Introduction
Worldwide, esophageal cancer is the fifth and ninth
leading cause of cancer mortality in males and females,
respectively (1). Esophageal
squamous cell carcinoma (ESCC) is the predominant histological
subtype in Asia, Africa, and South America (2).
SOX2, a member of the SOX family of transcription
factors (3), is critical for the
maintenance of pluripotency and self-renewal of embryonic stem
cells (ESCs) (4). In the esophagus,
SOX2 plays an important role in differentiation and morphogenesis
(5).
Our previous study (6), together with the study of others
(7), showed that the human
SOX2 gene, located at chromosome 3q26.3, is frequently
amplified in ESCC, and that the expression of the SOX2 mRNA
and the encoded protein is elevated in most primary ESCCs.
SOX2 recently was identified as a novel major oncogene,
recurrently amplified and activated in squamous cell carcinomas of
multiple organs and tissues, including the esophagus (6,7), lung
(8), and skin (9). Furthermore, we showed that SOX2
promotes ESCC cell proliferation in vitro and in vivo
via the AKT/mammalian target of rapamycin complex 1 (mTORC1)
signaling pathway (10). However,
the role of SOX2 in the carcinogenesis of ESCC is not fully
understood. Moreover, the mechanism by which SOX2 activates AKT
remains unknown.
AKT, a serine/threonine kinase, regulates many
biological processes, such as cell survival, proliferation, and
growth (11,12). AKT is activated by extracellular
signals through a process mediated by phosphatidylinositol 3-kinase
(PI3K) activation. PI3K produces phosphatidylinositol (3,4,5) -trisphosphate, which recruits two
protein kinases, AKT and phosphoinositide-dependent protein kinase
1 (PDK1), to the plasma membrane. PDK1 phosphorylates AKT at T308,
increasing AKT kinase activity. In turn, AKT phosphorylates
mitogen-activated protein kinase (MAPK) associated protein 1 (also
known as SAPK interacting protein 1; SIN1), a key component of
mTORC2, enhancing mTORC2 kinase activity and leading to
phosphorylation of AKT at S473 by mTORC2, thereby catalyzing full
activation of AKT (13). Conversely,
AKT activity is negatively regulated by phosphatase and tensin
homologue (PTEN).
Despite having an abundance of blood vessels, tumors
usually are hypoxic and nutrient-deprived because of dysfunction of
their vessels. Since AKT is involved in enhancing cell survival, we
hypothesized that SOX2, through AKT activation, may promote
survival of ESCC cells under poor nutrient conditions.
In the present study, we sought to investigate the
effect of SOX2 on ESCC cell survival and resistance to apoptosis
under serum starvation conditions. Furthermore, we examined the
underlying molecular mechanisms mediating SOX2's effects, focusing
on the role of AKT.
Materials and methods
Reagents and antibodies
Antibodies against SOX2 (#3579), total AKT (#9272),
phospho-AKT (p-AKT) (T308) (#13038), p-AKT (S473) (#4060), total
ERK (extracellular signal-regulated kinase) (#4695), p-ERK (#4370),
p-GSK-3β (glycogen synthase kinase-3β) (#5558), total GSK-3β
(#9832), and PARP (poly (ADP-ribose) polymerase) (#9542) were
purchased from Cell Signaling Technology. The antibody against
β-actin (#A1978) was purchased from Sigma-Aldrich Japan; Merck
KGaA. Doxorubicin was obtained from Toronto Research Chemicals.
MK2206 (S1078), AR-A014418 (S7435), GSK2334470 (S7087), and AZD8055
(S15555) were obtained from Selleck Chemicals. LY294002
(154447-36-6) was obtained from Cayman Chemical Company.
Cell culture
Four ESCC cell lines (KYSE30, KYSE140, TE4, and TE6)
were obtained from the American Type Culture Collection (ATCC) or
the JCRB Cell Bank. All cell lines were cultured at 37°C in
Dulbecco's modified Eagle's medium containing 10% fetal bovine
serum, except for KYSE140 cells, which was maintained in Ham's F-12
medium supplemented with 5% fetal calf serum.
Adenovirus vector
Recombinant adenovirus vectors expressing SOX2 or
control green fluorescent protein (GFP), which we here refer to as
Ad-SOX2 and Ad-control, respectively, were obtained from ViGene.
Cells were infected with Ad-SOX2 or Ad-control at a multiplicity of
infection (MOI) of 50.
Immunoblotting
Immunoblotting was performed as described previously
(14). Dilutions were as follows:
1:1,000 for antibodies against SOX2, AKT, p-AKT (T308), p-AKT
(S473), ERK, p-ERK, PARP, p-GSK3β, and PARP; and 1:5,000 for the
antibody against β-actin. For immunodetection, anti-rabbit IgG
(#7074) or anti-mouse IgG (#7076) (Cell Signaling Technology, Inc.)
was used as the secondary antibody at a dilution of 1:5,000 or
1:10,000, respectively. Antibody binding was detected using the ECL
system (Amersham Biosciences). Densitometric analysis was performed
using ImageJ software (version 1.53; National Institutes of
Health).
RNA interference
Two small interfering RNAs (siRNAs) targeting
SOX2 (#1 and #2; ID s13294 and s13296, respectively;
Ambion), as well as a control (non-silencing) siRNA, were delivered
into cells using Lipofectamine RNAiMAX (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Cell viability assay
Cell viability was determined using the WST-8 assay
(Cell Counting Kit-8; Dojindo) according to the manufacturer's
instructions.
Apoptosis assay
Apoptosis was evaluated based the level of cleaved
PARP, which was detected by immunoblotting.
