Introduction
Gastric cancer (GC) is the fourth most common cause
of cancer-related death worldwide (1). The incidence of GC increases with age,
and the median age at diagnosis is 70 years (2). However, ~10% of the gastric carcinomas
are detected at the age of 45 or younger (3). GC is a multifactorial disease, and both
environmental and genetic factors serve a role in its etiology
(2–4). GC in younger patients is termed
early-onset gastric cancer (EOGC), and is likely caused by a
predisposing genotype that facilitates the development of the
cancer due to various environmental triggers (4). It has been postulated that genetic
factors may be more important in EOGC than in elder patients, as
younger patients have a shorter duration of exposure to
environmental carcinogens (3).
Currently, there are two distinguished
classifications of GC, the World Health Organization (WHO) and the
Laurén classification system. According to the Laurén
classification, which is often used for diversification, there are
two histological subtypes: Diffuse and intestinal (5). Intestinal gastric carcinoma is more
common in elder patients and it is frequently accompanied by
multifocal atrophic gastritis. This is followed by intestinal
metaplasia and leads to cancer via dysplasia. Younger patients more
commonly exhibit a diffuse cancer, in which hereditary factors
serve a major role in the development.
DNA damage response genes are involved in the
preservation of a healthy genome. Defects in cell cycle checkpoints
and DNA repair genes, such as ATM, CHEK2, BRCA1, BRCA2 and
TP53, particularly mutations and/or aberrant downregulation,
may be associated with the development of gastric cancer (6).
The cell cycle checkpoint kinase 2 (CHEK2)
gene serves a significant role in the DNA damage response signaling
network, particularly in cancer development (7). Germline mutations in the gene have been
reported in families of patients with cancer; particularly for
breast cancer (8–10) and prostate cancer (11,12), but
also in other types of cancer (7,13,14),
including gastric cancer (15). The
most prevalent mutant alleles of CHEK2 are three mutant
alleles resulting in protein truncation (1100delC, IVS2G>A,
del5395), one which is a missense variant (I157T) (16).
In humans, the CHEK2 protein is a 65 kDa polypeptide
that consists of 543 amino acids and three different functional
domains. The N terminal SQ/TQ cluster domain contains a region rich
in serine-glutamine and threonine-glutamine motifs, which are
potential sites of phosphorylation by ATR and ATM kinases (part of
the PI3K family) (17). Under
physiological conditions, CHEK2 in retained in the nucleus in an
inactive, monomeric form. DNA damage initiates phosphorylation at
Thr68 and other sites of the SQ/TQ cluster domain. The
phosphorylated site binds to the forkhead-associated domain and
forms a dimer that is further activated by autophosphorylation at
the C-terminal serine/threonine kinase domain (18). As a result, the dimer dissociates
into fully active monomers. The Thr68 phosphorylated form of CHEK2
creates distinct nuclear foci as a reaction to ionizing radiation
(19). Only the activated form of
CHEK2 is present at sites of DNA double stranded breaks. These
observations suggest that CHEK2 is regulated at the site of the DNA
double stranded break, and that phosphorylated CHEK2 has an
important role in the DNA damage response. Our current study is
focused on both forms of CHEK2 to examine their role in the
development of age-dependent subtypes of gastric carcinoma.
The TP53 gene is located on the short arm of
chromosome 17 (17p13.1) (18).
Multiple studies have demonstrated the genetic association between
TP53 alterations and cancer susceptibility. The TP53
gene is the most frequently mutated gene (>50%) in human cancer,
suggesting that the TP53 gene serves a pivotal role in
preventing the formation of cancer (19). In gastric carcinoma the mutations are
located broadly across a gene, from exons 4–11 with hot spots of
mutations at codons 175, 248, 273, 282, 245 and 213. Additionally,
G:C>A:T transitions at CpG sites are the most popular types of
mutation (20).
The p53 protein serves an important role in
regulation or cancer progression through the regulation of
apoptosis, the cell cycle and genomic stability (21). p53 can stimulate DNA repair proteins
when DNA damage is persistent or serious. It can prevent
proliferation by arresting the cell cycle at the G1/S regulation
point following recognition of DNA damage. When a cell is arrested
at this checkpoint, the DNA repair proteins attempt a repair.
However, if the cells are arrested at this checkpoint for a
sufficient amount of time, the cell may instead undergo apoptosis
(22).
The present study focused on the frequency of
genetic alterations in the CHEK2 and TP53 genes, and
the expression patterns of the phosphorylated and
non-phosphorylated CHEK2 and p53 proteins in both conventional
(C)GC and early onset (EO)GC, to assess the effects of CHEK2 and
p53 in the development of the heterogeneous nature of GC.
Materials and methods
Patients
The tissue samples used in the present study
included 89 cases of CGC, diagnosed between 1993–2003 and 107
samples of EOGC, obtained from the Academic Medical Centre in
Amsterdam as well as collected from 24 institutions in the
Netherlands, Finland and Poland. Tumor samples were classified
according using the Laurén classification system (23) as either intestinal or diffuse gastric
adenocarcinomas (Table I). The
tissues were previously used for a previous analysis (24), where immunohistochemical labelling
was performed to determine the expression patterns of several
gastric cancer markers amongst EOGC and CGC. For sequencing
analysis, 53 GC tissues were selected, 35 with EOGC and 18 with
CGC, taking into consideration the DNA quality status of the
samples. The present study was approved by the Medical University
of Lublin Bioethical Committee and was performed in accordance with
the Declaration of Helsinki.
 | Table I.Histological types of GC samples
analyzed in the study (n=196). |
Table I.
Histological types of GC samples
analyzed in the study (n=196).
| Characteristic | Early-onset GC
(n=107) | Conventional GC
(n=89) |
|---|
| Age (range),
years | ≤45 (21–45) | >45 (47–86) |
| Histology, n
(%) |
|
|
|
Intestinal | 21 (20) | 42 (47) |
|
Diffuse | 78 (73) | 37 (42) |
|
Mixed | 8 (7) | 10 (11) |
DNA extraction
Tissues were obtained as previously described by
Sitarz et al (25). Genomic
DNA extraction from formalin-fixed paraffin-embedded (FFPE) tissues
was performed using the QIAamp DNA Mini kit (Qiagen GmbH) or a
Puregene DNA Isolation kit (Gentra, Inc.) in accordance with the
manufacturers' protocols. DNA was quantified using a Quantus™
Fluorometer with the QuantiFluor® dsDNA system according
to the manufacturer's protocol (Promega Corporation). DNA quality
was assessed using an Agilent 2200 TapeStation and Genomic DNA
ScreenTape (Agilent Technologies, Inc.). Further evaluation of the
suitability of the samples for sequencing was performed using an
Infinium HD FFPE QC assay protocol (Illumina, Inc.). The Real-Time
PCR using a CFX96 Touch™ Real-Time PCR Detection system (Bio-Rad
Laboratories, Inc.) was used to assess the amplification status of
each sample.
