Introduction
The glypican-3 (GPC3) protein has emerged as a
novel, promising target for cancer immunotherapy (1). GPC3 is a member of the membrane-bound
heparan sulphate proteoglycan (glypican) family (2). The C-terminal fragment of GPC3 is
anchored to the cell membrane via a glycosylphosphatidylinositol
(GPI) anchor, whereas its N-terminus can be released into the
extracellular matrix (3,4). This modular structure enables GPC3 to
function as a receptor interacting with several regulatory
molecules. The expression of GPC3 is relatively high during
embryonic development and is precisely regulated in a tissue- and
stage-specific manner (5),
suggesting a role for GPC3 in morphogenesis and embryonic
development. After birth, GPC3 is rarely detectable in healthy
tissue. Previous studies demonstrated that GPC3 was overexpressed
in hepatocellular carcinoma (HCC) and that its expression could
serve as a potential diagnostic marker and prognostic factor for
this disease (1,6–9). The
role of GPC3 in HCC pathogenesis and development is not fully
understood, and few underlying mechanisms have been proposed. Cell
membrane-bound GPC3 can interact with growth factors; for example,
it binds Wnt and stimulates Wnt/β-catenin signalling, leading to
HCC development (10). The
involvement of GPC3 in the Yap (Yes-associated protein) and
Hedgehog (Hh) signalling pathways was described in other cancer
types and developmental processes (11). Filmus and Capurro (12) proposed that GPC3 could stimulate cell
proliferation in tumours with a dominant influence of the Wnt
signalling and inhibit proliferation in tumours with predominant Hh
signalling. Evaluating the potential use of the GPC3 antigen would
provide further insight into the targeted therapy of other cancer
types. Aside from HCC, the overexpression has been observed in
several tumour types, especially in embryonic carcinoma, yolk sac
tumours, non-small cell lung cancer and thyroid cancer (13–25).
Conversely, in some tumours, the expression of GPC3 is decreased
compared with normal tissue (10,26–29).
In lung cancer, a major contributor to
cancer-associated deaths worldwide, the role of GPC3 may be
cell-type dependent and remains poorly understood. The presence of
GPC3 in healthy lung tissue has not been reported. GPC3 expression
is significantly increased in lung squamous cell carcinoma (LSCC),
both at the mRNA and the protein levels (24,30–33).
Typically, GPC3 presence is detected in more than half of analysed
specimens from patients with LSCC and LSCC cell lines (24,30–33).
Importantly, GPC3 levels correlate inversely with LSCC
differentiation grade, and positively with metastasis and disease
progression (24). Li et al
(33) demonstrated that GPC3 could
represent a rational target in immunotherapy for LSCC. These
authors developed a strategy based on (GPC3)-redirected chimeric
antigen receptor (CAR)-engineered T lymphocytes that is currently
under evaluation in a phase-I clinical trial (33,34). By
contrast, the GPC3 protein is rarely detected on the surface of
lung adenocarcinoma (LAD) cells, where it is expressed at low mRNA
levels (24,30,31). To
the best of our knowledge, there are no reports describing the role
of GPC3 in the exceptionally malignant small cell lung carcinoma
(SCLC). Therefore, the aim of the present study was to determine
whether the GPC3 protein could represent a potential target for
SCLC immunotherapy.
In this study, an effective and highly specific
PE38-based immunotoxin comprising the humanised mouse monoclonal
antibody hGC33 against a C-terminal epitope of GPC3 was used
(35). Recombinant immunotoxins
(RITs) are chimeric proteins composed of a portion of a monoclonal
antibody (mAb) fused to a portion of bacterial, plant or animal
toxin. Thus, the variable fragment (Fv) of the mAb directs the
toxin to the cells expressing the target antigen. As a result, the
cell surface-bound immunotoxin is internalised via
receptor-dependent endocytosis and translocates to the cytoplasm
where it causes cell death, mostly through protein synthesis
inhibition (36–38). Gao et al (39) developed immunotoxin variants based on
a P. aeruginosa exotoxin A fragments (PE38 variant) fused to
several different anti-GPC3 antibodies (39,40). The
results obtained in vitro and in mouse xenograft experiments
demonstrated that anti-GPC3 immunotoxins may become very potent
antitumor therapeutics for HCC therapy (39,40). The
aim of the present study was to evaluate the GPC3-directed
cytotoxicity on two SCLC cell lines, NCI-H510A and NCI-H446, chosen
for their relatively high GPC3 mRNA levels (41). The use of the GPC3 antigen as a
target for immunotoxin in the SCLC cell lines is described for the
first time. The present findings suggested a possible role for GPC3
in SCLC and indicated that this antigen might represent a useful
candidate for SCLC immunotherapy.
Materials and methods
Protein overexpression and
purification
The coding sequence of the hGC33-PE38 immunotoxin
was designed by linking two functional domains: i) the sequence
encoding the hGC33 antibody at the N-terminus; and ii) a truncated
exotoxin A fragment lacking its native binding moiety and a
fragment of the domain Ib (referred to as PE38) at the C-terminus
(42). The last, terminal codon for
lysine of PE38 was deleted resulting in the C-terminal REDL
sequence. The GPC3-binding domain sequence encoded the single-chain
Fv humanised mouse monoclonal antibody named hGC33 according to the
hGC33VHk/hGC33VLa_Arg variant created by Nakano et al
(35). Between the hGC33 antibody
and PE38, a short linker encoding the N-ASGGGGSGGGTSGGGGSA-C
sequence was inserted. In some experiments, the native PE38
exotoxin A (referred to as N-PE38 thereafter) was used as a
control. The production and purification of N-PE38 and hGC33-PE38
were performed in the same way.
The genes encoding the hGC33-PE38 immunotoxin and
N-PE38 were codon-optimised for expression in E. coli and
synthesised commercially by Invitrogen (Thermo Fisher Scientific,
Inc.). The synthetic coding fragments were cloned into the
pET28SUMO expression vector, which was previously produced in our
laboratory by the insertion of the SUMO protein coding sequence
into the pET28a (Novogene Co., Ltd.). As a result, the proteins of
interest were fused to a His-tagged SUMO. The constructs were
sequenced to confirm sequence identity and correct gene
orientation.
The NiCo21(DE3) chemocompetent E. coli strain
was transformed with expression vectors by heat shock and placed
onto Agar plates supplemented with 1% glucose and kanamycin. The
preculture was inoculated with a single colony and grown in TB
medium (Sigma-Aldrich; Merck KGaA) for 16 h at 37°C. Fresh TB
medium was warmed to 37°C and inoculated with seed culture at a
culture:medium ratio of 1:100. Protein overexpression was induced
with 0.5 mM IPTG when OD600 reached 0.4 and further
grown for 14 h at 23°C. At the end of the incubation, bacteria were
collected by centrifugation. The bacterial pellet was suspended in
lysis buffer (50 mM NaH2PO4; 300 mM NaCl; 20
mM imidazole; 10% glycerol; 0.5 mM PMSF; 5 mM β-mercaptoethanol; 1
mg/ml lysozyme; 0.05% Triton X-100; 5 U/ml Benzonase®;
pH 8.0). Re-suspended cells were sonicated and centrifuged at
15,000 × g at 4°C for 20 min. The supernatant containing the
protein of interest was collected and immediately processed.
Protein was purified on two connected chromatography columns, the
first containing chitin resin (New England Biolabs, Inc.) and the
second filled with NiNTA Superflow resin (Qiagen GmbH). Columns
were previously equilibrated using a lysis buffer (50 mM
NaH2PO4, 300 mM NaCl, 20 mM imidazole, 10%
glycerol, pH 8.0). The supernatant was loaded with a constant flow
rate of 0.1 ml/min. After protein binding, columns were washed
using 8 column volumes of lysis buffer and were disconnected
afterwards. The single NiNTA column was then washed with high-salt
buffer (50 mM NaH2PO4; 2000 mM NaCl; 20 mM
imidazole; 10% glycerol; pH 8.0) to remove non-specifically bound
material. The proteins were eluted in gradient-elution mode with
buffer (50 mM NaH2PO4; 300 mM NaCl; 500 mM
imidazole; 10% glycerol; pH 8.0). The collected fractions were
pooled and SUMO protease was used to remove the SUMO-tag. After
SUMO-tag removal, the protein of interest was filtered and loaded
on a size exclusion column (HiLoad Superdex 200; GE Healthcare Life
Sciences) and equilibrated with PBS (pH 7.4), containing 10%
glycerol. The fractions containing the hGC33-PE38 immunotoxin were
pooled and concentrated on a Vivaspin Turbo concentrator
(Sigma-Aldrich; Merck KGaA). Protein purity was assessed by
densitometry and through the use of a Bradford protein assay
(Bio-Rad Laboratories, Inc.). The integrity and molecular weight of
the immunotoxin were analysed by 10% SDS-PAGE in reducing
conditions and by western blotting. For western blotting, 50 ng
protein/lane was separated via 10% SDS-PAGE under reducing
conditions and then transferred onto a PVDF membrane using a
semi-dry electrophoretic transfer. Prior to immunodetection, the
membrane was blocked overnight with SuperBlock (TBS) Blocking
Buffer (Thermo Fisher Scientific, Inc.) at room temperature.
Subsequently, the membrane was incubated with an anti-Pseudomonas
exotoxin A-specific primary antibody (1:5,000; cat. no. P2318;
Sigma-Aldrich; Merck KGaA) for 1 h at room temperature with gentle
agitation. The primary antibody was detected using a goat
anti-rabbit HRP-conjugated secondary antibody (1:2,000; cat. no.
A6154; Sigma-Aldrich; Merck KGaA) by incubating for 1 h at room
temperature with gentle agitation. Bounded antibodies were
visualized using ECL Western Blotting Detection Reagents (GE
Healthcare Life Sciences) and chemiluminescent signals were
analyzed using a molecular imager (Bio-Rad Laboratories, Inc.).