Statistical analysis
Statistical analyses were performed using IBM SPSS
Statistics 24.0 (SPSS, Inc.). Comparisons were made using a
two-tailed unpaired Student's t-test. P-values <0.05 were
considered to indicate statistical significance.
Results
SOX2 overexpression activates AKT
To express SOX2 in ESCC cells using the adenoviral
vector, two cell lines (KYSE30 and TE4) that are known to express
SOX2 only at low levels (6) were
infected with Ad-SOX2 or Ad-control. Cells were cultured under
standard conditions and harvested at 48 h after infection.
Immunoblot analyses revealed that SOX2 protein was successfully
expressed in both of the lines infected with Ad-SOX2, and that
levels of p-AKT (S473), but not those of total AKT, were higher in
cells infected with Ad-SOX2 than in those infected with Ad-control
(Fig. 1A). However, p-ERK levels
were not increased in KYSE30 cells infected with Ad-SOX2 (Fig. 1A) compared to the same line infected
with Ad-control.
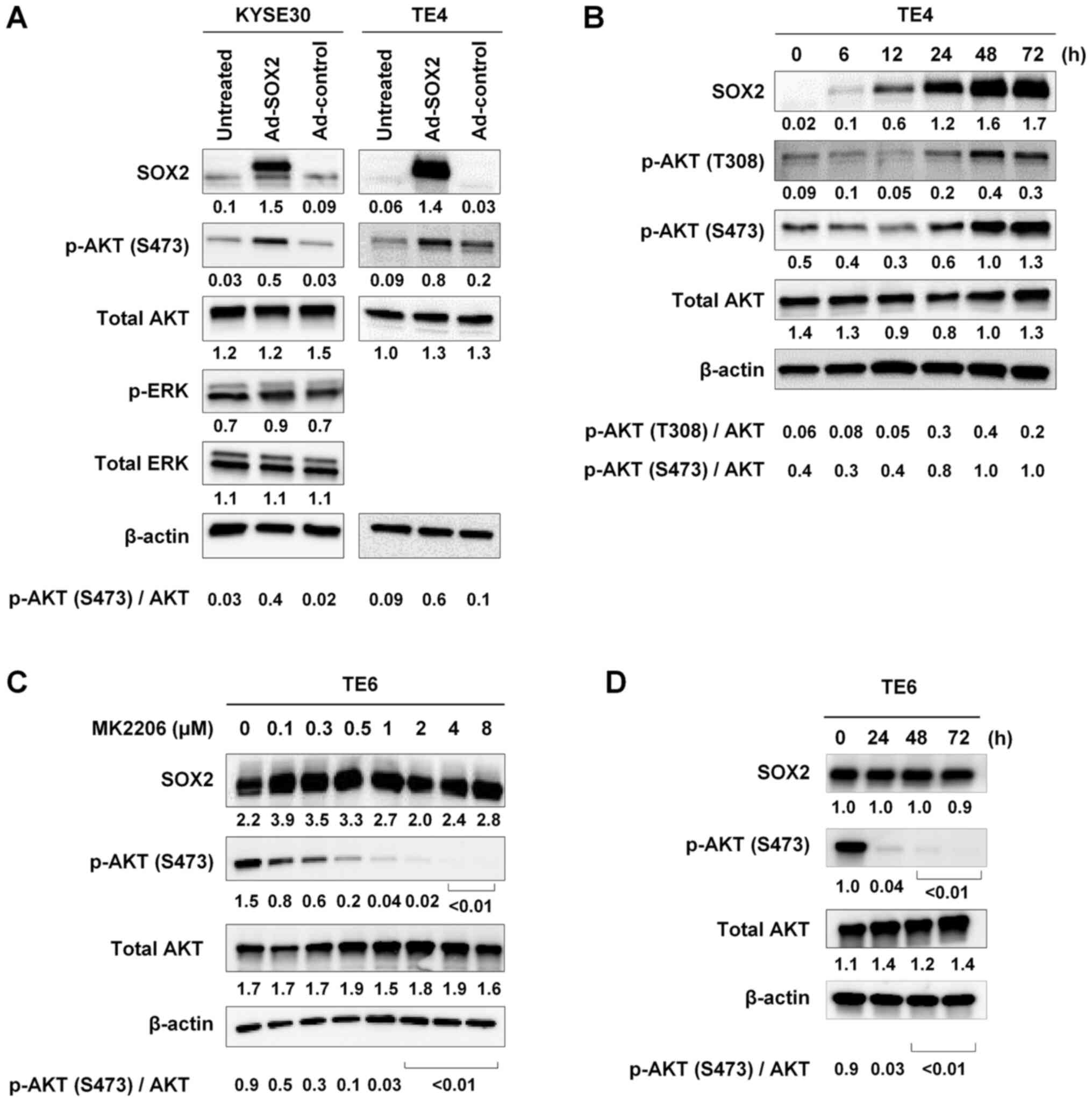 | Figure 1.Activation of AKT by adenoviral
vector-mediated overexpression of SOX2. (A) KYSE30 and TE4 cells,
which are known to express SOX2 at low levels, were infected with
Ad-SOX2 or Ad-control. Cells were harvested 48 h after infection
and subjected to immunoblot analysis of SOX2, p-AKT (S473), total
AKT, p-ERK and total ERK expression. (B) Time course of changes in
the levels of SOX2, p-AKT (T308), p-AKT (S473) and total AKT. TE4
cells were infected with Ad-SOX2 and harvested at the indicated
time points after infection, then subjected to immunoblot analysis.
(C) Effect of MK2206. TE6 cells, which express a high endogenous
level of SOX2, were treated for 48 h with MK2206, an AKT inhibitor,
at the indicated concentrations for 48 h, then subjected to
immunoblot analysis of SOX2, p-AKT (S473) and total AKT levels. (D)
TE6 cells were treated with 2 µM MK2206 for different time periods
and harvested at the indicated time points, then subjected to
immunoblot analysis of SOX2, p-AKT (S473) and total AKT expression.
