Introduction
Epstein-Barr virus (EBV), a large double-stranded
DNA virus, was discovered in 1964 from African Burkitt's lymphoma
(BL) cells (1). Like all
herpesviruses, EBV can infect cells in either their latent or lytic
forms. As a ubiquitous human virus, EBV causes infectious
mononucleosis and persists in the individual for life; however, it
is normally well controlled by the immune system (2). EBV is also associated with different
types of human cancer of both B-cell and epithelial origin,
including BL, nasopharyngeal carcinoma, lymphoproliferative disease
and gastric cancer (2,3). In addition, increasing evidence has
indicated that EBV reactivation occurs in a subset of individuals
with autoimmune diseases, including multiple sclerosis and
rheumatoid arthritis (4,5). It has been demonstrated that EBV is
reactivated under psychological stress, and chronic EBV
reactivation is an important mechanism in the pathogenesis of these
diseases (4).
Telomeres are located at the ends of chromosomes
composed of tandemly repeated G-rich DNA sequences, and they
predominantly function to maintain chromosomal integrity and genome
stability (6). The progression
through replication cycles leads to telomere-dependent pathways of
senescence and mortality (7).
Telomerase, a complex ribonucleoprotein, resolves this problem by
adding TTAGGG repeats to the ends of the chromosomes to promote the
capping of eukaryotic telomere ends (8). Human telomerase reverse transcriptase
(hTERT), the catalytic subunit of telomerase, plays an important
role in the rate-limiting step of activating telomerase (9). Transcriptional regulation is likely to
be the main mechanism for regulating hTERT expression (9). hTERT exhibits little or no expression
in normal somatic cells; however, it is abundantly present in stem
and germ cells (10). Dysregulation
of hTERT results in abnormal activation in ~85% of human cancers,
including cholangiocarcinoma, breast cancer and gastric cancer
(11). Notably, cancer cells have
acquired the ability to overcome and enhance tumor growth by
maintaining the length and activity of telomere, which makes hTERT
an attractive cancer biomarker in clinical practice (12). Recently, novel hTERT-based therapies
have been developed, such as immunotherapy and small molecule
interfering therapy (13,14). Thus, hTERT has been selected as a
target of small molecular drugs in human cancer (13).
Triptolide (TP), a diterpenoid triepoxide, is
extracted from the root of the Chinese herb Tripterygium
wilfordii (15). TP possesses a
broad-spectrum of bioactivity and cytotoxic functions, including
antitumor, anti-inflammatory, anti-fertility and immunosuppressive
properties (16,17). TP has been demonstrated to exert a
potent anticancer effect that involves multiple signaling pathways,
including the NF-κB signaling pathway, and targets in different
types of cancer, including pancreatic cancer, ovarian cancer,
cholangiocarcinoma and non-small cell lung carcinoma (18). TP also suppresses the growth of
breast cancer by decreasing HMGB1 expression (19). In addition, inhibiting Pol III
transcription with TP provides a new method for developing novel
treatments for colorectal cancer (20). TP inhibits the proliferation of
prostate cancer cells by decreasing SUMO-Specific Protease 1
expression (21). TP has
demonstrated prominent efficacy as an antitumor and
anti-inflammatory inhibitor compared with aclacinomycin, and has
entered clinical trials for the treatment of diabetic nephropathy
and nephritic syndrome (16).
Furthermore, good tolerance to a water-soluble pre-drug of TP has
been observed in patients with pancreatic cancer (18).
Despite several investigations, the effect of TP on
herpesvirus-related malignancies remains unclear (20,21). A
previous study demonstrated that TP inhibits the proliferation of
EBV-positive B lymphocytes by downregulating the viral protein,
LMP1 (22). The results of the
present study demonstrated that TP inhibited hTERT transcription
and translation in EBV-positive B lymphocytes. In addition,
inhibition of hTERT decreased the proliferation of EBV-positive B
lymphocytes. Mechanistically, TP inhibits hTERT transcription by
downregulating the transcription factors specificityprotein 1 (SP1)
and c-Myc, which bind to the promoter domain.
Materials and methods
Cell lines and reagents
B95-8 and P3HR-1 EBV-positive B lymphoma cell lines
were kindly provided by Professor Y. Cao at Central South
University (Changsha, China). 293T cells were purchased from the
American Type Culture Collection. EBV cells were maintained in
RPMI-1640 (HyClone; Cytiva) supplemented with 10% fetal bovine
serum (FBS; Thermo Fisher Scientific, Inc.), while 293T cells were
maintained in DMEM (HyClone; Cytiva) supplemented with 10% FBS at
37°C with 5% CO2. All cell lines were authenticated via
short tandem repeat analysis and cultured for 5 months following
resuscitation. TP, cycloheximide (both purchased from Merck KGaA),
and cidofovir (CDV; Selleck Chemicals) were dissolved in DMSO.
Plasmids
The pGL3.0 luciferase reporter vector was purchased
from Promega Corporation. Renilla plasmid was kindly
provided by Professor DeYin Guo from Wuhan University (Wuhan,
China). The hTERT promoter regions that span from −1126, −792, −461
or −277 to −47 were amplified from the BCBL-1 genome and
subsequently inserted into pGL3.0 (23). All constructs were purified using the
Plasmid Miniprep kit (cat. no. AP-MN-P-250; Corning Inc.),
according to the manufacturer's instructions.
Cell transfection
A total of 4×105 cells/well were placed
in 6-well plates. B95-8 and P3HR-1 cells were transiently
transfected with 0.5 µg/ml hTERT shRNA
(5′-GAACTTCCCTGTAGAAGACGA-3′) or 0.5 µg/ml non-targeting control
shRNA (5′-TACAACAGCCACAACGTCTAT-3′) (both Shanghai GenePharma, Co.,
Ltd.) using X-tremeGENE HP DNA Transfection Reagent (Roche
Diagnostics), according to the manufacturer's protocol. All cells
were maintained in RPMI-1640 medium supplemented with 10% FBS at
37°C for 48 h. At 48 h post-transfection, cells were collected for
subsequent experiments.