ADP-ribosylation assay
The ADP-ribosylation activities of the hGC33-PE38
and N-PE38 were measured using a solid-phase assay against S.
cerevisiae eEF2 as a substrate, as previously described
(43). The ADP-ribosylation capacity
was measured by rapid detection using the western blotting method
with HRP-conjugated streptavidin against biotin-labelled ADP-ribose
covalently bound to eEF2 as described by Borowiec et al
(43). The EC50 values
were calculated from dose-response curves using the GraphPad Prism
5 software (GraphPad Software, Inc.).
ELISA
A clear, flat-bottomed, polystyrene, 96-well plate
was coated overnight at 4°C with recombinant human glypican-3
protein (R&D Systems, Inc.) at increasing concentrations. As a
negative control, wells were also coated with 3% (w/v) Bovine Serum
Albumin (BSA) in wash buffer. The plate was subsequently washed
three times with PBS + 0.1% Tween-20 to remove unbound protein and
blocked with 3% BSA with 0.1% Tween-20 for 3 h at room temperature.
Then, the plate was washed three times with PBS + 0.1% Tween-20. In
the next step, 1 µg/ml immunotoxin was added to each well. The
plate was incubated for 1 h at room temperature, and subsequently
washed three times with PBS + 0.1% Tween-20. The plate was then
incubated with anti-P. aeruginosa exotoxin A-specific
antibodies (1:250,000; cat. no. P2318; Sigma-Aldrich; Merck KGaA)
for 1 h at room temperature and washed three times with PBS + 0.1%
Tween-20. The primary antibody was labelled using an anti-rabbit
HRP-conjugated secondary antibody (1:2,000; cat. no. P0448; Dako;
Agilent Technologies, Inc.) for 1 h at room temperature and
detected using Super Signal ELISA Pico Chemiluminescent Substrate
(cat. no. 37069; Thermo Fisher Scientific, Inc.). The incubation
with the substrate was performed for 30 min at room temperature.
The absorbance was then measured at 450 nm using a Sunrise Tecan
microplate reader (Tecan Group, Ltd.).
Cell lines
The human liver cancer cell lines HepG2, SNU-398 and
SNU-449; SCLC cell lines NCI-H510A (HTB-184), NCI-H446 (HTB-171)
and LAD A549, were purchased from American Type Culture Collection
(ATCC). The SNU-398, SNU-449, NCI-H446 cell lines were cultured in
RPMI-1640 medium (Thermo Fisher Scientific, Inc.) containing 10%
heat-inactivated FBS (Thermo Fisher Scientific, Inc.). The
NCI-H510A cell line was cultured in F-12K medium (ATCC)
supplemented with 10% FBS. The HepG2 and A549 cell lines were
maintained under standard conditions in Dulbecco's modified Eagle's
medium (Thermo Fisher Scientific, Inc.) supplemented with 10% FBS.
The cells were subcultured when they reached the exponential phase.
All cell cultures were free of mycoplasma which was confirmed by
routine test using the MycoAlert™ Mycoplasma Detection kit (Lonza
Group, Ltd.).
Internalisation studies
Alexa Fluor® 488 dye
staining and visualization
The fluorescent labelling of the hGC33-PE38
immunotoxin was performed using Alexa Fluor 488 Dye (Thermo Fisher
Scientific, Inc.) according to the manufacturer's recommendations.
A549 and HepG2 cells were incubated at 37°C with 1.5 µg/ml of
fluorescently labelled protein for 3 h. The cells were then fixed
in 4% formaldehyde for 10 min at room temperature, then stained for
20 min at room temperature with NucRed Live 647 ReadyProbes Reagent
(Thermo Fisher Scientific, Inc.) for nuclei visualisation, and
Alexa Fluor 594 Phalloidin (Thermo Fisher Scientific, Inc.) for
actin visualisation, both according to the manufacturer's protocol.
For fluorescence visualisation, the Nikon C1 confocal microscope
was used (Nikon Corporation; magnification, ×60).
ATTO 542 NHS-ester staining and
visualization
The hGC33-PE38 immunotoxin was covalently stained
with the fluorescent dye ATTO 542 NHS-ester (Atto-Tec GmbH)
according to manufacturer's protocol. NCI-H446 and NCI-H510A cells
were incubated with 4.5 µg/ml ATTO 542-stained hGC33-PE38
immunotoxin in complete medium for 1 h at 37°C. Subsequently, cells
were washed twice with complete medium for 10 min each and
resuspended in HBSS with 1 µM calcein-AM (Thermo Fisher Scientific,
Inc.) and 4 µM Hoechst 33342 (Thermo Fisher Scientific, Inc.).
After another 20-min incubation at 37°C, a portion of cells were
transferred onto the SensoPlate. The plate was centrifuged for 3
min at 300 × g at room temperature and the cells were then imaged.
Images were obtained using the Nikon Eclipse TE2000-S inverted
microscope with and Plan- Apochromat 60×/1.4 Oil DIC N2 objective
and a Nikon C1 confocal attachment (all from Nikon
Corporation).
Cytotoxicity assay
In order to evaluate the cytotoxicity of the
hGC33-PE38 immunotoxin, a neutral red uptake assay was performed
(44). For each line tested, cells
were seeded into 96-well plates in triplicate at a density of
1.5×104 cells/well. After 24 h, the cells were treated
with increasing concentrations of hGC33-PE38 and incubated for
another 48 h. Cells were also treated with 20 µg/ml of CHX as a
control to assess their sensitivity to the inhibition of protein
synthesis. SDS (200 µg/ml) was also used as a standard cell
membrane-damaging agent. After a 48-h incubation with a cytotoxic
agent, the medium was removed and cells were washed with cold PBS.
The cells were then incubated for 3 h with 50 µg/ml of neutral red
(Sigma-Aldrich; Merck KGaA) in HBSS. The neutral red solution was
removed, and the cells washed with PBS. Subsequently, the
cell-bound dye was extracted using a solution containing 50%
ethanol and 1% acetic acid by gentle shaking for 10 min at room
temperature. Absorbance at 550 nm was measured using a Sunrise
microplate reader (Tecan Group, Ltd.). The half-maximal inhibitory
concentration (IC50) values were calculated based on
linear dose-response curves using the GraphPad Prism 5
software.
Cell proliferation assay
For the determination of the effects of hGC33-PE38
immunotoxin on cell proliferation, a BrdU Cell Proliferation ELISA
kit (Abcam; cat. no. ab126556) was used. NCI-H446 cells were seeded
on clear 96-wells plates at a density of 1×104
cells/well. The following day, cells were treated with increasing
concentrations of hGC33-PE38 immunotoxin and incubated for an
additional 48 h. BrdU was added to wells 24 h before the end of the
experiment. The cells were then fixed at room temperature for 30
min using Fixing Solution (part of the aforementioned BrdU Cell
Proliferation ELISA kit), and the subsequent procedure was done
according to the manufacturer's protocol. The absorbance of 450 nm
was measured on an Infinite 200 PRO plate reader.
Microarray data analysis
The publicly available microarray results were
analysed for GPC3 expression by performing a search on the
Expression Atlas site (https://www.ebi.ac.uk/gxa/experiments/E-MTAB-2706)
using ‘GPC3’ as the gene name. The chosen diseases were ‘lung
adenocarcinoma’, ‘lung adenosquamous’, ‘small cell lung carcinoma’
and ‘squamous cell lung carcinoma’, which resulted in the automatic
selection of 67 cell lines. The applied expression value was 0.5.
The obtained results were viewed as fragments per kilobase of exon
model per million reads mapped (FPKM; normalised within each set of
biological replicates). Reads below the minimum quality threshold
were automatically discarded. The primary data are available
through the Array Express Archive (www.ebi.ac.uk/arrayexpress/) under the accession
number: E-MTAB-2706.
Reverse transcription-quantitative
(RT-q) PCR
Total RNA from NCI-H446 and NCI-H510A cells was
isolated using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instruction. Total RNA (5 µg) from each sample was reverse
transcribed for 10 min at 25°C followed by 15 min at 50°C using the
Maxima First Strand cDNA Synthesis kit for RT-qPCR (Thermo Fisher
Scientific, Inc.). The qPCR step was performed using
LightCycler® 480 SYBR Green I Master (Roche Diagnostics)
on the Light Cycler 480 Real-time PCR Instrument (Roche
Diagnostics). Reaction conditions were as follows: Initial
denaturation at 95°C for 5 min; 35 cycles at 95°C for 10 sec, 55°C
for 10 sec and 72°C for 20 sec; melting curve at 95°C for 30 sec,
72°C for 45 sec and 97°C continuous; and cooling at 40°C for 15
sec. The following primers were used: i) GPC3-forward,
5′-TGGAGTCAGGCTTGGGTAGT-3′ and reverse, 5′-ATTCAGAATGCTGCGGTTTT-3′;
ii) β-catenin-forward, 5′-CATTACAACTCTCCACAACC-3′ and reverse,
5′-CAGATAGCACCTTCAGCAC-3′ (45);
iii) hydroxymethylbilane synthase (HMBS)- forward,
5′-GGCAATGCGGCTGCAA-3′ and reverse, 5′-GGGTACCCACGCGAATCAC-3′; iv)
hypoxanthine phosphoribosyltransferase 1 (HPRT)-forward,
5′-TGACACTGGCAAAACAATGCA-3′ and reverse,
5′-GGTCCTTTTCACCAGCAAGCT-3′; v) ribosomal protein L13a (RPL13A)-
forward, 5′-CCTGGAGGAGAAGAGGAAAGAGA-3′ and reverse,
5′-TTGAGGACCTCTGTGTATTTGTCAA-3′. The HBMS, HPRT and RPL13A
housekeeping genes were used to normalise the Cq values. The qPCR
for each gene was carried out in triplicate. The obtained data was
analysed using the 2−∆∆Cq method (46). ∆Ct values were obtained by
subtracting Ct of the studied genes from Ct of the geometric mean
of reference genes (46). For
presentation, ∆Ct, were recalculated into relative copy
number values (number of copies of GPC3 or β-catenin per 1,000
copies of housekeeping genes).
Flow cytometry
HepG2, H446 and H510A cells were trypsinised into
single-cell suspensions and then stained with mouse anti-human
glypican-3 allophycocyanin-conjugated monoclonal antibody (10
µl/106 cells; cat. no. FAB2119A) or isotype control
antibody IgG2A (10 µl/106 cells; cat. no.