For all experiments, β-actin was detected as a loading control. The
numbers presented below the gels represent the expression levels of
each protein relative to those of β-actin. Values were normalized
so that β-actin expression in each well had a value of 1. The ratio
of p-AKT (S473 or T308) to total AKT is shown. Ad, adenovirus
vector; p-, phosphorylated. |
To examine the time courses of accumulation of SOX2
and p-AKT, TE4 cells were infected with Ad-SOX2 and harvested 6,
12, 24, 48 or 72 h later. SOX2 accumulation was detected starting
at 6 h; SOX2 protein levels gradually increased thereafter,
reaching a maximum at 48 h after infection. Levels of p-AKT (T308
and S473) increased at 24 h and reached a maximum at 48 h. The time
course analyses showed that the increase in SOX2 accumulation
preceded the increase in p-AKT levels (Fig. 1B). These results suggested that
overexpression of SOX2 increased the activation of AKT.
To examine whether, conversely, AKT activation
increased SOX2 accumulation in ESCC cells, TE6 cells, which express
a high endogenous level of SOX2 (6),
were treated with MK2206, an inhibitor of AKT, for 48 h. Exposure
to MK2206 resulted in decreased p-AKT (S473) levels in a
dose-dependent manner. However, the decrease in p-AKT (S473) levels
did not influence the level of SOX2 (Fig. 1C). The same results were obtained
when TE6 cells were treated with 2 µM of MK2206 for different
periods of time (0, 24, 48 and 72 h) and harvested for immunoblot
analysis (Fig. 1D).
SOX2 activates AKT under serum
starvation conditions
The activation of AKT by SOX2 was examined under
serum starvation conditions. KYSE30 cells were infected with
Ad-SOX2 or Ad-control. After a 24-h incubation, cells were cultured
in serum-free medium for 24 h. The cells then were left untreated
or treated with serum (10%) for 15 min. Immunoblot analysis showed
that, for cells maintained under serum starvation conditions, p-AKT
(S473) levels were higher in cells infected with Ad-SOX2 than in
those infected with Ad-control (Fig.
2A). However, increases in p-AKT (S473) levels did not appear
to differ when comparing serum-stimulated Ad-SOX2-infected cells to
serum-stimulated Ad-control infected cells (Fig. 2A).
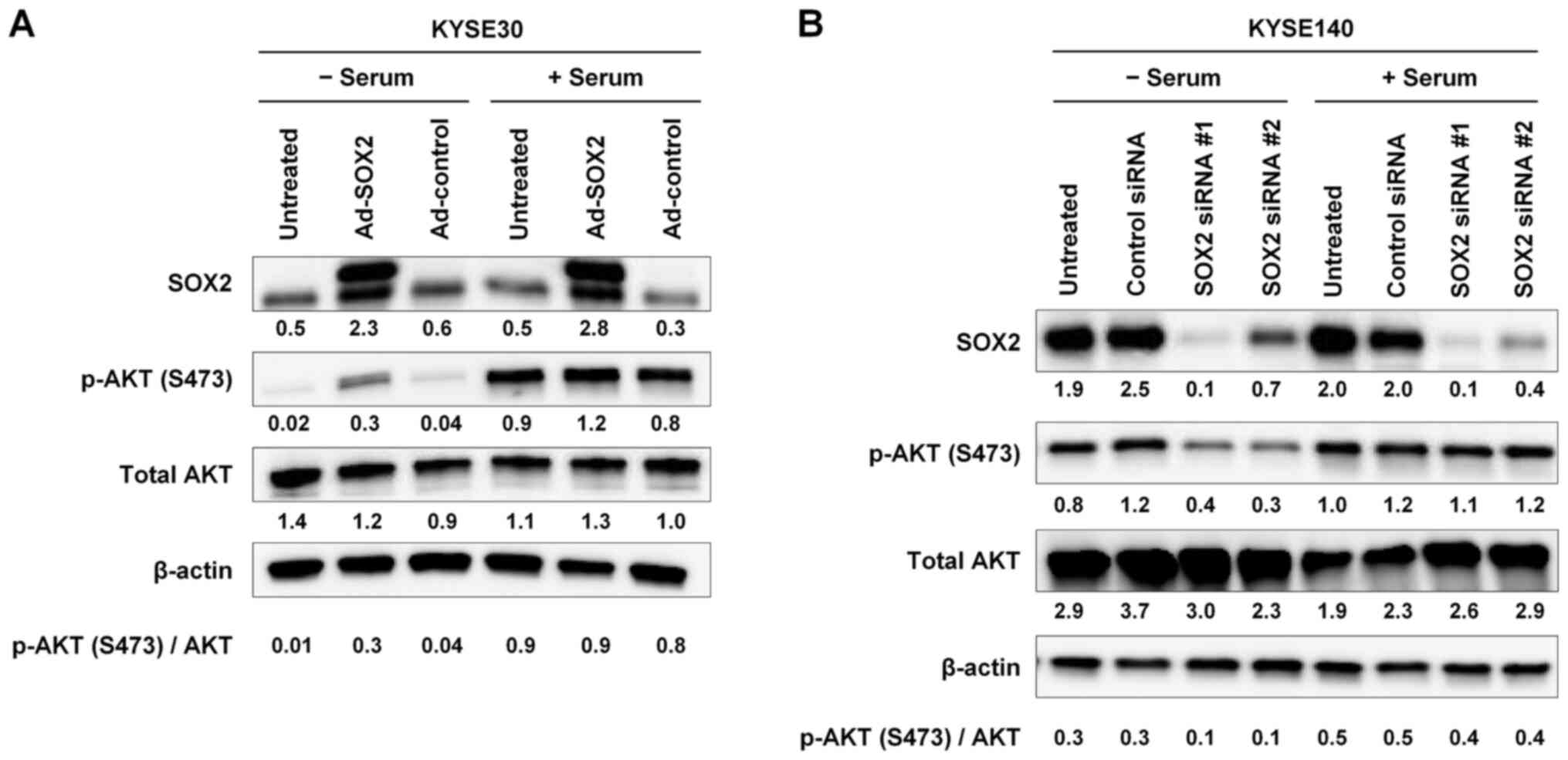 | Figure 2.Activation of AKT by SOX2 under serum
starvation conditions. (A) KYSE30 cells were infected with Ad-SOX2
or Ad-control. After a 24-h incubation, cells were cultured in
serum-free medium for 24 h. Subsequently, cells were incubated in
growth medium without or with serum (10%) for 15 min (− serum or +
serum, respectively), harvested and subjected to immunoblot
analysis of SOX2, p-AKT (S473) and total AKT expression. (B)
KYSE140 cells were transfected with one of two siRNAs targeting
SOX2 (SOX2 siRNA #1 and #2) or with a control siRNA. After a 48-h
incubation, cells were cultured in serum-free medium for 12 h.
