Introduction
Breast cancer is one of the most common female
malignant tumors in the world (1),
which has been proved to be the second leading cause of death by
cancer among women worldwide (2).
According to statistics, there were 1.67 million new breast cancer
cases and 500,000 mortalities worldwide in 2012 (3). Currently, the pathogenesis of breast
cancer is not completely understood, but the cause of breast cancer
is related to a number of factors including age and breast gland
density (4,5). The incidence of breast cancer in women
increases with age (6,7), and the incidence of breast cancer in
women with high breast density is 4–6 times higher than that in
women with low breast density (8,9).
Although the treatment of cancer has improved over the years, the
current treatment for breast cancer mainly including surgical
operation and chemotherapy cannot effectively ameliorate the
overall survival of patients with breast cancer because of the
metastasis of breast cancer (10).
The high mortality of breast cancer is due to the metastasis and
drug resistance of breast cancer cells (11,12).
About 30% of patients with breast cancer will have axillary lymph
node metastasis and distal organ (stone, liver, lung and brain)
metastasis, reducing the survival rate of patients (13,14).
During the course of chemotherapy treatment for triple negative
breast cancer (TNBC), the chemical resistance of cancer cells may
be caused by the disorder of DNA replication, the repair of key
enzymes, the change of apoptosis related genes and the abnormal
regulation of key signaling pathways, which decreases the
effectiveness of cancer treatment (14,15).
Therefore, the study of new treatment methods for breast cancer is
necessary and it is vital to explore the molecular mechanism of its
formation, which may provide improved treatment strategies for
breast cancer.
Epigenetic modifications including DNA modifications
(e.g. methylation) and histone modifications (e.g. phosphorylation
and methylation) and abnormalities of non-coding RNA affect
cellular function and transcriptional activity (16). A number of studies have shown that
cancer is often accompanied by various histone modifications, which
are related to the chromatin structure and the transcriptional
activity of proto-oncogene or tumor suppressor genes, affecting the
development of diverse cancers (17–19). For
example, DNA hypermethylation is associated with the pathogenesis
of colon cancer, lung cancer and prostate cancer (20–22).
Abdel-Hafiz and Horwitz (23)
showed that the level of histone methylation is associated with the
occurrence and development of metastatic breast cancer. In
addition, Liu et al (24)
found that the downregulation of microRNA-20a expression can
accelerate the progression of ovarian cancer. Other studies have
shown that signaling pathways can directly affect important
components of the epigenetic machinery (25). For instance, Avanzato et al
(26) suggested that the expression
of phosphorylated (p-) Akt in breast cancer tissues is
significantly higher than that in the paracancerous tissues. In
addition, and the PI3K/Akt pathway can positively regulates the
pro-apoptotic protein BAD (27).
Furthermore, Liu et al (28)
revealed that the protein bifunctional arginine demethylase and
lysyl-hydroxylase JMJD6 (JMJD6) promoted the development of TNBC
cells by phosphorylating the histone H2AX (H2A.X) at the Tyr39
(Y39) site, which regulates autophagy genes in order to stimulate
autophagy flux. In addition, the increase of autophagy flux can
inhibit cell proliferation in the middle and late stages of breast
cancer (28).
H2A.X plays an important role in regulating the
repair of double stranded DNA breaks (29). The phosphorylation of H2A.X at Ser139
(S139) binds well with each double stranded break and is considered
the most sensitive marker for examining the level of DNA damage and
the subsequent repair of DNA lesions (30). In cancer cells, when the DNA repair
mechanisms are dysfunctional, cells become dependent on the
remaining pathways, including the PI3K/Akt and ATM/γH2AX pathways
and this renders them more vulnerable to therapies that target
these specific pathways (31). The
loss of H2A.X in a human non-tumorigenic breast cell line leads to
the activation of epithelial-mesenchymal transition (EMT), thus
enhancing the ability of migration and invasion of breast cancer
cells (32). Ribosomal S6 kinase 2
(RSK2), an important regulator of cell survival, transcription,
growth and proliferation, is able to phosphorylate H2A.X at S139
and Ser16 (S16) to delay the process of breast cancer (33). RSK2 is known for mediating the growth
of osteosarcoma cells through regulating the Akt/mTOR signaling
pathway (34). In acid-tolerable
malignant mesothelioma, the phosphorylation of H2A.X is a biomarker
of DNA damage, and the Akt can negatively regulate the
phosphorylation of H2A.X (35). We
occasionally found p-Akt can negatively regulate the
phosphorylation of H2A.X in breast cancer cells, however, at
present, an interaction between Akt and H2A.X has not been reported
in breast cancer, and thus, the aim of the present study was to
explore this connection.
Materials and methods
Cells and animals
Human breast cancer cell lines MDA-MB-231 and MCF-7,
HGC-27, HeLa cell lines were purchased from the Cell Bank of Type
Culture Collection of the Chinese Academy of Sciences (Shanghai,
China). The human breast cancer cell line MDA-MB-157 was obtained
from the ATCC Global Bioresource Center. Cells were cultured in
complete Dulbecco's modified Eagle medium (DMEM) supplemented with
0.1 mg/ml streptomycin and 100 U/ml penicillin and 10% fetal bovine
serum (FBS; all purchased from Thermo Fisher Scientific, Inc.) in a
humidified incubator at 5% CO2 at 37°C for 1 week.
Antibodies
The primary and secondary antibodies used in this
study are as follows. The primary antibodies: Akt antibody (cat.
no. 9272; dilution 1:1,000), Phospho-Akt (Ser473) (193H12) Rabbit
mAb (p-Akt, cat. no. 4058; dilution 1:1,000), β-actin antibody
(cat. no. 4967; dilution 1:1,000), p-glycogen synthase kinase-3β
(p-GSK3β; cat. no. 9336, dilution 1:1,000), Phospho-GSK-3α/β
(Ser21/9) Antibody (t-GSK3β, cat. no. 9331, dilution 1:1,000),
Phospho-RSK2 (Ser227) (D53A11) Rabbit mAb (p-RSK2, cat. no. 3556;
dilution 1:1,000), RSK2 (D21B2) XP® Rabbit mAb (cat. no.
5528; dilution 1:1,000) were purchased from Cell Signaling
Technology, Inc., and anti-histone H2A.X antibody (cat. no.
ab229914; dilution 1:1,000), anti-HA-tag antibody (cat. no. ab9110;
dilution 1:1,0000), anti-Myc-tag [9E10] (cat. no. ab32; dilution
1:1,000), anti-histone H3 (acetyl K18) antibody (H2A.X S16ph; cat.
no. ab1191; dilution 1:1,000), anti-DDDDK-tag (Binds to
FLAG® tag sequence) antibody (Flag-tag, cat. no.
ab205606; dilution 1:50,000) and anti-GAPDH antibody (cat. no.
ab9485; dilution 1:2,500) were purchased from Abcam. All primary
antibodies were from Rabbit. The secondary antibodies: Goat
Anti-Rabbit IgG H&L (HRP) (cat. no. ab205718; dilution
1:5,000).
Western blotting
Cells were lysed using RIPA buffer (50 mM Tris, pH
7.4, 0.5% sodium deoxycholate, 1% NP-40, 0.5% sodium deoxycholate
and 150 mM NaCl), and the protein concentration was determined
using the bicinchoninic acid assay (BCA) method. Briefly, 200 µl of
the BCA working solution and 20 µl of diluted sample protein were
added per well into 96-well plates and the absorbance was measured
at 526 nm after incubation at 37°C for 30 min. Protein
concentration was calculated in relation to the standard curve.
Protein samples (50 µg) were then separated using 10% SDS-PAGE and
transferred to PVDF membranes, followed by blocking at 25°C for 1 h
in 5% non-fat milk powder. Membranes were then incubated at 4°C
overnight with primary antibodies. The membranes were then washed
with TBS-Tween buffer to remove the free protein and incubated with
anti-goat and anti-rabbit IgG H&L (HRP) (cat. no. ab205718;
dilution 1:5,000; Abcam) secondary antibodies at 25°C for 1 h. The
protein bands were visualized using the ECL Western blotting kit
(cat. no. WP20005; Thermo Fisher Scientific, Inc.), and the bands
were exposed to autoradiography film. β-actin was used as the
loading control. Each experiment was repeated three times.