IC003A) in Flow Cytometry Staining Buffer supplemented with BSA and
sodium azide for 30 min at room temperature (all from R&D
Systems, Inc.). Following incubation, excess antibody was removed
by washing the cells twice in 2 ml Flow Cytometry Staining Buffer.
Cell pellets were re-suspended in 200–400 µl Flow Cytometry
Staining Buffer for flow cytometric analysis using a BD LSR
Fortessa instrument (BD Biosciences).
Statistical analysis
Quantitative data are presented as the mean ± SD of
three independent replicates (or 95% confidence intervals for
ADP-ribosylation activity). Statistical significance among groups
was calculated using a one-way ANOVA followed by Dunnett's or
Tukey's post hoc test. All statistical analyses were performed
using GraphPad Prism 5 software (GraphPad Software, Inc.). P≤0.05
or P≤0.001 was considered to indicate a statistically significant
difference.
Results
Production of an active recombinant
hGC33-PE38 immunotoxin
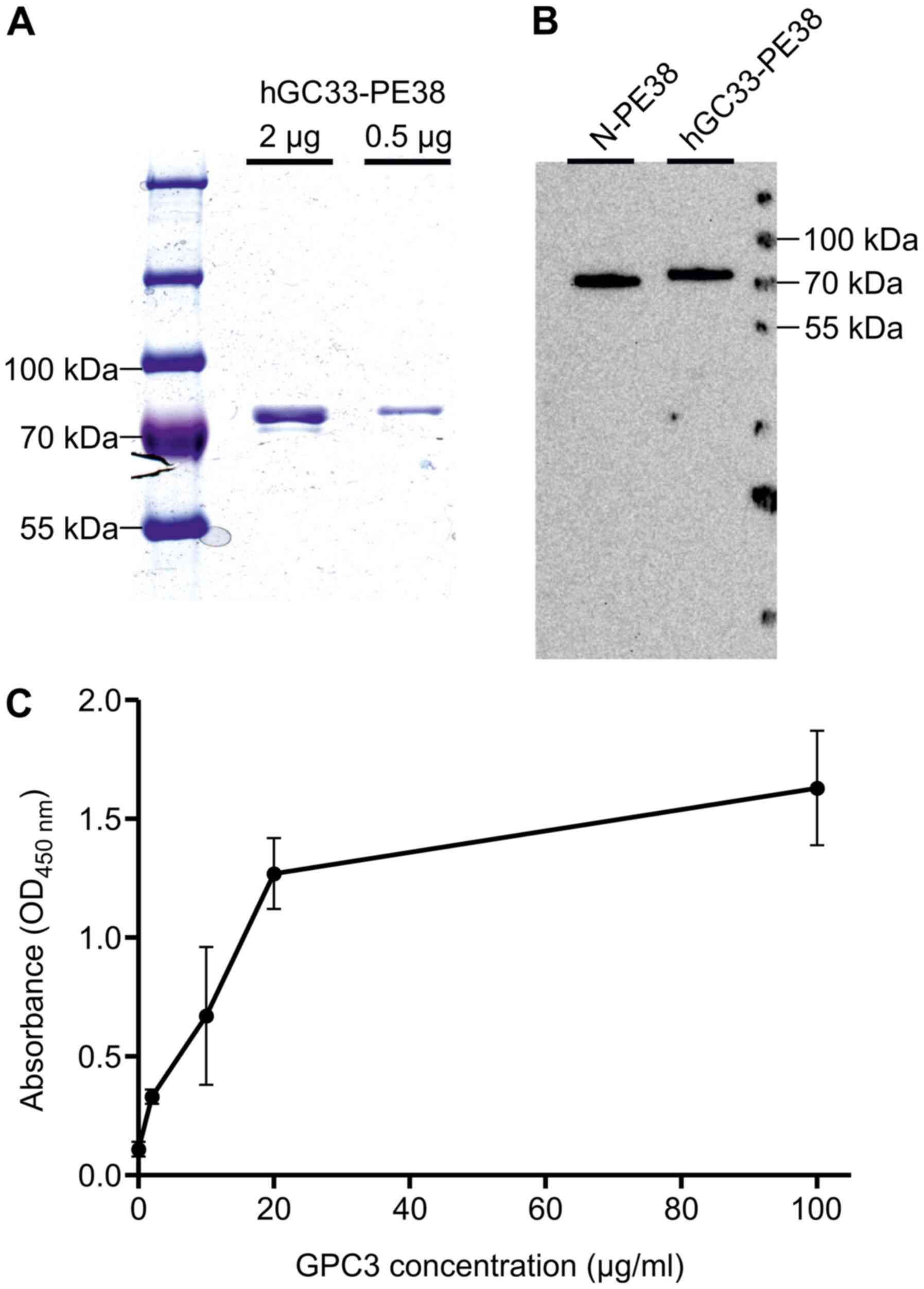
The recombinant hGC33-PE38 immunotoxin generated in
the present study displayed ~94% homogeneity and concentration of 9
mg/ml (Fig. 1).
An in vitro enzymatic activity of recombinant
proteins was evaluated using a solid-phase assay against S.
cerevisiae eEF2 as a substrate, as previously described
(43). The calculated
EC50 values for hGC33-PE38 and N-PE38 were nearly the
same (Table I), suggesting that the
enzymatic activity of the hGC33-PE38 was comparable to the wild
type of the N-PE38 toxin.
 | Table I.ADP-ribosylation activity of N-PE38
and hGC33-PE38. |
Table I.
ADP-ribosylation activity of N-PE38
and hGC33-PE38.
| Toxin | EC50,
pmol/ml | 95% CI |
|---|
| N-PE38 | 15.4 | 13.9–17 |
| hGC33-PE38 | 15.2 | 13.7–16.5 |
The affinity of the hGC33-PE38 immunotoxin to GPC3
was determined by ELISA. The immunoreactivity of hGC33-PE38 was
analysed for 1 µg/ml of protein against GPC3 antigen in five
different concentrations (2–100 µg/ml/well; Fig. 1). Observed absorbance values
(OD450nm) increased (0.22±0.03) even for the lowest
concentration of GPC3 tested (2 µg/ml) (Fig. 1), demonstrating that hGC33-PE38 can
bind to GPC3 in vitro.
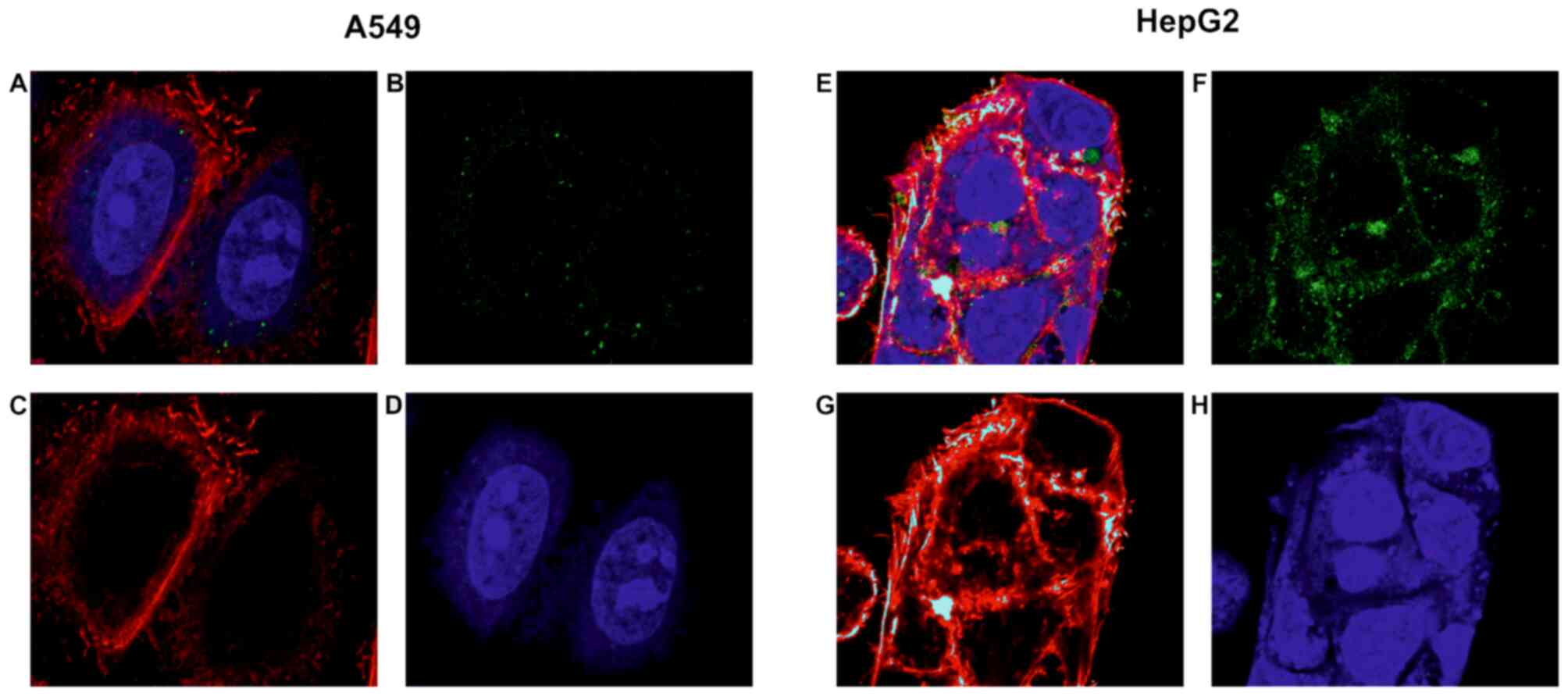
hGC33-PE38 internalisation
As a positive control for GPC3-directed immunotoxin
uptake, HepG2 liver cancer cells were used. HepG2 is known to
express GPC3 abundantly in the cell membrane and shows the ability
to internalise anti-GPC3 immunotoxins at a high rate (39). For the protein-binding specificity
evaluation, the A549 adenocarcinoma cell line was also tested,
chosen due to low/no GPC3 expression (30). As shown in Fig. 2, Alexa Fluor 488-labelled hGC33-PE38
is effectively internalised into HepG2 cells within 3 h.