Subsequently, cells were incubated in growth medium without or with
serum (10%) for 15 min (− serum or + serum, respectively),
harvested and subjected to immunoblot analysis of SOX2, p-AKT
(S473) and total AKT expression. For all experiments, β-actin was
detected as a loading control. The numbers presented below the gels
represent the expression levels of each protein relative to those
of β-actin. Values were normalized so that β-actin expression in
each well had a value of 1. The ratio of p-AKT (S473) to total AKT
is shown. Ad, adenovirus vector; p-, phosphorylated; siRNA, small
interfering RNA. |
To confirm this finding, KYSE140 cells, which have
high endogenous expression of SOX2 (6), were transfected with either of two
siRNAs (SOX2 siRNA #1 and #2) targeting SOX2 expression, or
with a control siRNA. After a 48-h incubation, cells were cultured
in serum-free medium for 12 h. Cells then were incubated for 15 min
in growth medium without or with serum (10%). SOX2 knockdown by
siRNAs #1 and #2 was confirmed by immunoblotting (Fig. 2B). Levels of p-AKT (S473) were
decreased following knockdown of SOX2 under serum starvation
conditions, but not following knockdown under serum-stimulated
conditions (Fig. 2B). Taken
together, these results suggested that SOX2 increases AKT
activation under serum starvation conditions. However, this effect
of SOX2 appears to be masked by serum stimulation.
SOX2 promotes cell survival under
serum starvation conditions
To examine the effect of SOX2 on cell viability
under serum starvation conditions, TE4 cells were infected with
Ad-SOX2 or Ad-control, and then cultured in growth medium in the
presence or absence of serum. A cell growth assay showed that cell
viability in serum-free medium was higher in cells infected with
Ad-SOX2 than in those infected with Ad-control. Notably, this
pattern was not seen in medium containing serum (Fig. 3A). However, the difference between
Ad-SOX2 and Ad-control in serum-free medium was no longer seen when
cells were treated with MK2206 (Fig.
3B), suggesting the involvement of AKT in the effect of SOX2 on
promotion of cell survival under serum starvation conditions.
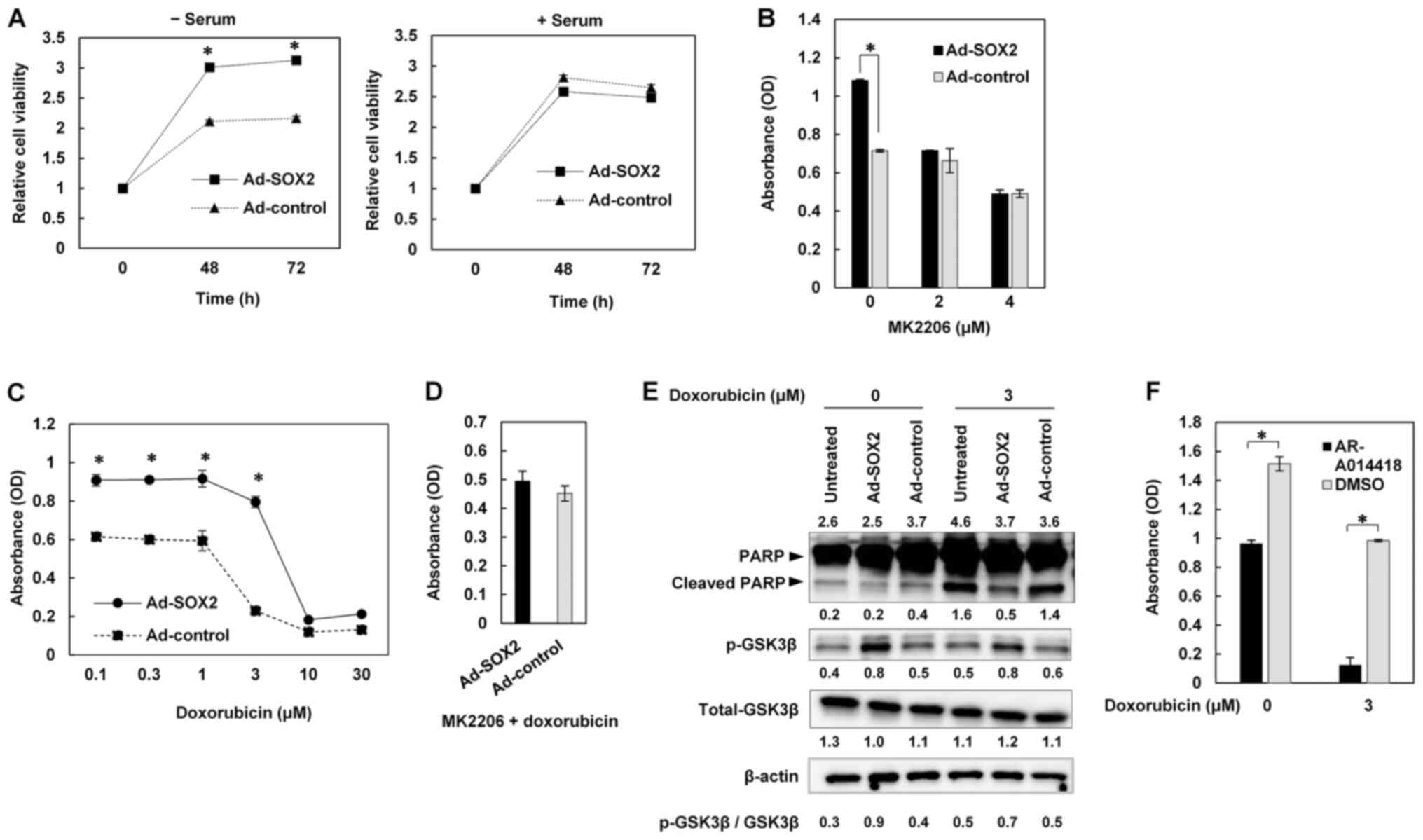 | Figure 3.Promotion of cell survival under serum
starvation conditions and resistance to apoptosis through
activation of the AKT/GSK-3β signaling pathway by SOX2. (A) TE4
cells were infected with Ad-SOX2 or Ad-control, and then cultured
in serum-free medium (− serum) or medium with serum (+ serum).
Relative cell viabilities were measured at the indicated time
points. (B) TE4 cells were infected with Ad-SOX2 or Ad-control,
then treated 24 h later with the indicated concentrations of MK2206
in serum-free medium. Cell viabilities were measured 48 h after
MK2206 treatment. (C) TE4 cells were infected with Ad-SOX2 or
Ad-control, then treated 24 h later with the indicated
concentrations of doxorubicin in serum-free medium. Cell
viabilities were measured 48 h after doxorubicin treatment. (D) TE4
cells were infected with Ad-SOX2 or Ad-control, then treated 24 h