Cell transfection and treatment
Akt siRNA (si-Akt), RSK2 siRNA (si-RSK2), negative
control siRNA (si-NC), RSK2S19A (Ser19 site of RSK2 was
replaced by Ala) plasmid and RSK2S19E (Ser19 site of
RSK2 was replaced by S19E) plasmid were purchased and constructed
by Hanbio Biotechnology Co., Ltd. The sequences of siRNA: si-Akt,
5′-CUGACCAAGAUGACAGCAU-3′ (sense), 5′-AUGCUGUCAUCUUGGUCAG-3′
(antisense); si-RSK2, 5′-AGUUUACUGAUGGAUAUGAAGUAAA-3′ (sense),
5′-AGAAGAAGAUGUCAAAUUCUACUUG-3′ (antisense); si-NC,
5′-UUCUCCGAACGUGUCACGUTT-3′ (sense), 5′-ACGUGACACGUUCGGAGAATT-3′
(antisense). DN-Akt (dominant negative Akt) and CA-Akt
(constitutively activated Akt) were purchased from (Addgene,
Inc.).MBA-MD-231 cells (106 cells/ml, 50 µl/well) were
cultured in the 6-well plates with complete DMEM for 24 h, before
being transfected with the plasmids (1 µg/ml) include the control
empty vector pEGFP-N1, and the generated plasmids DN-Akt, CA-Akt,
RSK2S19A and RSK2S19E. In addition, si-Akt,
si-RSK2 and the si-NCwere also transfected. Transfections were
performed using Lipofectamine 3,000 (Invitrogen; Thermo Fisher
Scientific, Inc.). After 48 h of transfection, the transfection
efficiency was determined by western blotting. As presented in
Fig. S1, the transfection
efficiency was high. When needed cells were stimulated with
insulin-like growth factor (IGF) 15 ng/ml for 30 min or treated
with the PI3K-Akt inhibitor LY294002 16 µM for 24 h.
Peptide competition assay
A polyclonal antibody was generated in-house to
specifically recognize an RSK2 peptide that harbored the
phosphorylated S19 (anti-RSK2-S19ph antibody). The specificity of
the anti-RSK2-S19ph antibody was examined by peptide competition
assay. The synthesized phospho-RSK2 and non-phospho-RSK2 peptides
were purchased from Medical & Biological Laboratories (Beijing,
China). Firstly, 10 µM anti-RSK2-S19ph antibody was pre-incubated
with phospho-RSK2 or non-phospho-RSK2 peptide for 20 min at 30°C.
Then, the total protein was extracted from MDA-MB-231 cells using
1× SDS buffer, and boiled at 95°C for 10 min to inactivate the
protein. Next, the total protein was separated on 15% SDS-PAGE, and
transferred to PVDF membranes. Then the membranes were incubated
with anti-RSK2-S19ph antibody (1:1,000) pre-treated with
phospho-RSK2 or non-phospho-RSK2 peptides for 1 h at 30°C for the
detection of p-RSK2. The membranes were also incubated anti-RSK2
antibody (1:1,000) with same condition for the detection of
RSK2.
MTT assay
MTT assay was used to detect the viability of
MDA-MB-231 cells. The MDA-MB-231 cells were cultured in MDEM with
penicillin-streptomycin and FBS for 7 days at 37°C. Then, the cells
were seeded into 96-well plates (5×103 cells/well) with
complete DMEM (100 µl per well). Next, the cells were transfected
with WT-/MUT (S19A and S19E)-RSK2 plasmids, and the blank vector as
the negative control. A total of 20 µl of a 5 mg/ml MTT solution
(Sigma-Aldrich, Merck KGaA) was added to each well after the cells
were cultured for 1, 2 and 3 days, and the plate was incubated for
4 h at 37°C. Then, the MTT was discarded, and DMSO (150 µl/well)
was added to each well. The plate was incubated for 10 min and the
absorbance values were measured at 570 nm using a microplate
reader.
Soft agar colony formation assay
First of all, we carried out a pre-experiment of
colony formation assay to measure the cell vitality by using acid
phosphatase method to detect the cellular ability of survival and
proliferation. Then, a cell suspension (MDA-MB-231 cells with DMEM,
FBS and penicillin-streptomycin) of 1,000 living cells/m and were
added to 3% (w/v) melted agar. Then, the cell-agar suspensions (1
ml/well) were added into 6-well plates and after solidification,
cells were maintained at 37°C. After 14 days, cells were fixed with
4% paraformaldehyde and stained with 1% crystal violet and the
colonies were counted manually.
Transwell migration assay
Cell migration was assayed using a Transwell chamber
(8 µm, 12 wells). A total of 1×104 cells in serum-free
DMEM were transferred to the upper compartment of the well inserts
and the cells were cultured for 24 h at 37°C. Meanwhile, the lower
compartment was filled with complete DMEM (10% FBS). After 24 h,
the cells on the top surface of the membrane were wiped off softly
with a cotton swab and the cells that migrated to the bottom of the
membrane were fixed with methanol and were stained with 5% crystal
violet for 10 min at room temperature and were visualized using a
Leica DC 300F microscope (magnification, ×400).
Kinase activity assay
The kinase activity of RSK2 was tested by measuring
the level of H2A phosphorylation after stimulation with RSK2. The
assay mixture included the Myc-tag wild type (WT) RSK2 plasmid
(addgen) or the mutant RSK2 (MUT-RSK2) plasmids (S19A and S19E),
with 10 µM CoA (Abcam) and 10 µg H2A (Abcam), which were incubated
with the kinase assay buffer (Thermo Fisher Scientific, Inc.) for
30 min at 30°C. The kinase activity of hemagglutinin (HA)-tagged
Akt (HA-Akt) phosphorylating Myc-tagged RSK2 (Myc-RSK2) was assayed
by western blotting by measuring the phosphorylated H2A in the
presence of H2A and acetyl-CoA. In addition, the protein tags,
including Myc and HA were attached to carrier and used to detect
the target protein more conveniently.
Chromatin immunoprecipitation
(ChIP)
A total of 3×106 cells/ml were
crosslinked in 1% formaldehyde for 10 min at 25°C, followed by
washing with ice-cold PBS for 3 times and cell lysis with SDS lysis
buffer (Upstate Biotechnology, Inc.). The lysates were then
sonicated to shear the DNA into 200–800 bp fragments and
centrifuged at 12,000 × g for 5 min at 4°C. The supernatant was
diluted with ChIP DB (1% Triton X-100; 1.2 mM EDTA; 16.7 mM
Tris-HCL, pH 0.8; 167 mM NaCl; 0.1 mM PMSF) and was cultured
overnight with the A/G agarose beads (Cell Signaling Technology,
Inc.) and primary antibody against RSK2, H2A.X and β-actin at 4°C.
The normal anti-Rabbit immunoglobulin G (IgG) antibody served as
the control. The beads were washed for 5 min with low-salt (150 mM
NaCl; 0.1% SDS; 1% Triton X-100; 2 mM EDTA; 20 mM Tris-HCL, pH 0.8)
high-salt (500 mM NaCl; 0.1% SDS; 1% Triton X-100; 2 mM EDTA; 20 mM
Tris-HCl, pH 0.8) and LiCl buffers at 4°C and were finally washed
with 1X Tris-EDTA at 25°C for 2 min. The DNA was eluted from the
beads by adding 1% SDS and NaCl was added to the eluant for 7 h at
65°C to reverse the crosslinks. Subsequently, the eluted DNA was
precipitated by ethanol at −20°C, following by treatment with 20 µg
proteinase K to digest proteins attached to DNA. DNA was purified
utilizing the QIAquick PCR Purification Columns (Qiagen China Co.,
Ltd.). Finally, the purified DNA was analyzed by RT-qPCR.
Co-immunoprecipitation analysis
MDA-MB-231 cells were washed three times with
ice-cold PBS and lysed in RIPA lysis buffer [50 mM Tris (pH 7.4),
150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate] for 30 min on ice.
After the lysates were centrifuged at 4°C for 30 min at 12,000 × g,
the supernatant was incubated with the primary antibodies: Anti-Akt
antibody (cat. no. 9272; dilution 1:1,000; Cell Signaling
Technology, Inc.) and anti-RSK2 antibody (cat. no. 5528; dilution
1:1,000; Cell Signaling Technology, Inc.) overnight at 4°C. The
normal anti-Rabbit IgG antibody (cat. no. 2729; dilution 1:1,000,
Cell Signaling Technology, Inc.) served as the negative control,
and the whole lysates as the positive control. On the following
day, A/G agarose beads (Cell Signaling Technology, Inc.) were added
to the lysates and incubated for a further 3 h, after which the
beads were washed with PBS and collected by centrifugation for 5
min at 3,000 × g at 4°C. The proteins were released by boiling the
samples and were analyzed by western blotting on a 15%
SDS-PAGE.
Reverse transcription-quantitative
(RT-q) PCR
The total RNA of RSK2 and the target genes of Akt
like PSAT-1 were extracted and purified using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions. Next, 5 µg total RNA was applied to
synthesize cDNA using the First Strand cDNA Synthesis kit (Thermo
Fisher Scientific, Inc.), according to the manufacturer's
instructions. Then, the mixture system including SYBR Green qPCR
Master Mix (500 nM), cDNA templates (50 ng) and primers (500 nM)
were applied to Real-time qPCR to detect the expression of RSK2 and
PSAT-1, according to standard methods (95°C, 3 min; 95°C, 30 sec,
30 circles; 55°C, 30 sec, and 72°C, 60 sec). Finally, the PCR
products was electrophoresed with 1.5% agarose gel, and the
electrophoretic bands were observed. The relative expression was
calculated by 2−ΔΔCq method (36). GAPDH were applied to as an internal
control of the target gene of Akt, and the β-actin as the control
of RSK2. The primer sequences used for qPCR are listed in Table I.
 | Table I.Primer sequences used for
quantitative PCR. |
Table I.