Library preparation using a target
enrichment workflow for next generation sequencing
Libraries for the sequencing experiments were
prepared using a Trusight Rapid Capture Preparation kit (Illumina,
Inc.) according to the manufacturer's protocol. DNA samples were
standardized to 50 ng and underwent a tagmentation step. The
produced libraries from input of genomic DNA were amplified and
adapter tagged multiple short library fragments of 220–350 base
pairs were prepared. Libraries for each sample were combined to
ensure proper complexity for a single run. Biotin labelled probes
were used to target regions of interest. Further hybridization
steps were performed using custom synthesized oligos that captured
genes with an assigned predisposition towards multiple cancer
types. The sequencing panel, TruSight Cancer Panel (Illumina,
Inc.), targeted 94 genes and 284 SNPs linked to common and rare
cancers. The complete list of target genes and SNPs is available
from illumina.com/techniques/popular-applications/genotyping.html.
Standard pools were enriched with regions of interest through
streptavidin coated beads that bound to the biotinylated probes.
Then, DNA fragments were eluted from the beads and another round of
hybridization and enrichment was performed. The final amplified
post-capture library concentrations were assessed using a Quantus™
Fluorometer according to the manufacturer's protocol. Libraries
were quantified by qPCR using a KAPA Library Quantification kit
(KAPA Biosystems, Inc.). The size of the obtained libraries
fragments was evaluated using an Agilent 2200 TapeStation and High
Sensitivity D1000 ScreenTape system (Agilent Technologies, Inc.).
Post capture enriched libraries were sequenced on a MiSeq
Sequencing Platform (Illumina, Inc.) using the manufacturer's
workflow. The concentration of loaded libraries amounted to 10 pM.
The sequencing experiments were performed using a MiSeq Reagent kit
version 2 (300 cycles).
Data processing
Results from each sample were mapped to the human
reference genome GRCh37, also known as hg19 (Human Genome version
19) using Burrows-Wheeler Aligner (BWA-mem, version 0.7.5). The
reads with low mapping quality scores, unmapped reads and
duplicates were filtered out using Samtools version 0.1.19
(26). Local realignment of reads
around indels (insertion or deletion) and detection of systematic
errors in base quality scores were performed using Genome Analysis
Toolkit (27). The reads mapped
outside the target region were removed. Variant calling for
germline SNPs and indels was performed using the GATK
HaplotypeCaller tool (28). The call
sets of SNPs and small insertions and deletions were separated for
further filtering. Hard filters applied to variant call sets for
SNPs were, QualByDepth (QD)<2.0, RMSMappingQuality (MQ)<40.0,
FisherStrand (FS)>60.0, HaplotypeScore>13.0,
QRankSum<-12.5 and ReadPosRankSum<-8.0; and for INDELs,
QD<2.0, ReadPosRankSum<-20.0, FS>200.0. Filtered variants
were concatenated into one record (VCF file) and the discovered
variants were annotated with SnpEff (version 4.2) using GEnome
MINIng (Gemini) version 0.18.3, and loaded into an SQLite database
(29).
Immunohistochemistry
CHEK2 immunohistochemical staining was performed
using a mouse monoclonal antibody for CHEK2 (Santa Cruz
Biotechnology, Inc.; cat. no. sc5278; 1:200; incubation period 1
h), a rabbit monoclonal antibody against p-CHEK2 (Thr68; Cell
Signaling Technology, Inc.; cat. no. 2197; 1:50; ARS pH 9; 72 h
incubation at 4°C). The sections (4 µm) were deparaffinized and
endogenous peroxidase activity was blocked by immersion in 0.3%
H2O2 and in methanol for 20 min. Antigen
retrieval was performed in sodium citrate/ARS buffer (0.01 M/pH
6.0) for 10 min at 120°C or in Tris/EDTA buffer (10 mM/1 mM; pH
9.0) for 10 min at 120°C. Subsequently, once the samples had cooled
for 10 min, they were washed in PBS and blocked using PBS with 5%
normal goat serum. Then the material was incubated with the
antibodies. Antibody binding was visualized using a Powervision +
poly-HRP detection system (Leica Microsystems; cat. no. PV6107)
with Bright DAB (Immunologic; cat. no. BS04-110) as the chromogen
for CHEK2, and PowerDAB (Immunologic; cat. no. BS03-25) used as the
chromogen for p-CHEK2. Sections were counterstained using
hematoxylin.
TP53 staining; sections (4-µm thick) were
deparaffinized and antigen retrieval was performed by boiling in 10
mM Tris/1 mM EDTA (pH 9.0) for 10 min. Subsequently, slides were
immersed in 0.3% hydrogen peroxide in methanol for 30 min and
nonspecific binding was blocked with 5% normal goat serum for 1 h
at room temperature. The sections were incubated for 1 h at room
temperature with an anti-p53 antibody (monoclonal antibody
combination of DO-7 and BP53-12; Neomarkers, Inc.; 1:2,000). The
Ultravision anti-polyvalent HRP detection system (Lab Vision Corp.)
was used to visualize antibody binding with 3,30-diaminobenzidine
used as the chromogen. Sections were counterstained using
hematoxylin.
Scoring
Scoring of immunohistochemistry was performed for
analysis of CHEK2 and p53 expression. Scoring was performed
independently by two researchers, any disagreements in score were
reanalyzed by the expert using a multiheaded microscopes in
accordance with his judgement. Cases where the staining was not
clearly abnormal were categorized into the normal group. No slides
exhibited increased negative staining around the edges, and the age
of the block had no impact on immunohistochemical staining.
Multiple scoring systems in the literature were considered to
ensure a system that adequately reflected the results generated was
used, and all cases were re-examined based on the agreed upon
scoring parameters.
