Introduction
Compound C (6-[4-(2-piperidin-1-yl-ethoxy)-phenyl]
3-pyridin-4-yl-pyrazolo[1,5-a] pyrimidine) is a small molecule with
cell-permeable and selective ATP-competitive functions that
inhibits AMP-activated protein kinase (AMPK) and interferes with
AMPK metabolic regulation (1).
Compound C has been reported to attenuate metformin-induced
apoptosis via AMPK inhibition (2).
In contrast, it has been suggested that compound C treatment
directly induces apoptosis in hepatoma, glioma and breast carcinoma
cells (2–4). In addition, compound C has been
described to inhibit AMPK-dependent autophagy in yeast, hepatocytes
and HeLa cells (5). Therefore,
controversially, compound C has dual roles in autophagic protection
against compound C-induced apoptosis in glioma cancer cells, and
autophagic cell death in colorectal cancer cells (6,7). This
evidence indicates that compound C has variable effects on
apoptosis and the type of autophagy in dose-, cell type- and/or
context-dependent manners.
The transcription factor early growth response-1
(EGR-1) is a member of the immediate-early family of genes
and encodes a 59-kDa phosphoprotein (8). Numerous factors, such as growth
factors, cytokines, radiation, injury and mechanical stress, can
induce EGR-1 protein expression (9).
There are five serum response elements (SREs) in the EGR-1
promoter region, and the level of EGR-1 transcription is
most commonly mediated by transcription factors in the Elk-1
family, which are activated by the MAP kinase family. CREB-binding
protein (CBP) and serum response factor (SRF) associate with Elk-1
to form the ternary complex factor that binds to SREs (9,10). There
are several specificity protein 1 (SP1) consensus sequences, a
putative AP-1-binding site, functional cAMP regulatory elements
(CREs) and a functional NF-κB-binding site in the EGR-1
promoter region (10). EGR1
transcription is also self-regulated via binding to functional
EGR-1 binding sites (EBS). The targeting of EGR-1 by
E26 transformation-specific transcription factors is involved in
hematopoiesis, angiogenesis and neoplasia (11). The EGR-1 promoter contains two
activating transcription factor 5 (ATF5) consensus sequences that
are activated by ATF5 in fibroblasts (10). Moreover, there are two functional
non-consensus binding sites for the tumor suppressor protein p53.
The binding of p53 to the EGR-1 promoter in response to DNA
damage leads to sustained expression of EGR-1 protein and induction
of apoptosis (12). The activity and
stability of the EGR-1 protein are regulated by post-translational
modifications. Acetylation improves EGR-1 protein stability and may
facilitate cell survival, whereas phosphorylation of EGR-1 protein
in response to stress may favor cell death (13). Sumoylation directs EGR-1 protein to
the nucleus (14), and the short
half-life of EGR-1 protein is a result of ubiquitination and
degradation by the proteasome (15).
EGR-1 protein is associated with cell proliferation
and the regulation of apoptosis as it belongs to a network of tumor
suppressor genes that include p53 and p73, which promote apoptosis
in cancer cells in response to stress and DNA damage (16–19). In
contrast, EGR-1 protein specifically promotes prostate cancer
progression. The mRNA expression of EGR-1 is higher in
prostate adenocarcinoma tissue compared with normal tissues. The
degree of differentiation of carcinoma cells is inversely
correlated with the levels of EGR-1 mRNA and protein
expression (20). Evidence indicates
that the role of EGR-1 protein in prostate cancer could be due, at
least in part, to an interaction with the androgen receptor
(21). Thus, while EGR-1 protein
mediates apoptosis in response to stress and DNA damage by
regulating a tumor suppressor network, it also promotes the
proliferation of prostate cancer cells via a mechanism that is not
fully understood. EGR-1 protein was reported to bind to the
promoter of the autophagy gene light chain 3B (LC3B), increasing
LC3B expression and promoting autophagy. Knocking down EGR-1
expression inhibits cigarette smoke extract-induced autophagy and
protects epithelial cells from cigarette smoke extract-induced
apoptosis (22).
In our previous study, we demonstrated that compound
C induced not only autophagy but also apoptosis in skin cancer
cells (23). The present study
showed that compound C could induce EGR-1 gene expression
through the reactive oxygen species (ROS)-mediated extracellular
signal-regulated kinase (ERK) activation pathway, which provided a
protective response against compound C-induced apoptosis. Thus,
targeting EGR-1 expression may increase the antitumor efficacy of
compound C in skin cancer.
Materials and methods
Reagents and antibodies
Compound C was purchased from Calbiochem; Merck
KGaA. Metformin, N-acetylcysteine (NAC) and U0126 were obtained
from Sigma-Aldrich; Merck KGaA. Cells were treated with 40 µM
compound C for 0, 1, 2, 4, 6, 8, 12, or 24 h, or 0, 1, 5, 10, 20,
or 40 µM compound C for 6 or 8 h. Cells were treated with 4 mM
metformin for 0, 1, 2, 4, 6, 8, 12, or 24 h, or 0, 1, 5, 10, 20, or
40 µM compound C for 6 or 8 h. Cells were treated with 2 mM NAC or
10 µM U0126 for 1 h before compound treatment. An antibody specific
for LC3 (cat. no. NB600-1384) was purchased from Novus Biologicals,
LLC. Antibodies specific for p53 (cat. no. 9282), cleaved caspase-3
(cat. no. 9661), caspase-9 (cat. no. 7237), poly ADP-ribose
polymerase (PARP) (cat. no. 9541), EGR-1 (cat. no. 4154), AMPKα
(cat. no. 2532), phosphorylated-AMPKα (Thr172) (cat. no. 2535),
phosphorylated-ERK (cat. no. 9101) and ERK (cat. no. 4695) were
purchased from Cell Signaling Technology, Inc. An antibody specific
for β-actin (cat. no. sc-47778) was purchased from Santa Cruz
Biotechnology, Inc.. All antibodies were 1000-fold dilution for
detection.
Cell culture
A basal cell carcinoma (BCC) cell line (BCC-1/KMC)
was cultured in Roswell Park Memorial Institute-1640 (RPMI-1640)
(Gibco; Thermo Fisher Scientific, Inc.) medium supplemented with
10% fetal bovine serum (FBS) (Gibco; Thermo Fisher Scientific,
Inc.). The human keratinocyte cell line HaCaT and human melanoma
cell lines C32 and A375 were obtained from Bioresource Collection
and Research Center. C32 and A375 cells were cultured in Minimum
Essential Medium (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS. HaCaT cells were cultured in Dulbecco's
modified Eagle's medium (DMEM) (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 10% FBS.
Cell viability assay
Cell viability was evaluated with a trypan blue
exclusion assay (Gibco; Thermo Fisher Scientific, Inc.). Cells were
seeded at 2×105 cells/well in a 6-well plate before
compound C treatment. After compound C treatment for 48 h, the
cells were collected and stained with trypan blue for 3 min at room
temperature to count the number of viable cells under a light
microscope (Carl Zeiss AF).
ROS detection
Cells were pretreated with 2 mM NAC for 1 h at 37°C
and then treated with compound C for 4 h. After compound C
treatment, the cells were stained with 1 µM CM-H2DCFDA
(Invitrogen; Thermo Fisher Scientific, Inc.) for 30 min at 37°C and
the stained cells were detected with a BD FACSCalibur™ cytometer
and analyzed with BD CellQuest™ Pro, Version 6.0 (Beckman Coulter,
Inc.).
Cell cycle analysis
Cells treated with compound C for 48 h were
harvested and fixed in 70% ethanol at 4°C for overnight. After
centrifugation (300 × g) at 4°C for 5 min, the cell pellets were
resuspended in a buffer containing phosphate-buffered saline (PBS),
0.05% RNase A and 40 µg/ml propidium iodide (PI) and incubated at
37°C for 30 min. After staining, the cells were collected and
resuspended in PBS. The fluorescence emitted from the PI-DNA
complexes following laser excitation of the fluorescent dye was
quantified using a BD FACSCalibur™ cytometer and analyzed with BD
CellQuest™ Pro version 6.0 (Beckman Coulter, Inc.).