later with 3 µM doxorubicin and 2 µM MK2206 in serum-free medium.
After a 48-h incubation, cells were subjected to cell viability
assays. (E) TE4 cells were infected with Ad-SOX2 or Ad-control,
then treated 24 h later with or without 3 µM doxorubicin in
serum-free medium. Cells were harvested 48 h after doxorubicin
treatment and subjected to immunoblot analysis of PARP, cleaved
PARP, p-GSK-3β and total GSK-3β expression. β-actin was detected as
a loading control. The numbers presented above and below the gel
for PARP and cleaved PARP represent the expression levels of PARP
and cleaved PARP relative to those of β-actin, respectively. The
numbers presented below the gels for p-GSK-3β and total GSK-3β
represent the expression levels of p-GSK-3β and total GSK-3β
relative to those of β-actin, respectively. Values were normalized
so that β-actin expression in each well had a value of 1. The ratio
of p-GSK-3β to total GSK-3β is shown. (F) TE4 cells were infected
with Ad-SOX2, and treated 24 h later with AR-A014418 or DMSO. The
cells were treated 24 h later with or without 3 µM doxorubicin in
serum-free medium. Cell viability was measured 72 h after
doxorubicin treatment. All data are presented as the mean ± SD
(n=3). *P<0.01 analyzed by two-tailed unpaired Student's t-test.
OD, optical density; Ad, adenovirus vector; p-, phosphorylated;
GSK-3β, glycogen synthase kinase-3β; PARP, poly (ADP-ribose)
polymerase. |
SOX2 promotes resistance to apoptosis
through activation of the AKT/GSK-3β pathway
We next investigated the effect of SOX2 on
resistance to apoptosis under serum starvation conditions. TE4
cells were infected with Ad-SOX2 or Ad-control, then treated with
doxorubicin, an anticancer drug, in serum-free medium. A cell
viability assay showed that cells infected with Ad-SOX2 were more
resistant to doxorubicin than those infected with Ad-control
(Fig. 3C), exhibiting 50% inhibitory
concentrations (IC50s) of 5.8 µM and 2.8 µM, respectively. However,
when TE4 cells were infected with Ad-SOX2 or Ad-control and then
treated with doxorubicin combined with MK2206, the difference in
doxorubicin resistance was no longer seen (Fig. 3D). This result suggested the
involvement of AKT in the effect of SOX2 on resistance to
doxorubicin.
Immunoblotting of cleaved PARP, a marker of
apoptosis, showed that TE4 cells infected with Ad-SOX2 were
attenuated for doxorubicin-induced apoptosis compared to
Ad-control-infected cells (Fig. 3E).
We screened among factors known to lie downstream of AKT to
identify those involved in resistance to apoptosis in cells
overexpressing SOX2. We found that the p-GSK-3β levels were
increased in TE4 cells infected with Ad-SOX2 compared to those
infected with Ad-control (Fig. 3E).
Similar results were seen when TE4 cells were treated with
doxorubicin after Ad-SOX2 or Ad-control infection (Fig. 3E). To test the role of GSK-3β, TE4
cells infected with Ad-SOX2 were treated with AR-A014418, an
inhibitor of GSK-3β, or dimethyl sulfoxide (DMSO), and then were
untreated or treated with doxorubicin in serum-free medium.
