Introduction
Lung cancer has been a common cause of mortality in Taiwan and other industrialized countries in the last decade (1–3). Lung cancer is classified into two major types: Small cell lung cancer and non-small cell lung cancer (NSCLC). NSCLC is further sub-divided into adenocarcinoma, squamous cell carcinoma and large cell carcinoma (4). NSCLC accounts for ~80% of all lung cancer cases (5). Despite advancements in clinical treatment, the prognosis of patients with lung cancer remains largely unsatisfactory, with a 5-year survival rate of only 15–20% (6). Thus, identifying valuable target genes and effective drugs remains critical for the clinical treatment of patients with NSCLC.
Previous studies have demonstrated that microRNAs (miRNAs/miRs), short non-coding RNAs that participate in silencing genes in a post-transcriptional manner, affect the expressions of several genes, such as PTEN, HOXD10, VEGFA, which are involved in cancer pathogenesis, from initiation and progression to metastasis and drug resistance (7–9). Given the characteristics of easy detection via liquid biopsy and in vivo stability (10,11), miRNAs have been identified as both novel therapeutic targets and effective tools for cancer treatment (12,13). Furthermore, the identification of miRNAs suitable for personalized treatment is an emerging topic in the field of cancer research (14,15).
Different sources of natural products that exhibit antitumor properties, and the search for anticancer drugs from natural substances containing active ingredients are areas of interest in the field of drug discovery (16,17). Withaferin A (WA), a steroidal lactone, has been identified as an active ingredient of root extract in the medical plant Withania somnifera, in Eastern India (18). WA has attracted notable attention due to its ability to regulate immunity and inhibit different types of cancer, including prostate (19), breast (20), melanoma (21), ovarian (22) and lung cancers (23). Previous studies have reported that WA targets NF-κB, heat shock proteins, Akt and estrogen receptors (24–26). Furthermore, Kyakulaga et al, demonstrated that WA inhibits transforming growth factor-β (TGF-β) and tumor necrosis factor-α (TNF-α), and induces epithelial-to-mesenchymal transition (EMT) of lung cancer cells by blocking SMAD and nuclear factor-kappa B (NF-κB) signaling (27). Groagan and Hsu et al reported that WA induces lung cancer apoptosis by downregulating the mTOR/STAT3 pathway (28,29). However, whether other molecules, particularly miRNAs, serve as novel targets of lung cancer cells engaging with WA remains unclear. The aim of the present study was to identify the miRNAs responsible for the inhibitory effects of WA in the lung cancer cells. Taken together, the results of the present study demonstrated that WA induced apoptosis of lung cancer cells, and decreased cell motility at different dosages by targeting miR-27a or miR-10b in a p53-dependent manner.
Materials and methods
Chemicals and reagents
WA was purchased from Sigma-Aldrich; Merck KGaA. Fetal bovine serum (FBS), glutamine and RPMI-1640 medium were purchased from Thermo Fisher Scientific, Inc. Antibodies against: β-actin, Bax, Bcl-2, E-cadherin, p53 and vimentin, and the p53 small interfering (si)RNA and SC siRNA were all purchased from Santa Cruz Biotechnology, Inc.
Cell culture
A549, A549 shRNA, A549-p53 short hairpin (sh)RNA, H460, H1355 and H1299 cell lines were provided by Dr Hsu Shih-Lan (Department of Medical Research, Taichung Veterans General Hospital, Taichung, Taiwan). All cells were maintained in RPMI-1640 medium supplemented with 10% FBS, penicillin and streptomycin (100 U/ml each), and 1% L-glutamine (Invitrogen; Thermo Fisher Scientific, Inc.), at 37°C in a humidified atmosphere with 5% CO2, and the culture medium was changed every 2 days. The WA was dissolved in 95% EtOH for the following experiments.
Cytotoxicity assay
A549, A549shRNA or A549-p53shRNA cells (5×104) were treated with different concentrations of WA (0, 0.5, 1 and 2 µM) for indicated time intervals (24 or 48 h) at 37°C in a humidified atmosphere suppling with 5% CO2. Two approaches were applied in determining the viability of cells under the treatment of WA. In the direct counting assay, the viable cells were counted under a phase-contrast microscopic using the trypan blue exclusion method as described previously (30). The vehicle control (0.1% of EtOH, v/v) exhibited no difference in viability and motility compared with the untreated cells (Fig. S1); thus the untreated cells were represented as control for the following experiments. In the MTT assay, the untreated or WA-treated H460 or H1355 cells (1×105 cells/well) were replaced with serum-free RPMI containing 20 ml MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) at 37°C for 2 h. The medium was then aspired and washed with 1×PBS twice. The cells were then added with 100 ml dimethyl sulfoxide (DMSO) and the absorbance of 590 nm were measured by a microplate reader.
Caspase activity assay
A549 cells were cultured with or without WA (1, 2, 3 µM) at 24 h. After treatment, both adherent and floating cells were harvested and washed once with ice-cold PBS, followed by lyzing in ice-cold lysis buffer (1% Triton X-100, 0.32 M sucrose, 5 mM ethylenediaminetetra-acetic acid (pH 8.0), 10 mM Tris-HCl (pH 8.0), 2 mM dithiothreitol, 1 µM phenylmethylsulfonylfluoride, 1 µg/ml leupeptin). The cell lysates (250 mg total protein) were then incubated with fluorogenic peptide substrate (DEVD-AFC specific for caspase-3, IETD-AFC specific for caspase-8, LEHD-AFC specific for caspase-9), incubated at 37°C for 4 h in the dark. Fluorescence intensity was measured with a Fluorescence plate reader (Fluoroskan Ascent; Labsystems) by exciting at 405 nm and emitting at 510 nm. For the caspase inhibitor assay, the cell lysates were incubated with 100 µM of different caspase inhibitors including z-DEVD-FMK (against caspase-3), z-IETD-FMK (against caspase-8) and z-LEHD-FMK (against caspase-9) (Abcam, Inc.) 1 h before the addition of indicated caspase substrate.
Protein extraction and western blotting
Cells were lysed using RIPA lysis buffer [150 mM NaCl, 50 mM Tris-HCl (pH 7.4), 0.25% Na Deoxycholate, 5 mM EDTA, 1% Triton X-100, 5 mM EGTA, and 1% protease inhibitor cocktail] on ice for 30 min. The lysates were centrifuged at 12,000 × g for 20 min at 4°C. The protein concentration of each sample was subsequently determined using the Bradford method (31). Equal amounts of protein samples (50 µg) were separated via 10%, 12% or 15% SDS-PAGE and subsequently transferred onto polyvinylidene difluoride membrane (EMD Millipore). The membranes were blocked with 5% BSA or milk, prior to incubation with the primary antibody. Membranes were rinsed three or four times with PBS and subsequently incubated with a horseradish peroxidase-conjugated secondary antibody. Indicated protein signals were then visualized using ECL Plus detection reagent.
