PCR was performed in a final volume of
25 µl containing 40 ng genomic DNA, 1X Master Mix
(GoTaq® Green Master Mix; Promega Corporation), and 5
pmol of forward and reverse primers (Table II). The following thermocycling
conditions were used: Initial denaturing step (95°C for 7 min)
followed by 40 cycles of 95°C for 1 min, annealing at 60°C for 90
sec, extension step at 72°C for 90 sec and final extension at 72°C
for 7 min. PCR was performed using an iCycler (Bio-Rad
Laboratories, Inc.). The PCR products were separated by 2% agarose
gel electrophoresis, visualized by ethidium bromide and the
products were purified using the Norgens PCR Purification kit (cat.
no. 45700; Norgen, Bioteck Corp.). GAPDH was used as the loading
control and for normalization. Sanger sequencing was performed in
both sense and antisense directions by using the BigDye Terminator
Cycle Sequencing kit version 3.1 (Applied Biosystems; Thermo Fisher
Scientific, Inc.), according to the manufacturers protocol.
Sequencing reactions were purified using the NucleoSEQ kit
(Macherey-Nagel GmbH) and final analysis performed using an ABI 310
DNA sequencer (Applied Biosystems; Thermo Fisher Scientific,
Inc.).
| Table II.Primer sequences used for PCR
amplification and Sanger sequencing. |
Table II.
Primer sequences used for PCR
amplification and Sanger sequencing.
| Gene | Exon | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| EXT1 | Exon 1 |
CGAGCGCAGGAGTAAACACC |
CGTTTTTTGGCCTGCATGTG |
|
| Exon 1 |
GAGCTGAAAGTGTTGATTGG |
GAGACTCTGCACCTTTGGATC |
|
| Exon 1 |
CCTCTTTGTCCTGAGTCTGG |
CCATCCCCCAACTTCACACC |
|
| Exon 2 |
CCCCACATTCGCAATGAGTC |
GAGAGGTGATAATGTTAAACCC |
|
| Exon 3 |
CTGATTGGAACAGCTTCTGCTG |
TGAAAGTTTGGACGGGGGCAGC |
|
| Exon 4 |
GTGCATCTCTTTGTTTTACAG |
GCTGAGAGAAGTGTATAAAGG |
|
| Exon 5 |
CCTTTCCAAATATCATCAGG |
GGCCTTTAGTTCTGTATGAC |
|
| Exon 6 |
GAGCAAGGAGGAGTAATTTTC |
ATAACAGGTAAGGAGGGCGG |
|
| Exon 7 |
AAGAGGCTTTGGGTTGGAGG |
AAGTGCCCCATGGAGAAAC |
|
| Exon 8 |
GGGAGAATTGTCCTGAAAAC |
ATCGTGCAACATGAGGTGAC |
|
| Exon 9 |
TTAGTGGGGAGAAGGTAATG |
TTCCTATTTATGCAGCAGCC |
|
| Exon 10 |
GTCTCAGAAGTCCACTTGTC |
ACGTGAGTCCTCATTACCTG |
|
| Exon 11 |
CCTTGCACTTCTCTCATCATTATCC |
GAAGAGAGAGCAGCTTGACC |
| EXT2 | Exon 1 |
GCCTGAATATAAGCACCTAC |
AAAAGCGGGCAGTCATTGTC |
|
| Exon 2 |
TCAAGTGTCATTTGCCATCC |
CCCTTCCCTTTAGTTCCCTG |
|
| Exon 3 |
GGCTTGGGGATCCTTGATAG |
ACTTCTAAATCTTCAGGAGG |
|
| Exon 4 |
ACTCTGTAAACGTTAGCTGG |
AGGACCCTACCCTGTAACTG |
|
| Exon 5 |
TCAGTGGAGGTGAAGACTGG |
CATAGGCCAAGCAGCTTTGC |
|
| Exon 6 |
GTATTGCTTGGCGTCAACCC |
GTAGTAGTTCTTGAACCAGG |
|
| Exon 7 |
GGATGTTGTTTCTGCTTGTG |
ACTCAGGCATTCAGCTCCTG |
|
| Exon 8 |
CCTGGAGTTGACTATGATAG |
TTATGCTGCCCTTATCAGGC |
|
| Exon 9 |
CATGTTTGGGTTTGCTGACG |
AAATGGAGGCATGCTGTCTC |
|
| Exon 10 |
GGATACAAGCTGATTCTCCC |
GCACACCTTTTGGACTCTAC |
|
| Exon 11 |
TGGAACATCTCCAGAATCCC |
AAGCCCTCTTGGCAGGTATG |
|
| Exon 12 |
TATGAGAGAAAGCTTGTCCC |
CCAATGTGACCGCATCAATC |
|
| Exon 13 |
CATGCAACATCTCAGCTTAC |
ACTATGGCTACCAGCTGCTG |
|
| Exon 14 |
CAGACTGTGGCTACTTGAGC |
AGTAGGTCAACCTTCCACCC |
The obtained sequences were compared with the normal
EXT1 (NM_000127) and EXT2 (NM_000401) genes reference
sequences and chromatograms were visualized by using the ChromasPro
1.34 (Technelysium Pty Ltd.) software package or Mutation Surveyor
(V4.07; SoftGenetics, LLC). Sequence nomenclatures for the coding
and noncoding variants were described in accordance with the Human
Genome Variation Society Nomenclature standards (http://www.hgvs.org/mutnomen). To assess and predict
the impact of newly identified missense variants, the Mutation
taster (http://www.mutationtaster.org/) and Polyphen2 programs
(http://genetics.bwh.harvard.edu/pph2/index.shtml)
were used.
Immunohistochemistry
Formalin fixed, paraffin embedded HMO tissues were
used in this study for immunohistochemical staining of EXT1 and
EXT2. Tissues were processed following the manufacturers
instructions. Immunostaining for EXT1 and EXT2 was performed
manually on the sample sections using two different commercially
available rabbit anti-human EXT1(dilution 1:200; cat. no.
abx100786; Abbexa Ltd.; and cat. no. HPA044394; Sigma-Aldrich;
Merck KGaA) and rabbit anti-human EXT2 (dilution 1:200; cat. no.
abx03435; Abbexa Ltd.; and cat. no. SAB2108124; Sigma-Aldrich;
Merck KGaA) antibodies. Tissue sections were observed under a light
microscopy at a magnification of ×40. Tissues known to express EXT1
and EXT2 (normal femoral head cartilage) were used as positive
control, and negative controls were created by omitting the primary
antibody step. The scoring criteria for EXT1 and EXT2 were as
follow: 0, 0–10%; 1, 10–30%; 2, 30–85%; and 3, >85%. The
intensity of the reaction was scored as: 0, negative; 1, weak; 2,
moderate; and 3, strong. The samples that were scored as 1 or more
were considered as positive.