Although a single treatment with AR-A014418 decreased cell
viability, combination treatment with AR-A014418 and doxorubicin
synergistically reduced cell viability (Fig. 3F). Taken together, these findings
suggested that SOX2 promotes resistance to apoptosis through
activation of the AKT/GSK-3β pathway.
The PTEN/PI3K/PDK1 and mTORC2 pathways
mediate phosphorylation of AKT in SOX2-overexpressing cells
In a further set of experiments, we examined the
role of the PTEN/PI3K/PDK1 and mTORC2 pathways in SOX2-mediated
activation of AKT by testing the effects of inhibitors of
components of these pathways or by knock-down of SOX2 expression.
KYSE140 and TE6 cells were treated with LY294002, an inhibitor of
PI3K, in serum-free medium. The levels of p-AKT (S473) were
decreased in a dose-dependent manner in the presence of LY294002,
as determined by immunoblot analysis (Fig. 4A).
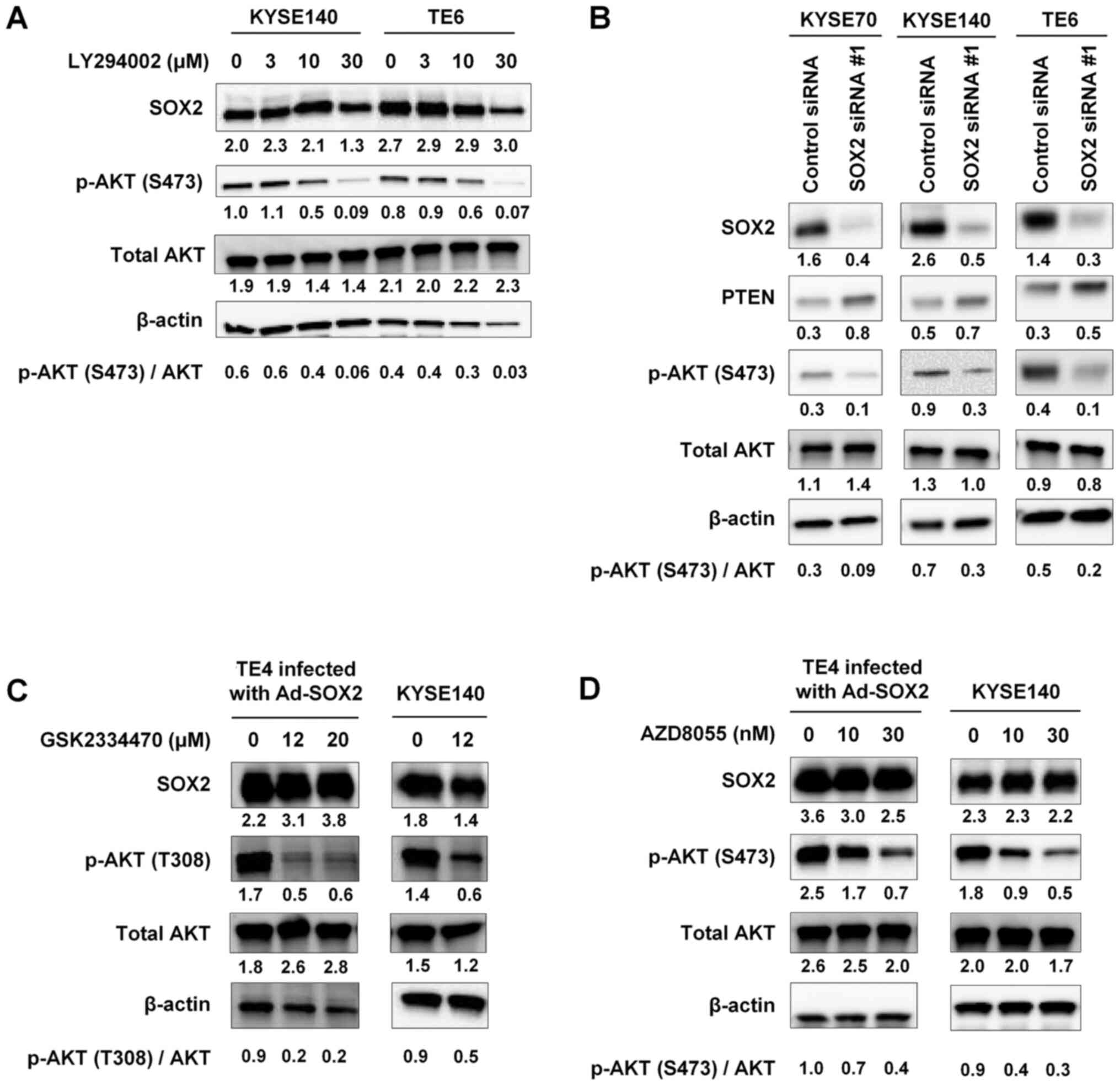 | Figure 4.PTEN/PI3K/PDK1- and mTORC2-mediated
phosphorylation of AKT in SOX2-overexpressing cells. (A) KYSE140
and TE6 cells were treated with the indicated concentrations of
LY294002, a PI3K inhibitor, in serum-free medium for 48 h,
harvested and subjected to immunoblot analysis of SOX2, p-AKT
(S473) and total AKT expression. (B) KYSE70, KYSE140 and TE6 cells
were transfected with either SOX2 siRNA#1 or with a control siRNA.
After a 48-h incubation in serum-free medium, cells were harvested
and subjected to immunoblot analysis of SOX2, PTEN, p-AKT (S473)
and total AKT expression. TE4 cells infected with Ad-SOX2 and
KYSE140 cells were treated for 1 h in serum-free medium containing
the indicated concentrations of (C) GSK2334470, a PDK1 inhibitor,
or (D) AZD8055, an mTORC2 inhibitor. Cells were harvested and
subjected to immunoblot analysis of SOX2, p-AKT (T308 or S473) and
total AKT expression. For all experiments, β-actin was detected as
a loading control. The numbers presented below the gels represent
the expression levels of each protein relative to those of β-actin.