RNA extraction and reverse transcription-quantitative (RT-qPCR) of miR-10b and miR-27a
The miRNAs of untreated, non-transfected or transfected or WA-treated A549, A549 shRNA and A549-p53 short hairpin (sh)RNA cells were extracted using the miRVANA® miRNA isolation kit (cat. no. AM1560, Thermo Fisher Scientific, Inc.), according to the manufacturer's instructions. Aliquots of 5 µl of the total miRNA were employed in the reverse transcription reaction with the miRNA reverse transcription kit (Thermo Fisher Scientific, Inc.), using a 5X miR-10b, miR-27a or RNU6B probe, respectively. RT-qPCR was performed with TaqMan PCR master mix kit (Thermo Fisher Scientific, Inc.), using a 20X miR-10b, miR-27a or RNU6B probe. Both the primers and probes of miR-10b (ID:002218), miR-27a (ID:002445) and RNU6B (ID:001093) were provided by the manufacture (Thermo Fisher Scientific, Inc.). RNU6B served as an internal control. The signals were read using a ABI PRISM 7900 Sequence Detector System. RNU6B reverse transcription and amplification were used as internal controls. Subsequently, the comparative 2−ΔΔCq method (32) was applied to quantify the gene expression levels.
Migration assay
For in vitro migration assay, A549, H460 and H1355 cells were trypsinized and a total of 5,000 cells were seeded in the upper chamber of the Transwell membrane (pore size: 8 µm; EMD Millipore) with RPMI containing 1% serum whereas RPMI containing 10% serum was added into the lower chambers as a chemoattractant at 37°C for 16 h. The medium was subsequently aspirated and the filters were washed twice with PBS, prior to fixation with methanol for 10 min at room temperature. Cells were then stained with Giemsa solution at room temperature to quantify the migrated cells under a phase-contrast light microscopy (magnification at 40X). The migrated cells from the WA-treated cells were normalized against the untreated cells as relative fold-change.
Cellular transfections
A total of 2 µg of p53-wild type plasmid (pLenti6/V5-p53_wt p53, Addgene) or vector pLenti6/V5 were transfected into H1299 cells using Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C for 12 h. A total of 20 µM of p53 siRNA or SC siRNA (Santa Cruz, Inc.) were transfected into A549 cells using Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C for 12 h. A total of 20 µM of miR-10b mimics (5′-UACCCUGUAGAACCGAAUUUGUGUU-3′) or miR-27a mimics (5′-AGGGCUUAGCUGCUUGUGAGCAUU-3′) were transfected into A549 cells using Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C for 12 h. A total of 20 µM of miR-10b or miR-27a antagomiRs (both were designated by MDBio, http://0800072222.tw/syn/RNA_GOmiR.php) were transfected into A549-p53shRNA cells using Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C for 12 h for the following experiments. The vector served as a plasmid transfection control and SC siRNA (20 µM) served as siRNA/miRNA transfection control. After 12 h, the transfected cells were replaced with new complete RPMI medium and incubated at 37°C for an another 12 h, the parental or transfected cells were subsequently applied in the following experiments.
The single-cell gel electrophoresis assay (Comet assay)
The comet assay was performed as previously described (33). Briefly, A549 cells were treated with 0, 0.5, 1 and 2 µM WA for 24 h followed by embedding in 0.3% of agarose in PBS and separated on microscope slides. The slides were incubated with lysis buffer at room temperature for 15 min, and in alkaline solution for another 10 min, electrophoresed for 20 min, immersed in neutralizing buffer for 5 min and then stained with ethidium bromide at room temperature for 10 min. Ethidium bromide-labeled nuclei were visualized using a fluorescence microscope (magnification, ×40). DNA damage was analyzed and scored using the CometScore software (version 1.5, Tritek Corporation) and the tail moment (tail length × %DNA in tail) of at least 100 cells of each experiment were quantified and normalized to control (untreated) as relative folds.
Determination of intracellular reactive oxygen species (ROS) level and mitochondrial membrane potential (ΔΨm)
To assess the intracellular ROS level, The A549 cells were incubated with WA for the indicated time periods, and then incubated with 10 µM of DCF-DA (2,7-dichlorodihydrofluorescein diacetate, Molecular Probes Inc., USA) or 10 µM of HE (dihydroethidium, Molecular Probes Inc., USA) for 30 min prior to harvest. For the analysis of mitochondrial membrane potential, lipophilic fluorochrome JC-1 (5, 5′, 6, 6′-tetrachloro −1,1′,3,3′-tetraethyl -benzamidazolyl carbocyanine iodide, Molecular Probes) was applied 30 min prior to harvest. The levels of fluorescence intensity of the cells were analyzed by flow cytometry (Becton Dickinson FACScan) under an excitation wavelength of 488 nm and emission wavelengths of 530 nm for green fluorescence and 585 nm for red fluorescence.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7.0 (GraphPad Software, Inc.). All experiments were performed in triplicate and data are presented as the mean ± standard deviation. Unpaired Student's t-test was used to compare differences between two groups, while one-way ANOVA followed by Tukey's post hoc test were used to compare differences between multiple groups. P<0.05 was considered to indicate a statistically significant difference.
Results
WA induces apoptosis in lung cancer cells
A549 cells were treated with different concentrations of WA (0, 0.5, 1, and 2 µM) for 24 and 48 h. The viability of the untreated control cells and WA-treated cells was determined via the trypan blue exclusion method. As presented in Fig. 1A, treatment with WA, below 1 µM for 24 h, only marginally decreased the viability of A549 cells (~5% of reduction), whereas for concentrations above 1 µM, WA significantly decreased the viability of A549 cells in a dose-dependent manner (1 µM, 20% reduction; 1.5 µM, 40% reduction; 2 µM, 70% reduction). Conversely, treatment with the same concentration of WA for a longer period of time (48 h) increased the mortality of A549 cells by 20–40% than that of 24 h. Similar results were observed in H460 and H1355 cells treated with WA (Fig. S2), suggesting that induced lung cancer cell death may be a common characteristic of WA treatment; however, the results also implied that WA may not rapidly induce A549 cell death (Fig. 1). The results demonstrated that administration of WA significantly induced the activities of both caspase 3 and 9 but not caspase 8, further addition of z-DEVE-FMK (the inhibitor of caspase-3) or caspase-9 (z-LHED-FMK) ameliorated the death of A549 cells induced by WA, whereas the inhibition of caspase 8 by z-IETD-FMK marginally protected the effects of A549 cells under the treatment of WA, these results indicated that WA induced A549 cells a caspase-dependent apoptosis (Fig. S3). Furthermore, the addition of WA induced the DNA damage of A549 cells in a dose dependent manner (Fig. S4A). The following experiments demonstrated that WA disrupted the membrane potential of mitochondrial in 8 h (Fig. S4B). Notably, the results of the present study demonstrated that WA induced the generation of superoxide anion radical rather than hydrogen peroxide in A549 cells (Fig. S4C, D). These results indicated that the generation of ROS, DNA damage and the disruption of mitochondria membrane potential induced by WA may be the early events that ultimately lead to the apoptosis of A549 cells. Besides, since we have proved that WA induced apoptosis of A54 cells, the expression of intrinsic apoptosis-related molecules of A549 cells in the presence of WA was subsequently analyzed. Western blot analysis demonstrated that the expression of both proapoptotic molecules, p53 and Bax increased following treatment with WA, in a dose-dependent manner, whereas the expression of the antiapoptotic molecule, Bcl-2 was downregulated (Fig. 1B). Taken together, these results suggest that WA induces intrinsic apoptosis of A549 cells, which is associated with upregulated p53 and Bax expression, and downregulated Bcl-2 expression.