Fluorescence is clearly visible in the cytoplasm with especially
high density of signal in globular clusters around the nuclei
(Fig. 2). Untreated HepG2 control
cells have not shown visible fluorescence in Alexa Fluor 488
specific channel (data not shown). No evident immunotoxin uptake
was observed in the case of A549 cells. Control untreated A549
cells have also not shown visible fluorescence in Alexa Fluor 488
specific channel (data not shown). As these cells are generally
capable of effectively internalising native exotoxin A and its
variant in analogous experiments (43), these observations demonstrated that
the internalisation of hGC33-PE38 was GPC3-dependent.
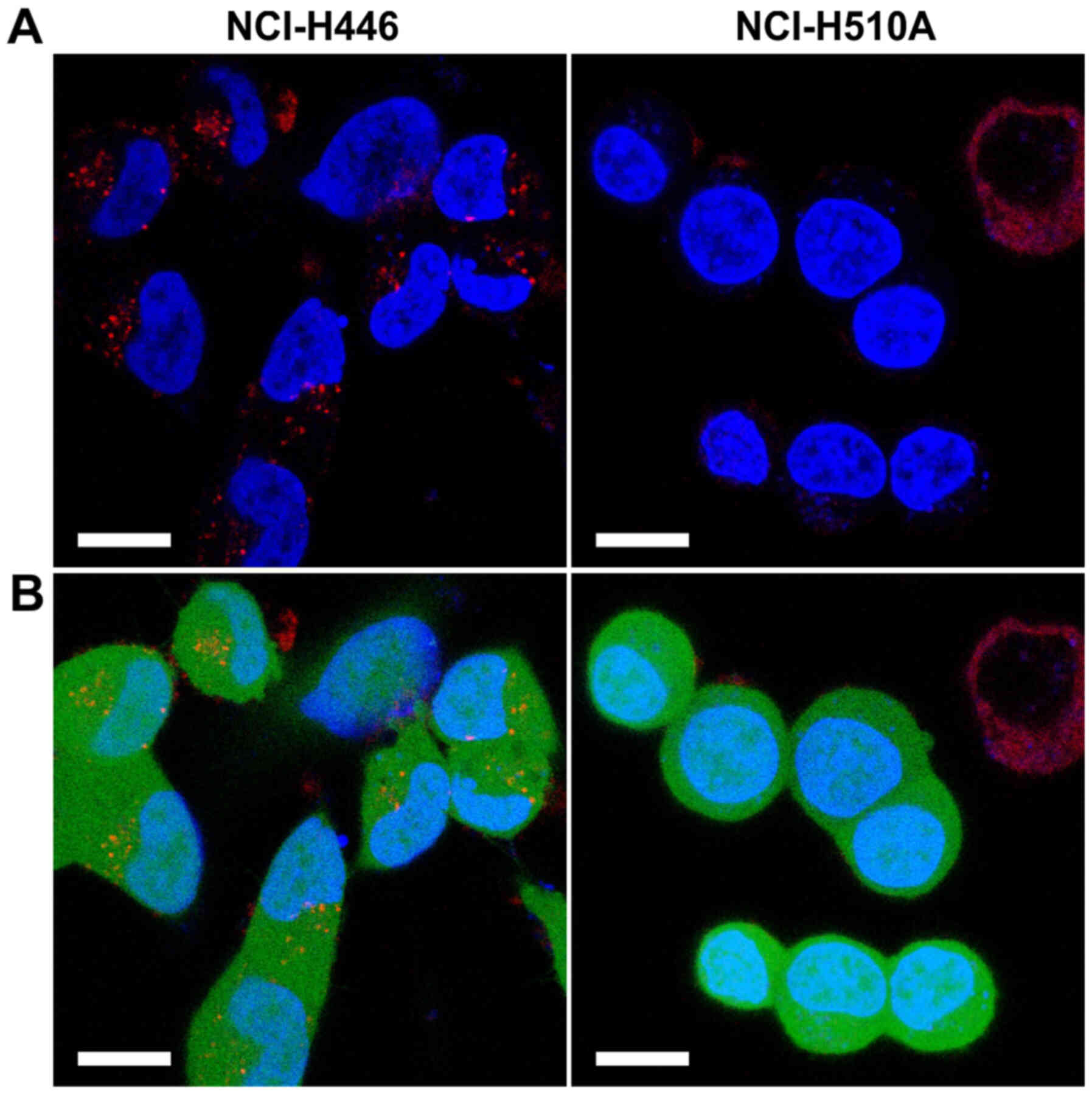
Additionally, the internalisation of ATTO
542-labelled hGC33-PE38 was tested on two SCLC lines, NCI-H510A and
NCI-H446. The immunotoxin uptake was evident in the case of H446
cells (Fig. 3), resulting in strong,
punctate/globular staining in the cytoplasm, while untreated
NCI-H510A control cells have not shown visible fluorescence in ATTO
542 specific channel (data not shown). In contrast, ATTO
542-labelled hGC33-PE38 internalisation was not detected in
NCI-H510A cells (Fig. 3) and
ATTO-542-specific fluorescence was also not visible in untreated
NCI-H510A control cells.
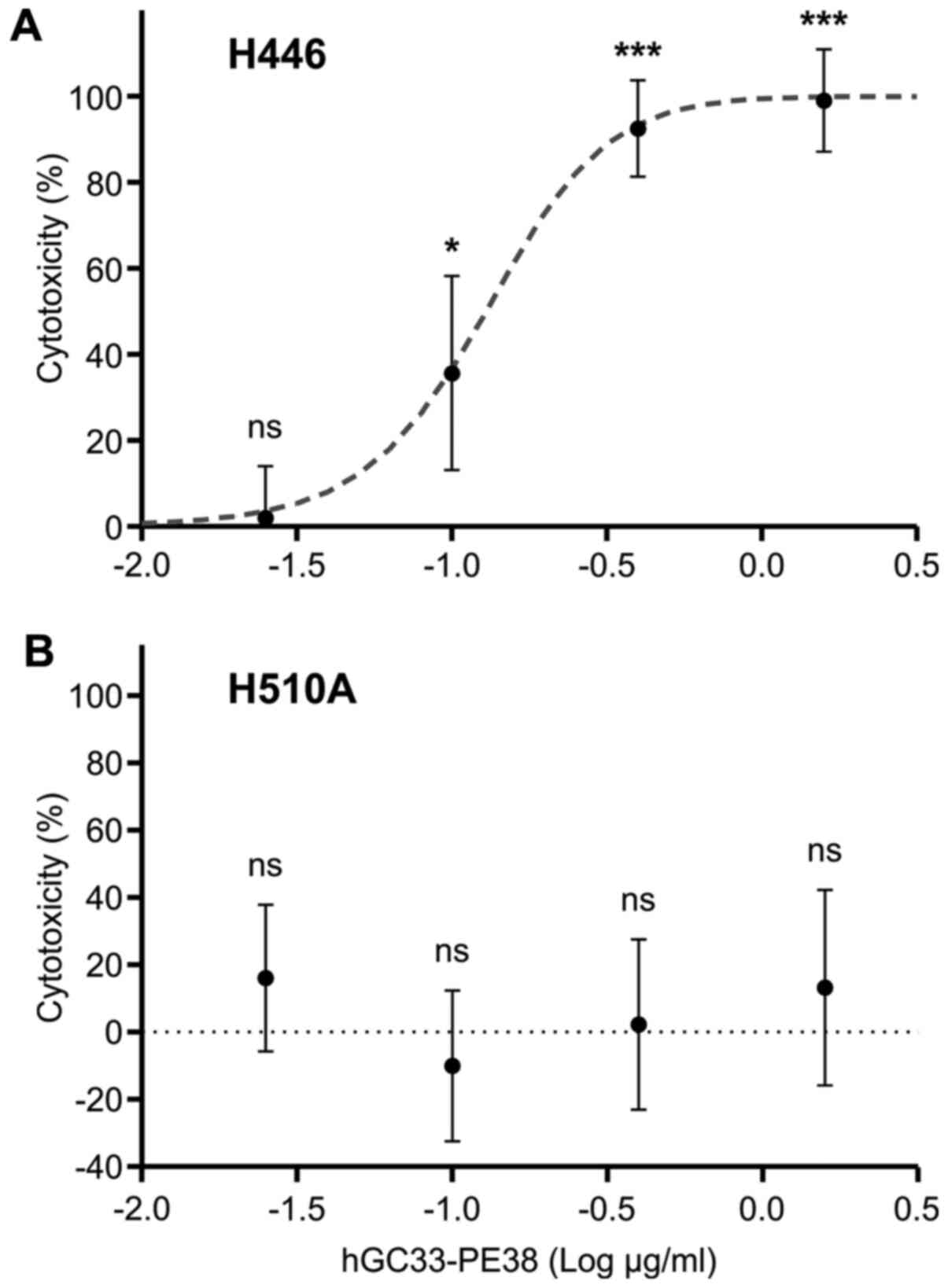
In vitro biological activity of
hGC33-PE38
The biological activity of hGC33-PE38 was evaluated
in vitro on cancer cell lines. Cytotoxicity was assessed
after 48-h incubation with hGC33-PE38 in various concentrations by
enumerating viable cells using a neutral red uptake assay (44). Preliminary analyses were performed on
liver cancer cell lines as a model for anti-GPC3 immunotoxin
potency validation. As a positive control, HepG2 cells, which
express GPC3 at high levels, were tested. hGC33-PE38 was cytotoxic
to HepG2 cells in a dose-dependent manner with an IC50
of 330±15 ng/ml (Table II). For
specificity evaluation, the SNU-398 (low GPC3 expression) and
SNU-449 (GPC3-null) lines were treated (47). As expected, no cytotoxicity in the
SNU-449 cell line was observed, and only small yet detectable
cytotoxicity (13% at the highest dose; data not shown) was seen in
SNU-398 cells. These findings suggested that the cytotoxicity of
hGC33-PE38 was specific and dependent on GPC3 expression levels on
the target cells. The SCLC cell lines tested were chosen based on
microarray results obtained previously by Klijn et al
(41). Two SCLC lines with the
highest levels of GPC3 expression were tested for immunotoxin
cytotoxicity, NCI-H510A and NCI-H446. Despite expressing similar
levels of GPC3 mRNA, the responses of these lines to hGC33-PE38
immunotoxin differed. The H446 line was sensitive to hGC33-PE38 in
a dose-dependent manner, and the IC50 for the
immunotoxin was 70.6±4.6 ng/ml (Table
II and Fig. 4). However, for
H510A, no cytotoxicity was observed even at the highest tested
concentration (1,562 ng/ml). Additionally, two lung cancer lines
with previously reported low or undetectable GPC3 expression were
analysed. The A549 LAD cells were insensitive to hGC33-PE38 even at
a 1,600 ng/ml dose (the highest tested) (Table II). The second GPC3-negative cell
line to be tested was the SK-MES-1 LSCC cell line (30). The SK-MES-1 cells were resistant to
hGC33-PE38 and remained viable at the highest concentration of
immunotoxin tested (1,650 ng/ml; Table
II).
| Table II.Cytotoxicity of hGC33-PE38 toward
liver and lung cancer cell lines. |
Table II.