Values were normalized so that β-actin expression in each well had
a value of 1. The ratio of p-AKT (S473 or T308) to total AKT is
shown. PI3K, phosphatidylinositol 3-kinase; Ad, adenovirus vector;
p-, phosphorylated; siRNA, small interfering RNA; PDK1,
phosphoinositide-dependent protein kinase 1; mTORC2, mammalian
target of rapamycin complex 2. |
KYSE70, KYSE140, and TE6 cells were transfected with
SOX2 siRNA #1 or control siRNA and then cultured in serum-free
medium. Immunoblot analysis showed that SOX2 knockdown resulted in
increased expression of PTEN and decreased expression of p-AKT
(S473) (Fig. 4B).
TE4 cells infected with Ad-SOX2 and KYSE140 cells
were treated with GSK2334470, a PDK1 inhibitor, in serum-free
medium. Immunoblot analyses indicated that exposure of these cells
to GSK2334470 diminished SOX2-mediated phosphorylation of AKT's
T308 residue (Fig. 4C).
TE4 cells infected with Ad-SOX2 and KYSE140 cells
growing in serum-free medium were treated with AZD8055, which
inhibits the phosphorylation of the mTORC2 substrate AKT (15). AZD8055 exposure provided
dose-dependent decreases in the levels of p-AKT (S473) in these
cells (Fig. 4D). Considered
together, these findings suggested that SOX2 may activate AKT
through the PTEN/PI3K/PDK1 and mTORC2 pathways under conditions of
serum starvation.
Discussion
In the present study, we used an adenoviral
vector-mediated expression system to demonstrate that SOX2
overexpression results in the phosphorylation and activation of
AKT, but not ERK, in ESCC cells. Although the AKT and MAP kinase
pathways, both of which are downstream of Ras, are major pathways
involved in the growth and proliferation of cancer cells, our
results suggest that SOX2 activates only the AKT pathway in
ESCC.
Previous studies have shown that the SOX2 protein
level is determined by AKT activity in embryonic stem cells (ESCs)
(16), non-small-cell lung cancer
(17), and breast cancer (18). However, our time course assay after
Ad-SOX2 infection showed that increased accumulation of SOX2
precedes the activation of AKT. Moreover, our experiments using the
AKT inhibitor MK2206 suggested that AKT activation does not
increase SOX2 accumulation in ESCC cells. Although the reason for
the discrepancy between those previous results and ours is unclear,
it may reflect the use of different types of cells and experimental
systems.
Our experiments showed that SOX2 overexpression
increased AKT activation under serum starvation conditions, but
this effect was masked by serum stimulation. Furthermore, SOX2's
promotion of cell survival was mediated through AKT activation
under serum starvation conditions, but not in medium containing
serum. These findings indicated that SOX2 facilitates ESCC cell
survival under poor nutrient conditions. Therefore, overexpression
of SOX2 may be beneficial for the survival of cancer cells
suffering from insufficient nutrient supply, as often faced by
solid tumors, and may be involved in the development and
progression of ESCC.
Resistance to apoptosis contributes not only to the
survival of cancer cells, but also to resistance to chemotherapy.
Our experiments indicated that overexpression of SOX2 led to
resistance to doxorubicin-induced apoptosis through AKT activation.
Moreover, our results suggested that GSK-3β, a factor that lies
downstream of AKT, may be involved in resistance to apoptosis in
cells that overexpress SOX2.
GSK-3β was first identified as a negative regulator
of glycogenesis and subsequently was found to regulate various
signaling pathways, including the Wnt/β-catenin pathway (19). GSK-3β phosphorylates multiple
substrates, including β-catenin, cyclin D1, MYC, BAX
(Bcl-2-associated X protein), and nuclear factor-kappa B (NF-κB),
and induces the degradation of the substrates or inhibition of
their enzymatic activities. GSK-3β is directly phosphorylated by
AKT and inhibitory phosphorylation of GSK-3β has numerous cellular
functions such as promoting glycogen metabolism, cell cycle
progression, and cell survival. In the context of cancer treatment,
GSK-3β inhibition has been studied as a possible therapeutic
strategy (20). Our results showed
the combinatorial effect of doxorubicin and AR-A014418, an
inhibitor of GSK-3β, in reducing cell viability in ESCC cells that
overexpress SOX2. Therefore, AKT and GSK-3β may be appropriate and
important targets for the development of novel therapies for
SOX2-overexpressing ESCC.
Finally, we explored the mechanism whereby SOX2
phosphorylates AKT under serum starvation conditions. Our findings
suggested that both the PTEN/PI3K/PDK1 and mTORC2 pathways may be
involved in the activation of AKT by SOX2.
Certain limitations should be considered in the
interpretation of our findings. First, although SOX2 activates the
AKT pathway, the direct target of SOX2 remains unclear. Second,
downstream factors of AKT other than GSK-3β may be involved in
apoptosis resistance in ESCC cells that overexpress SOX2. Third,
results of the present study need to be confirmed by further in
vivo studies.