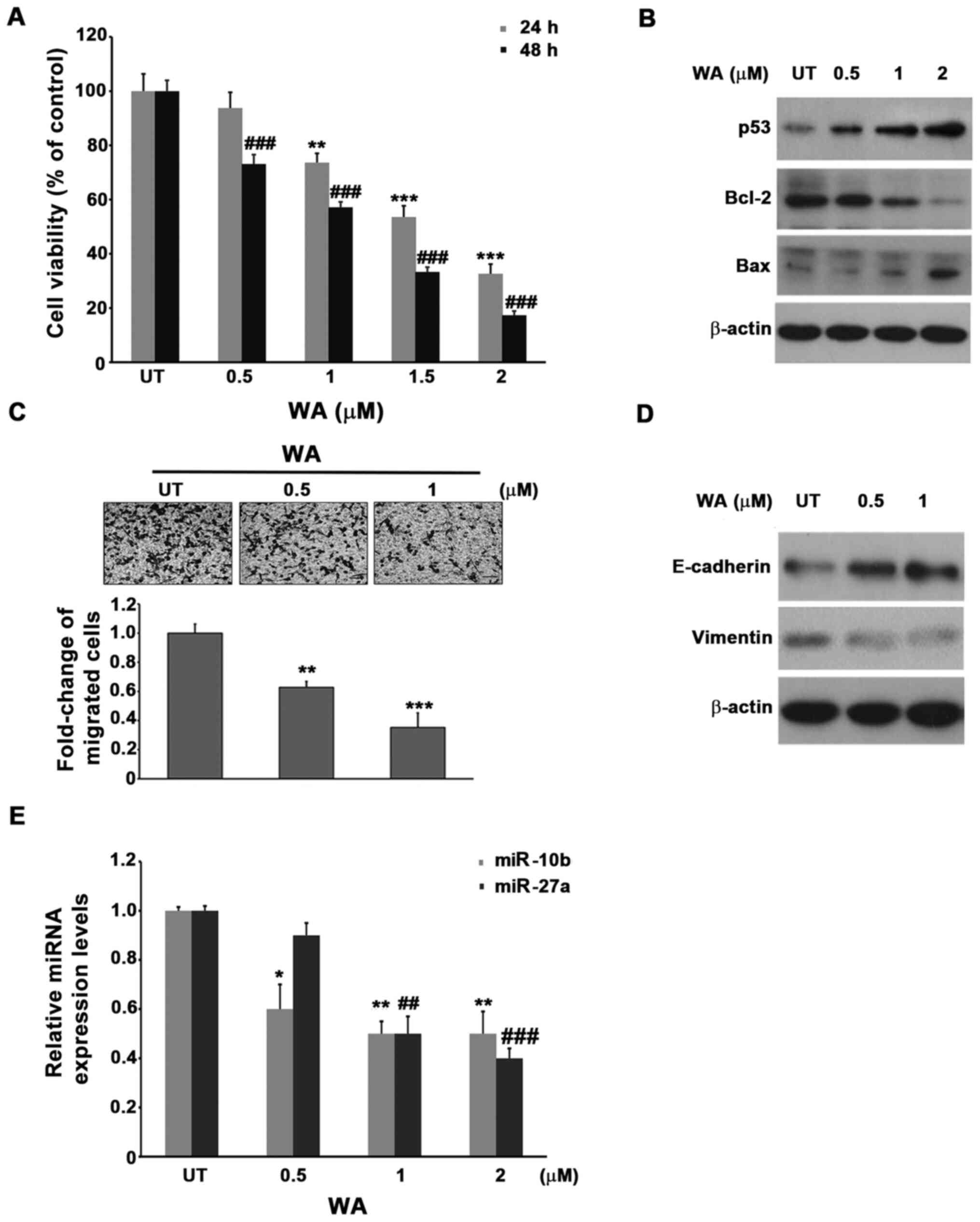 |
Figure 1.
miR-10b and miR-27a expression decreases following treatment with WA. (A) A549 lung cancer cells were treated with different concentrations of WA for 24 and 48 h, respectively. Cells were subsequently subjected to the trypan blue exclusion assay to determine cellular viability. UT cells were used as a control. **P<0.01 and ***P<0.001 vs. UT 24 h; ###P<0.001 vs. UT 48 h. (B) Expression levels of p53, Bax and Bcl-2 in A549 cells treated with different concentrations of WA for 48 h were detected via western blotting. (C) Cells were treated with different concentrations of WA for 24 h, and subjected to migration assay for another 16 h. UT cells were used as a control. **P<0.01 and ***P<0.001 vs. UT. Scale bar, 40 µm. (D) Cells treated with different concentrations of WA for 24 h were collected and subjected to western blot analysis using the indicated antibodies. (E) Cells were treated with different concentrations of WA and subjected to reverse transcription-quantitative PCR to detect the expression of miR-10b and miR-27a. UT cells were used as a control. *P<0.05 and **P<0.01 vs. UT miR-10b; ##P<0.01 and ###P<0.001 vs. UT miR-27a. UT, untreated; WA, withaferin A; miRNA/miR, microRNA.
|
WA decreases the motility of lung cancer cells
A549 cells were pretreated with a sublethal dose of WA (0.5 and 1 µM) for 24 h, followed by the migration assay. The results demonstrated that the addition of a low dose of WA inhibits the migration of A549 cells in a dose dependent manner, with up to 60% motility reduction with 1 µM of WA compared with the untreated control cells (Fig. 1C). Similar results were observed using H460 or H1355 cell lines, (Fig. S5). In addition, western blot analysis demonstrated that E-cadherin expression decreased following treatment with WA, whereas that of vimentin was induced. The results suggest that WA may act to inhibit epithelial-to-mesenchymal transition (Fig. 1D) (34), furthermore, since E-cadherin is known to involve in the motility of cancer cells, the results of the present study implied the association between WA and change in cellular motility (35). Collectively, these results demonstrated that a lower concentration of WA (0.5 µM), which do not induce cell death at 24 h, significantly inhibited the motility of A549, H460 and H1355 cells, these results suggest that WA may exhibit distinct antitumor functionality at different dosages.
WA decreases the expression of both miR-10b and miR-27a
Next, the present study aimed to identify miRNAs involved in the downregulation of viability and motility of A549 cells primed by WA. Following a thorough literature review, two candidate miRNAs were selected: miR-10b and miR-27a. miR-10b is a well-known oncomiR involved in the induction of motility and metastasis of several tumors, including lung cancer cells (36). Notably, one target gene of miR-10b is E-cadherin, which has been reported to be induced in the presence of WA (Fig. 1D). Conversely, the oncomiR miR-27a has been reported to decrease Bax expression (37), which was also observed to be upregulated in A549 cells following treatment with WA. These associations suggest that both miR-10b and miR-27a may be affected by WA. To test this hypothesis, A549 cells were treated with different concentrations of WA for 24 h and the levels of miR-10b and miR-27a were assessed via RT-qPCR analysis. The results demonstrated that treatment with WA decreased the expression of both miR-10b and miR-27a in a dose-dependent manner (Fig. 1E). Notably, WA decreased the expression of miR-10b more effectively than that of miR-27a at the lowest WA concentration (0.5 µM), where the expression of miR-10b was ~20% lower than that of miR-27a (Fig. 1E). These results link the association between the expression of both miR-10b and miR-27a, and the WA-mediated antitumor effect in the A549 lung cancer cell line.