Protein immunoblotting
Cells were harvested, and whole-cell extracts were
prepared with PRO-PREP protein extraction reagent (Level
Biotechnology, Inc.). Protein samples (30 µg/lane) were resolved by
electrophoresis with 15% SDS-acrylamide gel and transferred to
polyvinylidene difluoride membranes by electroblotting. Then, the
blots were blocking with 5% bovine serum albumin (BSA) (Gibco;
Thermo Fisher Scientific, Inc.) for 1 h at room temperature. After
blocking, the blots were incubated overnight at 4°C in PBS
supplemented with 0.05% Tween-20 (TBST) containing primary
antibodies as aforementioned. After washing three times (10
min/time at room temperature) with TBST, the blots were incubated
for 1 h at room temperature with a corresponding horseradish
peroxidase-coupled anti-rabbit or anti-mouse secondary antibody
(Pierce). After washing three times (10 min/time at room
temperature) with TBST, the blots were visualized with SuperSignal
West Pico ECL reagents (Pierce), and chemiluminescence was detected
by exposing the membranes to Kodak-X-Omat film (Sigma-Aldrich;
Merck KGaA). The levels of the β-actin signal were used to verify
equal protein loading among all lanes.
Reverse transcription PCR
Total RNA of whole cell extract was isolated using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.)
reagent. First-strand cDNA was synthesized from total RNA using the
Sprint PowerScript PrePrimed Single Shots kit (Takara Bio, Inc.).
PCR was performed using cDNA (25 cycles at 94°C for 30 sec, 55°C
for 30 sec and 68°C for 1 min) and Titanium Taq DNA polymerase
(Takara Bio, Inc.). The primer pairs were as follows: Human EGR-1
forward, 5′-CTGCGACATCTGTGGAAGAA-3′ and reverse,
5′-TGTCCTGGGAGAAAAGGTTG-3′; and GAPDH forward,
5′-ACCACAGTCCATGCCATCAC-3′ and reverse, 5′-TCCACCACCCTGTTGCTGT-3′.
All PCR products were separated by 2% agarose gel electrophoresis
and visualized with ethidium bromide.
Luciferase reporter assay
The human EGR-1 promoter sequence containing
the region from-714 bases to the translational start site was
amplified from the pDRIVE-hEGR-1 plasmid (InvivoGen) by PCR as
described in a previous report (24). This plasmid was co-transfected with
the pGL4.74 plasmid containing the Renilla luciferase gene (Promega
Corporation) into cells and incubated for 48 h. The transfected
cells were treated with compound C and then harvested. Supernatants
were collected for a Dual-Glo luciferase assay to evaluate promoter
activity. Luciferase activity was measured with a dual-luciferase
reporter assay system (Promega Corporation). Light units were
normalized to those corresponding to Renilla luciferase activity.
Final data were presented as the fold change in the EGR-1
luciferase activity compared with untreated group.
Immunocytochemistry
Cells (5×104 cells per chamber) were
cultured on two-chamber slides and then treated with compound C.
The treated cells were fixed in 4% formaldehyde in PBS overnight at
4°C. For intracellular staining, the cells were blocked with 2%
normal horse serum (Invitrogen; Thermo Fisher Scientific, Inc.) for
30 min at room temperature, incubated with antibodies against EGR-1
in PBS containing 0.2% Triton X-100 (PBST), and then incubated with
a goat fluorescein isothiocyanate-conjugated anti-rabbit IgG
antibody (Sigma-Aldrich; Merck KGaA). After washing with PBST, the
cells were mounted using an antifade, DAPI-containing, water-based
mounting medium (Vector Laboratories, Inc.). For Magic Red
staining, cells were seeded at 5×104 cells/well in a
6-well plate containing one 22×22-mm glass coverslip in each well
(Sigma-Aldrich; Merck KGaA), stained with a Magic Red assay kit
(ImmunoChemistry Technologies, LLC) at 37°C for 1 h and washed with
PBS before image capture. Before fluorescent image capture, the
coverslips were detached from the 6-well plate and sealed on glass
microscope slides. All fluorescent images were captured by confocal
microscopy (Olympus Corporation).
Construction of RNA interference
(RNAi) vectors and introduction of RNAi vectors into BCC cells
Human short hairpin shAMPKα1/2 and control shRNA
plasmids (Santa Cruz Biotechnology, Inc.) were transfected at 37°C
into BCC cells using jetPEI™ (Polyplus-transfection®)
for 48 h and then selected with 10 µg/ml puromycin to obtain stably
transfected clones. The stable clones were maintained with RMPI
medium containing 10 µg/ml puromycin. The level of AMPK-knockdown
in the stable clones was analyzed by immunoblotting with specific
AMPKα and phosphorylated-AMPKα (Thr172) antibodies. To construct an
EGR-1 RNAi vector, a DNA fragment containing a human U6 promoter
followed by an EGR-1-specific shRNA sequence
(5′-CACCGACCCTAAGCTGGAGGAGATGATTCAAGAGATCATCTCCTCCAGCTTAGGGTTTTTTTG-3′)
was retrieved from the CMV-/Hu6-RNAi plasmid by HindIII and
EcoRI double digestion. This DNA fragment was then inserted
into the mammalian expression vector pcDNA3, which resulted in a
plasmid carrying the U6 promoter driving the expression of the
EGR-1-specific shRNA. This allowed for the selection of stably
transfected clones. An EGFP RNAi vector was employed as the
negative control for EGR-1-knockdown. The EGR-1 RNAi and EGFP RNAi
vectors were subsequently transfected into BCC cells using jetPEI™,
followed by 800 µg/ml G418 (Invitrogen) treatment for 48 h
post-transfection to select stably transfected clones. The degree
of EGR-1 knockdown in these stable clones was verified by protein
immunoblotting analysis of endogenous EGR-1 protein expression
using anti-human EGR-1 antibodies after compound C treatment. The
method of protein immunoblotting analysis was performed as
aforementioned.
Oligodeoxynucleotide-based microarray
screening assay
The BCCs were treated with 40 µM compound C for 4 h,
and the control and compound C-treated cells were then harvested
for total RNA extraction. Total RNA was isolated using TRIzol
reagent. The concentration and purity of RNAs were checked to
confirm OD260/OD280 (>1.8) and OD260/OD230 (>1.6) using an
Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.). In total, 1
µg of RNA was prepared for fluorescent antisense RNA (aRNA) targets
using the OneArray® Amino Allyl aRNA Amplificant kit
(Phalanx Biotech Group) and Cy5 dye (Cyvita). The fluorescent
targets were hybridized to the Human Whole Genome
OneArray® with a Phalanx Hybridization system (Phalanx
Biotech Group) (25). The signal
intensity of each spot was analyzed with the Rosetta Resolver
system® (Rosetta Biosoftware), and the normalized
intensity of each spot was translated into gene expression by log2
ratios compared with the control. The distribution of 11,909
normalized plots, which were represented by 11,909 normalized
genes, was represented by scatter plots using the Nested graph
family in GraphPad Prism 8 (GraphPad Software, Inc.).
Ingenuity pathway analysis®
(IPA)
After the oligodeoxynucleotide-based microarray
screening assay, the resultant gene lists were used with a
decision-tree-based classifier method to analyze the microarray
data (26). The selected genes were
used to confirm the relationship with BCC cells. The genetic
pathways were analyzed by IPA (Ingenuity Systems; QIAGEN) with
species and confidence levels.
Statistical analysis
All statistical results are representative of three
independent experiments. Data are presented as mean ± SEM. Data
between were analyzed with one-way or two-way ANOVA tests for
multiple comparisons, and P<0.05 was considered to indicate a
statistically significant difference. The statistical analysis was
performed using GraphPad Prism 8 (GraphPad Software, Inc.).
Results
Compound C increases the expression of
EGR-1 in skin cancer cell lines
Several studies have indicated that the AMPK
inhibitor compound C induces apoptosis and autophagy in an
AMPK-independent manner (4,6,7,27). To evaluate the putative mechanisms
that regulate compound C-induced apoptosis and autophagy in cancer
cells, an oligodeoxynucleotide-based microarray screening assay was
used to identify compound C-induced gene expression. Then, 11,909
normalized genes were surveyed and the top five were found to be
FOS, EGR-1, BAAT, GDF15 and INSIG1 (Fig. 1A and Table SI.).