Dual-luciferase reporter assay
Dual-luciferase reporter assay was performed as
described previously (23). Briefly,
each luciferase reporter construct or pGL3.0 (0.5 µg) and pRL-TK
(0.05 µg) (kindly provided by Professor D. Guo of Wuhan University)
was co-transfected into 293T cells in triplicates, using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.) for
24 h at 37°C. Cells were incubated at 37°C with TP (200 nM) or
control (DMSO) for a further 24 h. Following incubation, luciferase
activities were detected using the Dual-Luciferase Assay kit (cat.
no. E1960; Promega Corporation), according to the manufacturer's
instructions. The activity of firefly luciferase was normalized to
Renilla luciferase control values and is shown as an average
of triplicates.
Immunoblotting
Immunoblotting was performed as previously described
(24). Briefly, B95-8 and P3HR-1
cells were lysed using RIPA buffer (Beyotime Institute of
Biotechnology) supplemented with 0.5% proteasome inhibitor cocktail
(Roche Diagnostics). Protein concentrations were determined using
the Bradford Protein Assay kit (cat. no. 5000201; Bio-Rad
Laboratories, Inc.) and 30–50 µg protein/lane was separated via 15%
SDS-PAGE. The separated proteins were subsequently transferred onto
PVDF membranes (Bio-Rad Laboratories, Inc.) and blocked with 5%
fat-free milk for 1 h at room temperature. The membranes were
incubated with primary antibodies against GAPDH (1:10,000; cat. no.
10494-1-AP; ProteinTech Group, Inc.), SP1 (1:1,000; cat. no. 9389;
Cell Signaling Technology, Inc.) and hTERT (1:1,000; cat. no.
sc-7212; Santa Cruz Biotechnology, Inc.) overnight at 4°C.
Following the primary incubation, membranes were incubated with
HRP-conjugated anti-rabbit IgG secondary antibodies (1:10,000; cat.
no. sc-2357; Santa Cruz Biotechnology, Inc.) for 1 h at room
temperature, and protein bands were detected using SuperSignal
Chemiluminescent Substrate (Bio-Rad Laboratories, Inc.).
Cell viability assay
B95-8 and P3HR-1 cells transfected with hTERT shRNA
were seeded at a density of 20,000 cells/well into 96-well plates
and maintained in RPMI-1640 media supplemented with 10% FBS at 37°C
for 24 h. Cells were subsequently treated with TP (200 nM) at 37°C
for 24 h. Cell viability was assessed via the Cell Counting Kit-8
(CCK-8; Dojindo Molecular Technologies, Inc.) assay by incubating
cells with the CCK-8 reagent for 30 min in the dark at 37°C,
according to the manufacturer's instructions. The optical density
was detected at a wavelength of 450 nm using a microplate reader
(BioTek China).
Analysis of hTERT degradation
B95-8 and P3HR-1 cells were treated with TP (0 or
200 nM) for 24 h, and Cycloheximide (CHX; 25 mg/ml; cat. no. C7698;
Sigma-Aldrich; Merck KGaA) for a further 12 h at 37°C before
harvest. hTERT protein expression was detected via immunoblotting,
as aforementioned. GAPDH was used as the loading control.
In silico analysis
To identify potential transcription factors
regulating hTERT expression, in silico analysis was
performed using the University of California Santa Cruz (UCSC)
genome browser gateway (http://genome.ucsc.edu/) for the human genomic
information of GRCh38/hg38. Potential transcription factors and
binding regions were obtained in the ChIP-seq clusters.
Progeny virus analysis
12-O-Tetradecanoylphorbol-13-Acetate (TPA; 20 ng/ml)
and sodium butyrate (NaB; 1.2 mM) were used to induce B95-8 and
P3HR-1 cells entering lytic replication. After 3 h of induction at
37°C, the cell medium was replaced and cells were cultured in the
absence or presence of TP (0, 100 or 200 nM) and CDV (20 µM) for
another 24 h at 37°C. EBV virion-associated DNA derived from the
B95-8 and P3HR-1 cell supernatants was collected as previously
described (25). Equal amounts of
viral lysate were used for reverse transcription-quantitative PCR
(RT-qPCR) of the EBV specific region, EBNA1. Intracellular viral
DNA of B95-8 and P3HR-1 cells was extracted and evaluated by
RT-qPCR analysis.
RT-qPCR
Total cellular RNA was extracted from B95-8 and
P3HR-1 cells using TRIzol® reagent (Thermo Fisher
Scientific, Inc.). RT-qPCR was performed as previously described
(25). Briefly, total RNA was
reverse transcribed into cDNA using the Reverse Transcription kit
(cat. no. 639505; Takara Bio, Inc.) according to the manufacturer's
instructions, and quantified using the CFX 96 Real-Time PCR
Detection System (Bio-Rad Laboratories, Inc.). The primer sequences
used for qPCR were as previously described (23). Relative mRNA expression levels were
calculated using the 2−ΔΔCq method (26) and normalized to the internal
reference gene GAPDH.