In conclusion, our results showed that SOX2 promotes
cell survival and enhances resistance to apoptosis under serum
starvation conditions in ESCC cells; these effects are mediated by
activation of the AKT/GSK-3β signaling pathway. SOX2 may
phosphorylate AKT though the PTEN/PI3K/PDK1 and mTORC2 pathways
under serum starvation conditions. We postulate that
molecular-targeted therapy against the SOX2/AKT/GSK-3β pathway may
be effective against ESCC cells that overexpress SOX2.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KT, KoY and YG contributed to the study conception,
data collection and data interpretation. KT contributed to
statistical analysis and manuscript preparation. KoY contributed to
manuscript preparation and finalization. NI, TS, TK, OD, HT, YS,
AU, TN, KaY, MM, HK, YN and YI contributed to acquisition of data,
or analysis and interpretation of data. KT and KoY confirmed the
authenticity of the data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Colombet M, Soerjomataram I,
Mathers C, Parkin DM, Piñeros M, Znaor A and Bray F: Estimating the
global cancer incidence and mortality in 2018: GLOBOCAN sources and
methods. Int J Cancer. 144:1941–1953. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rustgi AK and El-Serag HB: Esophageal
carcinoma. N Engl J Med. 371:2499–2509. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pevny LH and Lovell-Badge R: Sox genes
find their feet. Curr Opin Genet Dev. 7:338–344. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Masui S, Nakatake Y, Toyooka Y, Shimosato
D, Yagi R, Takahashi K, Okochi H, Okuda A, Matoba R, Sharov AA, et
al: Pluripotency governed by Sox2 via regulation of Oct3/4
expression in mouse embryonic stem cells. Nat Cell Biol. 9:625–635.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Que J, Okubo T, Goldenring JR, Nam KT,
Kurotani R, Morrisey EE, Taranova O, Pevny LH and Hogan BL:
Multiple dose-dependent roles for Sox2 in the patterning and
differentiation of anterior foregut endoderm. Development.
134:2521–2531. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gen Y, Yasui K, Zen Y, Zen K, Dohi O, Endo
M, Tsuji K, Wakabayashi N, Itoh Y, Naito Y, et al: SOX2 identified
as a target gene for the amplification at 3q26 that is frequently
detected in esophageal squamous cell carcinoma. Cancer Genet
Cytogenet. 202:82–93. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bass AJ, Watanabe H, Mermel CH, Yu S,
Perner S, Verhaak RG, Kim SY, Wardwell L, Tamayo P, Gat-Viks I, et
al: SOX2 is an amplified lineage-survival oncogene in lung and
esophageal squamous cell carcinomas. Nat Genet. 41:1238–1242. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hussenet T, Dali S, Exinger J, Monga B,
Jost B, Dembelé D, Martinet N, Thibault C, Huelsken J, Brambilla E,
et al: SOX2 is an oncogene activated by recurrent 3q26.3
amplifications in human lung squamous cell carcinomas. PLoS One.
5:e89602010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Maier S, Wilbertz T, Braun M, Scheble V,
Reischl M, Mikut R, Menon R, Nikolov P, Petersen K, Beschorner C,
et al: SOX2 amplification is a common event in squamous cell
carcinomas of different organ sites. Hum Pathol. 42:1078–1088.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gen Y, Yasui K, Nishikawa T and Yoshikawa
T: SOX2 promotes tumor growth of esophageal squamous cell carcinoma
through the AKT/mammalian target of rapamycin complex 1 signaling
pathway. Cancer Sci. 104:810–816. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Engelman JA, Luo J and Cantley LC: The
evolution of phosphatidylinositol 3-kinases as regulators of growth
and metabolism. Nat Rev Genet. 7:606–619. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang G, Murashige DS, Humphrey SJ and
James DE: A Positive Feedback Loop between Akt and mTORC2 via SIN1
Phosphorylation. Cell Rep. 12:937–943. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zen K, Yasui K, Nakajima T, Zen Y, Zen K,
Gen Y, Mitsuyoshi H, Minami M, Mitsufuji S, Tanaka S, et al: ERK5
is a target for gene amplification at 17p11 and promotes cell
growth in hepatocellular carcinoma by regulating mitotic entry.
Genes Chromosomes Cancer. 48:109–120. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chresta CM, Davies BR, Hickson I, Harding
T, Cosulich S, Critchlow SE, Vincent JP, Ellston R, Jones D, Sini
P, et al: AZD8055 is a potent, selective, and orally bioavailable
ATP-competitive mammalian target of rapamycin kinase inhibitor with
in vitro and in vivo antitumor activity. Cancer Res. 70:288–298.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jeong HC, Park SJ, Choi JJ, Go YH, Hong
SK, Kwon OS, Shin JG, Kim RK, Lee MO, Lee SJ, et al: PRMT8 Controls
the Pluripotency and Mesodermal Fate of Human Embryonic Stem Cells
By Enhancing the PI3K/AKT/SOX2 Axis. Stem Cells. 35:2037–2049.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Singh S, Trevino J, Bora-Singhal N,
Coppola D, Haura E, Altiok S and Chellappan SP: EGFR/Src/Akt
signaling modulates Sox2 expression and self-renewal of stem-like
side-population cells in non-small cell lung cancer. Mol Cancer.
11:732012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schaefer T, Wang H, Mir P, Konantz M,
Pereboom TC, Paczulla AM, Merz B, Fehm T, Perner S, Rothfuss OC, et
al: Molecular and functional interactions between AKT and SOX2 in
breast carcinoma. Oncotarget. 6:43540–43556. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Maurer U, Preiss F, Brauns-Schubert P,
Schlicher L and Charvet C: GSK-3 - at the crossroads of cell death
and survival. J Cell Sci. 127:1369–1378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Walz A, Ugolkov A, Chandra S, Kozikowski
A, Carneiro BA, O'Halloran TV, Giles FJ, Billadeau DD and Mazar AP:
Molecular Pathways: Revisiting Glycogen Synthase Kinase-3β as a
Target for the Treatment of Cancer. Clin Cancer Res. 23:1891–1897.
2017. View Article : Google Scholar : PubMed/NCBI
|