Primer sequences used for
quantitative PCR.
| Gene | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| RSK2 |
TCCGACTTAAATGAAAGGAG |
GCTCAGCATTGTAGGTGAC |
| CDKN2A |
GAAGAAAGAGGAGGGGTTGG |
GAAGAAAGAGGAGGGGTTGG |
| PUMA |
GCCAGATTTGTGAGACAAGAGG |
CAGGCACCTAATTGGGCTC |
| Dram 1 |
GCGCGCTCCGGAAATCCCCCGGGAG |
CCTCGCACCCCGAGGGCG |
| PTEN |
CCGAAAGGTTTTGCTACCATTCT |
AAAATTATTTCCTTTCTGAGCATTCC |
| ING3 |
ACCTGAGTGGAGGGAAGAGC |
CTGGTTTGCCAACTGAACCT |
| PSAT-1 |
CGGAATTCCGATGGACGCCCCCA |
GGAAGATCTCTCATAGCTGATGCATCTCCA |
| Bax |
TGCTTCAGGGTTTCATCCAGG |
TGGCAAAGTAGAAAAGGGCGA |
| RTK6 |
CGTACGAGGTAAACAGGAG |
AGCTTGTTCTCCTCGCTGTA |
| GAPDH |
TCGACAGTCAGCCGCATCTTCTTT |
GCCCAATACGACCAAATCCGTTGA |
| β-actin |
TCAGGTCATCACTATCGGCAAT | AAAGAA
AGGGTGTAAAACGCA |
Statistical analysis
All experiments were performed in triplicate and
data are presented as the mean ± standard deviation. Statistical
analysis between two groups was calculated using unpaired Student's
t-test, and the comparison among multiple groups was evaluated with
ANOVA with Sidak's multiple comparisons test. The statistical
graphs were drawn using GraphPad Prism (version 7.0; GraphPad
Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
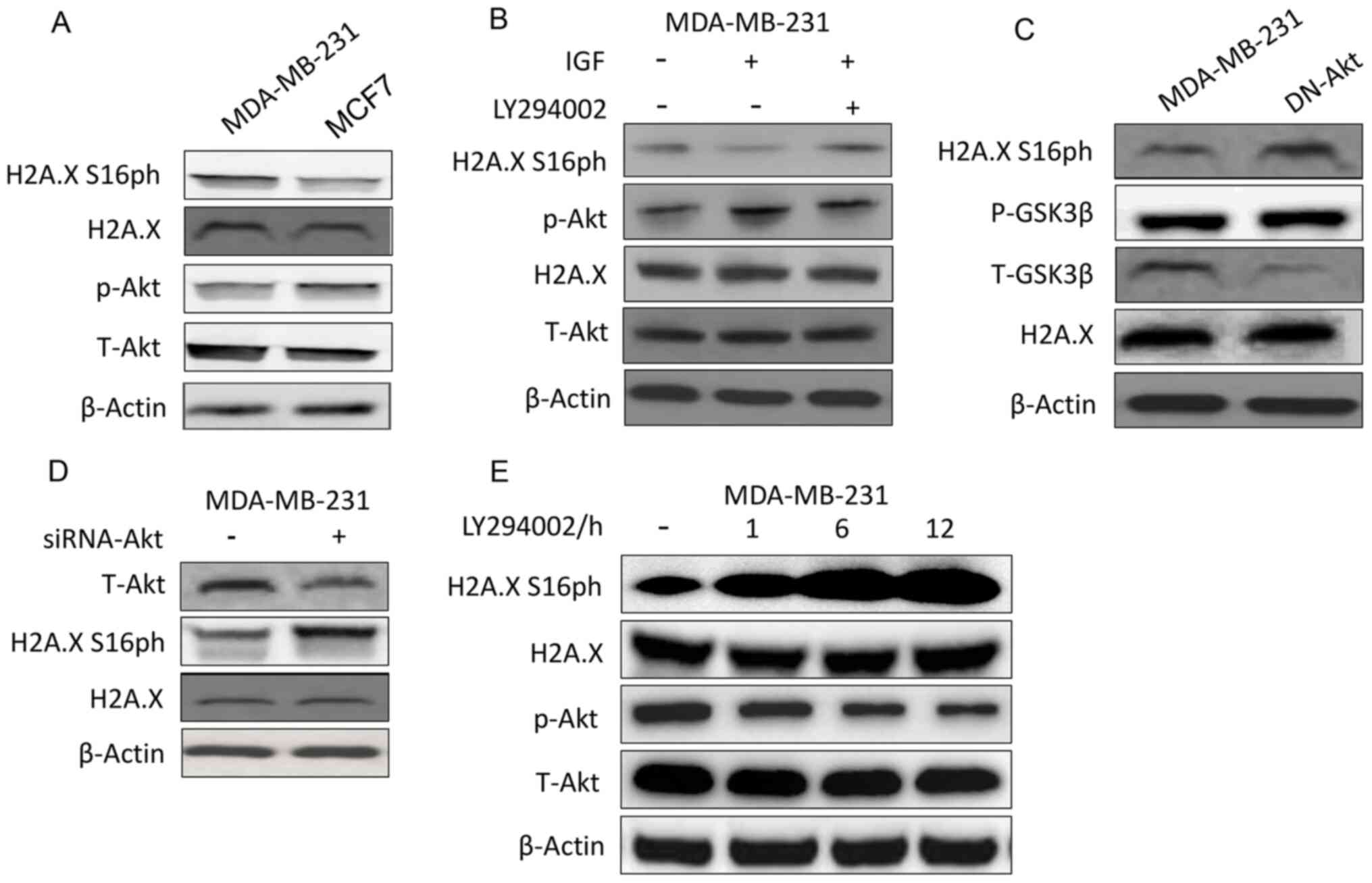
Akt inversely regulates H2A.X
S16ph
MDA-MB-231 was selected as the main experimental
cell line as it has a higher ability of invasion and migration
compared with the other breast cancer cell lines (37). Western blot results showed that
breast cancer cells with high levels of p-Akt generally had lower
levels of H2A.X S16ph (Fig. 1A).
This phenomenon prompted the investigation on whether H2A.X S16
phosphorylation was affected by Akt. Indeed, when the MDA-MB-231
cells were stimulated with IGF to increase the phosphorylation of
Akt, a decrease in H2A.X S16ph was observed, and this effect was
reversed after the introduction of the PI3K-Akt inhibitor LY294002
(Fig. 1B). Likewise, when the
MDA-MB-231 cells were transfected with DN-Akt (Fig. 1C and S1A) or when the expression of Akt was
knocked down by siRNA (Fig. 1D) the
phosphorylation of H2A.X S16 also increased. As presented in
Fig. 1C, GSK3β was a control of
p-Akt as it can be phosphorylated by Akt (38). Furthermore, the phosphorylation of
H2A.X S16 was negatively associated with the activity of Akt over
time (Fig. 1E).
Akt interacts with and phosphorylates
RSK2 on Ser19 (S19)
To examine how the phosphorylation of H2A.X S16 was
regulated by Akt signaling, the activity of the RSK2 enzyme that
regulates H2A.X S16ph (33) was
assessed. No change was observed after IGF was added (Fig. S2). Thus, Akt was examined to
understand if it can regulate RSK2 activity and therefore affect
the phosphorylation of H2A.X S16. Co-immunoprecipitation
experiments were performed to confirm the interaction between RSK2
and Akt. Results showed that Myc-RSK2 and HA-Akt could be detected
in the presence of each other, when CA-Akt was expressed but not
DN-Akt (Fig. 2A). Besides, the
association between the endogenous Akt and RSK2 was reduced after
treatment with LY294002 (Fig. 2B).
Thus, the results suggested that Akt and RSK2 interacted with each
other in vitro.