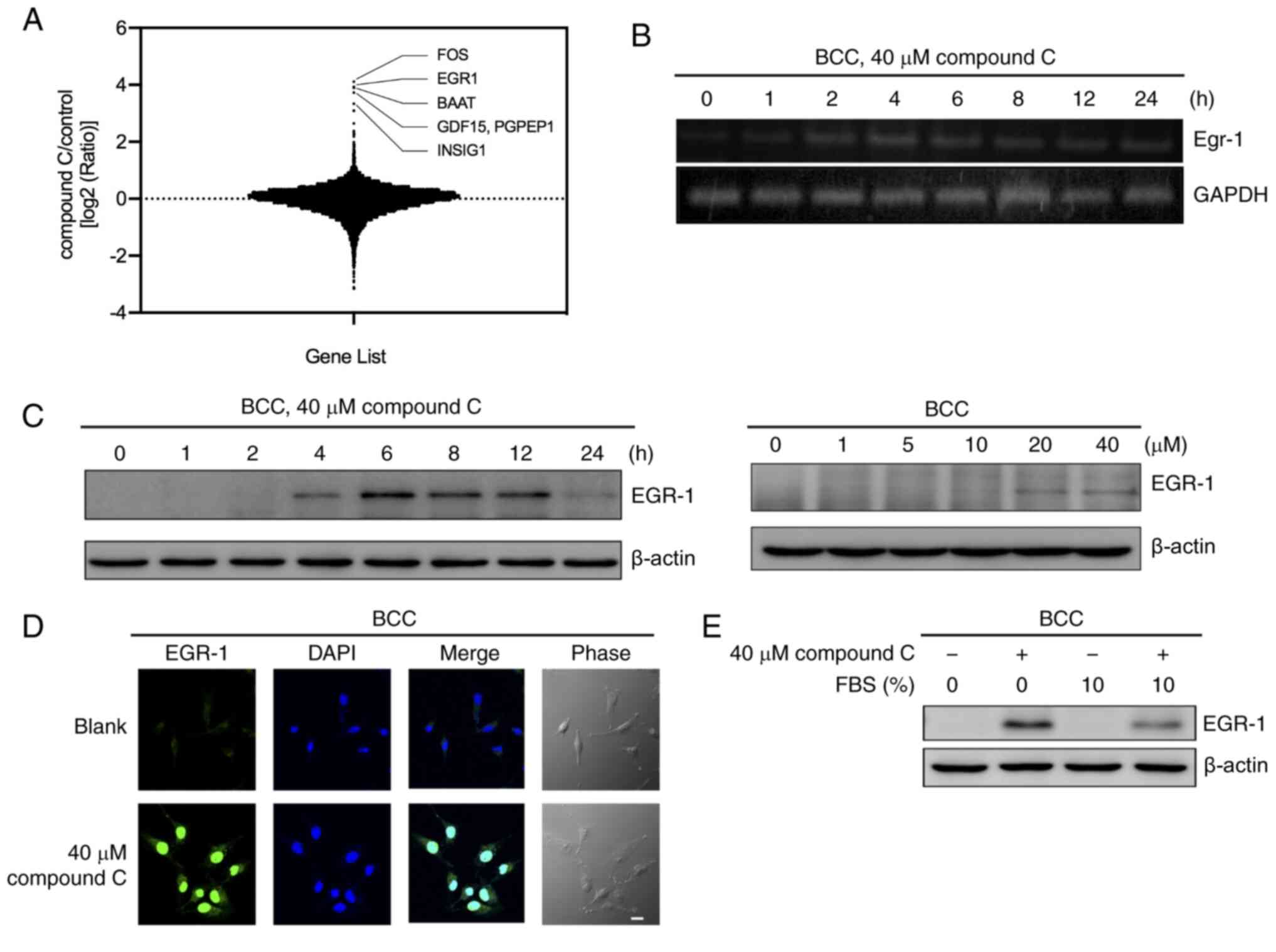 | Figure 1.Compound C induces EGR-1 expression
in BCC cells. (A) BCC cells were treated with 40 µM compound C for
4 h, and the mRNA expression levels of genes were detected with an
oligodeoxynucleotide-based microarray assay. (B) BCC cells were
treated with 40 µM compound C for 0, 1, 2, 4, 6, 8, 12 or 24 h, and
the mRNA expression levels of EGR-1 were detected by reverse
transcription PCR. (C) BCC cells were treated with 40 µM compound C
for 0, 1, 2, 4, 6, 8, 12 or 24 h or with 0, 1, 5, 10, 20 or 40 µM
compound C for 6 h. Protein expression levels of EGR-1 were
detected by immunoblotting. (D) BCC cells were treated with 0 or 40
µM compound C for 6 h. (E) BCC cells were treated with 40 µM
compound C and culture medium with or without 10% FBS for 6 h.
Scale bar, 20 µm. Protein expression level of EGR-1 was detected by
immunoblotting. EGR-1, early growth response-1; BCC, basal cell
carcinoma; FBS, fetal bovine serum. |
Next, the selected genes were input into IPA and
found that TP53-EGR-1 pathway was associated with compound C
treatment in BCC cells (Fig. S1A).
We had previously demonstrated that compound C-induced apoptosis in
skin cancer cells was p53-dependent (23). Hence, EGR-1 was selected as
the targeted gene for further investigation. The expression of the
EGR-1 gene was confirmed by RT-PCR compared with mock
DMSO-treated cells (0 h) (Fig. 1B).
After treatment with compound C, BCC cells showed enhanced
EGR-1 mRNA expression after 2 h, with expression peaking at
4 h and then gradually declining (Fig.
1B). The protein expression of EGR-1 was upregulated by
compound C treatment in a similar time- and dose-dependent manner,
although different kinetics were observed in the BCC, C32 and A375
cell lines (Figs. 1C and S1B). Furthermore, the protein expression
of EGR-1 was induced when treated with a high concentration of
compound C (40 µM) (Fig. 1C).
EGR-1 was also localized in the nucleus after
compound C treatment, as demonstrated by immunofluorescence
staining (Figs. 1D and S1C). Growth factors and cytokines have
been reported to induce the expression of EGR-1 (11). To rule out the possibility that
growth factors and cytokines involved in EGR-1 induction, the
expression of EGR-1 was examined in culture medium with or without
FBS. As shown in Fig. 1E, the EGR-1
expression in the BCC cell line was increased after compound C
treatment regardless of whether FBS was added to the culture
medium. This result indicated that FBS did not contribute to
compound C-mediated EGR-1 induction. Thus, it was concluded that a
high dose of compound C may induce EGR-1 mRNA and protein
expression in skin cancer cells.
Compound C-induced EGR-1 expression is
independent of AMPK activation
Compound C is a commonly used AMPK inhibitor, and
activation of AMPK also reportedly induces the expression of EGR-1
and its target dual specificity phosphatase 4 (28). To evaluate the role of AMPK
activation in the regulation of EGR1 expression, BCC cells were
treated with metformin, an AMPK activator, to evaluate whether the
activation of AMPK (p-AMPK) can control EGR-1 expression. Metformin
induced the activation of AMPK but had no effect on the expression
of EGR-1 in skin cancer cells (Figs.
2A and S2). Co-treatment with
metformin and compound C did not change the level of compound
C-induced EGR-1 expression in skin cancer cells (Figs. 2B and S2). To confirm whether AMPK is involved in
compound C-induced EGR-1 production, EGR-1 in stable AMPK-knockdown
BCC cells (shAMPK) treated with compound C were examined. In
control and AMPK-knockdown BCC cells without compound C treatment,
EGR-1 expression was not induced, indicating that AMPK may be
dispensable in the regulation of EGR-1 expression in un-stimulated
conditions (Fig. 2C). Expression of
EGR-1 was highly induced in AMPK-knockdown BCC cells treated with
compound C, revealing that compound C-induced EGR-1 expression may
be controlled by other pathways and further enhanced in the
AMPK-knockdown condition.