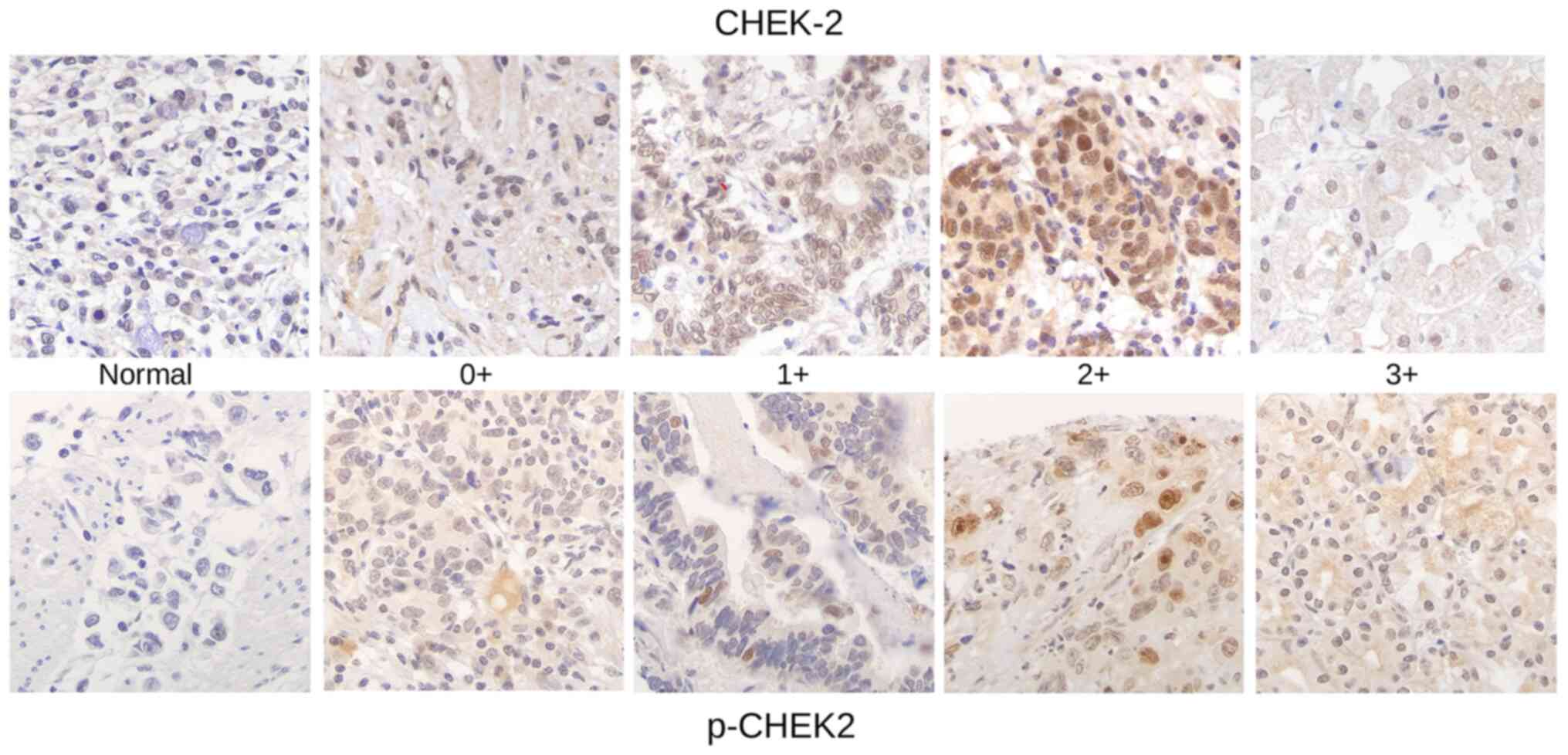
Immunohistochemical labelling revealed the patterns
of CHEK2 and p-CHEK2 expression in GC (Fig. 1). The scoring criteria for the
quantity of the tumor cells were: 1, No staining; 2, weak staining
(≤10% of the cancer cells); 3, moderate staining (10–50% of the
cancer cells); 4, strong staining (nuclear staining in >50% of
the cancer cells). For statistical analysis, scores 0 and 1 were
categorized as ‘CHEK2 low staining’ and scores 2 and 3 as ‘CHEK2
high staining’. The following scoring system was used for p53:
Cases scored as positive showed strong nuclear staining in >30%
of cells, whereas normal mucosa showed scant staining in the
proliferative zone only.
Statistical analysis
Statistical analysis (including immunohistochemistry
staining of CHEK2 and p-CHEK2 proteins, immunohistochemical
staining of p53 and association between the genetic alterations of
CHEK2 and TP53 genes and their protein expression
levels) were performed using the χ2 test. For this
purpose we used GraphPad programme and applied the 2×2 contingency
table. We chose chi-square test with the Yates' continuity
correction, which is designed to make the chi-square approximation
better. We calculated two-tailed (also called two-sided) P-values.
P<0.05 was considered to indicate a statistically significant
difference.
Results
CHEK2 genetic alterations amongst EOGC
and CGC
Next-generation sequencing results were generated
for 53 patients with GC, 35 patients with EOGC and 18 patients with
CGC. DNA from all samples was successfully amplified, libraries
were constructed and a minimum coverage amongst regions of interest
was obtained in all tested samples. Results from next-generation
targeted sequencing for CHEK2 are shown in Table II. A total of 5 different variants
in CHEK2 were found amongst 53 cases: 2 missense variants, 2
synonymous variants and 1 intron alteration. The c.1741G>T
missense alteration in CHEK2 was observed only in one case
and was described as a medium impact mutation, according to Gemini.
This variant seemed to be very rare, as it had not been previously
described in CHEK2 and has never been reported in NCBI
ClinVar or dbSNP (Table SI). Both
missense variants: c.1246A>G (medium) and the synonymous
variant: c.1245C>T (low) were detected in 5 out of 53 cases. The
CHEK2 c.252A>G synonymous variant, which according to
NCBI ClinVar database, is associated with a hereditary
predisposition to colorectal cancer and familial breast cancer, was
detected in 5 cases. Interestingly the intron variant
c.319+379A>G was detected in 47 cases, but according to Gemini,
its impact was low. The detected mutations are fairly uncommon for
gastric carcinoma; they were not found in Catalogue of Somatic
Mutations in Cancer (COSMIC).
| Table II.Presentation of variants identified
in CHEK2 gene in EOGC (n=35) and CGC (n=18) cases. |
Table II.