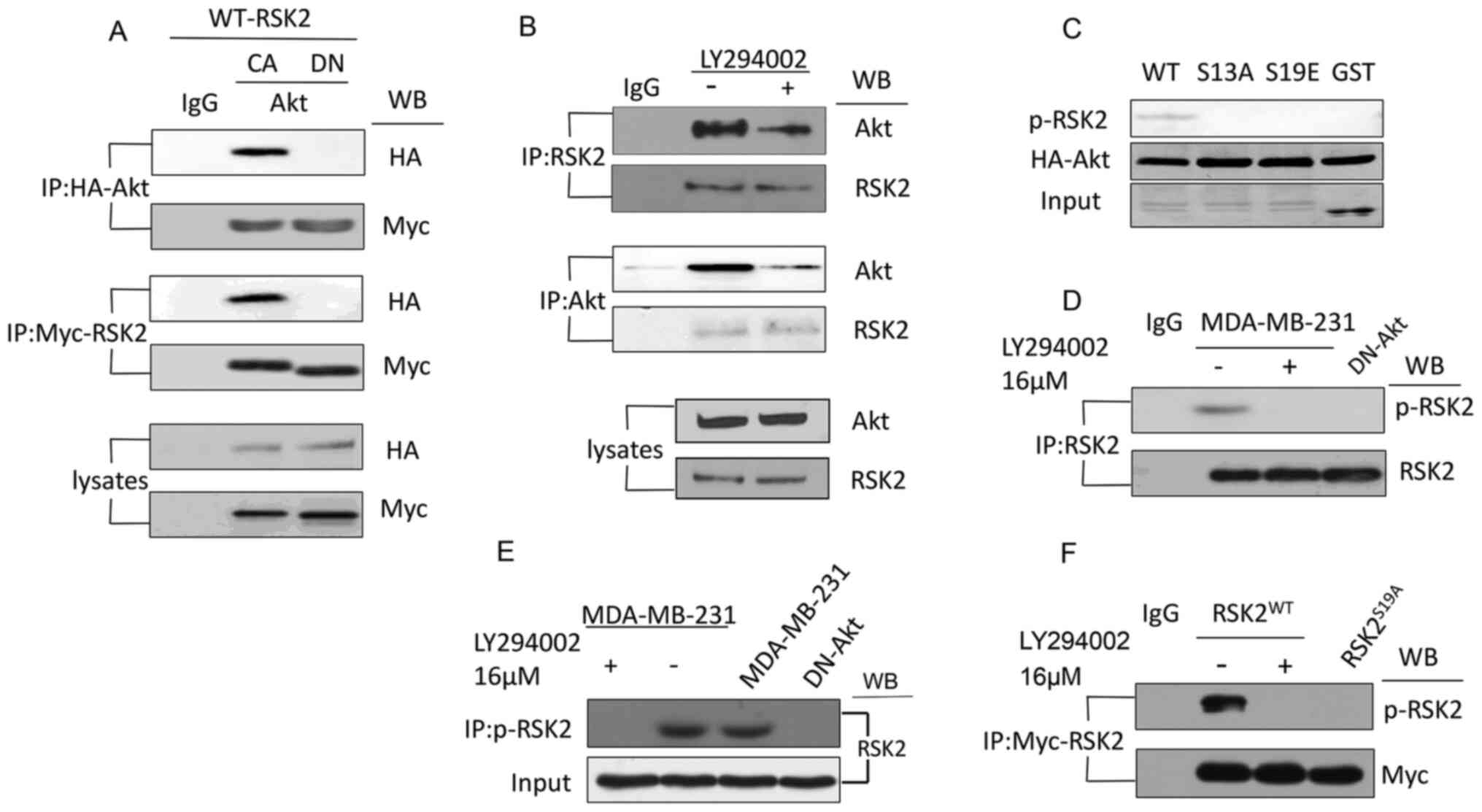 | Figure 2.Akt interacts with and phosphorylates
RSK2 at S19. (A) Co-IP was used to analyze the lysates from the
MDA-MB-231 cells transfected with the CA-Akt and DN-Akt plasmids
using antibodies targeting HA and Myc. (B) Co-IP was used to detect
the interaction of endogenous RSK2 and Akt in the MDA-MB-231 cells
with or without the PI3K-Akt inhibitor LY294002. (C) A western blot
was used to observe the phosphorylation of RSK2 in the MDA-MB-231
cells with WT- or mutant-RSK2 at the S19 site. (D) Endogenous RSK2
was immunoprecipitated using a RSK2-antibody or a p-RSK2 antibody
in the MDA-MB-231 cells treated with or without LY294002 or the
MDA-MB-231 cells transfected with DN-Akt. (E) The antibody
targeting p-RSK2 was used to immunoprecipitate the phosphorylated
endogenous RSK2, and the antibody targeting RSK2 was used for the
western blotting assay. (F) An IP was performed to assess RSK2 in
the cells transfected with the WT-RSK2 (in the presence or absence
of LY294002) or RSK2S19A plasmids, and the precipitates
were subjected to a western blot using the antibody targeting
p-RSK2. S19, Ser19; WB, western blotting; RSK2, ribosomal S6 kinase
2; RSK2S19A, RSK2 Ser19Ala; CA, constitutively
activated; DN, dominant negative; p-, phosphorylated; HA,
hemagglutinin; IP, immunoprecipitation; IgG, immunoglobulin G. |
Subsequently, a kinase assay was performed using
RSK2 mutants of possible phosphorylation sites to verify whether
the phosphorylation site was S19. The result showed that when the
S19 site of RSK2 was replaced by an Ala or Glu (S19A and S19E,
respectively) or by glutathione S-transferase (GST as a negative
control), RSK2 was not phosphorylated by Akt (Fig. 2C). Next, a polyclonal antibody was
generated in-house that specifically recognized an RSK2 peptide
that harbored the phosphorylated S19 (anti-RSK2-S19ph antibody) to
confirm that Akt phosphorylated RSK2 at S19 in vitro
(Fig. S3). The association of the
phosphorylation of RSK2 at S19 site and Akt was observed in
multiple cell lines including the gastric cancer cell line HGC-27
using the specially designed antibody (Fig. S4).
Subsequently, immunoprecipitation and immunoblotting
were used to investigate whether the phosphorylation of RSK2 at S19
was regulated by Akt using endogenous RSK2 from MDA-MB-231 cells
transfected with DN-Akt or treated with LY294002 (Fig. 2D). The result showed that the RSK2 in
the MDA-MB-231 cells was phosphorylated in the presence of LY194002
(inhibitor of Akt), but the RSK2 in cells transfected with DN-Akt
or in those treated with LY194002 was not phosphorylated (Fig. 2D). In addition, when performing the
immunoprecipitation using an antibody against p-RSK2, and western
blot using an antibody against RSK2, results showed that the levels
of p-RSK2 were reduced when Akt activity was blocked (Fig. 2E). Additionally, when the cells were
transfected with the WT- or S19A-RSK2 plasmid, a similar result was
observed (Fig. 2F). Altogether these
experiments suggested that Akt phosphorylated RSK2 at S19.
Akt suppresses RSK2 kinase
activity
To confirm whether the Akt-mediated phosphorylation
of RSK2 inhibit the phosphorylation of H2A.X S16, a western blot
was performed. The data revealed that CA-Akt decreased the
phosphorylation of H2A.X S16 but DN-Akt maintained the
phosphorylation of H2A.X S16 (Fig.
3A). In addition, the level of H2A.X S16ph in the cells
transfected with RSK2S19A was slightly higher than the
H2A.X S16ph in the cells transfected with RSK2S19E
(mimicking the phosphorylated state of RSK2) (Fig. 3B). In addition, the levels of H2A.X
S16ph decreased when RSK2 was knocked down using siRNA, and this
effect was not reversed by DN-Akt (Fig.
3C). In conclusion, p-RSK2, mediated by Akt, inhibited the
phosphorylation of H2A.X S16.
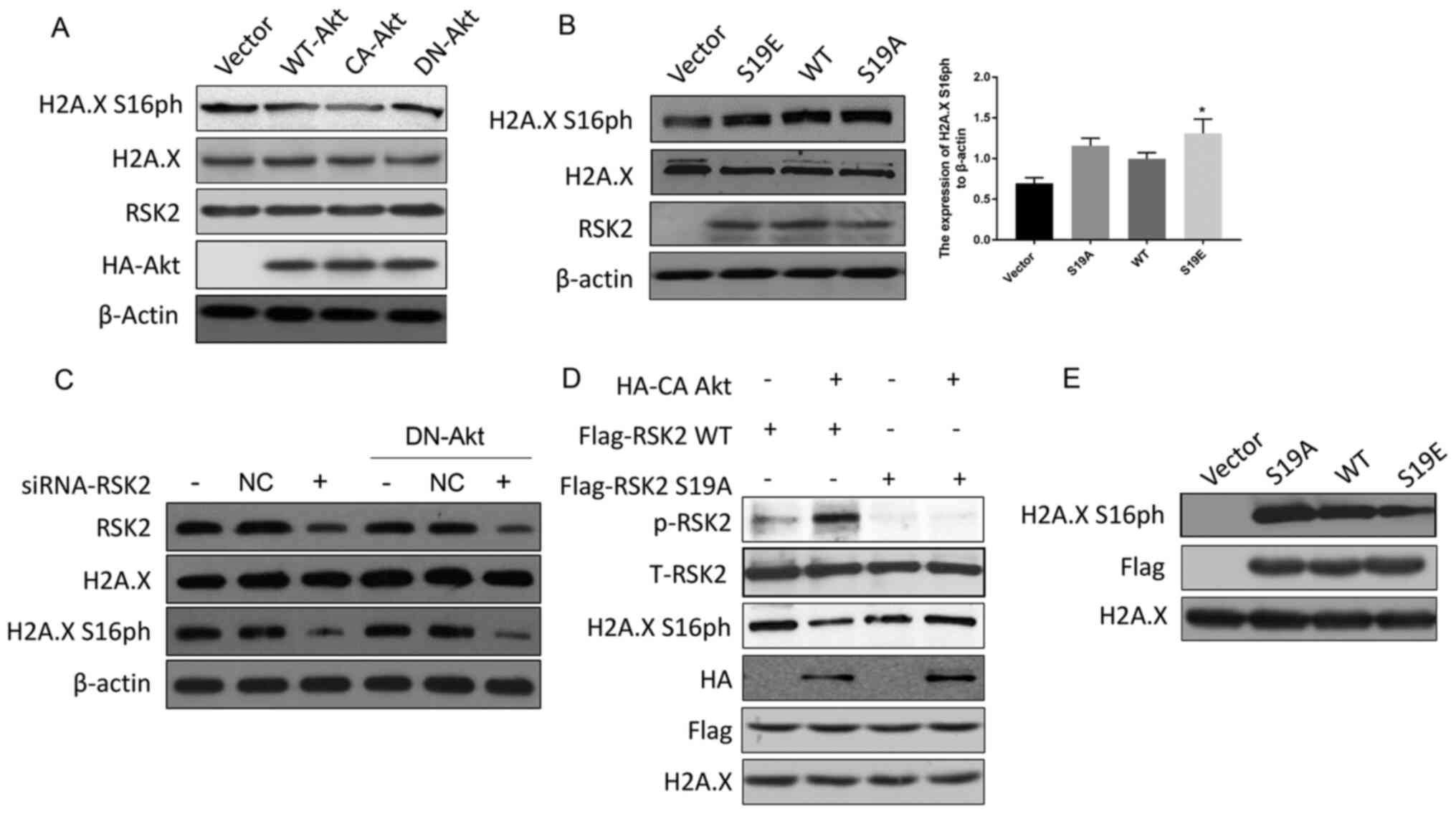 | Figure 3.Akt suppresses RSK2 kinase activity.