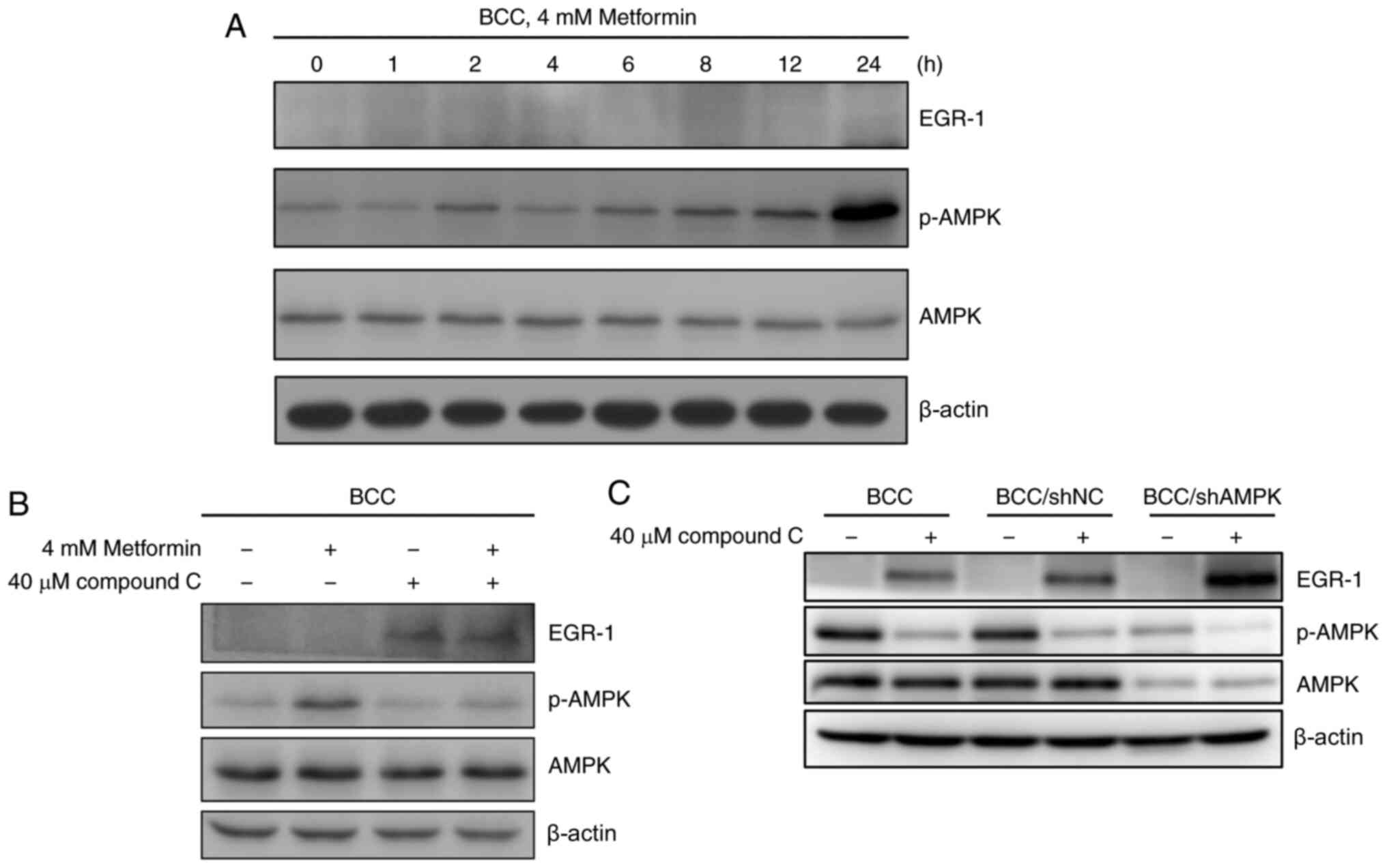 | Figure 2.AMPK activation is not involved in
compound C-induced EGR-1 expression in BCC cells. (A) BCC cells
were treated with 4 mM metformin for 0, 1, 2, 4, 6, 8, 12 or 24 h.
(B) BCC cells were cotreated with or without 4 mM metformin and 40
µM compound C for 6 h. (C) BCC, BCC/shNC and BCC/shAMPK cells were
treated with 40 µM compound C for 6 h. Expression levels of EGR-1,
AMPK, p-AMPK and β-actin were detected by immunoblotting. AMPK,
AMP-activated protein kinase; BCC, basal cell carcinoma; p-,
phosphorylated; NC, negative control; sh, short hairpin. |
Compound C upregulates EGR-1 mRNA
expression via the ROS-mediated ERK signaling pathway
Further investigation into whether compound C
induces EGR-1 expression through transcriptional regulation of the
EGR-1 gene was conducted. Using a luciferase reporter assay,
EGR1 promoter activity was shown to be increased by 1.3-, 1.6-,
2.3-, and 3.1-fold at 6, 8, 12 and 24 h, respectively, in cells
treated with compound C (Fig. 3A).
This result confirmed that compound C could induce EGR-1
expression at the mRNA level. As the transcription of EGR-1 is
reportedly regulated by the ERK signaling pathway in cancer cells
(20), it was evaluated whether
compound C-induced EGR-1 transcriptional regulation was mediated by
the ERK signaling pathway. First, compound C was demonstrated to
significantly induce ERK phosphorylation in skin cancer cells
(Figs. 3B and S3A). Next, the MEK/ERK inhibitor U0126 was
used to evaluate whether the ERK signaling pathway was involved in
compound C-induced EGR-1 expression in BCC, C32 and A375 cells.
Pretreatment with U0126 decreased the basal-level mRNA expression
of EGR-1 and inhibited the compound C-induced mRNA and
protein expression of EGR-1 and p-ERK (Fig. 3C and D) in BCC cells. Similarly,
U0126 pretreatment inhibited the compound C-induced protein
expression of EGR-1 and p-ERK in C32 cells (Fig. S3B). In the promoter activity assay,
pretreatment with U0126 not only decreased the promoter activity of
EGR-1 in the compound C-treated pEgr-1/-714 group compared
with only compound C-treated pEgr-1/-714 group, but also decreased
the promoter activity of EGR-1 in the U0126-treated
pEgr-1/-714 group compared with pEgr-1/-714 group (Fig. 3E). We previously demonstrated that
compound C-induced p53-dependent apoptosis in skin cancer cells is
mediated by compound C-induced ROS (23). Figs.
3F and S3C show that compound C
induced ROS production in BCC and C32 cells, but pretreatment with
NAC decreased the level of compound C-induced ROS production. Thus,
it was evaluated whether compound C-induced ROS also promoted ERK
activation and subsequent EGR-1 expression. As shown in Figs. 3G and S3D, the antioxidant NAC not only mitigated
compound C-induced ERK phosphorylation but also inhibited EGR-1
expression in BCC and C32 cells. Thus, it was concluded that
compound C upregulates EGR-1 mRNA expression via
ROS-mediated ERK activation.