Presentation of variants identified
in CHEK2 gene in EOGC (n=35) and CGC (n=18) cases.
| Variant type | Protein change | Molecular
consequence | Exon | CGC mutations | EOGC mutations |
|---|
| c.1741G>T | p. Val581Leu | Missense
variant | 16 | – | 1 HET |
| c.1246A>G | p. Lys416Glu | Missense
variant | 12 | 3 HET | 2 HET |
| c.1245C>T | p. Ser415Ser | Synonymous
variant | 12 | 3 HET | 2 HET |
| c.252A>G | p. Glu84Glu | Synonymous
variant | 2 | 3 HET | 2 HET |
|
c.319+379A>G | – | Intron variant | 2 | 7 HET + 10 ALT | 12 HET + 18
ALT |
TP53 genetic alterations amongst EOGC
and CGC
Results from next-generation sequencing for
TP53 are shown in Table
III. A total of 9 variant alterations in TP53 were
detected in 53 patients: 5 missense variants, 2 synonymous
variants, 1 frameshift variant and 1 premature stop codon. The
c.1129A>C missense variant was found in one case (EOGC, 1
HET-heterozygote) and was described as a medium impact mutation,
according to Gemini (Table SII),
and present in dbSNP and ClinVar where it was described to be
associated with Li-Fraumeni syndrome, but not present in the COSMIC
database. The c.1014C>T synonymous variant was present in 1 EOGC
case as heterozygous, was considered a low impact alteration,
described in clinical repositories as related to hereditary cancer
predisposing syndrome and Li-Fraumeni syndrome, and in COSMIC it
was described as being related to skin and upper aerodigestive
tract. A deletion (frameshift variant): c.851_852delCA was observed
in one CGC case, described as high impact, and was considered as
rare as it was not described in any of the databases. The missense
alteration c.844C>G was reported in 1 EOGC case, as
heterozygous, was considered medium impact and was associated with
hereditary cancer predisposing syndrome, and in COSMIC, it was
described as being associated with lung and breast cancer, as well
as hematopoietic and lymphoid, upper aerodigestive tract and
pancreatic neoplastic disease. This alteration was also found in
stomach cancer by Rugge et al (30) and by Xu et al (31).
| Table III.Presentation of variants detected in
TP53 gene in EOGC (n=35) and CGC (n=18) cases. |
Table III.
Presentation of variants detected in
TP53 gene in EOGC (n=35) and CGC (n=18) cases.
| Variant type | Protein change | Molecular
consequence | Exon | CGC mutations | EOGC mutations |
|---|
| c.1129A>C | p. Thr377Pro | Missense
variant | 11 | – | 1 HET |
| c.1014C>T | p. Phe338Phe | Synonymous
variant | 10 | – | 1 HET |
| c.851_852delCA | p. Thr284fs | Frameshift
variant | 8 | 1 DEL | – |
| c.844C>G | p. Arg282Gly | Missense
variant | 8 | – | 1 HET |
| c.818G>A | p. Arg273His | Missense
variant | 8 | 1 HET | – |
| c.734G>A | p. Gly245Asp | Missense
variant | 7 | 1 HET | – |
| c.639A>G | p. Arg213Arg | Synonymous
variant | 6 | 3 HET | 1 HET |
| c.586C>T | p. Arg196* | Nonsense
variant | 6 | 1 HET | – |
| c.215C>G | p. Pro72Arg | Missense
variant | 4 | 11 HET + 6 ALT | 12 HET + 17
ALT |
Missense variant c.818G>A was found in one
patient with CGC, as heterozygous, medium impact, related to
Li-Fraumeni syndrome, thyroid cancer and hereditary cancer
predisposing syndrome according to the ClinVar database; and
according to COSMIC, it was found in multiple studies in stomach
cancer, but predominantly in large intestine, breast, lung, central
nervous system and ovary cancer. Variant c.734G>A was a missense
alteration, found in one case (CGC, 1 HET) and was described as a
medium impact mutation, described in dbSNP and ClinVar as
associated with Li-Fraumeni syndrome and hereditary cancer
predisposing syndrome. In the COSMIC database, it was shown to be
found in large intestine, esophagus, hematopoietic and lymphoid,
lung and breast neoplasms. In cancers of the stomach, the variant
was previously reported by Gleeson et al (32) and Kim et al (33).
Interestingly, variant c.639A>G (synonymous) was
found in 3 CGC cases and 1 EOGC case as heterozygous. This was a
low impact variant, associated with hereditary cancer predisposing
syndrome and Li-Fraumeni syndrome, and was not found in stomach
cancer previously according to COSMIC. However it had been detected
in large intestine, soft tissue and central nervous system
neoplasms. The variant causing an introduction of premature stop
codon, c.586C>T was considered high impact and was detected in
one CGC case (HET), associated with hereditary cancer predisposing
syndrome and Li-Fraumeni syndrome; and in the COSMIC database was
described to be associated with large intestine, breast, esophagus,
skin and stomach cancer. The most frequent variant was the missense
c.215C>G variant, found in 17 cases of CGC (6 ALT and 11 HET)
and 29 EOGC cases (17 ALT and 12 HET), and was also present in
patients with hereditary cancer predisposing syndrome and
Li-Fraumeni syndrome. Based on the COSMIC database, it was found in
the large intestine and soft tissue cancers, but also in the
stomach according to the International Cancer Genome
Consortium.
Immunohistochemistry staining of CHEK2
and p-CHEK2 proteins
The analyzed group encompassed 196 patients, 89 with
CGC and 107 with EOGC (Table IV).
The first approach was to compare the results from
immunohistochemical staining in EOGCs and CGC cases. Two
categories, high and low expression, were distinguished.
Differential expression in the cytoplasmic CHEK2 and
nuclear-phosphorylated CHEK2 (P<0.001 and P=0.011, respectively)
was statistically significant between the analyzed subgroups. The
high expression of CHEK2 in the cytoplasm was predominant in the
CGC subtype, where it was observed in 63% of cases. Phospho-CHEK2
was highly expressed in the nuclei of CGC in 53%, whereas in EOGC,
it was observed in only 34% of samples. The differences in the
nuclear CHEK2 (CHEK2-nucleus) and cytoplasmic phosphorylated CHEK2
(phospho-CHEK2-cytoplasmic) expression patterns between CGC and
EOGC were not significant (P=0.178 and P=0.133 respectively).
| Table IV.Comparison of nuclear and cytoplasmic
CHEK2 and p-CHEK2 expression in CGC (n=89) and EOGC (n=107)
samples. |
Table IV.