(A) MDA-MB-231 cells were transfected with WT-Akt, CA-Akt and
DN-Akt plasmids, and the cell lysates were subjected to a western
blot assay 48 h after the transfection. (B) MDA-MB-231 cells were
transfected with WT-RSK2, RSK2S19A and
RSK2S19E plasmids and a western blot assay was performed
48 h after the transfection. *P<0.05. (C) A Western blot assay
was used to detect the levels of H2A.X S16ph in the MDA-MB-231
cells and the DN-Akt MDA-MB-231 cells transfected with the
siRNA-RSK2 or NC. (D) WT or S19A mutant Flag-RSK2 were mixed with
HA-CA Akt, which was immunoprecipitated from the untreated lysates,
and these were incubated for 30 min in the kinase assay buffer.
Then, the kinase activity was detected with the indicated
antibodies. (E) After the WT-RSK2, RSK2S19A and
RSK2S19E plasmids were transfected into the MDA-MB-231
cells, the level of H2A.X S16ph in vitro was detected by
co-immunoprecipitation with a Flag antibody. Then, the kinase
activity was assayed for the indicated antibodies. The empty
vectors were used as the negative control, and WT-RSK2 was used as
the positive control to eliminate the interference of foreign
sequences. S19A, Ser19Ala; S19E, Ser19Glu; RSK2, ribosomal S6
kinase 2; CA, constitutively activated; DN, dominant negative; HA,
hemagglutinin; H2A.X, histone H2AX; H2A.X S16ph, H2A.X
phosphorylated at the Ser16 site; NC, negative control; WT, wild
type; t-total; p, phosphorylated. |
In order to further prove that the phosphorylation
of RSK2 at S19 by Akt affected the kinase activity of RSK2, in
vitro kinase experiments were carried out with WT-RSK2,
RSK2S19A and CA-Akt obtained by immunoprecipitation. An
increase in the phosphorylation levels of RSK2 and a decrease in
RSK2 kinase activity (reflected by a decrease in H2A.X S16ph) were
observed for the WT-RSK2 but not the RSK2S19A mutant
(Fig. 3D). Flag-tag was used to
locate the protein (Fig. 3D). To
support this discovery, another kinase activity assay was performed
with RSK2 expressed in vitro, where the results showed that
H2A.X S16 was phosphorylated by the transfection of WT-RSK2 when
compared to the empty vector control, and this phosphorylation was
enhanced by the expression of RSK2S19A and was weakened
by the expression of RSK2S19E (Fig. 3E). These data suggested that the
phosphorylation of RSK2 at S19 regulated by Akt is responsible for
regulating the activity of the RSK2 kinase.
Akt-mediated phosphorylation of RSK2
changes substrate affinity
Co-immunoprecipitation assays were used to confirm
whether the phosphorylation of RSK2 affects its affinity to histone
H2A. The association of H2A with RSK2S19A was greater
than H2A with RSK2S19E and WT-RSK2 + LY294002 (Fig. 4A). Next, the binding of RSK2 with
soluble protein (S) and insoluble (chromatin-bound) nucleoplasm (C)
was evaluated by ChIP in cells for which Akt has been induced by
IGF or suppressed by the transfection of DN-Akt or treatment with
LY294002 inhibitor. The results demonstrated that the rate of the
integration of RSK2 with the chromatin-bound was high when Akt was
suppressed, but it was low when Akt was promoted (Fig. 4B-C). The results about the
integration rate of RSK2 with soluble demonstrated that the
integration rate was high when Akt was overexpressed; however, it
was low when Akt was inhibited (Fig. 4B
and C).
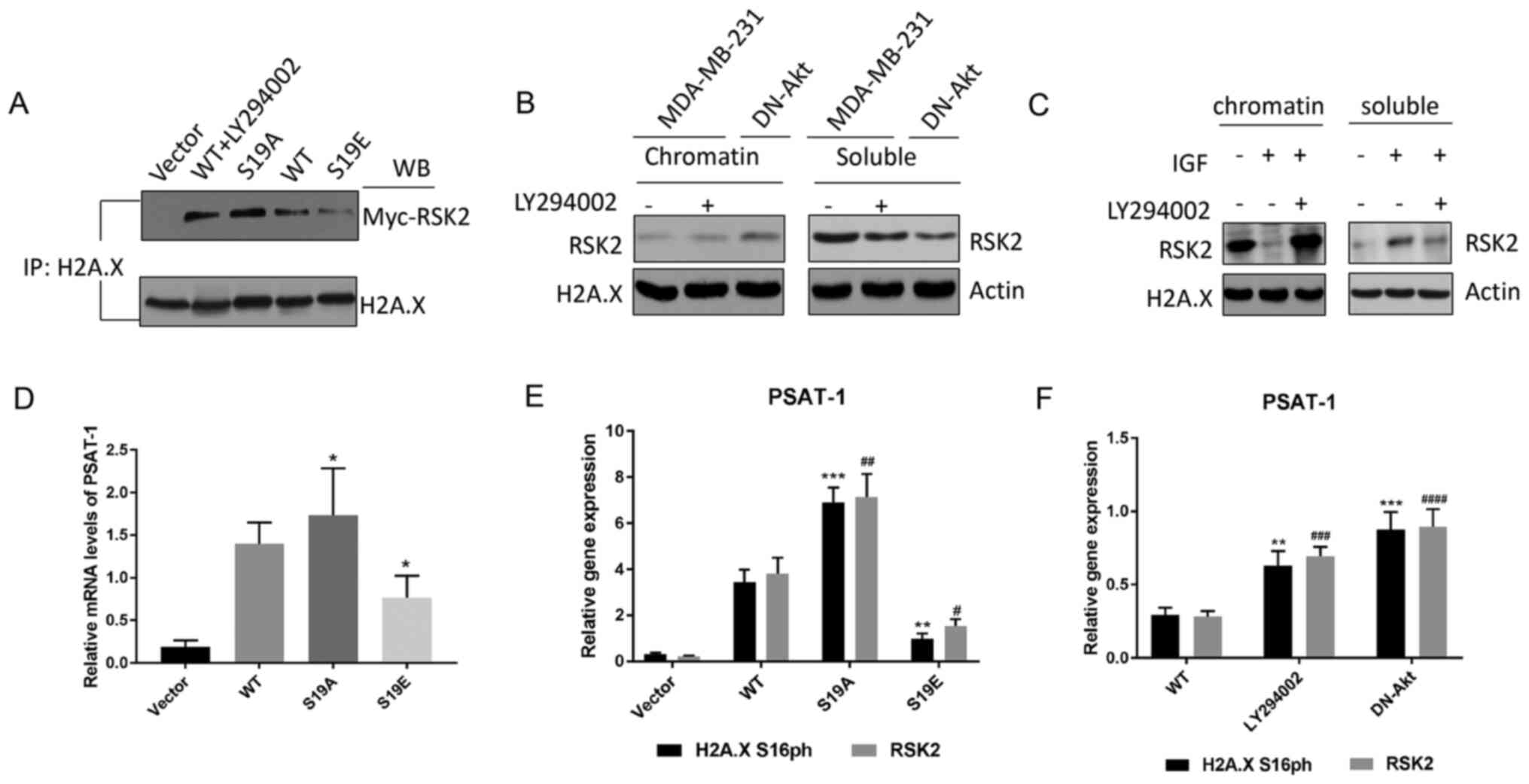 | Figure 4.Akt-mediated phosphorylation of RSK2
changes substrate affinity. (A) The endogenous histone H2A.X was
determined by co-IP in MDA-MB-231 cells transfected with WT-RSK2
(in the presence or absence of the PI3K-Akt inhibitor LY294002) or
MUT-RSK2 (S19A and S19E) plasmids. (B-C) Western blot of the
endogenous RSK2 in the c and s fractions from the (B) MDA-MB-231
cells (in the presence or absence of LY294002) and the DN-Akt
transfected MDA-MB-231 cells, and (C) the MDA-MB-231 cells treated
with IGF or a combination of IGF and LY294002. (D) The relative
PSAT-1 mRNA expression levels of in the MDA-MB-231 cells
transfected with vector, WT- or MUT-RSK2 (S19A and S19E) plasmids
were measured by RT-qPCR. *P<0.05 vs. WT. (E-F) The expression
of PSAT-1 in the (E) HeLa cells transfected with the WT- or
MUT-RSK2 (S19A and S19E) and (F) MDA-MB-231 cells (in the presence
or absence of LY294002) and the DN-Akt transfected MDA-MB-231 cells
was quantified via RT-qPCR analysis. **P<0.01; ***P<0.001 vs.