Results
Patients
A total of nine families (A-I) with 42 members were
included in the present study. These families included a total of
16 patients with HMO, and three families (B, E and F) only had one
affected member (Table III).
| Table III.Clinical severity class and
pathogenic EXT1 gene variants identified. |
Table III.
Clinical severity class and
pathogenic EXT1 gene variants identified.
| Family | Patient | Sex | Age at diagnosis
(years) | Clinical severity
class | Nucleotide change
(EXT1 gene) | Genomic
position | Protein level | Clinical
significance | Novel mutation | EXT1 protein IHC
staining | EXT2 protein IHC
staining |
|---|
| A | I.1 | M | 20 | IB | c.918del | g.119122368del | p.
(Lys306Asnfs*53) | Likely
pathogenic | Yes | RE | No |
|
| II.1 | F | 7 | IB |
| (Exon 1) |
|
|
|
|
|
|
| II.3 | F | 7 | IB |
|
|
|
|
|
|
|
| B | II.1 | M | 10 | IB | c.1019G>A |
g.118849384C>T | p. (Arg340His) | Pathogenic | Noa | RE | No |
|
|
|
|
|
|
| (Exon 2) | VUS |
|
|
|
|
|
|
|
|
|
| c.82T>A |
g.119123204A>T | p. (Phe28Ile) |
| Yes |
|
|
|
|
|
|
|
|
| (Exon 1) |
|
|
|
|
|
| C | II.2 | M | 18 | IIIB | c.1019G>A |
g.118849384C>T | p. (Arg340His) | Pathogenic | Noa | RE | No |
|
| III.1 | M | 12 | IA |
| (Exon 2) |
|
|
|
|
|
|
| III.2 | M | 15 | IA |
|
|
|
|
|
|
|
| D | I.1 | M | 6 | IA | c.96dup | g.119123190dup | p.
(Ser33Glufs*11) | Likely
pathogenic | Yes | RE | No |
|
| II.1 | M | 27 | IB |
| (Exon 1) |
|
|
|
|
|
| E | II.1 | M | 6 | IIA | c.1469del | g.118831982del | p.
(Leu490Argfs*9) | Pathogenic | Nob | RE | No |
|
|
|
|
|
|
| (Exon 6) |
|
|
|
|
|
| F | II.3 | M | 7 | IB |
c.1065_1066ins26 |
g.118847781_118847782ins26 | p.
(Val356Cysfs*12) | Likely
pathogenic | Yes | RE | No |
|
|
|
|
|
|
| (Exon 3) |
|
|
|
|
|
| G | I.1 | F | 10 | IIIA | c.962+1G>A |
g.119122323C>T | p.? | Pathogenic | Noc | RE | No |
|
| II.4 | M | 25 | IA |
| (Donor splice site
of intron 1) |
|
|
|
|
|
| H | I.1 | M | 24 | IB | None |
|
|
|
|
|
|
| I | 1.1 | F | 20 | IIIA | None |
|
|
|
|
|
|
|
| 11.1 | M | 9 | IIA | None |
|
|
|
|
|
|
These patients had a mean age of 13.9 years at their
initial diagnosis (range, 6–27 years). A total of 75% (12/16) of
the patients were males. The total number of tumors was 135, with
over half (58%) being located around the knee. According to HMO
severity classification by Mordenti et al (10) (Table
I), most (69%) (11/16) of these patients had a mild disease
(class I) (Table III). Moderate
(class II) and severe (class III) disease forms were recorded in 2
(13%) and 3 (19%) patients, respectively (Table III).
EXT1 and EXT2 genes mutational
analysis
Mutational analysis of the 16 patients from nine
families (A-I) and their family members for both EXT1 and
EXT2 genes revealed different heterozygous mutations in only
EXT1 gene. While 13 patients (77%) from seven unrelated
families harbored these EXT1 mutations, the remaining 3
patients (23%) from two families were negative for both EXT1
and EXT2 genetic variation in the targeted sequenced
regions. Seven different genetic variants were identified in the
EXT1 gene. These variants consisted of; two missense
variants [c.1019G>A, p. (Arg340His); and c.82T>A, p.
(Phe28Ile)], two deletions variants [c.918del, p.(Lys306Asnfs*53);
and c.1469del, p.(Leu490Argfs*9)], one insertion variant
[c.1065_1066ins26, p.(Val356Cysfs*12)], one duplication variant
[c.96dup, p.(Ser33Glufs*11)] and one splice site variant
(c.962+1G>A) (11,12). Three of the seven variants:
c.1019G>A, p.(Arg340His); c.962+1G>A; and c.1469del,
p.(Leu490Argfs*9) were novel since they were not found in known
databases such as ExAC (http://exac.broadinstitute.org/), GenomeAD (https://gnomad.broadinstitute.org) and dbSNP
(https://www.ncbi.nlm.nih.gov/snp). The
[c.1019G>A, p. (Arg340His] variant was identified in two
unrelated families (B-II.1, C-II.2, C-III.1 and C-III.2; Table III).