Comparison of nuclear and cytoplasmic
CHEK2 and p-CHEK2 expression in CGC (n=89) and EOGC (n=107)
samples.
| Expression | EOGC, n (%) | CGC, n (%) | P-value |
|---|
| CHEK2-nucleus
high | 93 (87) | 70 (79) | 0.178 |
| CHEK2-nucleus
low | 14 (13) | 19 (21) |
|
| CHEK2-cytoplasmic
high | 39 (36) | 56 (63) |
<0.001a |
| CHEK2-cytoplasmic
low | 68 (64) | 33 (37) |
|
| p-CHEK2-nucleus
high | 36 (34) | 47 (53) | 0.011a |
| p-CHEK2-nucleus
low | 71 (66) | 42 (47) |
|
| p-CHEK2-cytoplasmic
high | 29 (27) | 34 (38) | 0.133 |
| p-CHEK2-cytoplasmic
low | 78 (73) | 55 (62) |
|
Based on the result that nuclear CHEK2 expression
was upregulated in both subtypes of GC, this parameter was used to
distinguish a subgroup of samples with high nuclear CHEK2
expression, the ‘CHEK2-nucleus high subgroup’, and compared with
the other parameters (Table V). In
the CHEK2-nucleus high subgroup, there was a statistically
significant difference between the expression of phospho-CHEK2 in
the nucleus (phospho-CHEK2-nucleus) between CGC and ECOG (P=0.002);
in CGC, phospho-CHEK2 in the nucleus was upregulated in 64% of
cases whereas in EOGCs it was only upregulated only in 39% cases.
The levels of cytoplasmic CHEK2 expression (CHEK2-cytoplasmic) also
significantly varied in the two subtypes of GC (P<0.001). In CGC
cytoplasmic CHEK2 was high in 77% of cases, whereas in EOGC it was
only upregulated in 39% of the patients. Expression of
phospho-CHEK2 was significantly higher in CGC compared with EOGC
(P=0.029). The remaining subgroup of low nuclear expression of
CHEK2 (CHEK2-nucleus-low) was analyzed; however, none of the
evaluated parameters were considered to be significantly
associated.
| Table V.Comparison of CHEK2 and p-CHEK2
expression in the nucleus and cytoplasm among the CHEK2-nucleus
high subgroup between CGC (n=70) and EOGC (n=93) samples. |
Table V.
Comparison of CHEK2 and p-CHEK2
expression in the nucleus and cytoplasm among the CHEK2-nucleus
high subgroup between CGC (n=70) and EOGC (n=93) samples.
| Expression | CHEK2-nucleus high
subgroup in EOGC, n (%) | CHEK2-nucleus high
subgroup in CGC, n (%) | P-value |
|---|
| p-CHEK2-nucleus
high | 36 (39) | 45 (64) | 0.002a |
| p-CHEK2-nucleus
low | 57 (61) | 25 (36) |
|
| CHEK2-cytoplasmic
high | 36 (39) | 54 (77) |
<0.001a |
| CHEK2-cytoplasmic
low | 57 (61) | 16 (23) |
|
| p-CHEK2-cytoplasmic
high | 26 (28) | 32 (46) | 0.029a |
| p-CHEK2-cytoplasmic
low | 67 (72) | 38 (54) |
|
Immunohistochemical staining of
p53
The analyzed group on which p53 staining was
performed consisted of 181 patients, 90 with CGC and 91 with EOGC
(Table VI). Staining results for
p53 protein in both subtypes of GC were compared. Two scoring
criteria, positive and negative, were used for p53 expression
assessment. Differential expression in the p53-positive and
p53-negative was statistically significant between the two groups
(P=0.029). The expression of p53 was higher in the CGC group, where
it was observed in 49% of cases. The methodology and results of the
p53 staining was previously published by Milne et al
(24).
| Table VI.Comparison of p53-positive and
-negative expression between CGC (n=90) and EOGC (n=91)
samples. |
Table VI.
Comparison of p53-positive and
-negative expression between CGC (n=90) and EOGC (n=91)
samples.
| p53 expression | EOGC, n (%) | CGC, n (%) | P-value |
|---|
| p53-positive | 29 (32) | 44 (49) | 0.029a |
| p53-negative | 62 (68) | 46 (51) |
|
The EOGC p53-positive and CGC p53-positive
parameters were compared with subgroups of samples with
CHEK2-nucleus and CHEK2-cytoplasmic, phosphorylated and
non-phosphorylated, and high vs. low expression patterns,
respectively (Table VII). There
was a statistically significant difference between the expression
of CHEK2-cytoplasmic high and low, and the EOGC p53-positive and
CGC p53-positive patients (P=0.002). CHEK2-cytoplasmic was highly
expressed in the CGC p53-positive group (68%), whereas 72% of the
EOGC p53-positive patients had low CHEK2-cytoplasmic expression.
The remaining subgroups in Table
VII were analyzed but none of the parameters were found to be
significantly different between the subgroups.
| Table VII.Comparison of high and low CHEK2 and
p-CHEK2 expression in the nucleus and cytoplasm among EOGC
p53-positive (n=29) and CGC p53-positive (n=44) subgroups. |
Table VII.
Comparison of high and low CHEK2 and
p-CHEK2 expression in the nucleus and cytoplasm among EOGC
p53-positive (n=29) and CGC p53-positive (n=44) subgroups.
| Expression | EOGC p53-positive,
n (%) | CGC p53-positive, n
(%) | P-value |
|---|
| CHEK2-nucleus
high | 27 (93) | 38 (86) | 0.604 |
| CHEK2-nucleus
low | 2 (7) | 6
(14) |
|
| CHEK2-cytoplasmic
high | 8
(28) | 30 (68) | 0.002a |
| CHEK2-cytoplasmic
low | 21 (72) | 14 (32) |
|
| p-CHEK2-nucleus
high | 10 (34) | 24 (55) | 0.149 |
| p-CHEK2-nucleus
low | 19 (66) | 20 (45) |
|
| p-CHEK2-cytoplasmic
high | 5
(17) | 14 (32) | 0.264 |
| p-CHEK2-cytoplasmic
low | 24 (83) | 30 (68) |
|
The EOGC p53-negative and CGC p53-negative subgroups
were compared with CHEK2-nucleus and CHEK2-cytoplasmic, both
phosphorylated and non-phosphorylated, high and low expression,
respectively (Table VIII). The
EOGC p53-negative and CGC p53-negative patients had a significantly
different CHEK-2 cytoplasmic expression pattern (P=0.031).