Role of miR-10b in the WA-mediated reduction of motility
miR-10b mimics were transfected into A549 cells prior to treatment with WA for 24 h, followed by subjection to migration and western blotting assays. Transfection with miR-10b mimics alone increased the level of total miR-10b in A549 cells (Fig. S6A). The WA-treated A549 cells induced E-cadherin expression and downregulated miR-10b expression and motility compared with the untreated cells (Fig. 2A and B). However, the ectopic expression of miR-10b significantly doubled the ratio of migrated cells under the treatment of WA (Fig. 2A and B). These results demonstrate that miR-10b is the main target gene in the WA-mediated inhibition of cellular motility.
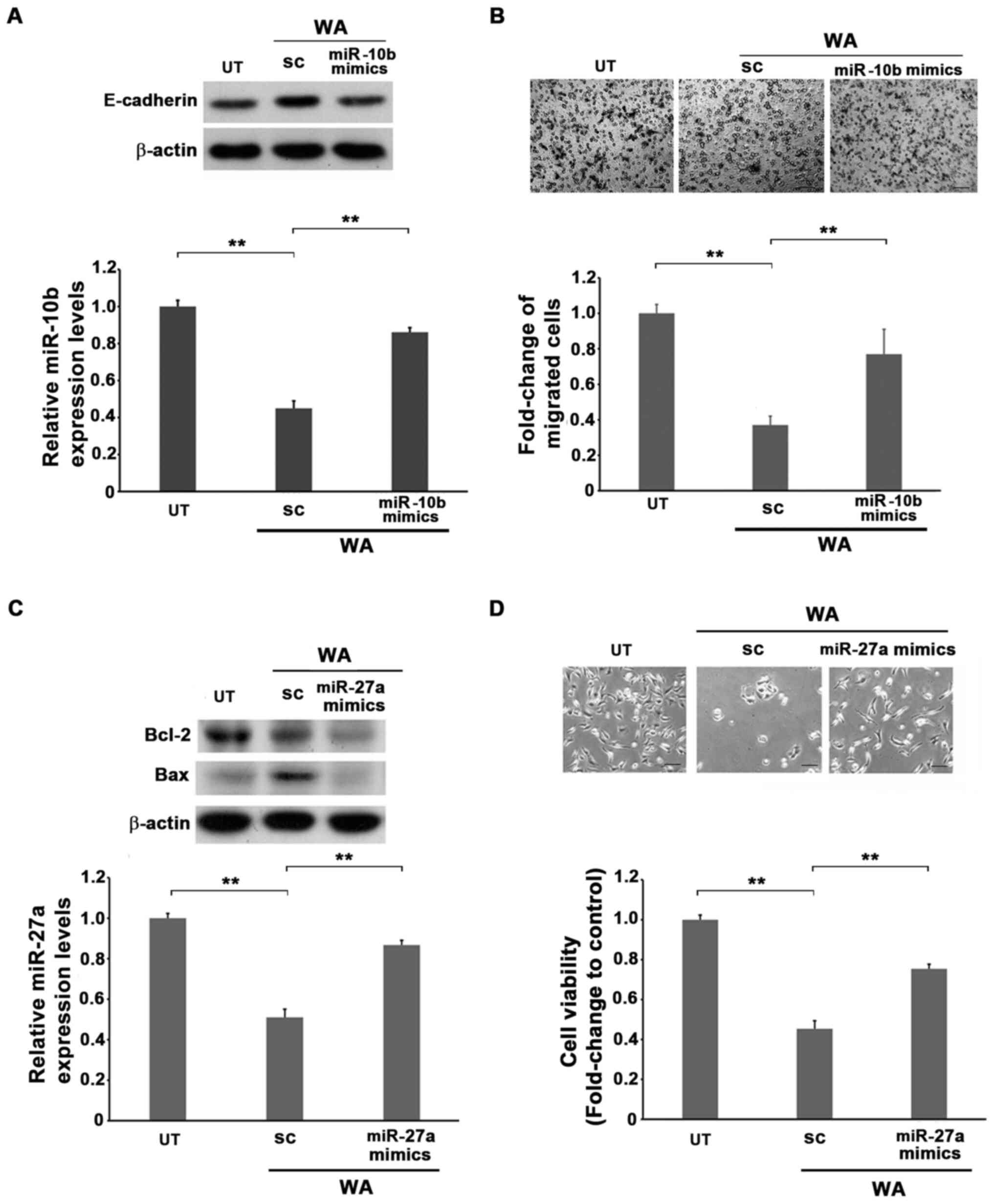 |
Figure 2.
Role of miR-10b and miR-27a in the WA-mediated attenuation of motility and viability of A549 lung cancer cells. (A) The untreated, SC or miR-10b mimic-transfected A549 cells were treated with 1 µM of WA for 24 h followed by western blotting or RT-qPCR analyses to detect the expression levels of E-cadherin or miR-10b, respectively. (B) The untreated, SC or miR-10b mimic A549 cells were treated with 1 µM of WA for 24 h followed by migration assay for a further 16 h. The migrated cells were collected and subjected to methanol fixation and giemsa staining. The number of migrated A549 cells from each experimental set was normalized to those of the UT cells as relative folds. Scale bar, 40 µm. (C and D) Non-transfected or miR-27a mimics transfected A549 cells were treated with WA for 24 h, and subjected to (C) western blot to detect the expression levels of Bax and Bcl-2 and RT-qPCR to uncover the levels of miR-27a or (D) viability assay with the MTT method to analyze cell viability. The optical density values of each experimental set of cells were then normalized to that of the parental cells as relative folds. Scale bar, 40 µm. **P<0.01. UT, untreated; SC, scramble RNA; RT-qPCR, reverse transcription-quantitative PCR; miR, microRNA; WA, withaferin A.
|
miR-27a: A target gene in WA-mediated lung cancer cell death
The role of miR-27a in WA-mediated cellular death was assessed. miR-27a mimics were transfected into A549 cells for 24 h prior to treatment with WA and subsequently subjected to viability and western blot assays. Transfection with miR-27a mimics alone induced the level of total miR-27a in A549 cells (Fig. S6A). Furthermore, the results demonstrated that increased miR-27a expression effectively decreased Bax expression originally induced in the presence of WA, resembling that of the untreated level, and recovered the viability originally attenuated by WA (Fig. 2C and D). The combination of these results suggested that the reduction of both miR-10b and miR-27a by WA may occur via a transcriptional manner, and further indicates that miR-10b and miR-27a are two potential molecules in the WA-mediated reduction of cellular motility and viability.