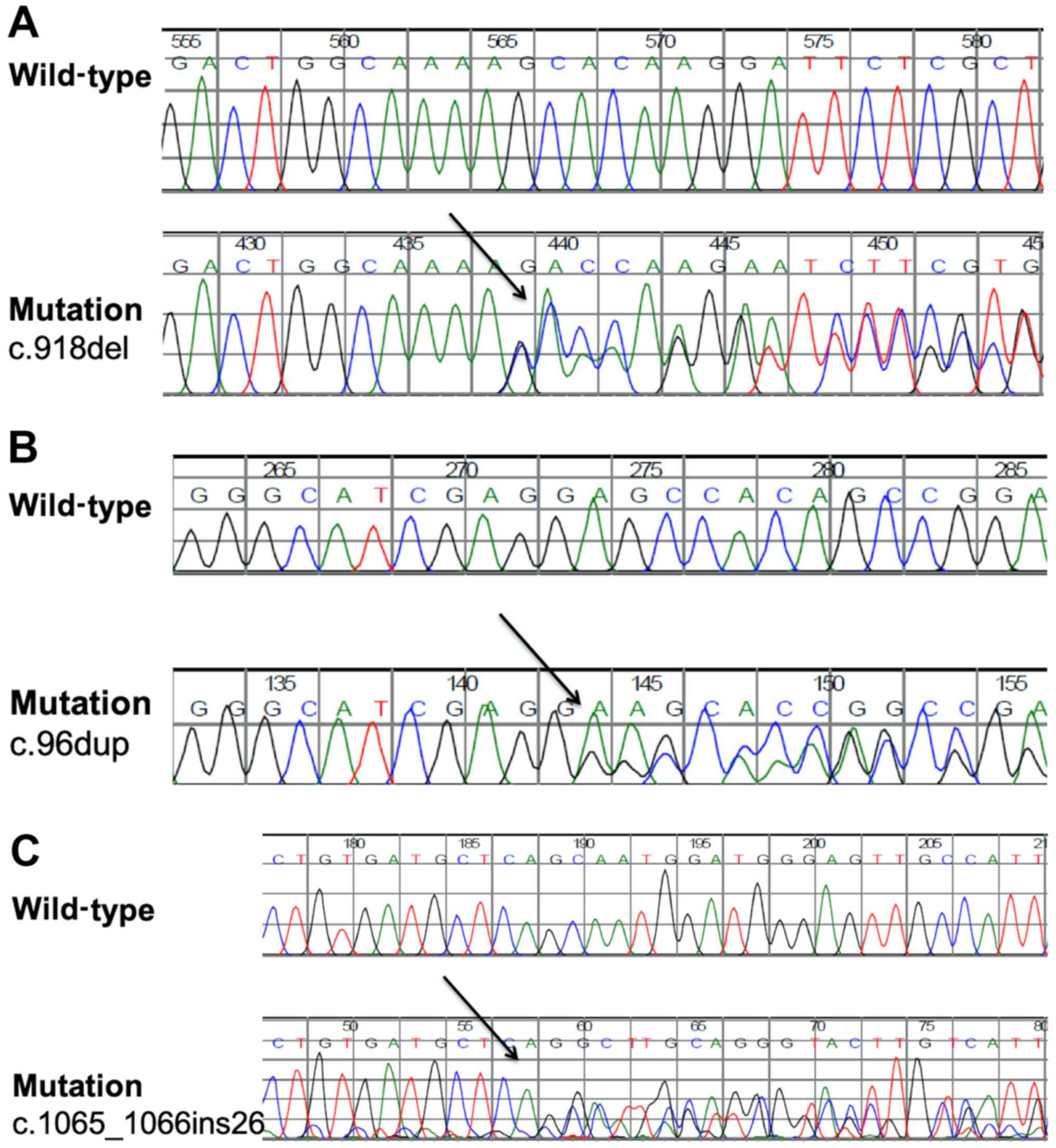
In family A, a novel heterozygous variant c.918del,
p. (Lys306Asnfs*53) was identified in three affected patients
(A-I.1, A-II.1 and A-II.3; Table
III and Fig. 1). This deletion
variant creates a frameshift starting at codon Lys306 and a new
reading frame ends in a new stop codon at position 53 downstream of
the mutation (Table III and
Fig. 2A). Unaffected individuals in
this family were wild type for this variant.
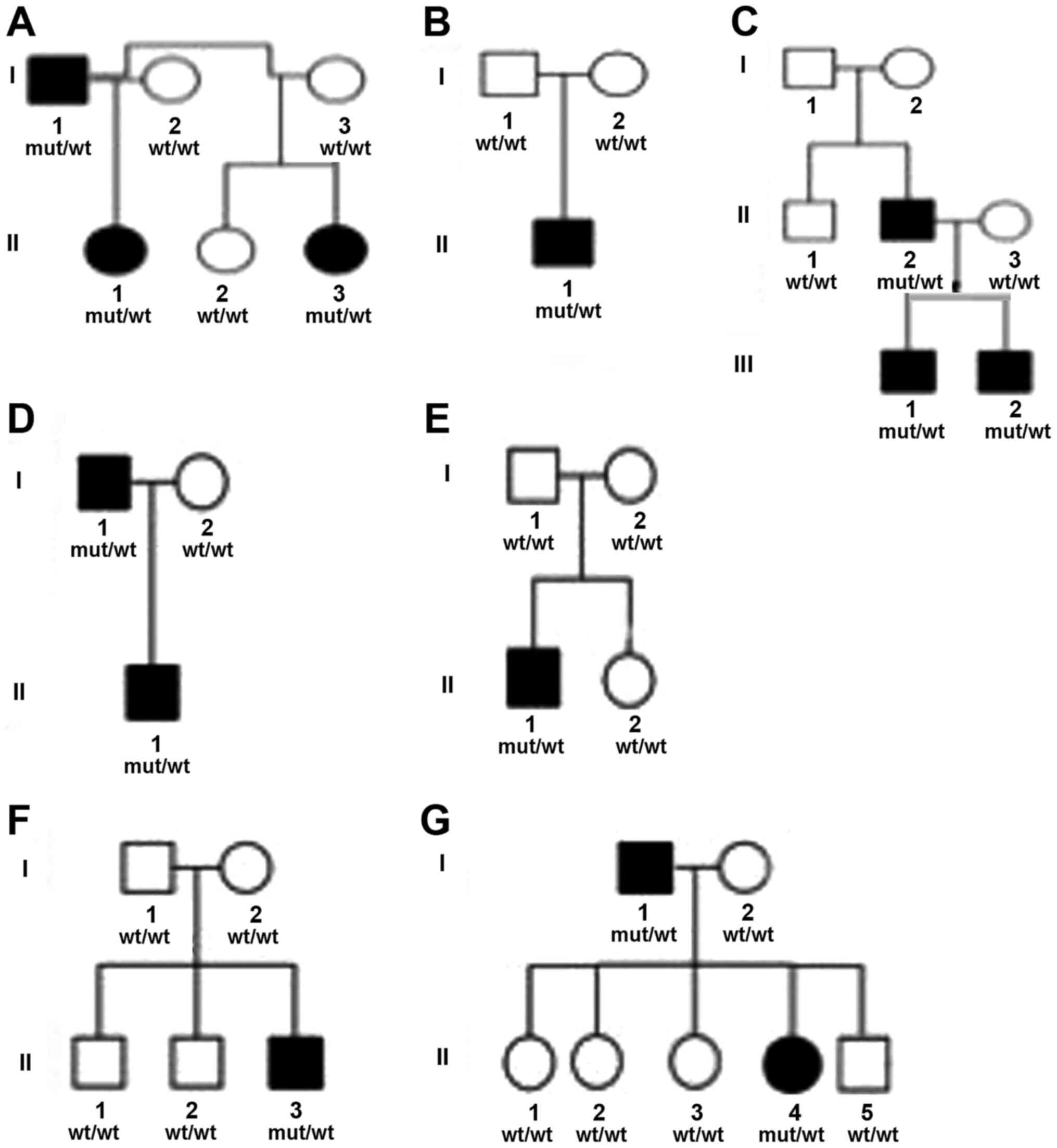 | Figure 1.Pedigrees of families (A-G) with
EXT1 genetic variants leading to HMO. (A) Family A, (B)
Family B, (C) Family C, (D) Family D, (E) Family E, (F) Family F,
(G) Family G. Squares, males; circles, females; solid symbols,
affected individuals with heterozygous mutations; open symbols,
unaffected individuals. EXT1 genotypes: Mu, mutant allele;
wt, wild-type allele. EXT, exostosin; HMO; hereditary multiple
osteochondromas. |
A second novel variant [c.96dup; p. (Ser33Glufs*11)]
was detected in family D (D-I.1 and D-II.1; Table III and Fig. 2B). This one-base-pair duplication
variant in exon 1 creates a frameshift starting at codon Ser33 and
the new reading frame is predicated to end in a stop codon at
position 11 (Table III and
Fig. 2B).