Cytotoxicity of hGC33-PE38 toward
liver and lung cancer cell lines.
| A, Liver
cancer |
|---|
|
|---|
| Cell line | IC50,
mean ± SD, ng/ml | P-value (vs.
H446) |
|---|
| HepG2 | 330±15 | P<0.0001 |
| SNU-398 | >1650 | P<0.0001 |
| SNU-449 | No effect | P<0.0001 |
|
| B, Lung
cancer |
|
| Cell
line | IC50,
mean ± SD, ng/ml | P-value (vs.
H446) |
|
| H446 | 70.6±4.6 | – |
| H510A | No effect | P<0.0001 |
| A549 | No effect | P<0.0001 |
| SK-MES1 | No effect | P<0.0001 |
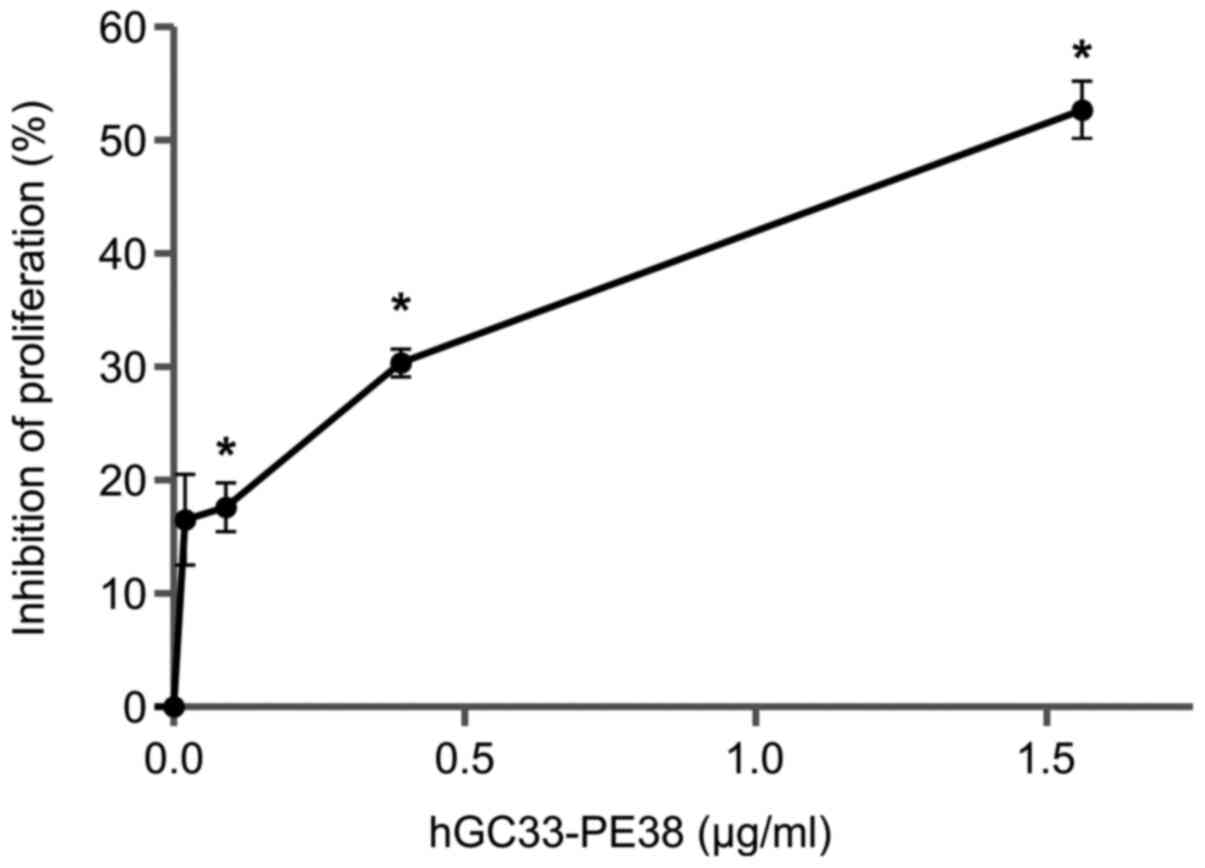
The anti-proliferative effect of hGC33-PE38
immunotoxin on NCI-H446 cells was evaluated using a BrdU assay.
Cells were analysed 48 h after immunotoxin treatment. As shown in
Fig. 5, hGC33-PE38 inhibits cell
proliferation in dose-dependent manner. A 48-h incubation with 1.5
µg/ml hGC33-PE38 resulted in inhibition of proliferation by ~50% in
NCI-H446 cell culture.
Comparison of GPC3 and β-catenin
expression in NCI-H446 and NCI-H510A cells
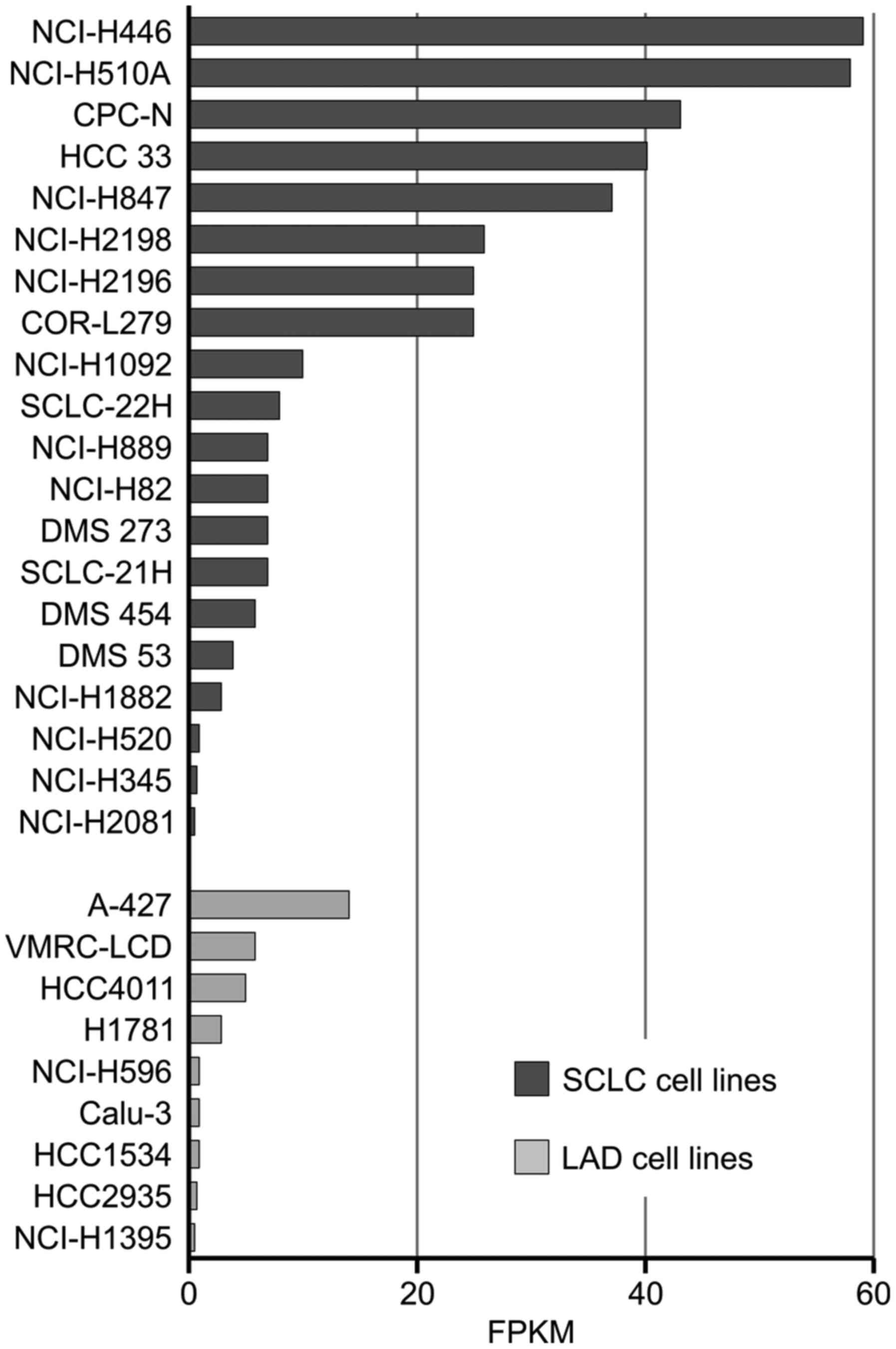
The SCLC cell lines in the present study were chosen
based on microarray results obtained by Klijn et al
(41). Analysis of the publicly
available microarray results was performed for lung adenocarcinoma,
lung adenosquamous, small cell lung carcinoma and squamous cell
lung carcinoma lines. Of the 67 cell lines analysed, 20 SCLC lines
and 9 LAD lines exhibited expression values above the cut-off
(Fig. 6). Almost the same mRNA
levels of GPC3 were reported for H446 and H510A lines (58 fragments
per kilobase of exon model per million reads mapped for H510A and
59 for H446).