CHEK2-cytoplasmic expression was low in EOGC p53-negative patients
(66%), whereas high expression was found in 57% of CGC p53-negative
patients. Phospho-CHEK2-cytoplasmic low expression was predominant
in the EOGC p53-negative group (76%), and high expression was
observed in the CGC p53-negative group (P=0.033). The other groups
in Table VIII were analyzed;
however, none of the assessed parameters were statistically
significant.
| Table VIII.Comparison of high and low CHEK2 and
p-CHEK2 expression in the nucleus and cytoplasm among EOGC
p53-negative (n=62) and CGC p53-negative (n=46) subgroups. |
Table VIII.
Comparison of high and low CHEK2 and
p-CHEK2 expression in the nucleus and cytoplasm among EOGC
p53-negative (n=62) and CGC p53-negative (n=46) subgroups.
| Expression | EOGC p53-negative,
n (%) | CGC p53-negative, n
(%) | P-value |
|---|
| CHEK2-nucleus
high | 53 (85) | 33 (72) | 0.131 |
| CHEK2-nucleus
low | 9
(15) | 13 (28) |
|
| CHEK2-cytoplasmic
high | 21 (34) | 26 (57) | 0.031a |
| CHEK2-cytoplasmic
low | 41 (66) | 20 (43) |
|
| p-CHEK2-nucleus
high | 20 (32) | 23 (50) | 0.096 |
| p-CHEK2-nucleus
low | 42 (68) | 23 (50) |
|
| p-CHEK2-cytoplasmic
high | 15 (24) | 21 (46) | 0.033a |
| p-CHEK2-cytoplasmic
low | 47 (76) | 25 (54) |
|
Correlation between the genetic
alterations of CHEK2 and TP53 genes and their protein expression
levels
Next, the EOGC and CGC cases were compared for the
different mutation status for SNPs most frequently occurring among
the analyzed groups in both genes: CHEK2 and TP53,
and their protein expression levels (Table SIII). Correlations between mutations
in CHEK2: c.319+379A>G, c.252A>G, c.1246A>G,
c.1245C>T and CHEK2 protein expression in the nucleus and
cytoplasm both in the phosphorylated and non-phosphorylated forms
was compared. There was a statistically significant correlation
between occurrence of the c.319+379A>G alteration in both CGC
and EOGC subtypes and phospho-CHEK2-nucleus expression levels
(P=0.003). The expression of phospho-CHEK2-nucleus was higher in
the CGC c.319+379A>G group (82% of cases). Between CGC and EOGC
with two SNPs in TP53 gene: c.215C>G, c.639A>G and
p53-positive and negative staining, there was no statistically
significant correlations.
Discussion
The primary function of CHEK2 is suppressing tumor
growth, and exhibits proapoptotic and mitotic functions. The
association between changes in CHEK2 gene expression and
cancer risk development have been demonstrated in numerous
case-controlled studies (34–36). The
CHEK2 gene encodes a serine/threonine kinase (CHEK2), which
is activated by ATM, following mobilization of the cascade of DNA
damage response molecules to double-stranded breaks. Additionally,
CHEK2 is widely phosphorylated at Thr68, resulting in its
activation, particularly during the development of precancerous
lesions and in the progression of cancer (37).
In the Polish population, three founder alleles were
found: A missense substitution of an isoleucine for a threonine in
exon 3: I157T and two alterations that truncate the CHEK2 protein:
IVS2+1G>A in exon 3 and 1100delC in exon 10, all of which have
been previously investigated with regard to their association with
predisposition to GC (16).
Li and Stern (38)
found that prior to DNA damage, CHEK2 is associated with chromatin,
and irradiation or topoisomerase inhibitors decrease this
association. They observed that phospho-CHEK2 was released from
chromatin following DNA damage, and accumulated in the soluble
cytoplasmic and soluble nuclear fraction. CHEK2 in human cells
exposed to DNA-damaging agents resulted in immediate redistribution
of the activated CHEK2 throughout the nucleus, rapidly spreading
the checkpoint arrest signal from localized sites of DNA damage to
the soluble mobile proteins, such as Cdc25 or p53 (34). Ćmielová et al (39) showed increased levels of
phospho-CHEK2 in the cytoplasmic fraction after 24 and 48 h of
mitoxantrone treatment. In the present study, only a weak,
insignificant signal was observed in the nuclear fraction at the
same time points.
The reports of the subcellular distribution of CHEK2
are inconsistent and the redistribution of CHEK2 after DNA damage
is very poorly described. An improved understanding of the DNA
damage response and protein distribution in the cells may assist in
improving the efficiency of treatment of several types of cancer
and may highlight specific novel therapeutic targets.
Sequencing results revealed 5 different SNPs within
the studied group of patients. Variants c.1246A>G (missense
variant), c.1245C>T (synonymous variant) and c.252A>G
(synonymous variant) were similarly distributed between the
subgroups. Amongst the SNPs detected, 3 were HET in CGC and 2 were
HET in EOGC. Interestingly, the intron variant c.319+379A>G was
present in a high number of cases; 94% of CGCs and 86% EOGCs cases.
Only variant c.252A>G has been previously shown to be associated
with several types of cancer, when searching NCBI ClinVar database.
However, in reference to GC, all the listed SNPs seem to be very
rare and thus far, have not been broadly described. Interestingly,
the intron variant c.319+379A>G, detected in the present study,
was previously shown to be associated with esophageal squamous cell
carcinoma risk, as reported by Li et al (40).
The tumor suppressor gene TP53 is one of the
most regularly mutated genes in human cancers, and TP53
alterations are present in ~77% of stomach cancers (41). Mutations of TP53 have been
detected in the early stages of gastric carcinoma, and their
frequency increases as the tumor progressed (42). TP53 has been termed ‘the
guardian of the genome’ (41).