A third novel heterozygous variant was identified in
family F (F-II.3) which was an insertion of 26 base pairs (bps) in
exon 3 [c.(1065_1066ins26); p. (Val356Cysfs*12)] (Table III and Fig. 2C). This variant was predicted to
create a frame shift mutation starting at codon Val356 and a new
reading frame ends in a new stop codon at downstream position
12.
Careful medical evaluation of all family members
(affected and unaffected individuals) of families B, E and F, who
only had one affected member each (Table III and Fig. 1) was performed. The tested mother and
father of these families were clinically normal with no signs of
HMO. Furthermore, genetic testing for the presence of a causative
genetic variant of the parents showed negative mutation results.
The affected individuals (B-II.1, E-II.1 and F-II.3) in these
families carry a de novo mutation and were the first ones
who gained the mutant allele.
Immunohistochemical staining of EXT1
and EXT2
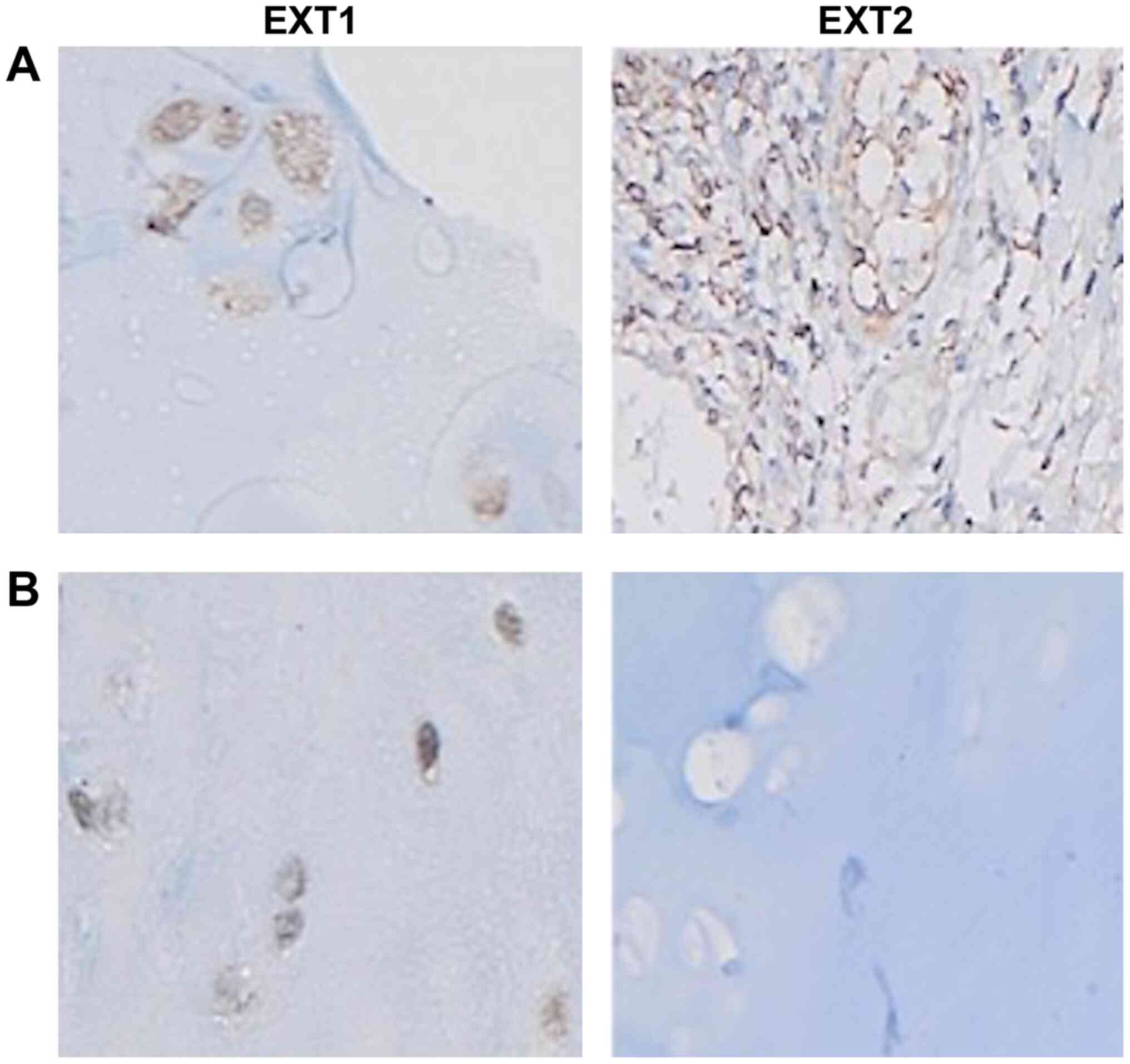
Osteochondroma tissue was available for
immunohistochemical staining from 12 of the 13 patients with
EXT1 gene mutations. EXT1 protein expression was found to be
significantly decreased (weak staining pattern; Fig. 3B) in all examined tissues, apart from
tissues obtained from the 2 proband patients of family D (D-I.1 and
D-II.1). Their tissues exhibited moderate staining pattern when
compared to EXT1 protein expression in normal chondrocytes
(Fig. 3A). A novel variant [c.96dup;
p. (Ser33Glufs*11)] was detected in this family. With regards to
the EXT2 gene, no protein expression was detected in any
osteochondroma tissues from the 12 tested patients (Fig. 3B).
Discussion
Several HMO studies investigated the associations of
the different clinical characteristics with the genetic findings in
different populations. The clinical characteristics of patients
with HMO in the present study, including the mean age at diagnosis
of 13.9 years, as well as male predilection, were similar to what
was reported in the literature (1,13). The
knee joint was the most common tumor location, which is also
consistent with the findings of other studies (14–16).
HMO severity was reported using various clinical
classifications (10). These
classifications were based on the clinical parameters of HMO
patients, including, age, tumor number, joint deformities, limb
length discrepancy, in addition to the morbidity associated with
HMO tumors. The clinical classification, used in the present study,
revealed that the majority of Jordanian patients with HMO have a
mild disease form (class 1). Since different HMO studies (14,17,18) used
different scoring systems to assess HMO severity, comparing HMO
severity among different populations can be difficult.