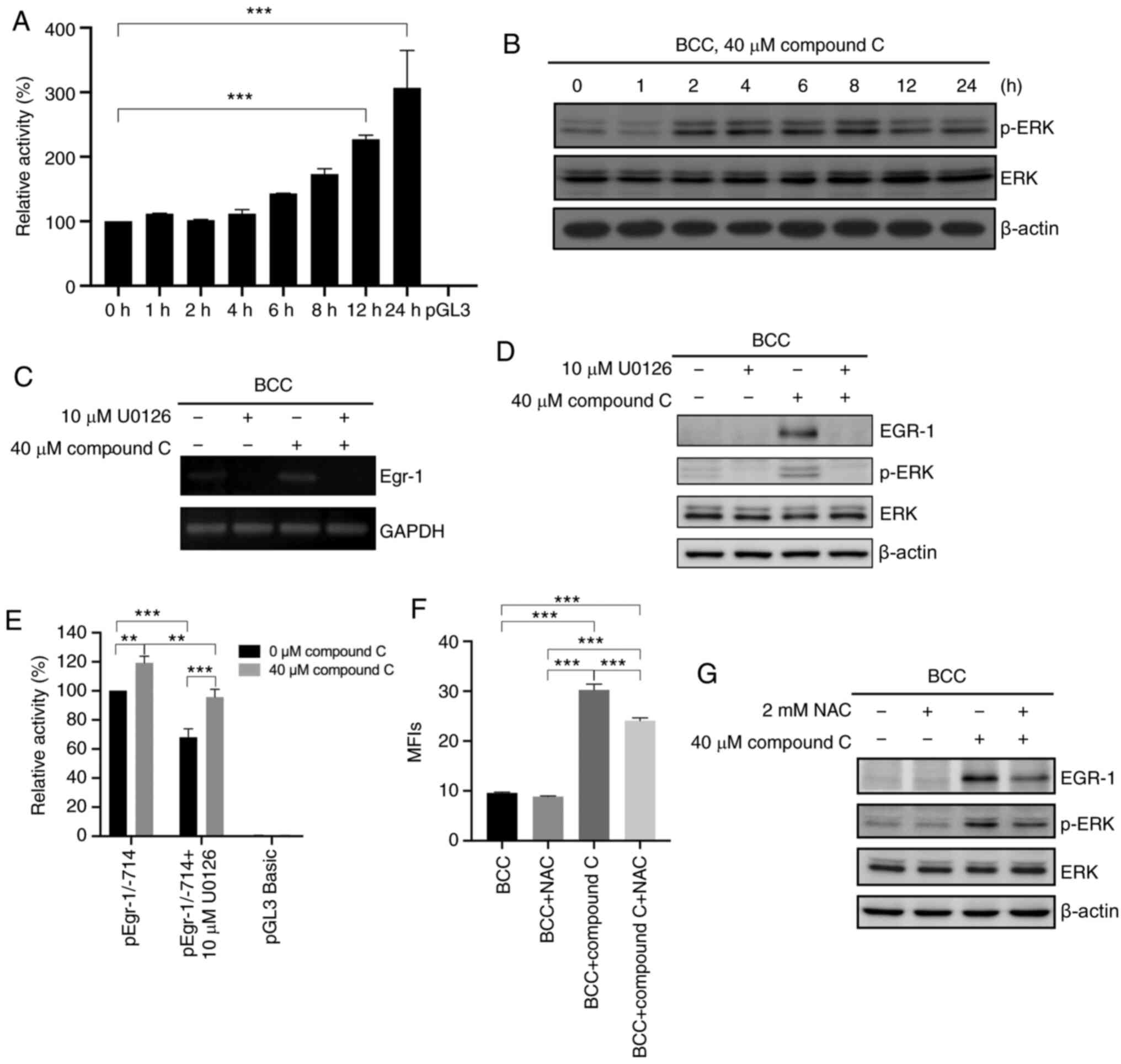 | Figure 3.Compound C upregulates EGR-1
mRNA expression via the ROS-mediated ERK signaling pathway. (A) BCC
cells were co-transfected with pGL3-basic-hEGR-1-pro-1/-714 and
pGL3 carrying the Renilla luciferase gene for 48 h and then
incubated in medium containing 0% FBS and 40 µM compound C for 0,
1, 2, 4, 6, 8, 12 or 24 h, and analyzed with one-way ANOVA. (B) BCC
cells were treated with 40 µM compound C for 0, 1, 2, 4, 6, 8, 12
or 24 h. Expression levels of p-ERK, ERK and β-actin were detected
by immunoblotting. (C and D) BCC cells were pretreated with 10 µM
U0126 for 1 h and then treated with 40 µM compound C for 6 h. mRNA
and protein expression levels of EGR-1 were detected by reverse
transcription PCR and immunoblotting, respectively. (E) BCC cells
were co-transfected with pGL3-basic-hEGR-1-pro-1/-714 and pGL4.74
carrying the Renilla luciferase gene for 48 h. Cells were
pretreated with 10 µM U0126 for 1 h in serum-free medium and then
treated with 0 or 40 µM compound C for 6 h, and analyzed with
two-way ANOVA. (F) BCC cells were pretreated with 2 mM NAC for 1 h
and then treated with 40 µM compound C for 4 or 6 h to evaluate the
ROS or protein expression level. This statistical result was
analyzed using one-way ANOVA. (G) Expression levels of EGR-1,
p-ERK, ERK and β-actin were detected by immunoblotting with
specific antibodies. Data are expressed as the mean ± SEM. of three
independent experiments. **P<0.01 and ***P<0.001 compared
with the control. EGR-1, early growth response-1; BCC, basal cell
carcinoma; NAC, N-acetyl-cysteine; p-, phosphorylated. |
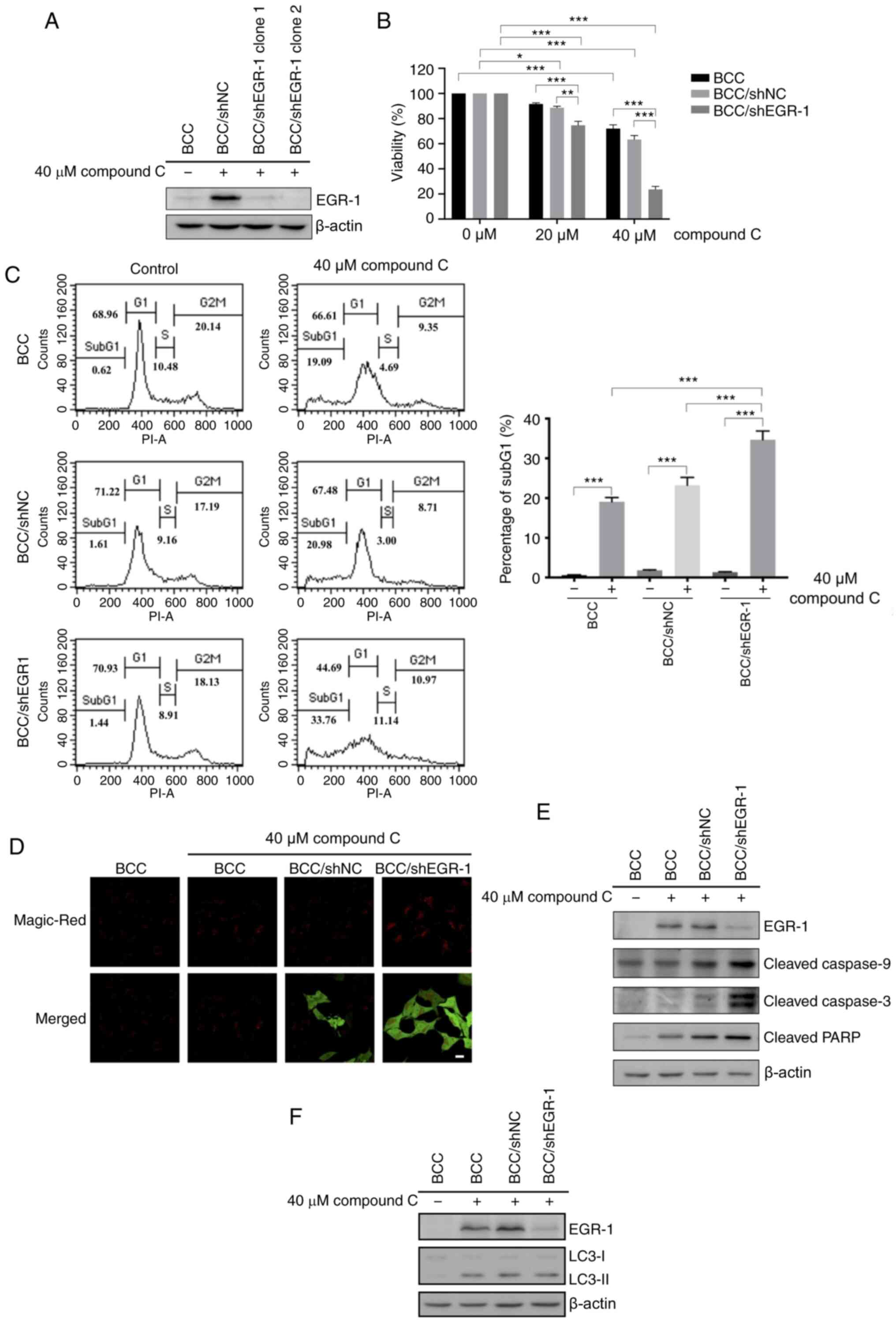
EGR-1 knockdown enhanced compound
C-induced apoptosis
Previous studies found that compound C efficiently
increases apoptotic and autophagic populations in skin cancer cells
(23). A rapid and transient
increase in EGR-1 expression was demonstrated during compound C
treatment. Therefore, it was hypothesized that the apoptotic and
autophagic effects of compound C may be regulated by EGR-1. To
investigate this hypothesis, the effect of compound C on BCC cells
with an EGR-1 functional deficiency was evaluated. RNAi was used to
reduce the level of endogenous EGR-1, which was sufficient to
abolish EGR-1 function. An EGR-1-specific shRNA vector carrying a
human U6 promoter was transfected into BCC cells, and the stably
transfected clones were screened by G418 selection. BCC cells
stably expressing an EGFP RNAi vector were used as a negative
control for EGR-1-knockdown, and the degree of EGR-1 knockdown in
cells was validated by EGR-1 immunoblotting. The endogenous EGR-1
level in BCC/EGR-1-shRNA stable clones dropped to a barely
detectable level after compound C treatment, which indicated the
efficiency of EGR-1 knockdown (Fig.