p53 regulates the expression of both miR-10b and miR-27a in the presence of WA
Wild-type p53 was transfected into H1299 cells and the expression of miR-10b and miR-27a was assessed. The results demonstrated that ectopic expression of wild-type p53 significantly decreased the expression of both miR-10b and miR-27a in a dose-dependent manner (Fig. 3A). Endogenous p53 of A549 cells was knocked down by transfecting p53 siRNA, followed by treatment with WA. The results demonstrated that the knockdown of p53 increased the levels of both miR-10b and miR-27a. Notably, the reduction of motility, viability and the levels of miR-10b and miR-27a in the presence of WA were significantly reversed by the reduction of p53 (Fig. 3B and C). This suggests that p53 may be an upstream inhibitor of miR-10b and miR-27a, and that treatment with WA induces the expression of p53, which in turn decreases the levels of miR-10b and miR-27a, thus attenuating the cellular motility or viability of lung cancer cells. A549 cells were used to generate p53 knockdown stable clones and confirm the causality between p53 and miR-10b or miR-27a in WA-mediated antitumor function. The results demonstrated that the levels of E-cadherin and Bax are much lower, whereas miR-10b and miR-27a were higher in p53shRNA stable clones compared with the A549-shRNA cells (Fig. 4A). Treatment with WA marginally decreased the motility and viability of A549-p53shRNA stable clones (Fig. 4B and C). A549-p53shRNA was transfected with antagomiR against either miR-10b or miR-27a to decrease the endogenous levels of miR-10b and miR-27a (Fig. S6B). Notably, the knockdown of miR-10b or miR-27a by antagomiRs significantly decreased the motility and viability of WA-treated A549-p53shRNA stable clones compared with untreated ones accompanied by increasing E-cadherin and Bax levels. (Fig. 4B and C). Collectively, these results suggest that p53 plays a central role in downregulating miR-10b and miR-27a, attenuating motility and viability primed by WA (Fig. 4D).
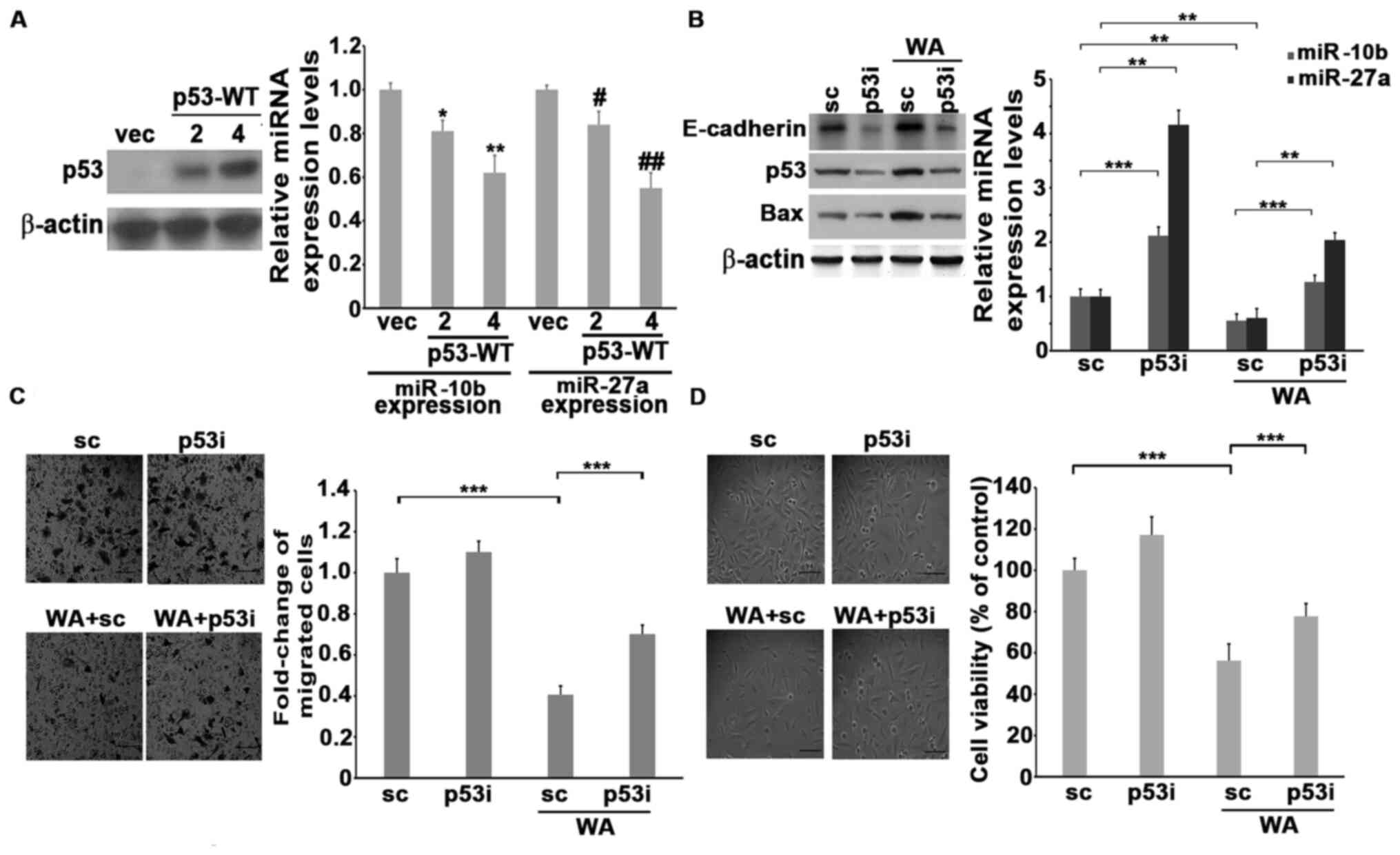 |
Figure 3.
p53 regulates the expression of miR-10b and miR-27a in the presence of WA. (A) The 2 or 4 µg of backbone vector or p53-WT vector were transfected into H1299 cells. After 24 h, the transfected cells were collected and subjected to western blotting or RT-qPCR analyses to detect the expression levels of p53, miR-10b and miR-27a, respectively. (B) The siRNA (scramble, designated as SC) or p53siRNA-transfected A549 cells were either left untreated or treated with 1 µM of WA and subjected to western blotting or RT-qPCR to analyze the levels of p53 or miR-10b and miR-27a, respectively. (C) The siRNA (scramble, designated as SC) or p53siRNA-transfected A549 cells were treated with 1 µM WA for 24 h, and subjected to a migration assay for a further 16 h. Scale bar, 40 µm. (D) The siRNA (scramble, designated as SC) or p53siRNA-transfected A549 cells were treated with 1 µM WA for 48 h, and cell viability was then determined by the trypan blue exclusion assay. *P<0.05 and **P<0.01 miR-10b differences between p53 transfections vs. vec. #P<0.05 and ##P<0.01 miR-27a differences between p53 transfections vs vec. Scale bar, 40 µm. SC, scramble RNA; RT-qPCR, reverse transcription-quantitative PCR; miRNA/miR, microRNA; WA, withaferin A; p53i, p53siRNA.