GPC3 mRNA levels were verified in both SCLC cell
lines via RT-qPCR. The expression levels of GPC3 were comparable
between NCI-H446 and NCI-H510A cells (Table III).
| Table III.Relative expression levels of the
GPC3 and β-catenin genes in NCI-H446 and NCI-H510A cells. |
Table III.
Relative expression levels of the
GPC3 and β-catenin genes in NCI-H446 and NCI-H510A cells.
| Cell line | GPC3 expression
mean ± SD | β-catenin
expression mean ± SD |
|---|
| H446 | 919±470 | 50±44 |
| H510A | 955±180; P=0.886
(vs. H446) | 26±9; P=0.629 (vs.
H446) |
The transcript abundance of the β-catenin (CTNNB1
gene) was also measured in both SCLC lines. The expression of
β-catenin was detectable in both cell types. However, β-catenin
levels were higher in NCI-H446, compared with NCI-H510A, although
this trend was not statistically significant (Table III).
Treatment of NCI-H446 and NCI-H510A cells with
hGC33-PE38 did not result in changes in expression of GPC3 and
β-catenin (data not shown).
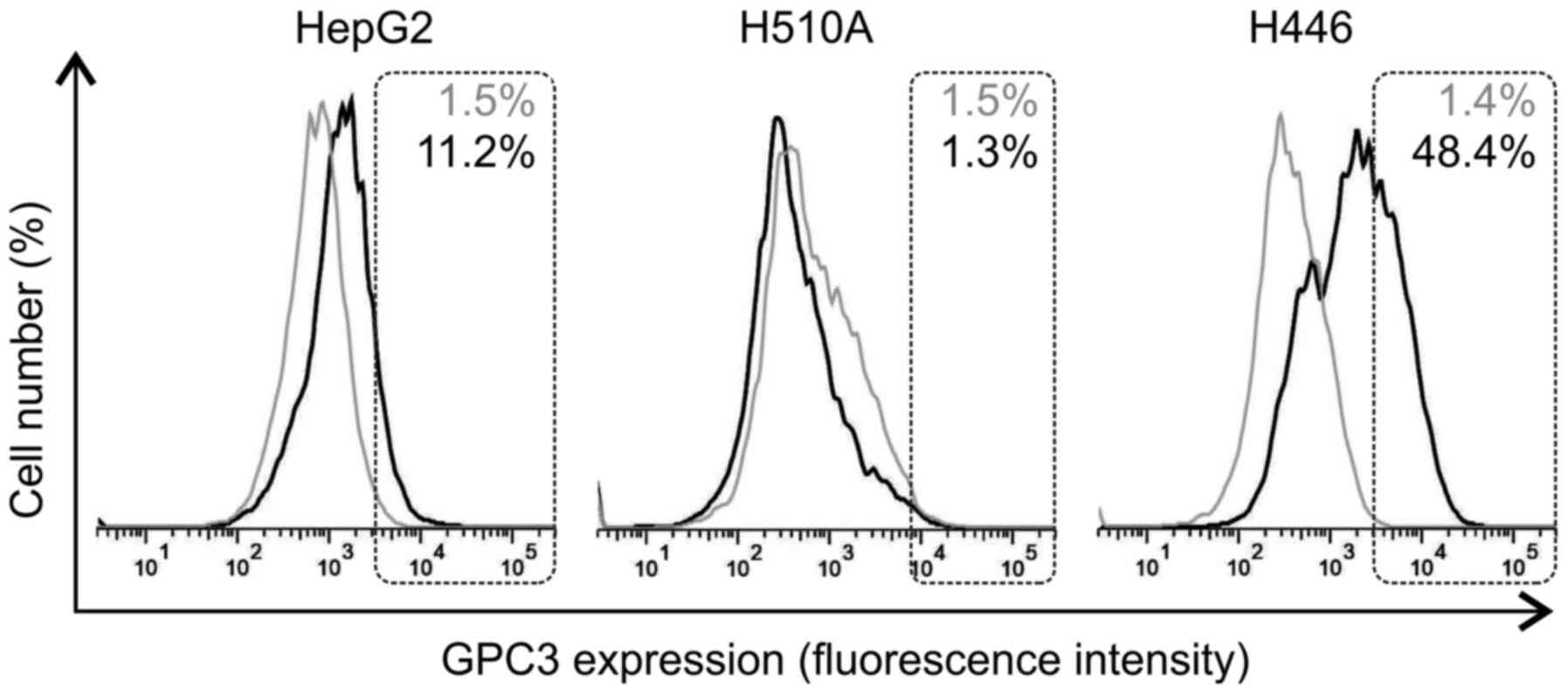
Surface-bound GPC3 detection
Due to the differences in hGC33-PE38 cytotoxicity on
SCLC lines, despite their similar levels of GPC3 expression, the
levels of cell surface-bound GPC3 were then examined in the H510A,
H446 and HepG2 cell lines using flow cytometry. The results
indicated varying levels of GPC3 surface expression in different
tested cell lines. While H446 cells exhibited a strong positive
staining with anti-GPC3 antibodies, H510A cells were GPC3-negative
(Fig. 7). As previously reported by
other authors, HepG2 cells showed GPC3-positive staining (Fig. 7). Interestingly, H446 cells exhibited
a higher level of GPC3 specific immunoreactivity on their surface
compared with HepG2 cells (Fig.
7).
Discussion
In the present study, a hGC33-PE38 immunotoxin
targeting GPC3 was generated using the recombinant humanized
monoclonal antibody hGC33. hGC33 has previously been demonstrated
to inhibit HCC tumour growth via antibody-dependent cellular
cytotoxicity (35,48,49).
Cytotoxicity of the hGC33-PE38 was first evaluated against a panel
of liver cancer cell lines employed as a model for GPC3-targeted
cancer immunotherapy. The results confirmed the hypothesis that the
hGC33-PE38 immunotoxin could kill GPC3-expressing cells (HepG2),
but not cancer cells lacking surface expression of this antigen
(SNU-449 line) and was only slightly cytotoxic to cells expressing
GPC3 at low levels (SNU-398 line). The specificity of hGC33-PE38
was additionally confirmed by treating known GPC3-negative lung
cancer cell lines. The hGC33 antibody could potentially serve as an
alternative to HN3, developed by Feng et al (50), to create effective anti-GPC3
immunotoxins. Although not fully of human origin, hGC33 was
well-tolerated in clinical trials (35,48,49). The
antitumour potential of hGC33-PE38 should be further evaluated
in vivo. However, the potential clinical use of hGC33-PE38
might be restricted due to the known immunogenicity of the PE38
molecule (51). Nonetheless, the
present findings further support the potential use of GPC3 as a
target in immunotherapy of SCLC.
The pathogenesis of SCLC is still unknown and the
underlying molecular mechanism remains unclear (52). The potential role of the GPC3 protein
in these processes might be of interest, due to its association
with the Hh and Wnt/β-catenin signalling pathways, which are
considered as contributing mechanisms in SCLC pathogenesis
(45,53–56).
Recently published data suggest the role of Wnt/β-catenin pathway
activation in chemotherapy resistance and SCLC relapse (56). Interestingly, Pan et al
(45) demonstrated the inhibitory
effect of XAV939 (a small-molecule inhibitor of the Wnt/β-catenin
signalling pathway) on the proliferation of NCI-H446 cells,
suggesting inhibition of Wnt signalling as a potential therapeutic
approach for advanced SCLC disease.
Previous studies on the pathogenesis of SCLC have
described this disease as dynamic and involving diverse cellular
and molecular processes (57–59). In
the present study, two cancer lines were tested, NCI-H510A and
NCI-H446. Both lines were isolated by Carney et al (60) in 1982 from male, adult patients, and
both were derived from metastatic sites: H510A from adrenal
metastasis and H446 from pleural effusion. On the basis of
biochemical markers and morphological differences, the H510A cell
line is a classic-SCLC line defined by upregulation of all four
APUD (amine precursor uptake and decarboxylation) biomarkers
(60,61). In contrast, H446 is a variant-SCLC
line expressing only two biomarkers and presenting atypical
morphology (60–63). Gazdor et al (62) suggested that the unique, variant
phenotype of the H446 line manifests early in in vitro
culture and reflects the morphology of the tumour of origin. Thus,
it is not a result of changes occurring during culture. It is known
that SCLC cells with variant morphologies have increased expression
of the c-myc oncogene, shorter doubling time, decreased or absent
expression of neuroendocrine cell features and can be resistant to
conventional therapy (62–65). In the present study, the relatively
high levels of GPC3 protein observed on the cell surface of the
NCI-H446 line suggested that GPC3 could potentially act as an
oncogene in these cells. The reason why GPC3 is almost undetectable
on the surface of H510A cells despite the high expression observed
at the mRNA level is unknown. The presence or absence of GPC3 on
the surface of the H446 and H510A cells directly corresponded to
their sensitivity to hGC33-PE38 immunotoxin. In the case of H446
cells, GPC3 acted as an antigen for the immunotoxin and enabled its
internalisation, resulting in cytotoxicity. The observed
cytotoxicity and anti-proliferative effects of the hGC33-PE38
immunotoxin on H446 cells were dose-dependent. To the best of our
knowledge, this is the first demonstration of the use of GPC3 as a
target for SCLC cell killing. Due to the known properties of GPC3,
differences may be expected between H446 and H510A lines in
signalling cascades that are important for SCLC initiation and
progression. Importantly, the mRNA level of β-catenin was higher in
H446 compared with H510A cells, although this trend was not
statistically significant (Table
III). Most probably, the H446 cell line represents the SCLC
subtype with the active canonical Wnt/β-catenin pathway enhanced by
overexpressed, cell surface-bound GPC3. Recently, Wang et al
(66) demonstrated that GPC3
promotes the progression of lung squamous cell carcinoma through
upregulation of β-catenin expression. It is also known that in
hepatocellular carcinoma, the GPI anchoring and the cell surface
localization of GPC3 is needed for Wnt/β-catenin signalling
activation and cell proliferation (10,67).