HMO is not only clinically heterogenous, but it is
also genetically heterogenous (6,17,18).
Mutational analysis studies reported variable frequencies of
EXT1 and EXT2 mutations in different ethnic groups
(13). Several authors reported
EXT1 mutations to be more common, particularly in Caucasians
(7,19–21).
Although the present study investigated a different ethnic group,
it revealed a similar predominance of EXT1 mutations in this
group of Jordanian patients with HMO. On the other hand, no
potential pathogenic genetic variants of EXT2 gene were
identified in the present study. This was inconsistent with other
studies which reported that EXT2 mutations to be present in
20–45% of the patients (7). This
inconsistency may be explained by the presence of mutations in
noncoding parts of the genes. In addition, the small number of the
included patients can be a contributing factor.
Several phenotype-genotype studies of HMO reported
that EXT1 gene mutations were more likely than EXT2
gene mutations to be associated with a more severe form of the
disease (14,15,18,20–23).
Other studies reported no difference in disease severity between
these two gene mutations (19,24). In
the present study, only 2 of the 13 patients with EXT1
mutations exhibited a severe form of the disease (class III). This
can be attributed to the variability of HMO severity, even among
patients with the same EXT gene mutations particularly EXT1
gene (18,25). In addition, ethnicity can be
considered as another influential factor. This is also consistent
with the findings of a previous study investigating osteochondroma
in Jordanian patients in whom a milder form of HMO was observed
compared with that of other populations (4).
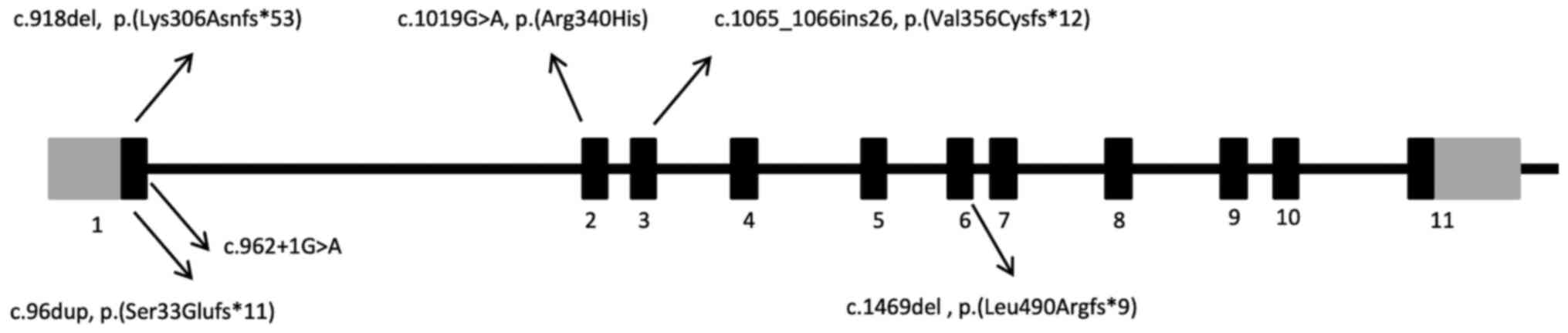
With regards to the mutational analysis of the
patients in the present study, EXT1 allelic heterogeneity
was observed and the identified mutations were shown to be
dispersed throughout the coding regions of the gene (Fig. 4). Furthermore, the truncated
mutations occurred in 66.6% of the tested families. These results
are similar to those reported from different studies in the
literature (9). A mutational
database from these studies is assembled in the Multiple
Osteochondromas Mutation Database (MOdb) (http://medgen.ua.ac.be/LOVDv.2.0/), with >600 and
200 different mutations in EXT1 and EXT2,
respectively. The majority of these mutations (80%) are nonsense,
whereas the remaining 20% are frameshift mutations and
splice-junction mutations, causing an early termination of
translation or partial/complete deletion of the gene and loss of
protein function (7).
In the present study, 19% (3/16) of patients with
HMO had no point mutations in the coding regions for either
EXT1 or EXT2 genes. This may be explained by variants
involving large rearrangements such as deletions, duplications,
inversions, translocations or somatic mosaicism, that include the
EXT1 and EXT2 genes. Deletion of a single or multiple
exons were previously detected in ~10% of all HMO cases (7,26,27).
Another study reported a complex rearrangement as causative
mechanism of the disease, which involved a 80.7 kb intronic
deletion of EXT1gene and a 68.9-kb duplication proximal of
EXT 1 (28). Furthermore,
genetic variants in the 5 and 3UTRs, deep intronic causing variants
or in the promoter regions were not determined in the present
study. In addition, several studies have reported that 10–15% of
patients with HMO have no mutations in either EXT1 or
EXT2 genes (29–31), suggesting that other genes may be
involved in the pathogenesis of the disease. Therefore, testing
this subgroup of patients by whole exome sequencing or whole genome
next generation sequencing will be an attractive approach to
identify other possible disease-causing gene(s) (32,33).
To the best of our knowledge, the majority of
phenotype-genotype research studies on HMO did not test for the
expression of EXT genes in the resected tumor tissues.
Immunohistochemistry studies revealed a decreased expression of
EXT1 and/or EXT2 corresponding to the EXT gene mutations
status (3,34,35). In
the present study, the patients with EXT1 mutations
exhibited a decreased expression of EXT1 protein. Surprisingly, the
same patients exhibited no expression of EXT2 proteins, although
none of them harbored EXT2 mutations. Previous studies
demonstrated that the presence of fully functional EXT1 and EXT2
proteins is required for their correct localization in the Golgi
complex (36,37). In addition, the present study
proposes a model (Fig. 5), in which
mutations in EXT1 result in a truncated product and/or
inactive form of the protein that can no longer bind to its EXT2
partner. The very low levels of EXT1, EXT2 and EXT1/2 alter the
stoichiometry of the complex and greatly diminish its
glycosyltransferase activity. Low levels of EXT1 can no longer
associate with its requisite partner EXT2 and, thus, EXT2 is
retained in the endoplasmic reticulum (ER) and is targeted through
the ER-associated protein degradation pathway for degradation. The
identification of EXT2 protein as a substrate for the Hrd1 E3
ligase and the identification of the lysine involved in ubiquitin
attachment to the protein (38,39) is
consistent with the aforementioned model, as explained in Fig. 5.