Statistical analysis
All experiments were repeated for three independent
cultures. Statistical analysis was performed using GraphPad Prism 5
(GraphPad Software, Inc.). Unpaired two-tailed Student's t-test was
used to compare differences between two groups, while two-way ANOVA
and Tukey's post hoc test were used to compare differences between
multiple groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
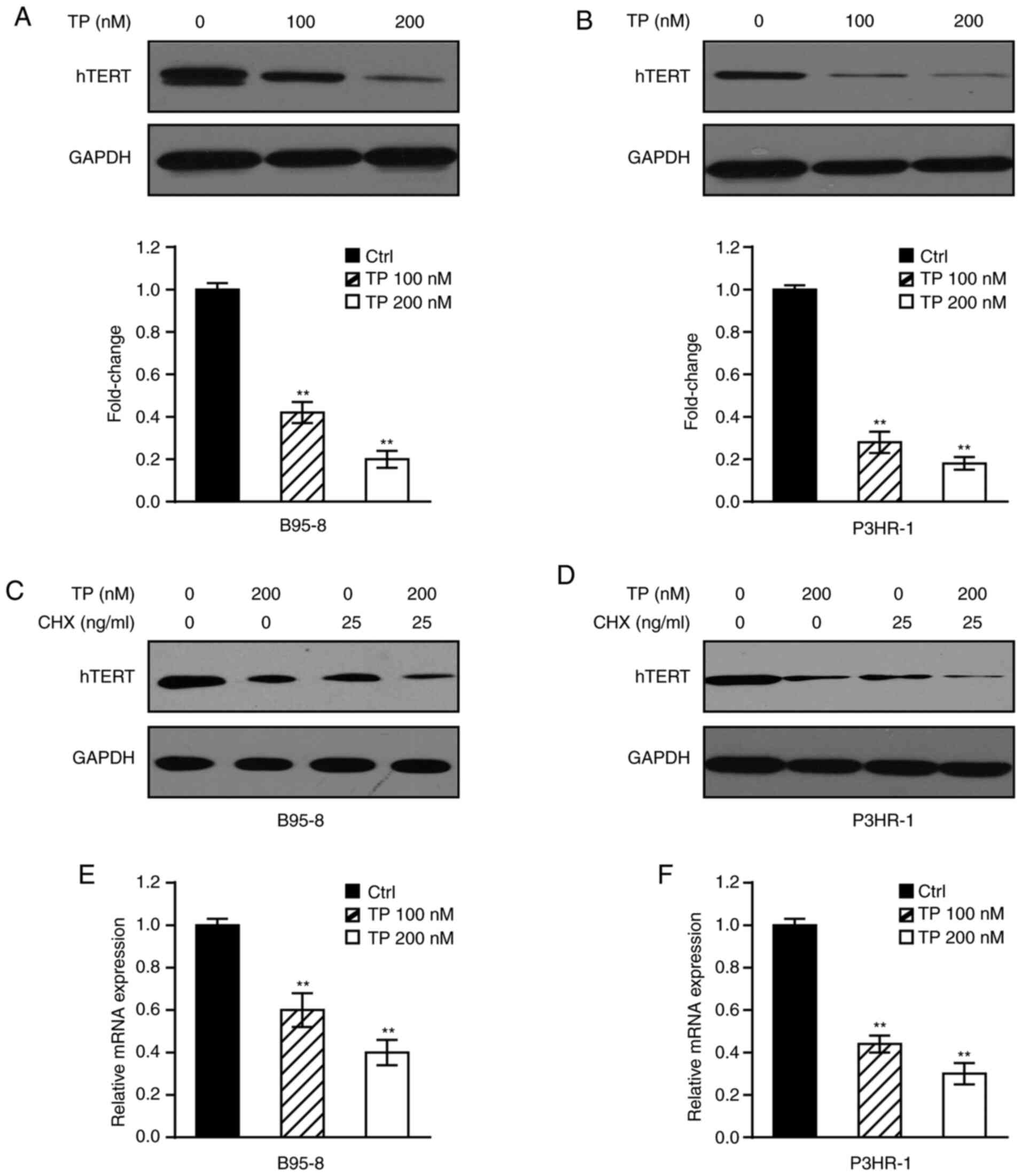
TP decreases hTERT transcription and
protein expression in EBV-positive B lymphoma cells
TP has previously been demonstrated to decrease
hTERT expression in primary effusion lymphoma (PEL) cells (23). To determine whether TP plays a
similar role in EBV-positive B lymphoma cells, immunoblotting was
performed in B95-8 and P3HR-1 cells treated with TP in a
dose-dependent manner. The results demonstrated that TP
significantly downregulated hTERT expression in B95-8 (Fig. 1A) and P3HR-1 (Fig. 1B) cells. Furthermore, B95-8 and
P3HR-1 cells were treated with TP in the presence or absence of CHX
to determine whether TP inhibits hTERT expression through hTERT
half-time. As presented in Fig. 1C and
D, the stability of hTERT decreased in both B95-8 and P3HR-1
cells in the presence of TP. These results indicated that TP mainly
attenuated hTERT protein expression, and partly the level of
protein stability. Mechanistically, RT-qPCR analysis was performed
in B95-8 and P3HR-1 cells treated with TP in a dose-dependent
manner to detect whether decreased hTERT expression affected mRNA
levels. As presented in Fig. 1E and
F, treatment with TP significantly decreased hTERT mRNA levels
in both B95-8 and P3HR-1 cells, particularly following the
administration of 200 nM TP. Taken together, these results suggest
that TP significantly inhibits mRNA and protein expression of hTERT
in EBV-positive B lymphoma cells.