|
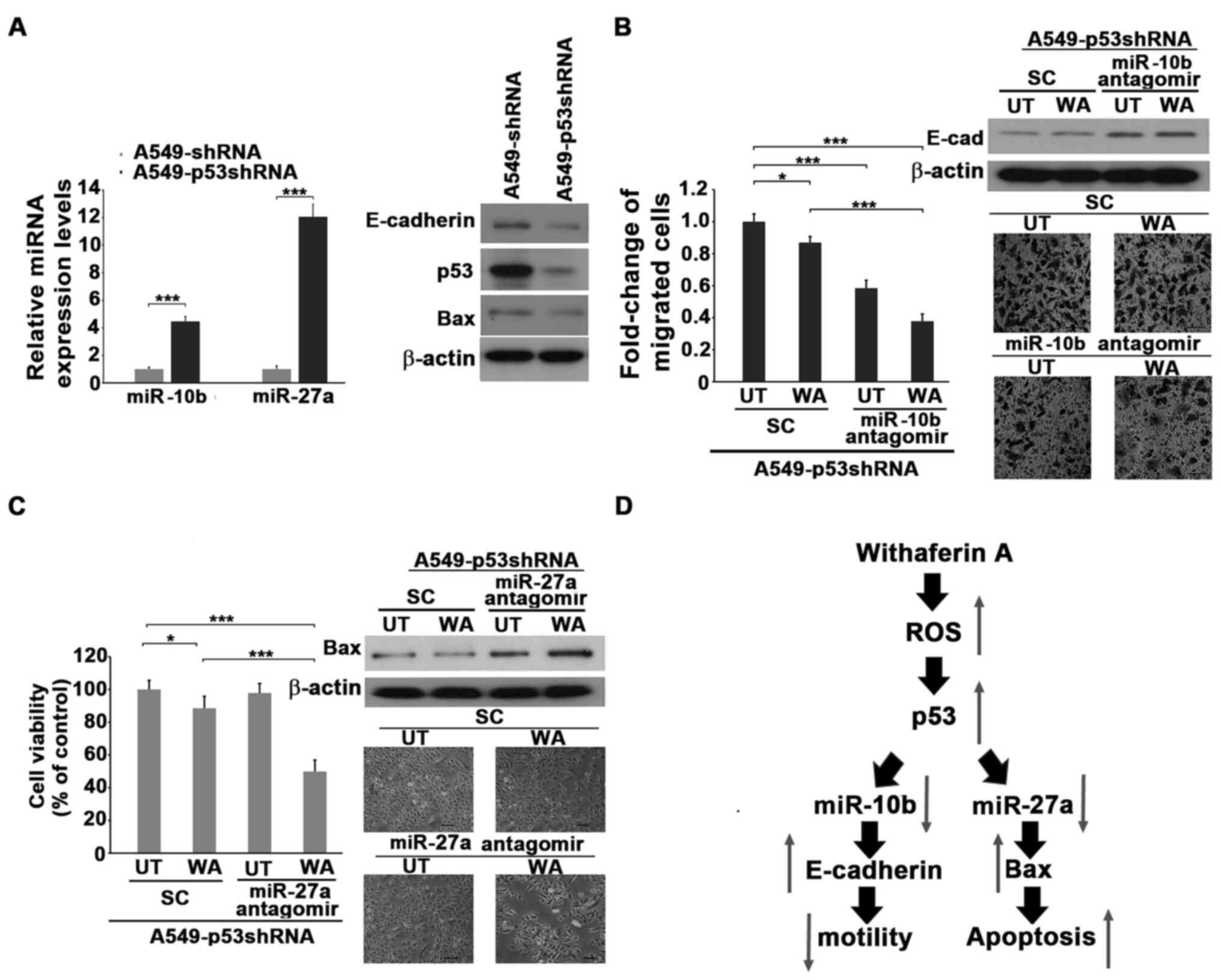 |
Figure 4.
Knockdown of miR-10b and miR-27a decreases the WA-resistant ability of A549-p53shRNA stable clone cells. (A) The A549, A549shRNA and A549-p53shRNA stable clone cells were subjected to western blotting or reverse transcription-quantitative PCR to reveal the expression of the indicated molecules. (B) The siRNA (scramble, designated as SC) or miR-10b antagomiR-transfected A549-p53shRNA cells were treated with 1 µM of WA (UT (untreated) cells were used as a control) for 24 h. The transfected cells were collected and subjected to western blotting or migration assay for another 16 h. The ratios of the migrated cells were determined. Scale bar, 40 µm. (C) The siRNA (scramble, designated as SC) or miR-27a antagomiR-transfected A549-p53shRNA cells were treated with 1 µM of WA for 48 h. UT cells were used as a WA untreated control. The cells were then collected and subjected to viability assay or western blotting. Scale bar, 40 µm. (D) Schematic representation of WA-induced apoptosis and WA-reduced motility via inhibiting both miR-10b and mi-27a in a p53-depedent manner. *P<0.05, ***P<0.001. UT, untreated; SC, scramble RNA; miRNA/miR, microRNA; WA, withaferin A.
|
Discussion
The results of the present study demonstrated that treatment with WA induced the expression of p53, which in turn decreased the expression of two oncomiRs, miR-10b and miR-27a, prior to the induction of E-cadherin and Bax expression, followed by a reduction in the viability and motility of lung cancer cells.
Several molecules are involved in WA-mediated antitumor functionality. For example, Oh et al (38) reported that the PI3K/Akt signaling module is inhibited in A549 cells treated with WA. Zhou et al (39) reported that p53, p21 and Bax are upregulated, whereas Bcl-2, cdk2 and cyclin D1 are downregulated in HCC cells treated with WA. WA also induces oxidative stress, DNA damage, and G2/M arrest in breast, osteosarcoma, and oral cancer cell lines (20,40,41). Furthermore, WA attenuates the motility of breast cancer cells by directly binding vimentin to block the assemble into lamellipodia (42). However, most reports mention only one malignant characteristic, such as growth ability or motility of cancer cells affected by WA. The present study further demonstrated that WA attenuated two malignant characteristics, including cell proliferation and motility at different concentrations by targeting p53 in lung cancer cells.
p53 is a well-known tumor suppressor, which predominantly acts as a transcriptional regulator in controlling target gene expression (43). It can either induce or decrease the expression levels of target genes depending on the binding sequence of the target gene promoter (44). The results of the present study demonstrated that p53 inhibited cellular motility and viability by targeting miR-10b or miR-27a, which in turn upregulated the expression levels of Bax and E-cadherin, respectively. Administration of WA decreased the expression of miR-10b at a lower dose, whereas higher doses of WA were necessary to inhibit the expression of miR-27a. The results of the present study are consistent with previous findings, where p53 has been reported to exhibit different levels of affinity towards distinct target genes, which may be determined by a combination of p53 expression levels, its localization and the binding sequence of the target gene promoters (45,46). p53 is an important regulator responsible for cell cycle arrest or apoptosis induced by genotoxic stress; and loss of p53 function occurs in nearly half of all patients with cancer (47). Furthermore, the status of p53 strongly affects the sensitivity of tumor cells to chemotherapeutic and/or radiotherapeutic agents, the p53 is required for the apoptosis of cancer cells to the treatment of both chemo- and radio-therapeutic agents (48,49). The results of the present study demonstrated that A549 cells with transient or stable knockdown of p53 exhibited higher viability in comparison with parental A549 cells treated with the same concentration of WA, suggesting that p53 may be a crucial molecule in WA-mediated antitumor activity.