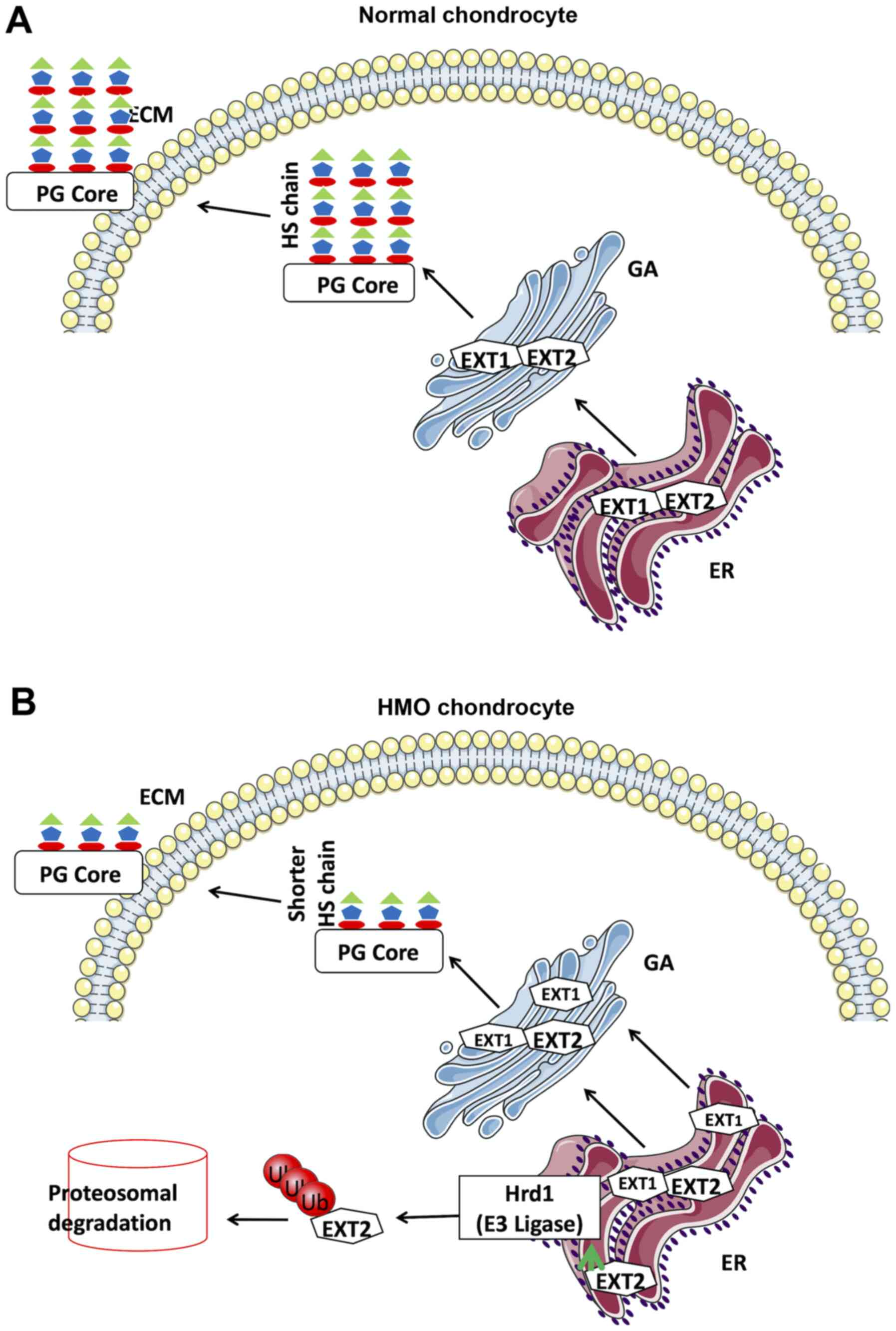 | Figure 5.Model of the function of the
EXT1/EXT2 complex in HS synthesis in (A) normal chondrocytes and
(B) HMO chondrocytes. (A) In normal chondrocytes, EXT1 and EXT2
form a complex, which is involved in the HS synthesis in the Golgi
apparatus. The EXT1/EXT2 complex, through its galactosyltransferase
activity, aids in the formation of HS proteoglycans HSPGs, which
are next exported to the cell exterior. (B) In HMO chondrocytes,
decreased levels of EXT1 disrupts the stoichiometry of the
EXT1/EXT2 complex, resulting in low HS synthesis and, thus,
diminished HSPGs on the cell exterior. Low levels of EXT1 render
EXT2 unable to be transported into the Golgi complex, which is
instead retained in the ER and thereby targeted for ER-associated
protein degradation. EXT2 is shown to be ubiquitinated at Lys 245
and is degraded though ubiquitin-proteasome system. HS, heparan
sulfate; HSPGs, HS proteoglycans; EXT, exostosin; HMO; hereditary
multiple osteochondromas; ER, endoplasmic reticulum; ECM,
extracellular matrix; GA, Golgi apparatus; PG, proteoglycans. |
Mutations in EXT1 and EXT2 are
associated with the pathogenesis of HMO; however, the mechanism
through which HS synthesis alteration leads to exostoses has yet to
be elucidated. Heterozygous EXT1 or EXT2 mutations
are common molecular changes identified in >80% of the
investigated exostoses (40). There
remains the question of whether osteochondromas arise via loss of
heterozygosity or haploinsufficiency mechanism (40,41). The
results of the present study and early biochemical studies
(36,37,42) of
the EXT1/2 complex suggest that haploinsufficiency for either EXT1
or EXT2 affects the ability of chondrocytes to synthesize HS, as
explained in Fig. 5. Although EXT1
and EXT2 are ubiquitously expressed, mutations in these genes are
only manifested in chondrocytes, suggesting that chondrocytes
require two fully active EXT1 and EXT2 proteins. However, Reijnders
et al (41) refuted the
haploinsufficiency theory and demonstrated that osteochondromas
arise via loss of heterozygosity and inactivation of both alleles
(Knudsons two-hit model) (43).
Further genetic analysis studies of large cohorts of patients with
HMO are required to determine the contribution of LOH and
haploinsufficiency in the molecular pathogenesis of the
disease.
The main limitation of the present study was the
lack of DNA samples from some members of family C (unaffected
individuals I.1 and I.2), and so genetic testing or further
investigations for mosaic mutations (pyrosequencing or
cloning of the suspected PCR products) could not be conducted.
In conclusion, the present study conducted a
phenotype-genotype study of HMO in 16 Jordanian patients from nine
families. These patients are representative of an ethnic group in
which the genetic background of HMO is infrequently investigated.