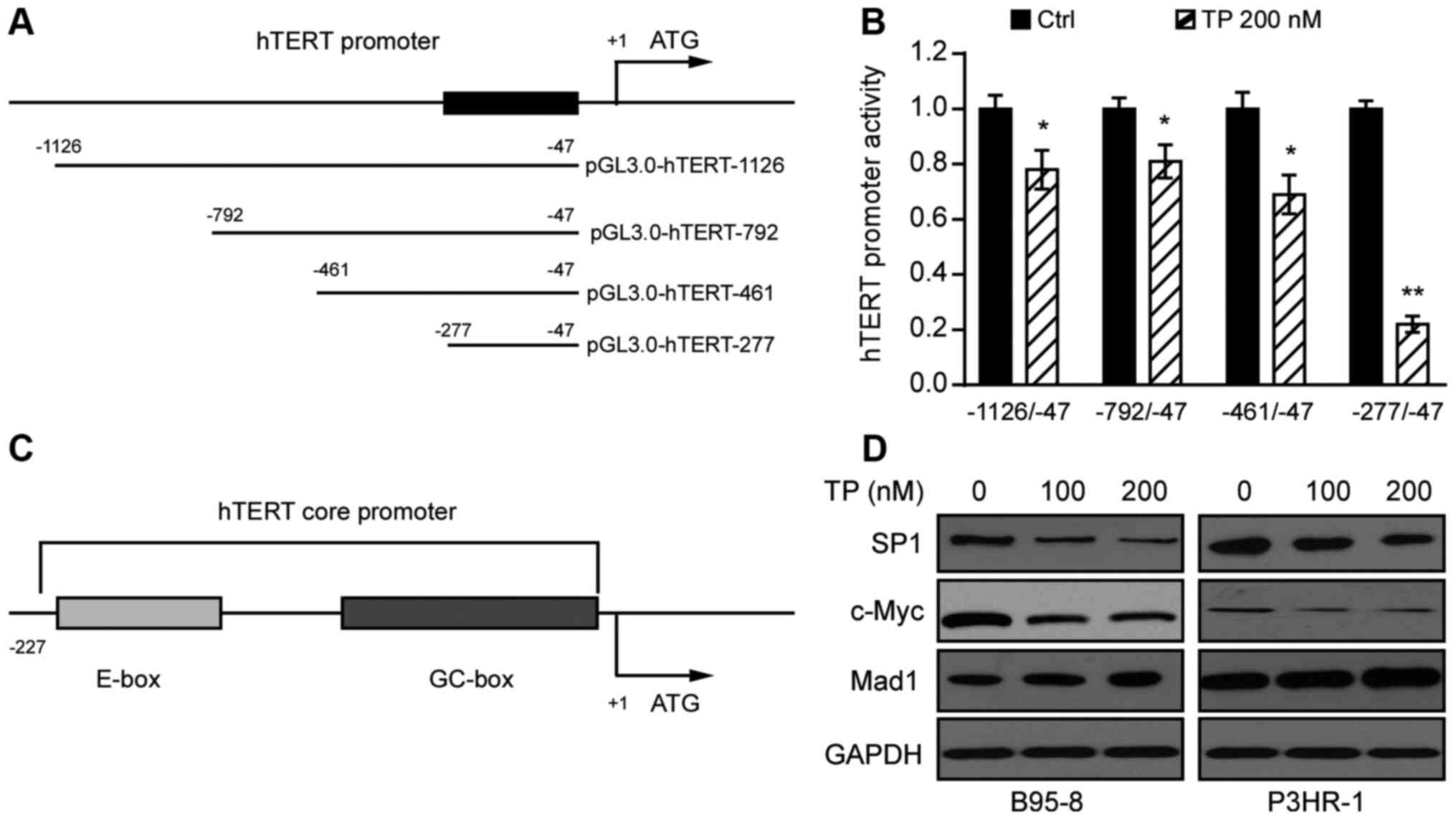
TP inhibits the promoter activity of
hTERT
Dysregulation of hTERT occurs in ~85% of human
cancers, with transcriptional regulation likely to be the major
mechanism regulating hTERT expression (11). To determine whether TP affects the
activity of the hTERT promoter, serial truncations of hTERT
promoter domains were inserted into the pGL3.0 vector (Fig. 2A). These constructs were subsequently
transfected into 293T cells with Renilla, followed by
treatment with TP 24 h post-transfection. The promoter activities
were determined using the Dual-GLU system after 24 h. As presented
in Fig. 2B, treatment with TP
significantly decreased the luciferase activities of hTERT
promoters, −1126/-47, −792/-47 and −461/-47, and particularly that
of the hTERT promoter −277/-47. Collectively, these results suggest
that the promoter domain, −277/-47 is likely to be the region most
responsive to TP.
TP decreases expression of the
transcription factors, SP1 and c-Myc in EBV-positive B lymphoma
cells
Transcription factor SP1, a member of the zinc
finger family, is considered a basal transcription factor that
regulates housekeeping genes (27).
A previous study demonstrated that the hTERT promoter sequence
contains several SP1 binding motifs (GC boxes), and plays an
important role in upregulating Kaposi's sarcoma-associated
herpesvirus-encoded latency-associated nuclear antigen (28). In addition, the super-transcription
factor c-Myc potentially regulates the transcription of at least
15% of the entire genome (29).
Thus, in silico analysis was performed in the UCSC genome
browser gateway for the human genomic information of GRCh38/hg38,
and the results demonstrated that the hTERT promoter sequence
contains the SP1 and c-Myc binding domains (Fig. 2C). To identify the effect of TP on
SP1, c-Myc, and Mad1, B95-8 and P3HR-1 cells were treated with TP
(0, 100 or 200 nM) for 24 h. As presented in Fig. 2D, treatment of B95-8 and P3HR-1 cells
with TP in a dose-dependent manner slightly enhanced Mad1
expression, and notably decreased SP1 and c-Myc expression levels.
Taken together, these results suggest that TP downregulates SP1 and
c-Myc expression in B95-8 and P3HR-1 cells.
TP increases lytic and latent
replication of EBV in EBV-positive B lymphoma cells
To determine the effect of TP on the antiviral
activity, B95-8 and P3HR-1 cells uninduced or induced by TPA (20
ng/ml) and NaB (1.2 mM) for 3 h were treated with TP (0, 100 or 200
nM). After 24 h, supernatants, cell lysates and genomic DNA were
prepared and incubated for a further 48 h. RT-qPCR analysis was
performed to detect the lytic and latent replication of EBV. As
presented in Fig. 3A and B,
treatment with TP increased the number of viral genome copies in
B95-8 and P3HR-1 cells. Production of progeny virion extracted from
the B95-8 and P3HR-1 cell supernatants was calculated according to
the standard curve derived from the EBNA1 construct (Fig. 3C). Results indicated that the
production of progeny virion was increased after TP treatment in
B95-8 and P3HR-1 cell supernatants (Fig.
3D). CDV, a positive control, efficiently decreased the lytic
replication of EBV, as previously described (30). Similar to the effect of TP on
intracellular production of progeny virion, extracellular
production of progeny virion was also increased in induced B95-8
and P3HR-1 cells (Fig. 3D). On the
contrary, CDV decreased intracellular and extracellular production
of progeny virion in induced B95-8 and P3HR-1 cells.