H2A.X S16ph. #P<0.05; ##P<0.01;
###P<0.001; ####P<0.0001 vs. WT-RSK2.
IP, immunoprecipitation; c, chromatin-bound fraction; s, soluble
fraction; H2A.X, histone H2AX; RSK2, ribosomal S6 kinase 2; DN,
dominant negative; IP, immunoprecipitation; WB, western blotting;
PSAT-1, phosphoserine aminotransferase 1; IGF, insulin-like growth
factor; MUT-RSK2, mutant-RSK2; S19A, Ser19Ala; S19E, Ser19Glu;
RT-qPCR, reverse transcription-quantitative PCR; WT, wild type. |
Moreover, to clarify whether p-RSK2 had a positive
effect on neoplasia, the influence of p-RSK2 on target genes from
the Akt pathway was assessed with MDA-MB-231 cells. The altered
transcription of these genes was confirmed by RT-qPCR (Fig. S5). Certain genes that are affected
by RSK2 have been reported to be associated with tumor progression,
such as phosphoserine aminotransferase 1 (PSAT-1) that is
known as an oncogene from an erythropoietin-producing hepatoma cell
line (39), and its expression was
promoted by RSK2S19A (RSK2S19A could not be
phosphorylated) and inhibited by RSK2S19E when compared
with the WT control (removes the influence of exogenous sequence)
(Fig. 4D). PCR analysis demonstrated
that H2A.X S16ph was highly expressed in the promoter of PSAT-1 of
the cells transfected with RSK2S19A (Fig. 4E). In addition, high levels of H2A.X
S16ph were observed in the promoter of PSAT-1 of the cells
transfected with DN-Akt (Fig. 4F).
All experimental results indicated that p-RSK2 may inhibit the
phosphorylation of H2A.X and promote the occurrence and development
of cancer.
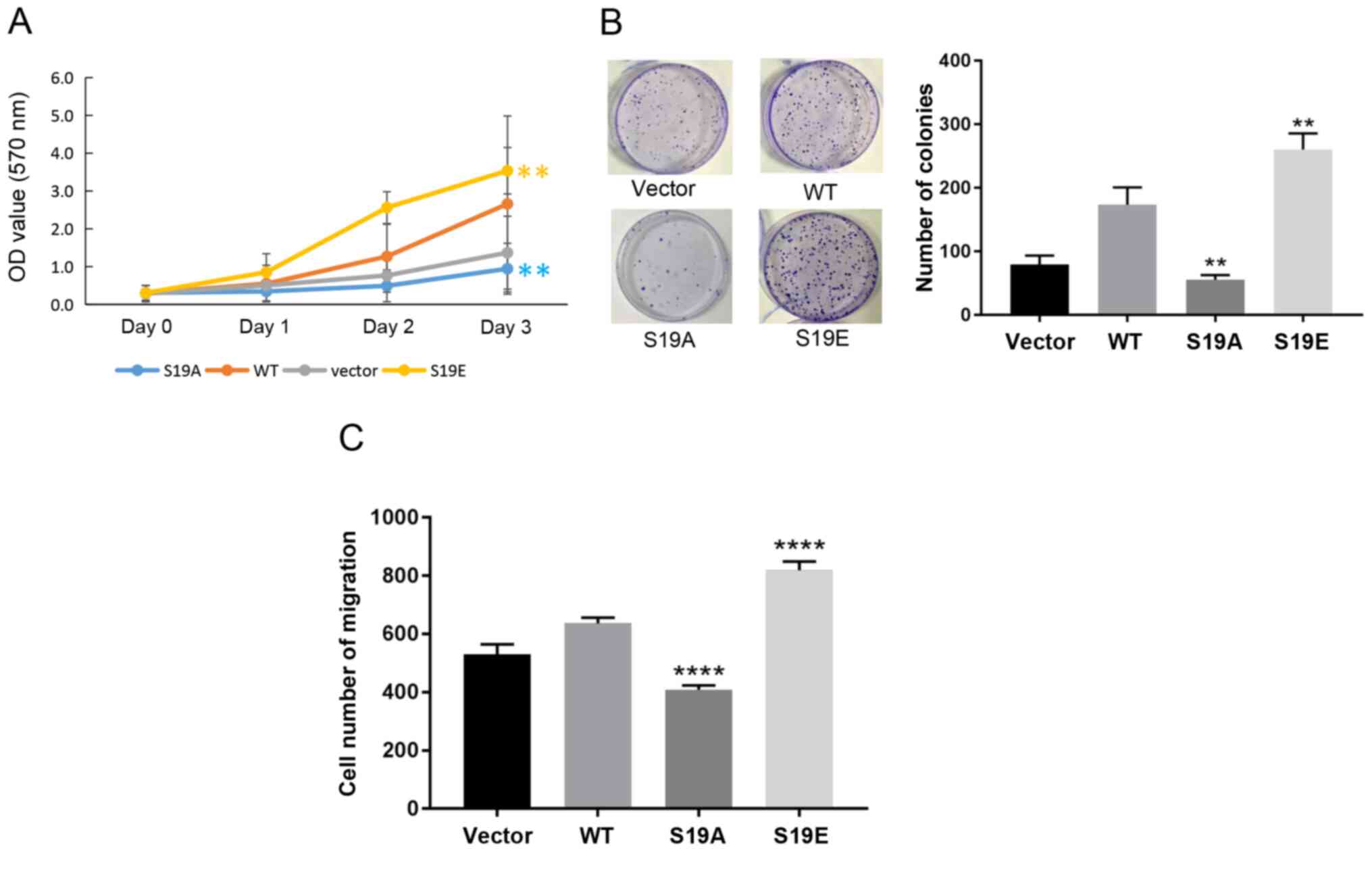
Phosphorylation of RSK2 at S19 by Akt
enhances its oncogenic activities
To further demonstrate that the phosphorylation of
RSK2 mediated by Akt induced oncogenic activity, MTT, soft agar
colony formation and Transwell migration assays were performed in
the MDA-MB-231 cells transfected with the WT-RSK2 or MUT-RSK2 (S19A
and S19E). The experimental results showed that RSK2S19E
and WT-RSK2 promoted cell viability and migration, but
RSK2S19A suppressed cell survival and migration when
compared with the empty vector control (Fig. 5A-C).
Discussion
In the present study, Akt was found to mediate the
phosphorylation of RSK2 at the S19 site, which inhibited the
phosphorylation of H2A.X at S16. In addition, it was revealed that
the interaction of Akt, RSK2 and H2A.X reduced the affinity between
RSK2 and the substrate histone and also inhibited the expression of
the cancer promoter, PSAT-1 to some extent, thereby promoting the
viability and migration of breast cancer cells.
Akt is a serine/threonine kinase that is located at
the center of the phosphatidylinositol-4,5-diphosphate 3-kinase
(PI3K)/Akt pathway (40). Akt is a
vital signaling node in cells, and it is also one of the most
widely utilized protein kinases for human physiological activities
(41). Akt plays an important role
in numerous cellular activities, including cell proliferation, cell
migration, angiogenesis, etc (42).
Additionally, the activation of Akt and its related pathways is
associated with tumorigenesis and poor prognosis (43). For instance, the amplification of
three subtypes of Akt is detected in a variety of tumor cells,
including colon, stomach, prostate and ovarian cancers (44,45).