It is thus conceivable that high surface expression
of GPC3 in H446 cells can represent some important characteristics
of late-stage SCLC, which is associated with high aggressiveness,
and chemo- and/or radio-therapy resistance. This warrants further
study of the potential oncogenic role of GPC3 in SCLC, particularly
in the context of different stages of the disease.
In conclusion, the present study described the
production and in vitro activity of hGC33-PE38, a PE38-based
immunotoxin targeting the GPC3 antigen. Similar to anti-GPC3
immunotoxins described elsewhere, the hGC33-PE38 was effectively
internalised and cytotoxic to HepG2 cells, and ineffective in the
case of GPC-3-negative cancer cells. A major focus of this study
was the evaluation of hGC33-PE38 toxicity against cell lines
representing cancer types that are not yet recognised as
GPC3-associated. SCLC was considered as such, based on previous
microarray results suggesting GPC3 gene upregulation. Two SCLC
lines were chosen and treated with hGC33-PE38 in order to test
GPC3-directed cytotoxicity. Despite similar GPC3 mRNA levels in
both cell lines, only the H446 cell line was sensitive to
hGC33-PE38, whereas H510A was resistant. This result was consistent
with the difference in the amount of cell surface-bound GPC3
detected by flow cytometry. Since these cell lines were SCLC
variants, the GPC3-associated phenotype reported here could reflect
some important differences in the molecular pathways involved in
diverse manifestations of the disease. Thus, cell surface-bound
GPC3 might be considered as a potential target for SCLC therapy
involving immunotoxins, other immunoconjugates or T cells. This
hypothesis is consistent with recently reported activation of the
Wnt/β-catenin pathway in the advanced, chemo-resistant form of
SCLC, and in line with ideas to treat advanced SCLC through
inhibition of Wnt signalling. These observations also suggest a
need for further studies of GPC3 cell-membrane abundance in
different stages of SCLC.
Acknowledgements
The authors would like to thank Mrs. Isabelle Garcin
and Professor Oliver Nüsse (University Paris-Sud, Orsay, France)
for helping with confocal microscope imaging, and Professor Terri
G. Kinzy (Robert Wood Johnson Medical School, Piscataway, NJ, USA)
for providing the TKY675 mutant yeast strain for eEF2
production.
Funding
This study was supported by The Polish National
Centre for Research and Development (grant no.
pbs1/a9/16/2012).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
AWD, MB, MG, JG, MiR, MaR and ER conducted the
research, prepared and visualized the data, and performed
statistical and computational analyses. ER formulated the research
goals. AWD, JD, KG, LR provided substantial contributions to the
design of the study. JD, KG and LR provided supervision and
leadership for their research teams. ER and AWD prepared the
original draft of manuscript. The manuscript review, corrections
and editing were performed by ER, JD, AWD, KG, MG, MaR and LR. All
authors read and approved the final version of manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
GPC3
|
glypican-3
|
|
HCC
|
hepatocellular carcinoma
|
|
SCLC
|
small cell lung carcinoma
|
|
LSCC
|
lung squamous cell carcinoma
|
|
LAD
|
lung adenocarcinoma
|
|
RIT
|
recombinant immunotoxin
|
|
Hh
|
Hedgehog
|
|
mAb
|
monoclonal antibody
|
|
PE38
|
Pseudomonas aeruginosa exotoxin A
38
|
References
|
1
|
Haruyama Y and Kataoka H: Glypican-3 is a
prognostic factor and an immunotherapeutic target in hepatocellular
carcinoma. World J Gastroenterol. 22:275–283. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fransson LA: Glypicans. Int J Biochem Cell
Biol. 35:125–129. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Filmus J, Shi W, Wong ZM and Wong MJ:
Identification of a new membrane-bound heparan sulphate
proteoglycan. Biochem J. 311:561–565. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
De Cat B, Muyldermans SY, Coomans C,
Degeest G, Vanderschueren B, Creemers J, Biemar F, Peers B and
David G: Processing by proprotein convertases is required for
glypican-3 modulation of cell survival, Wnt signaling, and
gastrulation movements. J Cell Biol. 163:625–635. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Iglesias BV, Centeno G, Pascuccelli H,
Ward F, Peters MG, Filmus J, Puricelli L and de Kier Joffé EB:
Expression pattern of glypican-3 (GPC3) during human embryonic and
fetal development. Histol Histopathol. 23:1333–1340.
2008.PubMed/NCBI
|
|
6
|
Hsu HC, Cheng W and Lai PL: Cloning and
expression of a developmentally regulated transcript MXR7 in
hepatocellular carcinoma: Biological significance and
temporospatial distribution. Cancer Res. 57:5179–5184.
1997.PubMed/NCBI
|
|
7
|
Ho M and Kim H: Glypican-3: A new target
for cancer immunotherapy. Eur J Cancer. 47:333–338. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li J, Gao JZ, Du JL and Wei LX: Prognostic
and clinicopathological significance of glypican-3 overexpression
in hepatocellular carcinoma: A meta-analysis. World J
Gastroenterol. 20:6336–6344. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu JW, Zuo XL and Wang S: Diagnosis
accuracy of serum Glypican-3 level in patients with hepatocellular
carcinoma and liver cirrhosis: A meta-analysis. Eur Rev Med
Pharmacol Sci. 19:3655–3673. 2015.PubMed/NCBI
|
|
10
|
Capurro MI, Xiang YY, Lobe C and Filmus J:
Glypican-3 promotes the growth of hepatocellular carcinoma by
stimulating canonical Wnt signaling. Cancer Res. 65:6245–6254.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kolluri A and Ho M: The role of glypican-3
in regulating Wnt, Yap, and Hedgehog in liver cancer. Front Oncol.
9:7082019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Filmus J and Capurro M: The role of
glypican-3 in the regulation of body size and cancer. Cell Cycle.
7:2787–2790. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Saikali Z and Sinnett D: Expression of
glypican 3 (GPC3) in embryonal tumors. Int J Cancer. 89:418–422.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ota S, Hishinuma M, Yamauchi N, Goto A,
Morikawa T, Fujimura T, Kitamura T, Kodama T, Aburatani H and
Fukayama M: Oncofetal protein glypican-3 in testicular germ-cell
tumor. Virchows Arch. 449:308–314. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yamanaka K, Ito Y, Okuyama N, Noda K,
Matsumoto H, Yoshida H, Miyauchi A, Capurro M, Filmus J and Miyoshi
E: Immunohistochemical study of glypican 3 in thyroid cancer.
Oncology. 73:389–394. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Baumhoer D, Tornillo L, Stadlmann S,
Roncalli M, Diamantis EK and Terracciano LM: Glypican 3 expression
in human nonneoplastic, preneoplastic, and neoplastic tissues: A
tissue microarray analysis of 4,387 tissue samples. Am J Clin
Pathol. 129:899–906. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zynger DL, Everton MJ, Dimov ND, Chou PM
and Yang XJ: Expression of glypican 3 in ovarian and extragonadal
germ cell tumors. Am J Clin Pathol. 130:224–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cao D, Li J, Guo CC, Allan RW and Humphrey
PA: SALL4 is a novel diagnostic marker for testicular germ cell
tumors. Am J Surg Pathol. 33:1065–1077. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang F, Liu A, Peng Y, Rakheja D, Wei L,
Xue D, Allan RW, Molberg KH, Li J and Cao D: Diagnostic utility of
SALL4 in extragonadal yolk sac tumors: An immunohistochemical study
of 59 cases with comparison to placental-like alkaline phosphatase,
alpha-fetoprotein, and glypican-3. Am J Surg Pathol. 33:1529–1539.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ikeda H, Sato Y, Yoneda N, Harada K,
Sasaki M, Kitamura S, Sudo Y, Ooi A and Nakanuma Y:
α-Fetoprotein-producing gastric carcinoma and combined
hepatocellular and cholangiocarcinoma show similar morphology but
different histogenesis with respect to SALL4 expression. Hum
Pathol. 43:1955–1963. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aydin O, Yildiz L, Baris S, Dundar C and
Karagoz F: Expression of Glypican 3 in low and high grade
urothelial carcinomas. Diagn Pathol. 10:342015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nguyen T, Phillips D, Jain D, Torbenson M,
Wu TT, Yeh MM and Kakar S: Comparison of 5 immunohistochemical
markers of hepatocellular differentiation for the diagnosis of
hepatocellular carcinoma. Arch Pathol Lab Med. 139:1028–1034. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Foda AA, Mohammad MA, Abdel-Aziz A and
El-Hawary AK: Relation of glypican-3 and E-cadherin expressions to
clinicopathological features and prognosis of mucinous and
non-mucinous colorectal adenocarcinoma. Tumour Biol. 36:4671–4679.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu X, Li Y, Chen SW, Shi Y and Xu F:
Differential expression of glypican-3 (GPC3) in lung squamous cell
carcinoma and lung adenocarcinoma and its clinical significance.
Genet Mol Res. 14:10185–10192. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ortiz MV, Roberts SS, Glade Bender J,
Shukla N and Wexler LH: Immunotherapeutic targeting of GPC3 in
pediatric solid embryonal tumors. Front Oncol. 9:1082019.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Murthy SS, Shen T, De Rienzo A, Lee WC,
Ferriola PC, Jhanwar SC, Mossman BT, Filmus J and Testa JR:
Expression of GPC3, an X-linked recessive overgrowth gene, is
silenced in malignant mesothelioma. Oncogene. 19:410–416. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xiang YY, Ladeda V and Filmus J:
Glypican-3 expression is silenced in human breast cancer. Oncogene.
20:7408–7412. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Peters MG, Farías E, Colombo L, Filmus J,
Puricelli L and Bal de Kier Joffé E: Inhibition of invasion and
metastasis by glypican-3 in a syngeneic breast cancer model. Breast
Cancer Res Treat. 80:221–232. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Valsechi MC, Oliveira AB, Conceição AL,
Stuqui B, Candido NM, Provazzi PJ, de Araújo LF, Silva WA Jr,
Calmon MF and Rahal P: GPC3 reduces cell proliferation in renal
carcinoma cell lines. BMC Cancer. 14:6312014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim H, Xu GL, Borczuk AC, Busch S, Filmus
J, Capurro M, Brody JS, Lange J, D'Armiento JM, Rothman PB, et al:
The heparan sulfate proteoglycan GPC3 is a potential lung tumor
suppressor. Am J Respir Cell Mol Biol. 29:694–701. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aviel-Ronen S, Lau SK, Pintilie M, Lau D,
Liu N, Tsao MS and Jothy S: Glypican-3 is overexpressed in lung
squamous cell carcinoma, but not in adenocarcinoma. Mod Pathol.