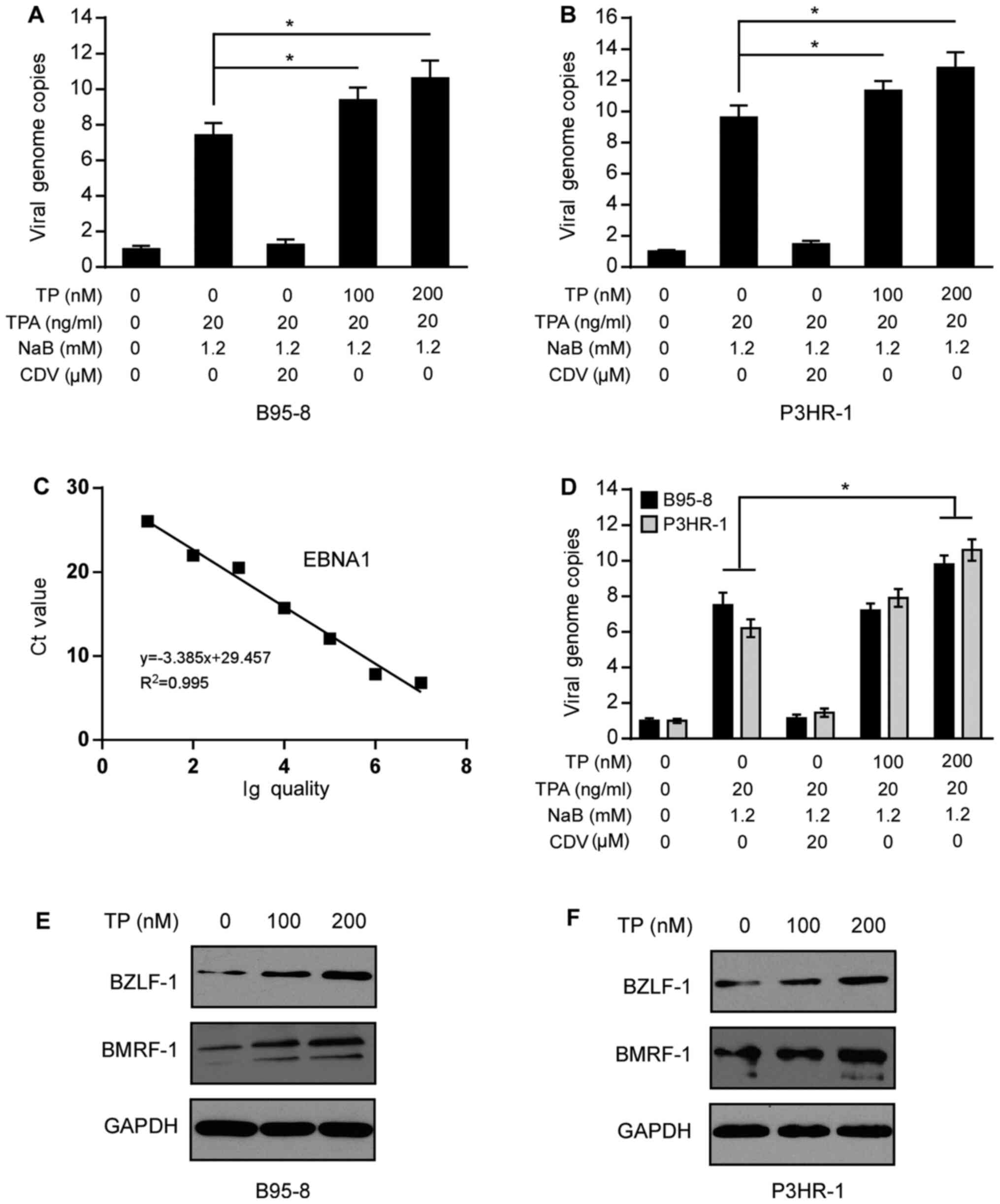 | Figure 3.TP promotes the virus production and
lysis of EBV in B95-8 and P3HR-1 cells. Uninduced and induced (A)
B95-8 and (B) P3HR-1 cells were treated with TP for 24 h, and cell
supernatants, genomic DNA and lysates were prepared. The copy
numbers of the intracellular viral DNA were detected via RT-qPCR
analysis and normalized to the internal reference gene GAPDH. (C)
Standard curve for virions quantification used to calculate the
number of EBV virions in the supernatants of B95-8 and P3HR-1
cells. (D) Virion production extracted from equivalent B95-8 and
P3HR-1 cell supernatants was determined by RT-qPCR, and the viral
copy numbers were calculated using the standard curve derived from
the EBNA1 construct. Immunoblotting was performed to detect BZLF-1
and BMRF-1 protein expression following treatment of (E) B95-8 and
(F) P3HR-1 cells with TP (0, 100 or 200 nM) for 24 h. GAPDH was
used as the loading control. *P<0.05. TP, triptolide; EBV,
Epstein-Barr virus; RT-qPCR, reverse transcription-quantitative
PCR; TPA, 12-O-Tetradecanoylphorbol-13-Acetate; NaB, sodium
butyrate; CDV, cidofovir. |
The transition from the latent to the lytic state is
triggered by two viral transcription factors, BZLF-1 (also known as
ZEBRA, Zta or EB1) and BMRF-1 (also known as Rta or R) (31). The promoters regulating BZLF-1 and
BMRF-1 expression are tightly repressed during latency (30). It was speculated that TP promotes the
entry of EBVs into the lytic period. Thus, B95-8 and P3HR-1 cells
were treated with TP (0, 100 or 200 nM) for 24 h and harvested for
immunoblotting analysis. As presented in Fig. 3E and F, treatment with TP markedly
increased BZLF-1 and BMRF-1 expression in B95-8 and P3HR-1 cells.
Collectively, these results suggest that TP promotes the lytic
state, and increases the production of EBV progeny virions in
EBV-positive B lymphoma cells.