The majority of these patients were males, diagnosed the age of ~14
years, and exhibited a mild clinical disease form. Genetic analysis
revealed mutations exclusively involving EXT1 gene and none
involved EXT2 gene. These mutations were not necessarily
associated with a severe clinical disease. Three of the identified
mutations were novel. Three patients did not show any mutations for
either EXT1 or EXT2 genes. Upon immunohistochemical
testing, osteochondroma tissue resected form all patients with
EXT1 mutations exhibited decreased expression of EXT1
protein. Surprisingly, EXT2 protein was not detected in these
patients although none had EXT2 mutations. Therefore, a
model may be suggested that questions the role of EXT2 gene
in HMO pathogenies. HMO continues to represent a clinically and
genetically heterogenous disease among different ethnic groups.
Therefore, further genetic and immunohistochemical studies are
required to further elucidate the pathogenesis of HMO. In addition,
mutational analysis studies can be helpful in screening for
patients with HMO, particularly those who may be at a risk of
developing a severe form of the disease, which will have a
significant impact on the clinical management and follow up of
patients with HMO.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Jordan
University of Science and Technology (grant no. 20180318).
Availability of data and materials
Sanger sequencing data are available at https://www.ebi.ac.uk/eva with the following
accessions: Project, PRJEB41290; analyses, ERZ1673834. The
following links can also be used: https://www.ebi.ac.uk/ena/data/view/PRJEB41290,
https://www.ebi.ac.uk/eva/?eva-study=PRJEB41290 and
https://wwwdev.ebi.ac.uk/eva/?eva-study=PRJEB41290.
Authors contributions
ZM performed the collection and analysis of data,
study design, writing and editing of the manuscript. KB performed
the data analysis, study design, writing and editing of the
manuscript. MA was responsible for the study design and writing of
the manuscript. RA performed the data collection, study design and
writing of the manuscript. MAA performed the data collection, study
design and writing of the manuscript. ABK cotributed to the data
collection, study design and writing of the manuscript. KAB
performed the data analysis and writing of the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Approval for this research was obtained from the
Human Research Ethics Committee and IRB at Jordan University of
Science and Technology (approval no. 22/116/2018). All individuals
included in this study and/or their legal guardians provided
written informed consent for participating in this study.
Patient consent for publication
All participants and/or their legal guardians
provided a written informed consent regarding the publication of
case details and any associated images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Darienzo A, Andreani L, Sacchetti F,
Colangeli S and Capanna R: Hereditary multiple exostoses: Current
insights. Orthop Res Rev. 11:199–211. 2019.PubMed/NCBI
|
|
2
|
Hakim DN, Pelly T, Kulendran M and Caris
JA: Benign tumours of the bone: A review. J Bone Oncol. 4:37–41.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bovée JV: Multiple osteochondromas.
Orphanet J Rare Dis. 3:32008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mohaidat ZM, Saleh AA, Al-Omari MH,
Obeidat AA and Khasawneh RA: Osteochondroma in Jordanian Patients:
Clinical manifestations and management. J Clin Diagnostic Res.
12:RC11–RC15. 2018.
|
|
5
|
Beltrami G, Ristori G, Scoccianti G,
Tamburini A and Capanna R: Hereditary multiple exostoses: A review
of clinical appearance and metabolic pattern. Clin Cases Miner Bone
Metab. 13:110–118. 2016.PubMed/NCBI
|
|
6
|
de Souza AM and Bispo Júnior RZ:
Osteochondroma: Ignore or investigate? Rev Bras Ortop. 49:555–564.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jennes I, Pedrini E, Zuntini M, Mordenti
M, Balkassmi S, Asteggiano CG, Casey B, Bakker B, Sangiorgi L and
Wuyts W: Multiple osteochondromas: Mutation update and description
of the multiple osteochondromas mutation database (MOdb). Hum
Mutat. 30:1620–1627. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zak BM, Crawford BE and Esko JD:
Hereditary multiple exostoses and heparan sulfate polymerization.
Biochim Biophys Acta. 1573:346–355. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Busse-Wicher M, Wicher KB and
Kusche-Gullberg M: The extostosin family: Proteins with many
functions. Matrix Biol. 35:25–33. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mordenti M, Ferrari E, Pedrini E, Fabbri
N, Campanacci L, Muselli M and Sangiorgi L: Validation of a new
multiple osteochondromas classification through Switching Neural
Networks. Am J Med Genet Part A. 161A:556–560. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fusco C, Nardella G, Fischetto R, Copetti
M, Petracca A, Annunziata F, Augello B, DAsdia MC, Petrucci S,
Mattina T, et al: Mutational spectrum and clinical signatures in
114 families with hereditary multiple osteochondromas: Insights
into molecular properties of selected exostosin variants. Hum Mol
Genet. 28:2133–2142. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bali DS, Goldstein JL, Banugaria S, Dai J,
Mackey J, Rehder C and Kishnani PS: Predicting cross-reactive
immunological material (CRIM) status in Pompe disease using GAA
mutations: Lessons learned from 10 years of clinical laboratory
testing experience. Am J Med Genet Part C Semin Med Genet.
160C:40–49. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Santos SCL, Rizzo IMPO, Takata RI,
Speck-Martins CE, Brum JM and Sollaci C: Analysis of mutations in
EXT1 and EXT2 in Brazilian patients with multiple osteochondromas.
Mol Genet Genomic Med. 6:382–392. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jäger M, Westhoff B, Portier S, Leube B,
Hardt K, Royer-Pokora B, Gossheger G and Krauspe R: Clinical
outcome and genotype in patients with hereditary multiple
exostoses. J Orthop Res. 25:1541–1551. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Porter DE, Lonie L, Fraser M, Dobson-Stone
C, Porter JR, Monaco AP and Simpson AH: Severity of disease and
risk of malignant change in hereditary multiple exostoses. A
genotype-phenotype study. J Bone Joint Surg Br. 86:1041–1046. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Saglik Y, Altay M, Unal VS, Basarir K and
Yildiz Y: Manifestations and management of osteochondromas: A
retrospective analysis of 382 patients. Acta Orthop Belg.
72:748–755. 2006.PubMed/NCBI
|
|
17
|
Li Y, Wang J, Wang Z, Tang J and Yu T: A
genotype-phenotype study of hereditary multiple exostoses in
forty-six Chinese patients. BMC Med Genet. 18:1262017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Francannet C, Cohen-Tanugi A, Le Merrer M,
Munnich A, Bonaventure J and Legeai-Mallet L: Genotype-phenotype
correlation in hereditary multiple exostoses. J Med Genet.
38:430–434. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Delgado MA, Martinez-Domenech G, Sarrión
P, Urreizti R, Zecchini L, Robledo HH, Segura F, De Kremer RD,
Balcells S, Grinberg D and Asteggiano CG: A broad spectrum of
genomic changes in latinamerican patients with EXT1/EXT2-CDG. Sci
Rep. 4:64072014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pedrini E, Jennes I, Tremosini M, Milanesi
A, Mordenti M, Parra A, Sgariglia F, Zuntini M, Campanacci L,
Fabbri N, et al: Genotype-phenotype correlation study in 529
patients with multiple hereditary exostoses: Identification of
‘protective’ and ‘risk’ factors. J Bone Joint Surg Am.