Moreover, the overactivation of the PI3K/Akt pathway increased the
number of cancer stem cells, promotes cancer cell metastasis and
induces drug resistance (46,47). In
addition, the activity of the Akt/PI3K pathway regulates the
growth, proliferation and survival of cells affecting the response
of tumor cells to radiotherapy (48).
Histone H2A.X is a mutant of histone H2A and is
widely expressed in the genome and is highly conserved among
species (49). In the absence of
histone H2A.X, gene stability is decreased and the susceptibility
of cells to become cancerous increases, suggesting that H2A.X may
be a tumor suppressor (50). Weyemi
et al (50) showed that the
presence of H2A.X in colon cancer cells confers a strong ability to
metastatic colonization, making the tumor invasive, and a high
level of phosphorylation of H2A.X is one sign of a premalignant
lesion and the early stages of cancer. Zhu et al and Dong
et al (33,51) found that the combination of a high
phosphorylation of H2A.X at the Tyr142 or S139 sites and the
RAS/ERK pathway in gastric cancer cells enhanced the apoptosis
response caused by DNA damage, and increased the susceptibility of
cancer cells to drug treatment. In addition, Livin/Baculoviral IAP
repeat-contaning protein 7 induces the proliferation and migration
of colon cancer cells by phosphorylating H2A.X at the Y39 site by
the degradation of JMJD6 (28).
Livin also increases the phosphorylation of H2A.XY39 in
lung cancer and breast cancer cells (29,52). S16
is a newly reported phosphorylation site for H2A.X, and the
phosphorylation of this site prevents the ubiquitination of the
histone (33). Based on the
aforementioned properties of Akt and H2A.X, the present study
attempted to explore the relationship between these two molecules
in breast cancer. Ultimately, it was demonstrated that knocking out
or inhibiting Akt lead to an increase in H2A.X phosphorylation at
S16 and that Akt was negatively associated with H2A.X.
To examine this interaction, the present work
focused on RSK2, which is an important kinase of H2A.X. RSK2
controls several processes related to tumorigenesis, including
proliferation, viability and motility (33). In addition, RSK2 promotes the
invasion and metastasis of liver cancer and colon cancer,
contributes to the survival of multiple myeloma cells and
participates in the proliferation of prostate cancer cells and the
development of c-fos-dependent osteosarcoma (53–55). Zhu
et al (33) also suggested
that RSK2-mediated phosphorylation of H2A.X increased the stability
of H2A.X, thereby reducing the rate of cell transformation and
inhibiting the onset of cancer. Thus, we hypothesized that Akt and
RSK2 interacted with each other.
Supporting this hypothesis, Akt was found to
phosphorylate RSK2 at S19, which inhibited H2A.X phosphorylation at
S16. Then, it was established that Akt interacted with RSK2 and
H2A.X. The Akt-mediated phosphorylation of RSK2 altered its
substrate affinity to H2A and the expression level of certain
transcription factors, including PSAT-1. This is important because
the PSAT-1 factor was inhibited by the decrease of H2A.X S16ph that
was mediated by the Akt-RSK2 pathway, and PSAT-1 is known to
promote the survival and proliferation of cancer cells (56). Moreover, the present study further
indicated that the survival and migration of breast cancer cells
was promoted by the Akt-induced phosphorylation of RSK2 at S19,
which inhibited the phosphorylation of H2A.X at S16.
In conclusion, the findings of the present study
demonstrated that the interaction between Akt, RSK2 and H2A.X
serves an important role in regulating the development of breast
cancer. Specifically, Akt targets the phosphorylation of RSK2 at
S19 to regulate the phosphorylation of histone H2A.X in breast
cancer, which in turn, affects the survival and migration of breast
cancer cells. Therefore, these molecules might be effective
molecular targets for the treatment of breast cancer, providing
another avenue for understanding the pathogenesis of breast
cancer.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
ZFG designed experiments, drafted the initial
manuscript and approved the manuscript. FLK performed the
experiments and analyzed the experimental results. Both authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yu Y, Luo W, Yang ZJ, Chi JR, Li YR, Ding
Y, Ge J, Wang X and Cao XC: miR-190 suppresses breast cancer
metastasis by regulation of TGF-β-induced epithelial-mesenchymal
transition. Mol Cancer. 17:702018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu BW, Yu ZH, Chen AX, Chi JR, Ge J, Yu Y
and Cao XC: Estrogen receptor-α-miR-1271-SNAI2 feedback loop
regulates transforming growth factor-β-induced breast cancer
progression. J Exp Clin Cancer Res. 38:1092019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
van de Water W, Bastiaannet E, Dekkers OM,
de Craen AJ, Westendorp RG, Voogd AC, van de Velde CJ and Liefers
GJ: Adherence to treatment guidelines and survival in patients with
early-stage breast cancer by age at diagnosis. Br J Surg.
99:813–820. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Duffy SW, Morrish OWE, Allgood PC, Black
R, Gillan MGC, Willsher P, Cooke J, Duncan KA, Michell MJ, Dobson
HM, et al: Mammographic density and breast cancer risk in breast
screening assessment cases and women with a family history of
breast cancer. Eur J Cancer. 88:48–56. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Derks MGM, Bastiaannet E, van de Water W,
de Glas NA, Seynaeve C, Putter H, Nortier JWR, Rea D, Hasenburg A,
Markopoulos C, Dirix LY, et al: Impact of age on breast cancer
mortality and competing causes of death at 10 years follow-up in
the adjuvant TEAM trial. Eur J Cancer. 99:1–8. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McPherson K, Steel CM and Dixon JM: ABC of
breast diseases. Breast cancer-epidemiology, risk factors, and
genetics. BMJ. 309:1003–1006. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ursin G, Ma H, Wu AH, Bernstein L, Salane
M, Parisky YR, Astrahan M, Siozon CC and Pike MC: Mammographic
density and breast cancer in three ethnic groups. Cancer Epidemiol
Biomarkers Prev. 12:332–338. 2003.PubMed/NCBI
|
|
9
|
Vachon CM, van Gils CH, Sellers TA, Ghosh
K, Pruthi S, Brandt KR and Pankratz VS: Mammographic density,
breast cancer risk and risk prediction. Breast Cancer Res.
9:2172007. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li X, Huang R, Ma L, Liu S and Zong X:
Locoregional surgical treatment improves the prognosis in primary
metastatic breast cancer patients with a single distant metastasis
except for brain metastasis. Breast. 45:104–112. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mamounas EP, Anderson SJ, Dignam JJ, Bear
HD, Julian TB, Geyer CE Jr, Taghian A, Wickerham DL and Wolmark N:
Predictors of locoregional recurrence after neoadjuvant
chemotherapy: Results from combined analysis of National Surgical
Adjuvant Breast and Bowel Project B-18 and B-27. J Clin Oncol.
30:3960–3966. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Holohan C, Van Schaeybroeck S, Longley DB
and Johnston PG: Cancer drug resistance: An evolving paradigm. Nat
Rev Cancer. 13:714–726. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kapoor PM, Lindstrom S, Behrens S, Wang X,
Michailidou K, Bolla MK, Wang Q, Dennis J, Dunning AM, Pharoah PDP,
et al: Assessment of interactions between 205 breast cancer
susceptibility loci and 13 established risk factors in relation to
breast cancer risk, in the Breast Cancer Association Consortium.
Int J Epidemiol. 49:216–232. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nguyen LV, Vanner R, Dirks P and Eaves CJ:
Cancer stem cells: An evolving concept. Nat Rev Cancer. 12:133–143.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen W, Qin Y, Wang D, Zhou L, Liu Y, Chen
S, Yin L, Xiao Y, Yao XH, Yang X, et al: CCL20 triggered by
chemotherapy hinders the therapeutic efficacy of breast cancer.
PLoS Biol. 16:e20058692018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sueoka T, Koyama K, Hayashi G and Okamoto
A: Chemistry-driven epigenetic investigation of histone and DNA
modifications. Chem Rec. 18:1727–1744. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Z, Long H, Chang C, Zhao M and Lu Q:
Crosstalk between metabolism and epigenetic modifications in
autoimmune diseases: A comprehensive overview. Cell Mol Life Sci.
75:3353–3369. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dziaman T, Gackowski D, Guz J, Linowiecka
K, Bodnar M, Starczak M, Zarakowska E, Modrzejewska M, Szpila A,
Szpotan J, et al: Characteristic profiles of DNA epigenetic
modifications in colon cancer and its predisposing
conditions-benign adenomas and inflammatory bowel disease. Clin
Epigenetics. 10:722018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bhol CS, Panigrahi DP, Praharaj PP,
Mahapatra KK, Patra S, Mishra SR, Behera BP and Bhutia SK:
Epigenetic modifications of autophagy in cancer and cancer
therapeutics. Semin Cancer Biol. 66:22–33. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Su J, Huang YH, Cui X, Wang X, Zhang X,
Lei Y, Xu J, Lin X, Chen K, Lv J, et al: Homeobox oncogene
activation by pan-cancer DNA hypermethylation. Genome Biol.