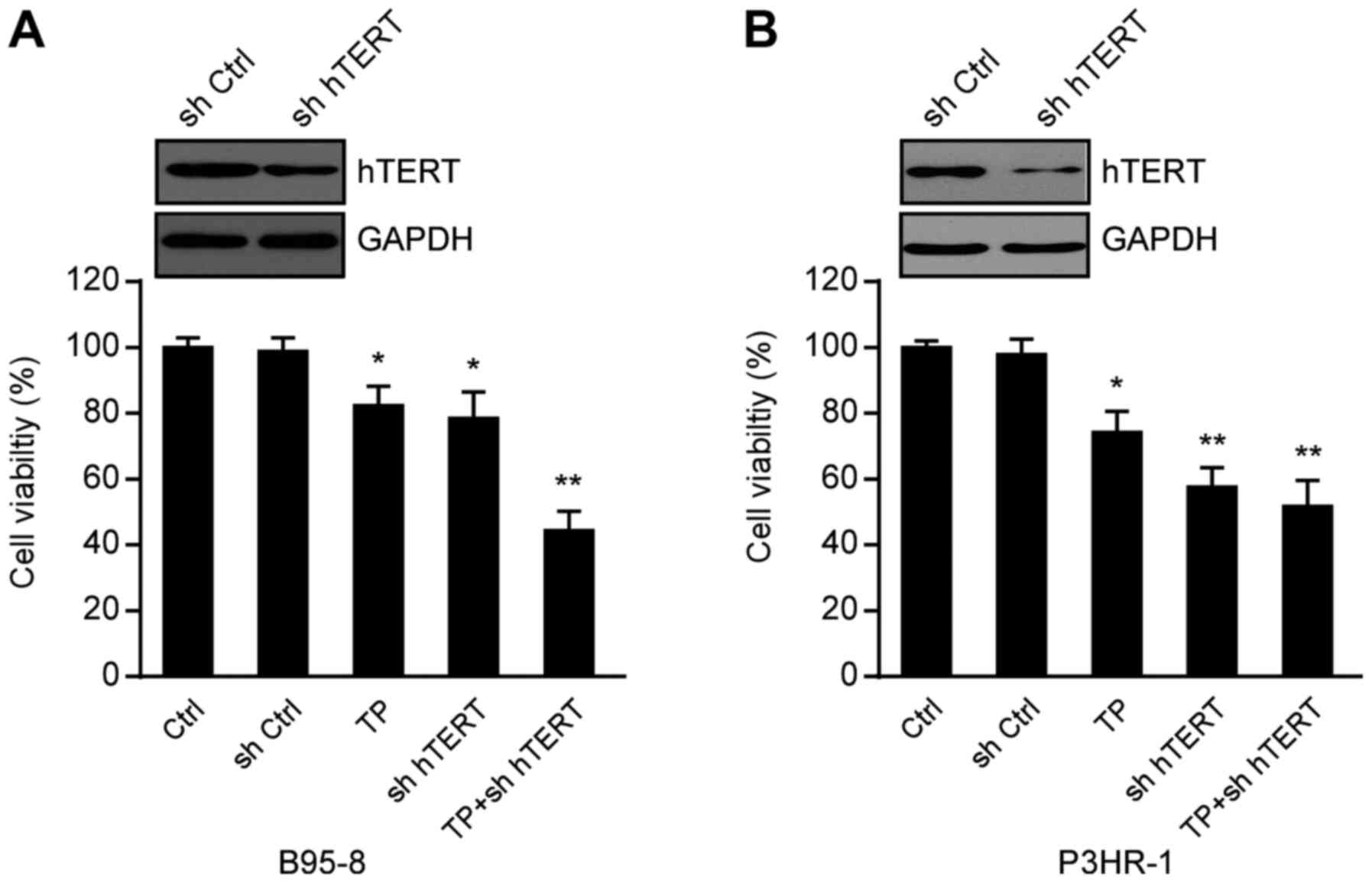
Inhibition of hTERT enhances the
suppression of TP on the proliferation of EBV-positive B lymphoma
cells
To determine the effect of hTERT inhibition and TP
on cell viability, B95-8 and P3HR-1 cells transfected with or
without hTERT shRNA and control shRNA were treated with TP (0 and
200 nM). After 24 h, cell viability was assessed via the CCK-8
assay. As presented in Fig. 4A and
B, both TP- and hTERT-knockdown inhibited B95-8 and P3HR-1 cell
viability, respectively. In addition, the combination of TP- and
hTERT-knockdown synergistically decreased cell viability. Taken
together, these results suggest that TP- and hTERT-knockdown
inhibit cell proliferation, particularly when combined.
Discussion
Telomerase reactivation is a critical hallmark of
>90% of cancers (12). However,
the molecular mechanism regulating hTERT expression in tumors
remains elusive. Thus, understanding how hTERT is activated and how
it contributes to tumor continues to be a major research focus. The
present study used EBV-positive B lymphoma cells as a model system,
and the results confirmed that TP prominently inhibited hTERT at
the transcriptional and translational levels in vitro.
Mechanistically, the present study suggested that TP decreased the
expression levels of the transcription factors, SP1 and c-Myc in
EBV-positive B lymphoma cells. Notably, the results of the present
study demonstrated that TP increased the lytic and latent
replication of EBV in B95-8 and P3HR-1 cells, suggesting that TP
may be effective as an hTERT-selective cancer therapeutic.
hTERT expression is typically detectable in
cancerous tissues and immortalized cells (13). KSHV-encoded LANA1 has been
demonstrated to enhance the transactivation of hTERT promoter by
interacting with SP1 (28). A
previous study demonstrated that TP decreases LANA1 expression in
KSHV- and herpesvirus-related PEL cells (25). Another study reported that TP
inhibits the transcription of hTERT by downregulating SP1 in PEL
cells (23). The results of the
present study demonstrated that hTERT knockdown inhibited cell
viability, particularly in combination with TP, which expanded the
therapeutic tumor types. Furthermore, the results of the promoter
truncation assay indicated that TP decreased the luciferase
activity of a serial truncation hTERT promoter construct. This
mechanism suggested that the hTERT promoter activity inhibited by
TP led to a reduction in the transcription levels of hTERT,
particularly in the −277/-47 promoter domain.