Both miR-10b and miR-27a are well-known oncomiRs and are involved in the upregulation of several malignant characteristics, including migration/invasion and anti-apoptosis of different cancer cells (50,51). It has been reported that Myc may potentiate the expression of miR-27a via the regulation of its promoter (52), whereas the upstream regulator of miR-10b has not yet been identified. The results of the present study demonstrated that p53 has a potential upstream effect on both miR-10b and miR-27a, which further reveals potential malignancy induction under the loss function of this tumor suppressor. Notably, neither miR-10b or miR-27a promoters have been demonstrated to contain a classic p53-binding element composed of four half-sites (RRRCW or WGYYY) (53). However, some non-canonical p53-binding sequences identified previously (46) are located in both miR-10b and miR-27a promoters. Since the misregulation of target genes by p53 is critical to cellular transformation and malignancy, including lung cancer (54), these findings indicate that whether p53 does indeed repress the expression levels of both miR-10b and miR-27a via binding to the non-canonical promoter sequences may be worthy of investigation to clarify the underling mechanisms of lung tumorigenesis mediated by p53.
The limitation of the present study is the restricting numbers of lung cancer cell lines assessed for WA antitumor ability. However, in conclusion, the present study successfully identified two oncomiRs, miR-10b and miR-27a, that serve as novel targets of WA in a p53-dependent manner to attenuate the malignant characteristics of lung cancer cells. The combined effects of WA with cisplatin or taxol in treating patients with lung cancer may be worthy to investigate in prospective studies.
Supplementary Material
Supporting Data
Acknowledgements
The authors of the present study would like to thank Dr Shih-Lan Hsu (Department of Medical Research, Taichung Veterans General Hospital, Taichung) for providing cell lines, experimental materials, and article construction.
Funding
The present study was supported by research grants from Chung Shan Medical University (grant no. CSMU-INT-105) and Taichung Veterans General Hospital (grant no. TCVGH-1083206C).
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author upon reasonable request.
Author's contributions
CCW devised the main conceptual ideas, proof outlined and drafted the initial manuscript. CCL and TYY CCL and TYY performed all cellular biology experiments. CKW and HJL participated in the miRNA assay comet assay, MMP assay and ROS experiments. CCW and SLH verified the results and proposed interpretations in discussions with CCL, TYY and CKW. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
1
|
Islami F, Torre LA and Jemal A: Global trends of lung cancer mortality and smoking prevalence. Transl Lung Cancer Res. 4:3272015.PubMed/NCBI
|
|
2
|
Lin HT, Liu FC, Wu CY, Kuo CF, Lan WC and Yu HP: Epidemiology and survival outcomes of lung cancer: A population-based study. Biomed Res Int. 2019:81481562019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel RL, Miller KD and Jemal A: Cancer statistics, 2020. CA Cancer J Clin. 70:7–30. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schabath MB and Cote ML: Cancer progress and priorities: Lung cancer. Cancer Epidemiol Biomarkers Prev. 28:1563–1579. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Molina JR, Yang P, Cassivi SD, Schild SE and Adjei AA: Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clinic Proceedings. 83:584–594. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Torre LA, Siegel RL and Jemal A: Lung cancer statistics. Adv Exp Med Biol. 893:1–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xue X, Liu Y, Wang Y, Meng M, Wang K, Zang X, Zhao S, Sun X, Cui L, Pan L and Liu S: MiR-21 and MiR-155 promote non-small cell lung cancer progression by downregulating SOCS1, SOCS6, and PTEN. Oncotarget. 7:84508–84519. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ma L, Teruya-Feldstein J and Weinberg RA: Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 449:682–688. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hu Y, Qiu Y, Yagüe E, Ji W, Liu J and Zhang J: miRNA-205 targets VEGFA and FGF2 and regulates resistance to chemotherapeutics in breast cancer. Cell Death Dis. 7:e22912016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mraz M, Malinova K, Mayer J and Pospisilova S: MicroRNA isolation and stability in stored RNA samples. Biochem Biophys Res Commun. 390:1–4. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Glinge C, Clauss S, Boddum K, Jabbari R, Jabbari J, Risgaard B, Tomsits P, Hildebrand B, Kääb S, Wakili R, et al: Stability of circulating blood-based microRNAs-pre-analytic methodological considerations. PLoS One. 12:e01679692017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rupaimoole R and Slack FJ: MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 16:2032017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bajan S and Hutvagner G: RNA-based therapeutics: From antisense oligonucleotides to miRNAs. Cells. 9:1372020. View Article : Google Scholar
|
|
14
|
Fabbri M: MicroRNAs and cancer: Towards a personalized medicine. Curr Mol Med. 13:751–756. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Florczuk M, Szpechcinski A and Chorostowska-Wynimko J: miRNAs as biomarkers and therapeutic targets in non-small cell lung cancer: Current perspectives. Target Oncol. 12:179–200. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rayan A, Raiyn J and Falah M: Nature is the best source of anticancer drugs: Indexing natural products for their anticancer bioactivity. PLoS One. 12:e01879252017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sharifi-Rad J, Ozleyen A, Boyunegmez Tumer T, Oluwaseun Adetunji C, El Omari N, Balahbib A, Taheri Y, Bouyahya A, Martorell M, Martins N and Cho WC: Natural products and synthetic analogs as a source of antitumor drugs. Biomolecules. 9:6792019. View Article : Google Scholar
|
|
18
|
Yu SM and Kim SJ: Production of reactive oxygen species by withaferin A causes loss of type collagen expression and COX-2 expression through the PI3K/Akt, p38, and JNK pathways in rabbit articular chondrocytes. Exp Cell Res. 319:2822–2834. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roy RV, Suman S, Das TP, Luevano JE and Damodaran C: Withaferin A, a steroidal lactone from Withania somnifera, induces mitotic catastrophe and growth arrest in prostate cancer cells. J Nat Prod. 76:1909–1915. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stan SD, Zeng Y and Singh SV: Ayurvedic medicine constituent withaferin a causes G2 and M phase cell cycle arrest in human breast cancer cells. Nutr Cancer. 60 (Suppl 1):S51–S60. 2008. View Article : Google Scholar
|
|
21
|
Mayola E, Gallerne C, Degli Esposti D, Esposti DD, Martel C, Pervaiz S, Larue L, Debuire B, Lemoine A, Brenner C and Lemaire C: Withaferin A induces apoptosis in human melanoma cells through generation of reactive oxygen species and down-regulation of Bcl-2. Apoptosis. 16:1014–1027. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fong MY, Jin S, Rane M, Singh RK, Gupta R and Kakar SS: Withaferin A synergizes the therapeutic effect of doxorubicin through ROS-mediated autophagy in ovarian cancer. PLoS One. 7:e422652012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cai Y, Sheng ZY, Chen Y and Bai C: Effect of withaferin A on A549 cellular proliferation and apoptosis in non-small cell lung cancer. Asian Pac J Cancer Prev. 15:1711–1714. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Oh JH, Lee TJ, Kim SH, Choi YH, Lee SH, Lee JM, Kim YH, Park JW and Kwon TK: Induction of apoptosis by withaferin A in human leukemia U937 cells through down-regulation of Akt phosphorylation. Apoptosis. 13:1494–1504. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yu Y, Hamza A, Zhang T, Gu M, Zou P, Newman B, Li Y, Gunatilaka AA, Zhan CG and Sun D: Withaferin A targets heat shock protein 90 in pancreatic cancer cells. Biochem Pharmacol. 79:542–551. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hahm ER, Lee J, Huang Y and Singh SV: Withaferin a suppresses estrogen receptor-α expression in human breast cancer cells. Mol Carcinog. 50:614–624. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kyakulaga AH, Aqil F, Munagala R and Gupta RC: Withaferin a inhibits epithelial to mesenchymal transition in non-small cell lung cancer cells. Sci Rep. 8:157372018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Grogan PT, Sleder KD, Samadi AK, Zhang H, Timmermann BN and Cohen MS: Cytotoxicity of withaferin A in glioblastomas involves induction of an oxidative stress-mediated heat shock response while altering Akt/mTOR and MAPK signaling pathways. Invest New Drugs. 31:545–557. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hsu JH, Chang PM, Cheng TS, Kuo YL, Wu AT, Tran TH, Yang YH, Chen JM, Tsai YC, Chu YS, et al: Identification of withaferin A as a potential candidate for anti-cancer therapy in non-small cell lung cancer. Cancers (Basel). 11:10032019. View Article : Google Scholar
|
|
30
|
Strober W: Trypan blue exclusion test of cell viability. Curr Protoc Immunol. 21:A. 3B. 1–A. 3B. 2. 1997.