4A). As expected, 40 µM compound C triggered a slight reduction
in the number of cells transfected with the BCC/sh negative control
(NC) (Fig. 4B). However, compound C
decreased the BCC/EGR-1-shRNA clone cell numbers in a
dose-dependent manner (Fig. 4B),
suggesting that compound C-induced cell death was enhanced by EGR-1
knockdown.
Then, apoptosis was characterized by a DNA content
assay, revealing that ~32% of the apoptosis observed for the
BCC/EGR-1-shRNA clone was caused by compound C (Fig. 4C). Caspase-3 activity was also
increased in the BCC/EGR1-shRNA clone compared with BCC control
cells after compound C treatment (Fig.
4D). Consistently, the cleaved caspase-9, caspase-3 and PARP
levels were increased in the BCC/EGR1-shRNA clone after compound C
treatment (Fig. 4E). However, there
was no difference in the protein expression of the autophagy
induction marker light chain 3-II (LC3-II) between the BCC/shNC and
BCC/EGR-1-shRNA cells (Fig. 4F).
Collectively, these data revealed that EGR-1 can protect against
compound C-induced apoptosis but not against compound C-induced
autophagy in skin cancer cells.
Discussion
In our previous study, we demonstrated that compound
C-induced apoptosis in human skin cancer cells is p53-dependent
(23). The present
oligodeoxynucleotide-based microarray assay showed that the
EGR-1 gene was one of highest upregulated genes in BCC cells
after compound C treatment. The results of IPA indicated that
TP53 and EGR-1 expression might be connected in BCC
cells with compound C treatment. The report that p53 gene
expression is under EGR-1 regulation may indicate the possibility
that EGR-1 is involved in compound C-induced p53-dependent
apoptosis (17). However, according
to the result of Figure S4,
compound C treatment still induced p53 expression in
EGR-1-knockdown BCC cells. This result indicated that p53 gene
expression cannot be controlled by EGR-1 in compound C-treated BCC
cells, and therefore EGR-1 may play another role in compound
C-induced apoptosis in BCC cells. Thus, understanding the role of
EGR-1 in cancer could improve cancer therapeutic methods. The
expression of EGR-1 increased after compound C treatment in
BCC, C32 and A375 cells, and the EGR-1 protein was also localized
in the nucleus. These results indicated that compound C-induced
EGR-1 expression and localization are dose-dependent in different
skin cancer cells. The kinetic expression patterns of EGR-1 may be
related to the different genetic backgrounds in different cells
(13–15,29–33). The
present study demonstrated that the expression level of EGR-1 after
compound C treatment was higher in serum-free medium compared with
in serum. EGR-1 is a zinc-finger transcription factor with EBS,
with a CRE-binding site, serum response elements/erythroblast
transformation-specific-binding site and NF-κB-like-binding site,
ATF5 consensus sequences, an AP1 site and an SP1 site upstream of
the promoter region (11). Growth
factors activate the Ras/Raf/MAP kinase signaling pathway and
facilitate formation of the Elk-1/CBP/SRF complex, which binds to
the SRE functional element of the EGR-1 promoter to induce
EGR-1 transcription (20).
When cells are in the absence of growth factors, EGR-1 is induced,
and the activities of JNK/SAPK and p38 are increased (22,34–36).
Collectively, these results indicate that compound C is capable of
inducing EGR-1 expression and regulating EGR-1 transcription
in an environmental context-dependent manner in cancer cells.
In the absence of energy, AMPK activation induces
EGR-1 expression in hepatocytes (28). A high-glucose state activates the
protein kinase C (PKC)-ERK signaling pathway, inducing EGR-1
expression; however, valsartan, a Food and Drug Administration
approved antihypertensive drug, activates the liver kinase
B1-AMP-activated protein kinase signaling pathway and inhibits
EGR-1 expression independent of the PKC-ERK signaling pathway in
THP1 cells (37). These results
indicate that the role of AMPK in EGR-1 expression might be
dependent on cell type and environmental conditions. Compound C is
a widely used AMPK inhibitor that has the ability to regulate
apoptosis and autophagy in an AMPK-independent manner (1,4,6,7,16,27). The
present results indicated that treatment with metformin did not
induce EGR-1 expression. In addition, co-treatment with metformin
and compound C did not decrease compound C-induced EGR-1
expression. These results showed that compound C-induced EGR-1
expression is AMPK activation-independent in skin cancer cells.
However, the level of compound C-induced EGR-1 induction in
AMPK-knockdown cells was higher compared with that in control
cells. However, EGR-1 expression was not induced in control and
AMPK-knockdown BCC cells without compound C treatment. This
indicated that AMPK inhibition or knockdown is not enough to solely
induce EGR-1 expression, and that compound C-induced EGR-1
expression may be controlled by other pathways and only enhanced by
AMPK-knockdown conditions in skin cancer cells.
MAPK proteins, such as ERK1/2, JNK, and p38-MAPK,
induce EGR-1 activation (20). In
the present study, compound C can induce EGR-1 expression at
the mRNA level. According to RT-PCR analysis and reporter assay
evaluating the EGR-1 promoter, the present study
hypothesized that ERK may regulate compound C-induced EGR-1
expression. Pretreatment with the ERK inhibitor U0126 decreased
compound C-induced promoter activity and the mRNA and protein
expression of EGR-1. Notably, pre-treatment with the ERK inhibitor
U0126 decreased the promoter activity of the control group and
compound C treatment group. This result implied that ERK might also
be involved in EGR-1 mRNA expression at the
post-transcriptional level. Additionally, it is possible that the
regulatory sequences that are responsible for endogenous
EGR-1 mRNA induction may not be contained in the cloned
promoter of the luciferase reporter plasmid. These results
consequently indicated that EGR-1 mRNA expression was
upregulated by compound C via the ERK signaling pathway. In
malignant tumor progression, cellular adhesion is lost and
eventually promotes tumor cell migration or invasion into
surrounding tissues (38).
Hepatocyte growth factor can facilitate cellular mobility by
triggering the ERK/EGR-1 signaling pathway to promote HepG2 cell
scattering (39,40). U0126 and PD98059, ERK inhibitors, can
inhibit the EGR-1 expression induced by neuron growth factor in
PC-12 cells (41). In addition to
the ERK signaling pathways involved in EGR-1 expression, the
PI3K/Akt signaling pathway has been shown to modulate EGR-1
expression (36,42,43).
Protein kinase A (PKA) activates CREB and cAMP response
element-binding protein, binding to CREs to promote the
transcription of target genes (44,45), and
exendin-4 induces the expression of EGR-1 through the cAMP/PKA/CREB
signaling pathway in type II diabetes (46,47).
These studies demonstrate that numerous signaling pathways are
involved in the modulation of EGR-1 expression. Whether these other
signaling pathways, in addition to the ERK pathway, regulate
compound C-induced EGR-1 expression requires further
investigation.
ROS constitutes one of numerous extracellular and
intracellular stimuli that activate the MAPK pathways for cell
proliferation, differentiation, survival and death; such as the
Raf-MEK-ERK axis activated in response to growth factor receptor
stimulation by oxidative stress. ROS can also activate EGF and
platelet derived growth-factor (PDGF) receptors to stimulate the
Ras and ERK pathways in cancer cells (48,49). Our
previous study demonstrated that compound C induced ROS production
and caused apoptosis in skin cancer cells in a p53-dependent manner
(23). The data from the present
study further suggested that compound C-activated EGR-1 may act
through ROS-mediated ERK activation in cancer cells.