The −277/-47 promoter domain of hTERT contains five
GC boxes, which are binding sites for zinc finger transcription
factor SP1 (32). As a basal
transcription factor, SP1 recognizes GC boxes and regulates a
series of signaling pathways by interacting with other
transcriptional activators (27).
Thus, SP1 binds to the GC boxes and functions synergistically to
maintain the activity of hTERT. In the present study, the
pharmacological inhibition of SP1 by TP decreased hTERT expression.
In addition, as a super-transcription factor, Myc potentially
regulates the transcription of at least 15% of the entire genome,
and is deregulated in >50% of human cancers (29,33).
Previous studies have demonstrated that c-Myc acts as a key
regulator of hTERT during tumorigenesis by binding to the E-box
(11,34). In the present study, TP significantly
deceased c-Myc protein expression, partially accounting for the
attenuation of hTERT activity in TP treatment. It has been reported
that hTERT activity and expression is associated with the
Myc/Mad/Max network in colon cancer cells (35). Conversely, in the present study the
expression of Mad1, a transcriptional repressor of hTERT activity,
was not affected by treatment with TP in EBV-positive B lymphoma
cells. The hTERT core promoter is highly specific to cancer cells,
and there may be other transcription factors that are affected by
TP, leading to the inhibition of hTERT expression.
EBV has two modes of infection, latent and lytic
(36). EBV, with a predominantly
latent form, infects almost all humans during their lifetime, and
following the lytic phase, persists for the remainder of the
individuals life (37). The
transition from the latent to the lytic state is regulated by the
expression of two transcription factors, BZLF-1 and BRLF-1
(31,38). BZLF-1 is the master regulator of the
EBV lytic cycle, whereby overexpression of BZLF-1 in latent cells
can initiate the lytic cycle and drive it to completion (39). In addition, the primary role of
BZLF-1 is to stimulate the BRLF-1 promoter during lytic
reactivation (40). Given that most
cells in EBV-associated tumors harbor latent virus, several studies
have attempted to reactivate the lytic cycle for realizing
oncolytic treatment repertoire (41,42).
Thus, BZLF-1 and BRLF-1 were selected and analyzed in the present
study. TP inhibition of interferon production sensitizes prostate
cancer PC3 cells to vesicular stomatitis virus replication and
virus-mediated apoptosis (43). The
results of the present study demonstrated that TP increases latent
replication and promotes the lytic cycle of EBV in EBV-positive B
lymphoma cells, improving the number of lytic reactivated cells and
inhibiting cell viability. Overall, the results of the present
study suggest that TP-based therapeutics may be used to treat
EBV-positive B lymphoma. It was demonstrated that TP inhibited
hTERT by downregulating the transcription factors SP1 and c-Myc in
EBV-positive B lymphocytes. The toxicity of TP in vivo and
other mechanisms induced by TP treatment should be further
investigated in EBV-positive B lymphocytes in future studies.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural
Science Foundation of Hubei Province (grant no. 2017CFB738 awarded
to CL).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FG and YJ conceived and designed the present study.
CL and FG supervised the present study. CL and YJ drafted the
initial manuscript. CL, QBX and LD performed most of the
experiments. CL, QBX, LY, WJ, FG and YJ analyzed the data. LY and
WJ provided technical assistance for immunoblotting experiments.
All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Epstein MA, Achong BG and Barr YM: Virus
particles in cultured lymphoblasts from burkitt's lymphoma. Lancet.
1:702–703. 1964. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Farrell PJ: Epstein-barr virus and cancer.
Annu Rev Pathol. 14:29–53. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Klein G: Tumor associations of
EBV-historical perspectives. Curr Top Microbiol Immunol. 390:17–22.
2015.PubMed/NCBI
|
|
4
|
Niller HH, Wolf H and Minarovits J:
Regulation and dysregulation of Epstein-Barr virus latency:
Implications for the development of autoimmune diseases.
Autoimmunity. 41:298–328. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cohen JI: Primary immunodeficiencies
associated with EBV disease. Curr Top Microbiol Immunol.
390:241–265. 2015.PubMed/NCBI
|
|
6
|
Blackburn EH: Switching and signaling at
the telomere. Cell. 106:661–673. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Campisi J, Kim SH, Lim CS and Rubio M:
Cellular senescence, cancer and aging: The telomere connection. Exp
Gerontol. 36:1619–1637. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zvereva MI, Shcherbakova DM and Dontsova
OA: Telomerase: Structure, functions, and activity regulation.
Biochemistry (Mosc). 75:1563–1583. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gladych M, Wojtyla A and Rubis B: Human
telomerase expression regulation. Biochem Cell Biol. 89:359–376.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wright WE, Piatyszek MA, Rainey WE, Byrd W
and Shay JW: Telomerase activity in human germline and embryonic
tissues and cells. Dev Genet. 18:173–179. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kyo S, Takakura M, Fujiwara T and Inoue M:
Understanding and exploiting hTERT promoter regulation for
diagnosis and treatment of human cancers. Cancer Sci. 99:1528–1538.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jafri MA, Ansari SA, Alqahtani MH and Shay
JW: Roles of telomeres and telomerase in cancer, and advances in
telomerase-targeted therapies. Genome Med. 8:692016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lü MH, Liao ZL, Zhao XY, Fan YH, Lin XL,
Fang DC, Guo H and Yang SM: hTERT-based therapy: A universal
anticancer approach (Review). Oncol Rep. 28:1945–1952. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mizukoshi E and Kaneko S:
Telomerase-targeted cancer immunotherapy. Int J Mol Sci.