Genomic instability causes genetic variability and accelerates the
rate at which a cell acquires and accumulates mutations, which may
underlie the initiation of tumorigenesis (43). In the present study, 9 different
genetic alterations of the TP53 gene were detected. Most of
these occurred sporadically; however, variant c.215C>G was
present in 17 cases of CGC (6 ALT and 11 HET) and 29 cases of EOGC
cases (17 ALT and 12 HET), whereas variant c.639A>G was present
in 3 CGC cases and 1 EOGC cases (all HET). The clinical
significance of the detected variants was assessed using dbSNP and
ClinVar. Most of these were associated with the Li-Fraumeni
syndrome and hereditary cancer predisposing syndrome. According to
COSMIC, five of the detected variants were previously shown to be
associated with stomach cancer (c.844C>G, c.818G>A,
c.734G>A, c.586C>T and c.215C>G). In the present study, it
was demonstrated that the TP53 mutations may accumulate over
a very long period of time (lifetime) (44). With aging, genetic damage may
increase, increasing the importance of the function of TP53,
and therefore making an individual more vulnerable to loss of
function mutations.
Immunohistochemistry analysis may have additional
value for the assessment of the ATM>CHEK2>p53 pathway.
When CHEK2 is phosphorylated at Thr68, it is activated. Therefore,
expression of the phosphorylated protein may be used as a potential
marker of active CHEK2 status (45).
The constitutive activation of the DNA damage checkpoint pathway
may be associated with an increased level of p53 alterations in
cancer, taking into consideration that p53 is a downstream target
of ATM and CHEK2. The immunohistochemical analyses performed in the
present study included analysis of cytoplasmic and nuclear
expression of CHEK2 and phospho-CHEK2.
The levels and distribution of expression of these
proteins appeared to vary between CGC and EOGC. Interestingly, the
levels of p-CHEK2 increased with age, and a statistical difference
between the two subtypes with regard to nuclear p-CHEK2 expression
was observed. Additionally, high nuclear CHEK2 expression was also
more prevalent in CGC, following stratification based on levels of
nuclear CHEK2 expression. Cytoplasmic phospho-CHEK2 expression was
increased. The phosphorylated forms of both CHEK2 types may be an
indicator of GC development. The levels of CHEK2 protein in the
nuclei was elevated in both subtypes of GC, and this outcome
supports further study into the potential therapeutic value of
CHEK2.
p53 staining was performed on 181 tumor samples, in
both age dependent subtypes of GC; expression of the p53 protein
increased with age. Immunohistochemistry analysis of p53 may
explain the biology of the tumor. Multiple studies have used
immunohistochemistry analysis to assess p53 expression. In lung
cancer, a meta-analysis showed that positive immunohistochemistry
staining of p53 may be used as a prognostic biomarker (46). The results of the present study
showed that p53 expression could be used to distinguish age
dependent onset of GC.
The EOGC p53-positive and CGC p53-positive groups
were compared based on phosphorylation of CHEK2. There was a
statistically significant difference between the expression of
CHEK2-cytoplasmic high and low, and the EOGC p53-positive and CGC
p53-positive patients. The expression of CHEK2 in the cytoplasm was
increased (68%) in the CGC p53-positive group. Statistically
important differences between CHEK2-cytoplasmic high and low, and
the EOGC and CGC p53-negative patients were also observed.
CHEK2-cytoplasmic expression was increased in CGC p53-negative
patients (57%), and its expression was higher in the CGC
p53-positive subgroup (68%). Phospho-CHEK2-cytoplasmic was also
higher amongst the CGC TP53-negative group. Together, these results
suggests a link between the expression of p53 and CHEK2 cytoplasmic
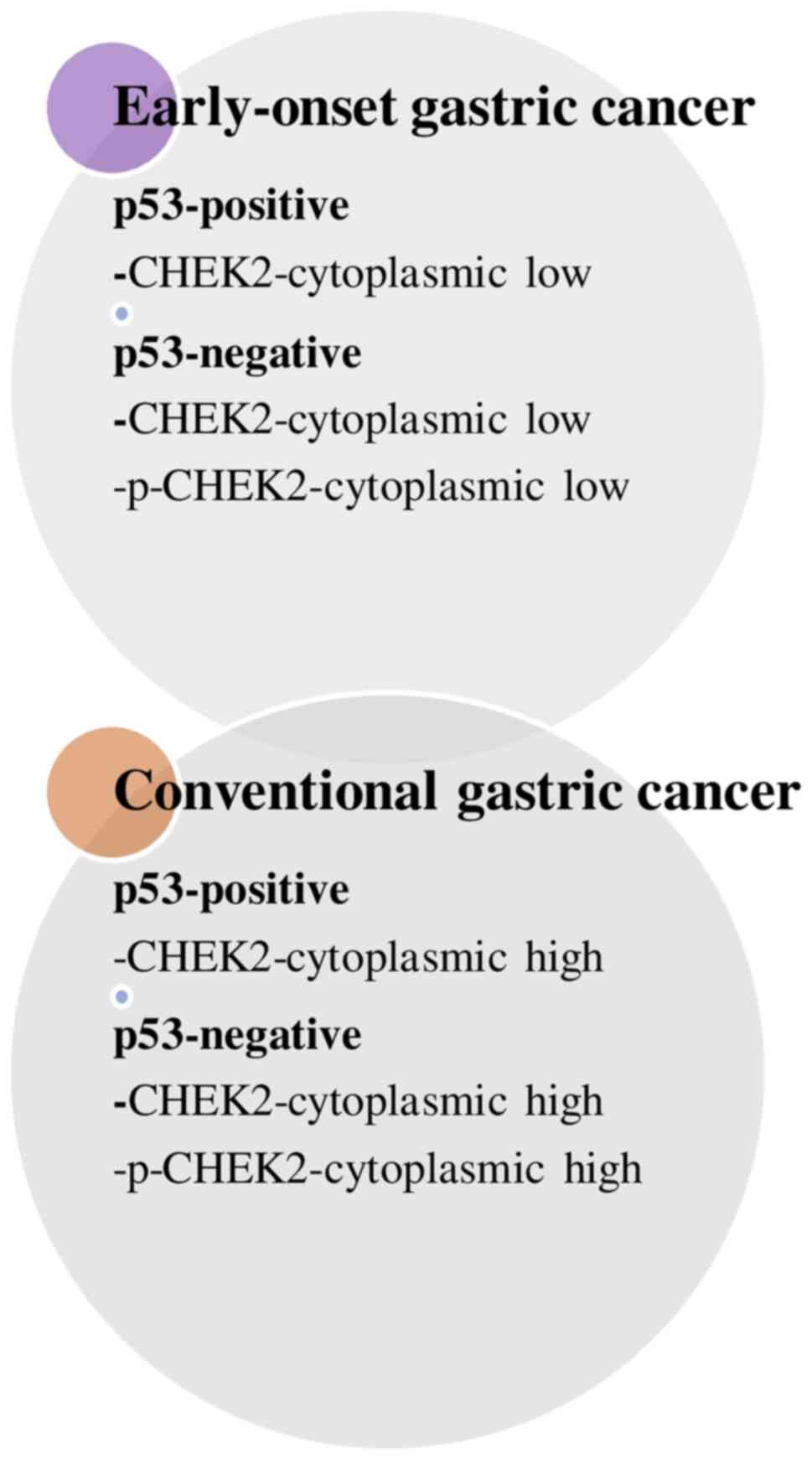
proteins in GC development based on age. Fig. 2 summarizes the primary differences
between EOGC and CGC with respect to CHEK2 and p53 staining.