|
|
31
|
Ernst O and Zor T: Linearization of the Bradford protein assay. J Vis Exp. 19182010.
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu CC, Huang KF, Yang TY, Li YL, Wen CL, Hsu SL and Chen TH: The topoisomerase 1 inhibitor austrobailignan-1 isolated from Koelreuteria Henryi induces a G2/M-phase arrest and cell death independently of p53 in non-small cell lung cancer cells. PLoS One. 10:e01320522015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Loh CY, Chai JY, Tang TF, Wong WF, Sethi G, Shanmugam MK, Chong PP and Looi CY: The E-cadherin and N-cadherin switch in epithelial-to-mesenchymal transition: Signaling, therapeutic implications, and challenges. Cells. 8:11182019. View Article : Google Scholar
|
|
35
|
Liang G, Ding M, Lu H, Cao NA, Niu Y, Gao Y and Lu J: Metformin upregulates E-cadherin and inhibits B16F10 cell motility, invasion and migration. Oncol Lett. 10:1527–1532. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ma L, Reinhardt F, Pan E, Soutschek J, Bhat B, Marcusson EG, Teruya-Feldstein J, Bell GW and Weinberg RA: Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat Biotechnol. 28:341–347. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang R, He Y, Zhang X, Xing B, Sheng Y, Lu H and Wei Z: Estrogen receptor-regulated microRNAs contribute to the BCL2/BAX imbalance in endometrial adenocarcinoma and precancerous lesions. Cancer Lett. 314:155–165. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Oh JH and Kwon TK: Withaferin A inhibits tumor necrosis factor-α-induced expression of cell adhesion molecules by inactivation of Akt and NF-kappaB in human pulmonary epithelial cells. Int Immunopharmacol. 9:614–619. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhou YF, Yu XT, Yao JJ, Xu CW, Huang JH, Wan Y and Wu MJ: Withaferin A inhibits hepatoma cell proliferation through induction of apoptosis and cell cycle arrest. Int J Clin Exp Pathol. 9:12381–12389. 2016.
|
|
40
|
Chang HW, Li RN, Wang HR, Liu JR, Tang JY, Huang HW, Chan YH and Yen CY: Withaferin A induces oxidative stress-mediated apoptosis and DNA damage in oral cancer cells. Front Physiol. 8:6342017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lv TZ and Wang GS: Antiproliferation potential of withaferin A on human osteosarcoma cells via the inhibition of G2/M checkpoint proteins. Exp Therap Med. 10:323–329. 2015. View Article : Google Scholar
|
|
42
|
Thaiparambil JT, Bender L, Ganesh T, Kline E, Patel P, Liu Y, Tighiouart M, Vertino PM, Harvey RD, Garcia A and Marcus AI: Withaferin A inhibits breast cancer invasion and metastasis at sub-cytotoxic doses by inducing vimentin disassembly and serine 56 phosphorylation. Int J Cancer. 129:2744–2755. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mello SS and Attardi LD: Deciphering p53 signaling in tumor suppression. Curr Opin Cell Biol. 51:65–72. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Braithwaite A and Prives C: p53: More research and more questions. Cell Death Differ. 13:877–880. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wu M, Ye H, Tang Z, Shao C, Lu G, Chen B, Yang Y, Wang G and Hao H: p53 dynamics orchestrates with binding affinity to target genes for cell fate decision. Cell Death Dis. 8:e31302017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yang TY, Teng CLJ, Lin TCC, Chen KC, Hsu SL and Wu CC: Transcriptional repression of Aurora-A gene by wild-type p53 through directly binding to its promoter with histone deacetylase 1 and mSin3a. Int J Cancer. 142:92–108. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Muller PA and Vousden KH: p53 mutations in cancer. Nat Cell Biol. 15:2–8. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lu C and El-Deiry WS: Targeting p53 for enhanced radio-and chemo-sensitivity. Apoptosis. 14:597–606. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
He C, Li L, Guan X, Xiong L and Miao X: Mutant p53 gain of function and chemoresistance: The role of mutant p53 in response to clinical chemotherapy. Chemotherapy. 62:43–53. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sheedy P and Medarova Z: The fundamental role of miR-10b in metastatic cancer. Am J Cancer Res. 8:1674–1688. 2018.PubMed/NCBI
|
|
51
|
Li X, Xu M, Ding L and Tang J: MiR-27a: A novel biomarker and potential therapeutic target in tumors. J Cancer. 10:2836–2848. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Li X, Liu X, Xu W, Zhou P, Gao P, Jiang S, Lobie PE and Zhu T: c-MYC-regulated miR-23a/24-2/27a cluster promotes mammary carcinoma cell invasion and hepatic metastasis by targeting Sprouty2. J Biol Chem. 288:18121–18133. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Riley T, Sontag E, Chen P and Levine A: Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 9:402–412. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Takahashi T, Nau MM, Chiba I, Birrer MJ, Rosenberg RK, Vinocour M, Levitt M, Pass H, Gazdar AF and Minna JD: p53: A frequent target for genetic abnormalities in lung cancer. Science. 246:491–494. 1989. View Article : Google Scholar : PubMed/NCBI
|