20:18232019. View Article : Google Scholar
|
|
15
|
Kupchan SM, Court WA, Dailey RG Jr,
Gilmore CJ and Bryan RF: Triptolide and tripdiolide, novel
antileukemic diterpenoid triepoxides from Tripterygium
wilfordii. J Am Chem Soc. 94:7194–7195. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wong KF, Yuan Y and Luk JM:
Tripterygium wilfordii bioactive compounds as anticancer and
anti-inflammatory agents. Clin Exp Pharmacol Physiol. 39:311–320.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hou W, Liu B and Xu H: Triptolide:
Medicinal chemistry, chemical biology and clinical progress. Eur J
Med Chem. 176:378–392. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Noel P, Von Hoff DD, Saluja AK, Velagapudi
M, Borazanci E and Han H: Triptolide and its derivatives as cancer
therapies. Trends Pharmacol Sci. 40:327–341. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jiang W, Chen M, Xiao C, Yang W, Qin Q,
Tan Q, Liang Z, Liao X, Mao A and Wei C: Triptolide suppresses
growth of breast cancer by targeting HMGB1 in vitro and in vivo.
Biol Pharm Bull. 42:892–899. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liang X, Xie R, Su J, Ye B, Wei S, Liang
Z, Bai R, Chen Z, Li Z and Gao X: Inhibition of RNA polymerase III
transcription by Triptolide attenuates colorectal tumorigenesis. J
Exp Clin Cancer Res. 38:2172019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang W, He T, Chai C, Yang Y, Zheng Y,
Zhou P, Qiao X, Zhang B, Liu Z, Wang J, et al: Triptolide inhibits
the proliferation of prostate cancer cells and down-regulates
SUMO-specific protease 1 expression. PLoS One. 7:e376932012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou H, Guo W, Long C, Wang H, Wang J and
Sun X: Triptolide inhibits proliferation of Epstein-Barr
virus-positive B lymphocytes by down-regulating expression of a
viral protein LMP1. Biochem Biophys Res Commun. 456:815–820. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Long C, Wang J, Guo W, Wang H, Wang C, Liu
Y and Sun X: Triptolide inhibits transcription of hTERT through
down-regulation of transcription factor specificityprotein 1 in
primary effusion lymphoma cells. Biochem Biophys Res Commun.
469:87–93. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang J, Jiang J, Chen H, Wang L, Guo H,
Yang L, Xiao D, Qing G and Liu H: FDA-approved drug screen
identifies proteasome as a synthetic lethal target in MYC-driven
neuroblastoma. Oncogene. 38:6737–6751. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Long C, Guo W, Zhou H, Wang J, Wang H and
Sun X: Triptolide decreases expression of latency-associated
nuclear antigen 1 and reduces viral titers in Kaposi's
sarcoma-associated and herpesvirus-related primary effusion
lymphoma cells. Int J Oncol. 48:1519–1530. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Beishline K and Azizkhan-Clifford J: Sp1
and the ‘hallmarks of cancer’. FEBS J. 282:224–258. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Verma SC, Borah S and Robertson ES:
Latency-associated nuclear antigen of Kaposi's sarcoma-associated
herpesvirus up-regulates transcription of human telomerase reverse
transcriptase promoter through interaction with transcription
factor Sp1. J Virol. 78:10348–10359. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dang CV, O'Donnell KA, Zeller KI, Nguyen
T, Osthus RC and Li F: The c-Myc target gene network. Semin Cancer
Biol. 16:253–264. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pica F, Serafino A, Garaci E and Volpi A:
Cidofovir on HHV-8 in BCBL-1 cells. Antivir Ther. 9:823–825.
2004.PubMed/NCBI
|
|
31
|
Miller G, El-Guindy A, Countryman J, Ye J
and Gradoville L: Lytic cycle switches of oncogenic human
gammaherpesviruses. Adv Cancer Res. 97:81–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang Y, Sun M, Shi W, Yang Q, Chen C,
Wang Z and Zhou X: Arsenic trioxide suppresses transcription of
hTERT through down-regulation of multiple transcription factors in
HL-60 leukemia cells. Toxicol Lett. 232:481–489. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Meyer N and Penn LZ: Reflecting on 25
years with MYC. Nat Rev Cancer. 8:976–990. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cong YS, Wen J and Bacchetti S: The human
telomerase catalytic subunit hTERT: Organization of the gene and
characterization of the promoter. Hum Mol Genet. 8:137–142. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Toaldo C, Pizzimenti S, Cerbone A,
Pettazzoni P, Menegatti E, Daniela B, Minelli R, Giglioni B,
Dianzani MU, Ferretti C and Barrera G: PPARgamma ligands inhibit
telomerase activity and hTERT expression through modulation of the
Myc/Mad/Max network in colon cancer cells. J Cell Mol Med.
14:1347–1357. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tsurumi T, Fujita M and Kudoh A: Latent
and lytic Epstein-Barr virus replication strategies. Rev Med Virol.
15:3–15. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kerr JR: Epstein-Barr virus (EBV)
reactivation and therapeutic inhibitors. J Clin Pathol. 72:651–658.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kanda T: EBV-encoded latent genes. Adv Exp
Med Biol. 1045:377–394. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
McKenzie J and El-Guindy A: Epstein-barr
virus lytic cycle reactivation. Curr Top Microbiol Immunol.
391:237–261. 2015.PubMed/NCBI
|
|
40
|
Bhende PM, Seaman WT, Delecluse HJ and
Kenney SC: BZLF1 activation of the methylated form of the BRLF1
immediate-early promoter is regulated by BZLF1 residue 186. J
Virol. 79:7338–7348. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tikhmyanova N, Paparoidamis N,
Romero-Masters J, Feng X, Mohammed FS, Reddy PAN, Kenney SC,
Lieberman PM and Salvino JM: Development of a novel inducer for EBV
lytic therapy. Bioorg Med Chem Lett. 29:2259–2264. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Murata T and Tsurumi T: Switching of EBV
cycles between latent and lytic states. Rev Med Virol. 24:142–153.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ben Yebdri F, Van Grevenynghe J, Tang VA,
Goulet ML, Wu JH, Stojdl DF, Hiscott J and Lin R:
Triptolide-mediated inhibition of interferon signaling enhances
vesicular stomatitis virus-based oncolysis. Mol Ther. 21:2043–2053.
2013. View Article : Google Scholar : PubMed/NCBI
|