93:2294–2302. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sarrión P, Sangorrin A, Urreizti R,
Delgado A, Artuch R, Martorell L, Armstrong J, Anton J, Torner F,
Vilaseca MA, et al: Mutations in the EXT1 and EXT2 genes in Spanish
patients with multiple osteochondromas. Sci Rep. 3:13462013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Heinritz W, Hüffmeier U, Strenge S,
Miterski B, Zweier C, Leinung S, Bohring A, Mitulla B, Peters U and
Froster UG: New mutations of EXT1 and EXT2 genes in German patients
with multiple osteochondromas. Ann Hum Genet. 73(Pt 3):283–291.
2009. View Article : Google Scholar
|
|
23
|
Alvarez C, Tredwell S, De Vera M and
Hayden M: The genotype-phenotype correlation of hereditary multiple
exostoses. Clin Genet. 70:122–130. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jennes I, Entius MM, Van Hul E, Parra A,
Sangiorgi L and Wuyts W: Mutation screening of EXT1 and EXT2 by
denaturing high-performance liquid chromatography, direct
sequencing analysis, fluorescence in situ hybridization, and a new
multiplex ligation-dependent probe amplification probe set in
patients with multiple osteochondromas. J Mol Diagn. 10:85–92.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cousminer DL, Arkader A, Voight BF,
Pacifici M and Grant SFA: Assessing the general population
frequency of rare coding variants in the EXT1 and EXT2 genes
previously implicated in hereditary multiple exostoses. Bone.
92:196–200. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vink GR, White SJ, Gabelic S, Hogendoorn
PC, Breuning MH and Bakker E: Mutation screening of EXT1 and EXT2
by direct sequence analysis and MLPA in patients with multiple
osteochondromas: Splice site mutations and exonic deletions account
for more than half of the mutations. Eur J Hum Genet. 13:470–474.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Szuhai K, Jennes I, De Jong D, Bovée JV,
Wiweger M, Wuyts W and Hogendoorn PC: Tiling resolution array-CGH
shows that somatic mosaic deletion of the EXT gene is causative in
EXT gene mutation negative multiple osteochondromas patients. Hum
Mutat. 32:E2036–E2049. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Waaijer CJ, Winter MG, Reijnders CM, de
Jong D, John Ham S, Bovée JV and Szuhai K: Intronic deletion and
duplication proximal of the EXT1 gene: A novel causative mechanism
for multiple osteochondromas. Genes Chromosomes Cancer. 52:431–436.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hong G, Guo X, Yan W, Li Q, Zhao H, Ma P
and Hu X: Identification of a novel mutation in the EXT1 gene from
a patient with multiple osteochondromas by exome sequencing. Mol
Med Rep. 15:657–664. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang H, Ping XL, Lee PK, Wu XL, Yao YJ,
Zhang MJ, Silvers DN, Ratner D, Malhotra R, Peacocke M and Tsou HC:
Role of PTCH and p53 genes in early-onset basal cell carcinoma. Am
J Pathol. 158:381–385. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu Y, Xing X, Xu S, Ma H, Cao L, Wang S
and Luo Y: Novel and recurrent mutations in the EXT1 and EXT2 genes
in Chinese kindreds with multiple osteochondromas. J Orthop Res.
31:1492–1499. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu H, Wu S, Duan L, Zhu W, Zhang S, Hu X,
Jia W, Yang G, Liu C, Li W, et al: Identification of a novel EXT1
mutation in patients with hereditary multiple exostosis by exome
sequencing. Oncol Rep. 33:547–552. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang F, Liang J, Guo X, Zhang Y, Wen Y,
Li Q, Zhang Z, Ma W, Dai L, Liu X, et al: Exome sequencing and
functional analysis identifies a novel mutation in EXT1 gene that
causes multiple osteochondromas. PLoS One. 8:e723162013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cheung PK, McCormick C, Crawford BE, Esko
JD, Tufaro F and Duncan G: Etiological point mutations in the
hereditary multiple exostoses gene EXT1: A functional analysis of
heparan sulfate polymerase activity. Am J Hum Genet. 69:55–66.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hameetman L, David G, Yavas A, White SJ,
Taminiau AH, Cleton-Jansen AM, Hogendoorn PC and Bovée JV:
Decreased EXT expression and intracellular accumulation of heparan
sulphate proteoglycan in osteochondromas and peripheral
chondrosarcomas. J Pathol. 211:399–409. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
McCormick C, Duncan G, Goutsos KT and
Tufaro F: The putative tumor suppressors EXT1 and EXT2 form a
stable complex that accumulates in the Golgi apparatus and
catalyzes the synthesis of heparan sulfate. Proc Natl Acad Sci USA.
97:668–673. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kobayashi S, Morimoto K, Shimizu T,
Takahashi M, Kurosawa H and Shirasawa T: Association of EXT1 and
EXT2, hereditary multiple exostoses gene products, in Golgi
apparatus. Biochem Biophys Res Commun. 268:860–867. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee KA, Hammerle LP, Andrews PS, Stokes
MP, Mustelin T, Silva JC, Black RA and Doedens JR: Ubiquitin ligase
substrate identification through quantitative proteomics at both
the protein and peptide levels. J Biol Chem. 286:41530–41538. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ernst R, Claessen JH, Mueller B, Sanyal S,
Spooner E, van der Veen AG, Kirak O, Schlieker CD, Weihofen WA and
Ploegh HL: Enzymatic blockade of the ubiquitin-proteasome pathway.
PLoS Biol. 8:e10006052011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hall CR, Cole WG, Haynes R and Hecht JT:
Reevaluation of a genetic model for the development of exostosis in
hereditary multiple exostosis. Am J Med Genet. 112:1–5. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Reijnders CM, Waaijer CJ, Hamilton A,
Buddingh EP, Dijkstra SP, Ham J, Bakker E, Szuhai K, Karperien M,
Hogendoorn PC, et al: No haploinsufficiency but loss of
heterozygosity for EXT in multiple osteochondromas. Am J Pathol.
177:1946–1957. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Busse M and Kusche-Gullberg M: In vitro
polymerization of heparan sulfate backbone by the EXT proteins. J
Biol Chem. 278:41333–41337. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Knudson AG Jr: Mutation and cancer:
Statistical study of retinoblastoma. Proc Natl Acad Sci USA.
68:820–823. 1971. View Article : Google Scholar : PubMed/NCBI
|