19:1082018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zummeren MV, Kremer WW, Leeman A, Bleeker
MCG, Jenkins D, Sandt MV, Doorbar J, Heideman DAM, Steenbergen RDM,
Snijders PJF, et al: HPV E4 expression and DNA hypermethylation of
CADM1, MAL, and miR124-2 genes in cervical cancer and precursor
lesions. Mod Pathol. 31:1842–1850. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Angulo JC, Andres G, Ashour N,
Sanchez-Chapado M, Lopez JI and Ropero S: Development of castration
resistant prostate cancer can be predicted by a DNA
hypermethylation profile. J Urol. 195:619–626. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Abdel-Hafiz HA and Horwitz KB: Role of
epigenetic modifications in luminal breast cancer. Epigenomics.
7:847–862. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu Y, Han S, Li Y, Liu Y, Zhang D, Li Y
and Zhang J: MicroRNA-20a contributes to cisplatin-resistance and
migration of OVCAR3 ovarian cancer cell line. Oncol Lett.
14:1780–1786. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Safdari Y, Khalili M, Ebrahimzadeh MA,
Yazdani Y and Farajnia S: Natural inhibitors of PI3K/AKT signaling
in breast cancer: Emphasis on newly-discovered molecular mechanisms
of action. Pharmacol Res. 93:1–10. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Avanzato D, Pupo E, Ducano N, Isella C,
Bertalot G, Luise C, Pece S, Bruna A, Rueda OM, Caldas C, et al:
High USP6NL levels in breast cancer sustain chronic AKT
phosphorylation and GLUT1 stability fueling aerobic glycolysis.
Cancer Res. 78:3432–3444. 2018.PubMed/NCBI
|
|
27
|
Khor TO, Gul YA, Ithnin H and Seow HF:
Positive correlation between overexpression of phospho-BAD with
phosphorylated Akt at serine 473 but not threonine 308 in
colorectal carcinoma. Cancer Lett. 210:139–150. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu Y, Long YH, Wang SQ, Zhang YY, Li YF,
Mi JS, Yu CH, Li DY, Zhang JH and Zhang XJ: JMJD6 regulates histone
H2A.X phosphorylation and promotes autophagy in triple-negative
breast cancer cells via a novel tyrosine kinase activity. Oncogene.
38:980–997. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu Y, Long YH, Wang SQ, Li YF and Zhang
JH: Phosphorylation of H2A.XTyr39 positively regulates
DNA damage response and is linked to cancer progression. FEBS J.
283:4462–4473. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sharma A, Singh K and Almasan A: Histone
H2AX phosphorylation: A marker for DNA damage. Methods Mol Biol.
920:613–626. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
O'Connor MJ: Targeting the DNA damage
response in cancer. Mol Cell. 60:547–560. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Weyemi U, Redon CE, Sethi TK, Burrell AS,
Jailwala P, Kasoji M, Abrams N, Merchant A and Bonner WM: Twist1
and Slug mediate H2AX-regulated epithelial-mesenchymal transition
in breast cells. Cell Cycle. 15:2398–2404. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu F, Zykova TA, Peng C, Zhang J, Cho YY,
Zheng D, Yao K, Ma WY, Lau AT, Bode AM and Dong Z: Phosphorylation
of H2AX at Ser139 and a new phosphorylation site Ser16 by RSK2
decreases H2AX ubiquitination and inhibits cell transformation.
Cancer Res. 71:393–403. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qiu Q, Jiang J, Lin L, Cheng S, Xin D,
Jiang W, Shen J and Hu Z: Downregulation of RSK2 influences the
biological activities of human osteosarcoma cells through
inactivating AKT/mTOR signaling pathways. Int J Oncol.
48:2508–2520. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee YJ, Bae JH, Kim SA, Kim SH, Woo KM,
Nam HS, Cho MK and Lee SH: Cariporide Enhances the DNA damage and
apoptosis in acid-tolerable malignant mesothelioma H-2452 cells.
Mol Cells. 40:567–576. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR.
Methods. 25:402–408. 2002. View Article : Google Scholar
|
|
37
|
Bassett JJ, Bong A HL, Janke EK,
Robitaille M, Roberts-Thomson SJ, Peters AA and Monteith GR:
Assessment of cytosolic free calcium changes during
ceramide-induced cell death in MDA-MB-231 breast cancer cells
expressing the calcium sensor GCaMP6m. Cell Calcium. 72:39–50.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang Q, Wen L, Meng Z and Chen Y: Blockage
of endoplasmic reticulum stress attenuates nilotinib-induced
cardiotoxicity by inhibition of the Akt-GSK3β-Nox4 signaling. Eur J
Pharmacol. 822:85–94. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ravez S, Spillier Q, Marteau R, Feron O
and Frederick R: Challenges and opportunities in the development of
serine synthetic pathway inhibitors for cancer therapy. J Med Chem.
60:1227–1237. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang Q, Jiang W and Hou P: Emerging role
of PI3K/AKT in tumor-related epigenetic regulation. Semin Cancer
Biol. 59:112–124. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zheng CH, Wang JB, Lin MQ, Zhang PY, Liu
LC, Lin JX, Lu J, Chen QY, Cao LL, Lin M, et al: CDK5RAP3
suppresses Wnt/β-catenin signaling by inhibiting AKT
phosphorylation in gastric cancer. J Exp Clin Cancer Res.
37:592018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Searle EJ, Telfer BA, Mukherjee D, Forster
DM, Davies BR, Williams KJ, Stratford IJ and Illidge TM: Akt
inhibition improves long-term tumour control following radiotherapy
by altering the microenvironment. EMBO Mol Med. 9:1646–1659. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Revathidevi S and Munirajan AK: Akt in
cancer: Mediator and more. Semin Cancer Biol. 59:80–91. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Altomare DA and Testa JR: Perturbations of
the AKT signaling pathway in human cancer. Oncogene. 24:7455–7464.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Engelman JA, Luo J and Cantley LC: The
evolution of phosphatidylinositol 3-kinases as regulators of growth
and metabolism. Nat Rev Genet. 7:606–619. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Guerrero-Zotano A, Mayer IA and Arteaga
CL: PI3K/AKT/mTOR: Role in breast cancer progression, drug
resistance, and treatment. Cancer Metastasis Rev. 35:515–524. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Xie S, Yu X, Li Y, Ma H, Fan S, Chen W,
Pan G, Wang W, Zhang H, Li J and Lin Z: Upregulation of lncRNA
ADAMTS9-AS2 promotes salivary adenoid cystic carcinoma metastasis
via PI3K/Akt and MEK/Erk signaling. Mol Ther. 26:2766–2778. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Starska K, Forma E, Lewy-Trenda I,
Stasikowska-Kanicka O, Skora M and Brys M: Fibroblast growth factor
receptor 1 and 3 expression is associated with regulatory PI3K/AKT
kinase activity, as well as invasion and prognosis, in human
laryngeal cancer. Cell Oncol (Dordr). 41:253–268. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Redon CE, Nakamura AJ, Martin OA, Parekh
PR, Weyemi US and Bonner WM: Recent developments in the use of
γ-H2AX as a quantitative DNA double-strand break biomarker. Aging
(Albany NY). 3:168–174. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Weyemi U, Redon CE, Choudhuri R, Aziz T,
Maeda D, Boufraqech M, Parekh PR, Sethi TK, Kasoji M, Abrams N, et
al: The histone variant H2A.X is a regulator of the
epithelial-mesenchymal transition. Nat Commun. 7:107112016.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Dong C, Sun J, Ma S and Zhang G:
K-ras-ERK1/2 down-regulates H2A.XY142ph through WSTF to
promote the progress of gastric cancer. BMC Cancer. 19:5302019.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ge Y, Liu BL, Cui JP and Li SQ: Livin
promotes colon cancer progression by regulation of
H2A.XY39ph via JMJD6. Life Sci. 234:1167882019.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yao K, Peng C, Zhang Y, Zykova TA, Lee MH,
Lee SY, Rao E, Chen H, Ryu J, Wang L, et al: RSK2 phosphorylates
T-bet to attenuate colon cancer metastasis and growth. Proc Natl
Acad Sci USA. 114:12791–12796. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lara R, Mauri FA, Taylor H, Derua R, Shia
A, Gray C, Nicols A, Shiner RJ, Schofield E, Bates PA, et al: An
siRNA screen identifies RSK1 as a key modulator of lung cancer
metastasis. Oncogene. 30:3513–3521. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kang S, Elf S, Lythgoe K, Hitosugi T,
Taunton J, Zhou W, Xiong L, Wang D, Muller S, Fan S, et al: p90
ribosomal S6 kinase 2 promotes invasion and metastasis of human
head and neck squamous cell carcinoma cells. J Clin Invest.
120:1165–1177. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ravez S, Spillier Q, Marteau R, Feron O
and Frédérick R: Challenges and opportunities in the development of
serine synthetic pathway inhibitors for cancer therapy. J Med Chem.
60:1227–1237. 2017. View Article : Google Scholar : PubMed/NCBI
|