21:817–825. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lin Q, Xiong LW, Pan XF, Gen JF, Bao GL,
Sha HF, Feng JX, Ji CY and Chen M: Expression of GPC3 protein and
its significance in lung squamous cell carcinoma. Med Oncol.
29:663–669. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li K, Pan X, Bi Y, Xu W, Chen C, Gao H,
Shi B, Jiang H, Yang S, Jiang L, et al: Adoptive immunotherapy
using T lymphocytes redirected to glypican-3 for the treatment of
lung squamous cell carcinoma. Oncotarget. 7:2496–2507. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jiang Z, Jiang X, Chen S, Lai Y, Wei X, Li
B, Lin S, Wang S, Wu Q, Liang Q, et al: Anti-GPC3-CAR T Cells
Suppress the Growth of Tumor Cells in Patient-Derived Xenografts of
Hepatocellular Carcinoma. Front Immunol. 7:6902017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nakano K, Ishiguro T, Konishi H, Tanaka M,
Sugimoto M, Sugo I, Igawa T, Tsunoda H, Kinoshita Y, Habu K, et al:
Generation of a humanized anti-glypican 3 antibody by CDR grafting
and stability optimization. Anticancer Drugs. 21:907–916. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Brinkmann U: Recombinant immunotoxins:
Protein engineering for cancer therapy. Mol Med Today. 2:439–446.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pastan I, Hassan R, Fitzgerald DJ and
Kreitman RJ: Immunotoxin therapy of cancer. Nat Rev Cancer.
6:559–565. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shapira A and Benhar I: Toxin-based
therapeutic approaches. Toxins (Basel). 2:2519–2583. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gao W, Tang Z, Zhang YF, Feng M, Qian M,
Dimitrov DS and Ho M: Immunotoxin targeting glypican-3 regresses
liver cancer via dual inhibition of Wnt signalling and protein
synthesis. Nat Commun. 6:65362015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang C, Gao W, Feng M, Pastan I and Ho M:
Construction of an immunotoxin, HN3-mPE24, targeting glypican-3 for
liver cancer therapy. Oncotarget. 8:32450–32460. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Klijn C, Durinck S, Stawiski EW, Haverty
PM, Jiang Z, Liu H, Degenhardt J, Mayba O, Gnad F, Liu J, et al: A
comprehensive transcriptional portrait of human cancer cell lines.
Nat Biotechnol. 33:306–312. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Weldon JE and Pastan I: A guide to taming
a toxin--recombinant immunotoxins constructed from Pseudomonas
exotoxin A for the treatment of cancer. FEBS J. 278:4683–4700.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Borowiec M, Gorzkiewicz M, Grzesik J,
Walczak-Drzewiecka A, Salkowska A, Rodakowska E, Steczkiewicz K,
Rychlewski L, Dastych J and Ginalski K: Towards Engineering Novel
PE-Based Immunotoxins by Targeting Them to the Nucleus. Toxins
(Basel). 8:3212016. View Article : Google Scholar
|
|
44
|
Repetto G, del Peso A and Zurita JL:
Neutral red uptake assay for the estimation of cell
viability/cytotoxicity. Nat Protoc. 3:1125–1131. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pan F, Shen F, Yang L, Zhang L, Guo W and
Tian J: Inhibitory effects of XAV939 on the proliferation of
small-cell lung cancer H446 cells and Wnt/β-catenin signaling
pathway in vitro. Oncol Lett. 16:1953–1958. 2018.PubMed/NCBI
|
|
46
|
Vandesompele J, De Preter K, Pattyn F,
Poppe B, Van Roy N, De Paepe A and Speleman F: Accurate
normalization of real-time quantitative RT-PCR data by geometric
averaging of multiple internal control genes. Genome Biol.
3:research0034.1. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sun Ck, Chua Ms, Wei W and So S:
Glypican-3-Mediates Autophagy and Promotes Self-Renewal and Tumor
Initiation of Hepatocellular Carcinoma Cells. Biol J Stem Cell Res
Ther. 4:92014.
|
|
48
|
Ishiguro T, Sugimoto M, Kinoshita Y,
Miyazaki Y, Nakano K, Tsunoda H, Sugo I, Ohizumi I, Aburatani H,
Hamakubo T, et al: Anti-glypican 3 antibody as a potential
antitumor agent for human liver cancer. Cancer Res. 68:9832–9838.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yen CJ, Daniele B, Kudo M, Merle P, Park
JW, Ross P, Péron JO, Ebert O, Chan S, Poon RT, et al: Randomized
phase II trial of intravenous RO5137382/GC33 at 1600 mg every other
week and placebo in previously treated patients with unresectable
advanced hepatocellular carcinoma. J Clin Oncol. 32 (Suppl
5):4102a2014. View Article : Google Scholar
|
|
50
|
Feng M, Gao W, Wang R, Chen W, Man YG,
Figg WD, Wang XW, Dimitrov DS and Ho M: Therapeutically targeting
glypican-3 via a conformation-specific single-domain antibody in
hepatocellular carcinoma. Proc Natl Acad Sci USA. 110:E1083–E1091.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
FitzGerald DJ, Wayne AS, Kreitman RJ and
Pastan I: Treatment of hematologic malignancies with immunotoxins
and antibody-drug conjugates. Cancer Res. 71:6300–6309. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Schulze AB, Evers G, Kerkhoff A, Mohr M,
Schliemann C, Berdel WE and Schmidt LH: Future Options of
Molecular-Targeted Therapy in Small Cell Lung Cancer. Cancers
(Basel). 11:6902019. View Article : Google Scholar
|
|
53
|
Watkins DN, Berman DM, Burkholder SG, Wang
B, Beachy PA and Baylin SB: Hedgehog signalling within airway
epithelial progenitors and in small-cell lung cancer. Nature.
422:313–317. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Park KS, Martelotto LG, Peifer M, Sosml,
Karnezis AN, Mahjoub MR, Bernard K, Conklin JF, Szczepny A, Yuan J,
et al: A crucial requirement for Hedgehog signaling in small cell
lung cancer. Nat Med. 17:1504–1508. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Szczepny A, Rogers S, Jayasekara WSN, Park
K, McCloy RA, Cochrane CR, Ganju V, Cooper WA, Sage J, Peacock CD,
et al: The role of canonical and non-canonical Hedgehog signaling
in tumor progression in a mouse model of small cell lung cancer.
Oncogene. 36:5544–5550. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wagner AH, Devarakonda S, Skidmore ZL,
Krysiak K, Ramu A, Trani L, Kunisaki J, Masood A, Waqar SN, Spies
NC, et al: Recurrent WNT pathway alterations are frequent in
relapsed small cell lung cancer. Nat Commun. 9:37872018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Borromeo MD, Savage TK, Kollipara RK, He
M, Augustyn A, Osborne JK, Girard L, Minna JD, Gazdar AF, Cobb MH,
et al: ASCL1 and NEUROD1 Reveal Heterogeneity in Pulmonary
Neuroendocrine Tumors and Regulate Distinct Genetic Programs. Cell
Rep. 16:1259–1272. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Poirier JT, Gardner EE, Connis N, Moreira
AL, de Stanchina E, Hann CL and Rudin CM: DNA methylation in small
cell lung cancer defines distinct disease subtypes and correlates
with high expression of EZH2. Oncogene. 34:5869–5878. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Esteller L, Hernández S, Lopez-Rios F and
Remon J: Could WNT inhibitors really knock on the treatment door of
small cell lung cancer? J Thorac Dis. 11 (Suppl 3):S381–S384. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Carney DN, Gazdar AF, Bepler G, Guccion
JG, Marangos PJ, Moody TW, Zweig MH and Minna JD: Establishment and
identification of small cell lung cancer cell lines having classic
and variant features. Cancer Res. 45:2913–2923. 1985.PubMed/NCBI
|
|
61
|
Carney DN, Gazdar AF, Nau M and Minna JD:
Biological heterogeneity of small cell lung cancer. Semin Oncol.
12:289–303. 1985.PubMed/NCBI
|
|
62
|
Gazdar AF, Carney DN, Nau MM and Minna JD:
Characterization of variant subclasses of cell lines derived from
small cell lung cancer having distinctive biochemical,
morphological, and growth properties. Cancer Res. 45:2924–2930.
1985.PubMed/NCBI
|
|
63
|
Zhang Z, Zhou Y, Qian H, Shao G, Lu X,
Chen Q, Sun X, Chen D, Yin R, Zhu H, et al: Stemness and inducing
differentiation of small cell lung cancer NCI-H446 cells. Cell
Death Dis. 4:e6332013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhang W, Girard L, Zhang YA, Haruki T,
Papari-Zareei M, Stastny V, Ghayee HK, Pacak K, Oliver TG, Minna
JD, et al: Small cell lung cancer tumors and preclinical models
display heterogeneity of neuroendocrine phenotypes. Transl Lung
Cancer Res. 7:32–49. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Carney DN, Mitchell JB and Kinsella TJ: In
vitro radiation and chemotherapy sensitivity of established cell
lines of human small cell lung cancer and its large cell
morphological variants. Cancer Res. 43:2806–2811. 1983.PubMed/NCBI
|
|
66
|
Wang D, Gao Y, Zhang Y, Wang L and Chen G:
Glypican-3 promotes cell proliferation and tumorigenesis through
up-regulation of β-catenin expression in lung squamous cell
carcinoma. Biosci Rep. 39:BSR201811472019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Gao W and Ho M: The role of glypican-3 in
regulating Wnt in hepatocellular carcinomas. Cancer Rep. 1:14–19.
2011.PubMed/NCBI
|