An EGR-1-specific shRNA was used to knock down EGR-1
expression to evaluate whether the apoptotic and autophagic effects
of compound C are regulated by EGR-1. Knocking down EGR-1
expression caused more compound C-induced cell death. Furthermore,
caspase-3 activity and the expression of cleaved caspase-9,
caspase-3 and PARP were significantly increased, but there was no
difference in the protein expression of LC3-II between BCC/shNC
control cells and BCC/EGR-1-shRNA cells. These results implied that
the protective role of EGR-1 in compound C-induced cell death
affects apoptosis but not autophagy in skin cancer cells. In HaCaT
cells, cigarette smoke extracts and UVB exposure induces EGR-1
expression and then increases TNF-α secretion (50,51).
UVB-induced EGR-1 expression is involved in the expression of DNA
repair proteins that repair DNA damage and protect cells against
apoptosis (52). The localization of
EGR-1 in the nucleus after compound C treatment in BCC and HaCaT
cells implied that EGR-1 may be related to DNA repair in compound
C-induced apoptosis. Thus, the mechanism involving EGR-1 in the DNA
repair system needs to be further investigated.
The present study found that the ROS-mediated ERK
activation pathway may be a mechanism by which compound C induces
EGR-1 expression. In EGR-1-knockdown cells, treatment with compound
C increased caspase-3 activity and the expression of cleaved
caspase-9, caspase-3 and PARP. Loss of EGR-1 causes pancreatic
β-cell apoptosis induced by palmitic acid treatment (53). Overexpression of EGR-1 has been
reported to promote cell proliferation and prevent cycle arrest and
apoptosis in prostate carcinoma cells (54). Thus, one function of EGR-1 in various
cell types is to protect against stress-induced death. The present
study's results indicated that EGR-1 played a protective role in
compound C-induced apoptosis. It was concluded that compound C has
potential in skin cancer therapy and that EGR-1 may be a promising
therapeutic target for increasing compound C sensitivity in skin
cancer cells.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thanks Mrs. Mei-Chun Liu
(Instrument Center of the Department of Medical Research of
Taichung Veterans General Hospital) for helping confocal imaging
analysis.
Funding
This work was supported by grants from The Taichung
Veterans General Hospital/National Chung Hsing University Joint
Research Program (grant no. TCVGH-NCHU-1027603) and The Ministry of
Science and Technology (grant no. MOST-108-2320-B-005-005-MY3). The
funders had no role in the study design, data collection and
analysis, decision to publish, or preparation of the
manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
KCC, MHT and JJS conceived the study. KCC, JSS and
MHT curated the data. KCC, FWC, MHT and JJS did the formal
analysis. JSS acquired the funding and was the project
administrator. KCC and FWC did the investigation for the study and
wrote the methodology. KCC, FWC, MHT and JJS provided the
resources. MHT and JJS supervised the study. JJS validated the
study. KCC and JJS visualized the study. KCC wrote and prepared the
original draft. JJS reviewed the writing and edited. MHT and JJS
confirm the authenticity of all raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhou G, Myers R, Li Y, Chen Y, Shen X,
Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al: Role of
AMP-activated protein kinase in mechanism of metformin action. J
Clin Invest. 108:1167–1174. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bae EJ, Cho MJ and Kim SG: Metformin
prevents an adaptive increase in GSH and induces apoptosis under
the conditions of GSH deficiency in H4IIE cells. J Toxicol Environ
Health A. 70:1371–1380. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vucicevic L, Misirkic M, Janjetovic K,
Harhaji-Trajkovic L, Prica M, Stevanovic D, Isenovic E, Sudar E,
Sumarac- Dumanovic M, Micic D and Trajkovic V: AMP-activated
protein kinase-dependent and -independent mechanisms underlying in
vitro antiglioma action of compound C. Biochem Pharmacol.
77:1684–1693. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jin J, Mullen TD, Hou Q, Bielawski J,
Bielawska A, Zhang X, Obeid LM, Hannun YA and Hsu YT: AMPK
inhibitor compound C stimulates ceramide production and promotes
Bax redistribution and apoptosis in MCF7 breast carcinoma cells. J
Lipid Res. 50:2389–2397. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Meley D, Bauvy C, Houben-Weerts JH,
Dubbelhuis PF, Helmond MT, Codogno P and Meijer AJ: AMP-activated
protein kinase and the regulation of autophagic proteolysis. J Biol
Chem. 281:34870–34879. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vucicevic L, Misirkic M, Janjetovic K,
Vilimanovich U, Sudar E, Isenovic E, Prica M, Harhaji-Trajkovic L,
Kravic-Stevovic T, Bumbasirevic V and Trajkovic V: Compound C
induces protective autophagy in cancer cells through AMPK
inhibition-independent blockade of Akt/mTOR pathway. Autophagy.
7:40–50. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jang JH, Lee TJ, Yang ES, Min do S, Kim
YH, Kim SH, Choi YH, Park JW, Choi KS and Kwon TK: Compound C
sensitizes Caki renal cancer cells to TRAIL-induced apoptosis
through reactive oxygen species-mediated down-regulation of c-FLIPL
and Mcl-1. Exp Cell Res. 316:2194–2203. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Milbrandt J: A nerve growth factor-induced
gene encodes a possible transcriptional regulatory factor. Science.
238:797–799. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gashler A and Sukhatme VP: Early growth
response protein 1 (Egr-1): Prototype of a zinc-finger family of
transcription factors. Prog Nucleic Acid Res Mol Biol. 50:191–224.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Aicher WK, Sakamoto KM, Hack A and Eibel
H: Analysis of functional elements in the human Egr-1 gene
promoter. Rheumatol Int. 18:207–214. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pagel JI and Deindl E: Early growth
response 1-a transcription factor in the crossfire of signal
transduction cascades. Indian J Biochem Biophys. 48:226–235.
2011.PubMed/NCBI
|
|
12
|
Yu J, Baron V, Mercola D, Mustelin T and
Adamson ED: A network of p73, p53 and Egr1 is required for
efficient apoptosis in tumor cells. Cell Death Differ. 14:436–446.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu J, de Belle I, Liang H and Adamson ED:
Coactivating factors p300 and CBP are transcriptionally
crossregulated by Egr1 in prostate cells, leading to divergent
responses. Mol Cell. 15:83–94. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yu J, Zhang SS, Saito K, Williams S,
Arimura Y, Ma Y, Ke Y, Baron V, Mercola D, Feng GS, et al: PTEN
regulation by Akt-EGR1-ARF-PTEN axis. EMBO J. 28:21–33. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bae MH, Jeong CH, Kim SH, Bae MK, Jeong
JW, Ahn MY, Bae SK, Kim ND, Kim CW, Kim KR and Kim KW: Regulation
of Egr-1 by association with the proteasome component C8. Biochim
Biophys Acta. 1592:163–167. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim HS, Hwang JT, Yun H, Chi SG, Lee SJ,
Kang I, Yoon KS, Choe WJ, Kim SS and Ha J: Inhibition of
AMP-activated protein kinase sensitizes cancer cells to
cisplatin-induced apoptosis via hyper-induction of p53. J Biol
Chem. 283:3731–3742. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Krones-Herzig A, Mittal S, Yule K, Liang
H, English C, Urcis R, Soni T, Adamson ED and Mercola D: Early
growth response 1 acts as a tumor suppressor in vivo and in vitro
via regulation of p53. Cancer Res. 65:5133–5143. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Das A, Chendil D, Dey S, Mohiuddin M,
Mohiuddin M, Milbrandt J, Rangnekar VM and Ahmed MM: Ionizing
radiation down-regulates p53 protein in primary Egr-1−/−
mouse embryonic fibroblast cells causing enhanced resistance to
apoptosis. J Biol Chem. 276:3279–3286. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baron V, Adamson ED, Calogero A, Ragona G
and Mercola D: The transcription factor Egr1 is a direct regulator
of multiple tumor suppressors including TGFbeta1, PTEN, p53, and
fibronectin. Cancer Gene Ther. 13:115–124. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gitenay D and Baron VT: Is EGR1 a
potential target for prostate cancer therapy? Future Oncol.
5:993–1003. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang SZ and Abdulkadir SA: Early growth
response gene 1 modulates androgen receptor signaling in prostate
carcinoma cells. J Biol Chem. 278:39906–39911. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lu C, Shi Y, Wang Z, Song Z, Zhu M, Cai Q
and Chen T: Serum starvation induces H2AX phosphorylation to
regulate apoptosis via p38 MAPK pathway. FEBS Lett. 582:2703–2708.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang SW, Wu CY, Wang YT, Kao JK, Lin CC,
Chang CC, Mu SW, Chen YY, Chiu HW, Chang CH, et al: p53 modulates
the AMPK inhibitor compound C induced apoptosis in human skin
cancer cells. Toxicol Appl Pharmacol. 267:113–124. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Datta R, Rubin E, Sukhatme V, Qureshi S,
Hallahan D, Weichselbaum RR and Kufe DW: Ionizing radiation
activates transcription of the EGR1 gene via CArG elements. Proc
Natl Acad Sci USA. 89:10149–10153. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kao JK, Wang SC, Ho LW, Huang SW, Lee CH,
Lee MS, Yang RC and Shieh JJ: M2-like polarization of THP-1
monocyte-derived macrophages under chronic iron overload. Ann
Hematol. 99:431–441. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tsai MH, Wang HC, Lee GW, Lin YC and Chiu
SH: A decision tree based classifier to analyze human ovarian
cancer cDNA microarray datasets. J Med Syst. 40:212016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang WL, Perillo W, Liou D, Marambaud P
and Wang P: AMPK inhibitor compound C suppresses cell proliferation
by induction of apoptosis and autophagy in human colorectal cancer
cells. J Surg Oncol. 106:680–688. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Berasi SP, Huard C, Li D, Shih HH, Sun Y,
Zhong W, Paulsen JE, Brown EL, Gimeno RE and Martinez RV:
Inhibition of gluconeogenesis through transcriptional activation of
EGR1 and DUSP4 by AMP-activated kinase. J Biol Chem.
281:27167–27177. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu DX, Qian D, Wang B, Yang JM and Lu Z:
p300-Dependent ATF5 acetylation is essential for Egr-1 gene
activation and cell proliferation and survival. Mol Cell Biol.
31:3906–3916. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cao X, Mahendran R, Guy GR and Tan YH:
Protein phosphatase inhibitors induce the sustained expression of
the Egr-1 gene and the hyperphosphorylation of its gene product. J
Biol Chem. 267:12991–12997. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang RP, Fan Y, deBelle I, Ni Z, Matheny
W and Adamson ED: Egr-1 inhibits apoptosis during the UV response:
Correlation of cell survival with Egr-1 phosphorylation. Cell Death
Differ. 5:96–106. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Manente AG, Pinton G, Tavian D,
Lopez-Rodas G, Brunelli E and Moro L: Coordinated sumoylation and
ubiquitination modulate EGF induced EGR1 expression and stability.
PLoS One. 6:e256762011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Varshavsky A: The ubiquitin system. Trends
Biochem Sci. 22:383–387. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kyriakis JM, Banerjee P, Nikolakaki E, Dai
T, Rubie EA, Ahmad MF, Avruch J and Woodgett JR: The
stress-activated protein kinase subfamily of c-Jun kinases. Nature.
369:156–160. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lim CP, Jain N and Cao X: Stress-induced
immediate-early gene, egr-1, involves activation of p38/JNK1.
Oncogene. 16:2915–2926. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sarker KP and Lee KY: L6 myoblast
differentiation is modulated by Cdk5 via the PI3K-AKT-p70S6K
signaling pathway. Oncogene. 23:6064–6070. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ha YM, Park EJ, Kang YJ, Park SW, Kim HJ
and Chang KC: Valsartan independent of AT1 receptor
inhibits tissue factor, TLR-2 and −4 expression by regulation of
Egr-1 through activation of AMPK in diabetic conditions. J Cell Mol
Med. 18:2031–2043. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Birchmeier W and Birchmeier C:
Epithelial-mesenchymal transitions in development and tumor
progression. EXS. 74:1–15. 1995.PubMed/NCBI
|
|
39
|
Grotegut S, von Schweinitz D, Christofori
G and Lehembre F: Hepatocyte growth factor induces cell scattering
through MAPK/Egr-1-mediated upregulation of Snail. EMBO J.
25:3534–3545. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vande Woude GF, Jeffers M, Cortner J,
Alvord G, Tsarfaty I and Resau J: Met-HGF/SF: Tumorigenesis,
invasion and metastasis. Ciba Found Symp. 212:119–132, 148-154.
1997.PubMed/NCBI
|
|
41
|
Harada T, Morooka T, Ogawa S and Nishida
E: ERK induces p35, a neuron-specific activator of Cdk5, through
induction of Egr1. Nat Cell Biol. 3:453–459. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Guillemot L, Levy A, Raymondjean M and
Rothhut B: Angiotensin II-induced transcriptional activation of the
cyclin D1 gene is mediated by Egr-1 in CHO-AT(1A) cells. J Biol
Chem. 276:39394–39403. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cabodi S, Morello V, Masi A, Cicchi R,
Broggio C, Distefano P, Brunelli E, Silengo L, Pavone F, Arcangeli
A, et al: Convergence of integrins and EGF receptor signaling via
PI3K/Akt/FoxO pathway in early gene Egr-1 expression. J Cell
Physiol. 218:294–303. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mayr B and Montminy M: Transcriptional
regulation by the phosphorylation-dependent factor CREB. Nat Rev
Mol Cell Biol. 2:599–609. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jhala US, Canettieri G, Screaton RA,
Kulkarni RN, Krajewski S, Reed J, Walker J, Lin X, White M and
Montminy M: cAMP promotes pancreatic beta-cell survival via
CREB-mediated induction of IRS2. Genes Dev. 17:1575–1580. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kang JH, Kim MJ, Ko SH, Jeong IK, Koh KH,
Rhie DJ, Yoon SH, Hahn SJ, Kim MS and Jo YH: Upregulation of rat
Ccnd1 gene by exendin-4 in pancreatic beta cell line INS-1:
Interaction of early growth response-1 with cis-regulatory element.
Diabetologia. 49:969–979. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kang JH, Kim MJ, Jang HI, Koh KH, Yum KS,
Rhie DJ, Yoon SH, Hahn SJ, Kim MS and Jo YH: Proximal cyclic AMP
response element is essential for exendin-4 induction of rat EGR-1
gene. Am J Physiol Endocrinol Metab. 292:E215–E222. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
León-Buitimea A, Rodriguez-Fragoso L,
Lauer FT, Bowles H, Thompson TA and Burchiel SW: Ethanol-induced
oxidative stress is associated with EGF receptor phosphorylation in
MCF-10A cells overexpressing CYP2E1. Toxicol Lett. 209:161–165.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lei H and Kazlauskas A: Growth factors
outside of the platelet-derived growth factor (PDGF) family employ
reactive oxygen species/Src family kinases to activate PDGF
receptor alpha and thereby promote proliferation and survival of
cells. J Biol Chem. 284:6329–6336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jeong SH, Park JH, Kim JN, Park YH, Shin
SY, Lee YH, Kye YC and Son SW: Up-regulation of TNF-alpha secretion
by cigarette smoke is mediated by Egr-1 in HaCaT human
keratinocytes. Exp Dermatol. 19:e206–e212. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kim JN, Kim HJ, Jeong SH, Kye YC and Son
SW: Cigarette smoke-induced early growth response-1 regulates the
expression of the cysteine-rich 61 in human skin dermal
fibroblasts. Exp Dermatol. 20:992–997. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Thyss R, Virolle V, Imbert V, Peyron JF,
Aberdam D and Virolle T: NF-kappaB/Egr-1/Gadd45 are sequentially
activated upon UVB irradiation to mediate epidermal cell death.
EMBO J. 24:128–137. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cheong MW, Kuo LH, Cheng YN, Tsai PJ, Ho
LC, Tai HC, Chiu WT, Chen SH, Lu PJ, Shan YS, et al: Loss of Egr-1
sensitizes pancreatic β-cells to palmitate-induced ER stress and
apoptosis. J Mol Med (Berl). 93:807–818. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Parra E, Ferreira J and Ortega A:
Overexpression of EGR-1 modulates the activity of NF-κB and AP-1 in
prostate carcinoma PC-3 and LNCaP cell lines. Int J Oncol.
39:345–352. 2011.PubMed/NCBI
|