Non-coding regions possess regulatory functions for
the coding regions. Thus coding regions are the primary actors, but
their function is regulated by non-coding regions. Thus any
modifications to the non-coding may have an impact on the
expression and regulation of the expressed genes. There was a
statistically significant correlation between occurrence of the
c.319+379A>G intronic alteration in both CGC and EOGC subtypes
and phospho-CHEK2-nuclear expression, which increased with age.
The present study has some limitations. First, the
number of biopsies from patients was a limiting factor. The
selected subgroups were small, and thus it is difficult to observe
the differences in SNP frequencies in the CHEK2 gene. Use of a
benign gastric disease control group may better allow for
determination of whether the protein expression of these two genes
may be used as biomarkers of early gastric cancer development.
In vitro studies are required to determine the functional
effects of the detected SNPs in more detail. It is also probable
that different allele frequency amongst individuals of different
ethnicities may have an impact on the obtained data. Thus, a more
diverse cohort may allow for identification of differences in
frequencies of SNPs in different populations around the world.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was partly supported by a grant
from the Polish Ministry of Science and Higher Education (grant no.
NN402423838).
Availability of data and materials
The NGS datasets generated and/or analyzed during
the current study are available in the Open Science Framework
Repository. Outcome for the 94 genes and 284 SNPs presented in the
NGS panel are available at http://osf.io/9483c/(snps.variants-all.csv) and
http://osf.io/zxwg9/(genes.variants-all.csv).
Authors' contributions
PW and RS conceptualized the study. JM, FM, GJAO and
RS developed the methodology. PK and PW performed the data
processing. JM, PK, AB and RS analyzed the results. JM, PK, MS, AB,
FM, PW, RM, GJAO and RS performed the experiments. PW and RS
confirmed the authenticity of the data. JM, MS, AB and RS wrote the
original draft. JM, PK, MS, AB, FM, PW, RM, GJAO and RS reviewed
and edited the manuscript. PW, GJAO and RS supervised the study. RM
and RS acquired funding. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All procedures involving human participants were in
accordance with the ethical standards of the Medical University of
Lublin Bioethical Committee (Lublin, Poland; approval no.
KE-0254/322/2019) and with the 1964 Declaration of Helsinki and its
later amendments or comparable ethical standards. Due to the
retrospective nature of the study, patient consent was waived by
the ethics committee.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zou Z and Jemal A: Cancer statistics,
2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Howlader N, Noone AM, Krapcho M, Neyman N,
Aminou R, Waldron W, Altekruse SF, Kosary CL, Ruhl J, Tatalovich Z,
et al: SEER Cancer Statistics Review, 1975–2008, National Cancer
Institute. Bethesda; MD: pp. 192011
|
|
3
|
Milne AN, Sitarz R, Carvalho R, Carneiro F
and Offerhaus GJ: Early onset gastric cancer: On the road to
unraveling gastric carcinogenesis. Curr Mol Med. 7:15–28. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Milne AN, Carneiro F, O'Morain C and
Offerhaus GJ: Nature meets nurture: Molecular genetics of gastric
cancer. Hum Genet. 126:615–628. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Milne AN, Offerhaus GJ and Carneiro F:
Histopathology of familial and early-onset gastric cancer.
Diagnostic Histopathol. 17:62–68. 2011. View Article : Google Scholar
|
|
6
|
Slavin T, Neuhausen SL, Rybak C, Solomon
I, Nehoray B, Blazer K, Niell-Swiller M, Adamson AW, Yuan YC, Yang
K, et al: Genetic gastric cancer susceptibility in the
international clinical cancer genomics community research network.
Cancer Genet. 216–217:111–119. 2017. View Article : Google Scholar
|
|
7
|
Cybulski C, Górski B, Huzarski T, Masojć
B, Mierzejewski M, Debniak T, Teodorczyk U, Byrski T, Gronwald J,
Matyjasik J, et al: CHEK2 is a multiorgan cancer susceptibility
gene. Am J Hum Genet. 75:1131–1135. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Al-Rakan MA, Hendrayani SF and Aboussekhra
A: CHEK2 represses breast stromal fibroblasts and their paracrine
tumor-promoting effects through suppressing SDF-1 and IL-6. BMC
Cancer. 16:5752016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Desrichard A, Bidet Y, Uhrhammer N and
Bignon YJ: CHEK2 contribution to hereditary breast cancer in
non-BRCA families. Breast Cancer Res. 13:R1192011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang N, Ding H, Liu C, Li X, Wei L, Yu J,
Liu M, Ying M, Gao W, Jiang H and Wang Y: A novel recurrent CHEK2
Y390C mutation identified in high-risk Chinese breast cancer
patients impairs its activity and is associated with increased
breast cancer risk. Oncogene. 34:5198–5205. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dong X, Wang L, Taniguchi K, Wang X,
Cunningham JM, McDonnell SK, Qian C, Marks AF, Slager SL, Peterson
BJ, et al: Mutations in CHEK2 associated with prostate cancer risk.
Am J Hum Genet. 72:270–280. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hale V, Weischer M and Park JY: CHEK2 (*)
1100delC mutation and risk of prostate cancer. Prostate Cancer.
2014:2945752014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cybulski C, Masojc B, Oszutowska D,
Jaworowska E, Grodzki T, Waloszczyk P, Serwatowski P, Pankowski J,
Huzarski T, Byrski T, et al: Constitutional CHEK2 mutations are
associated with a decreased risk of lung and laryngeal cancers.
Carcinogenesis. 29:762–765. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kilpivaara O, Alhopuro P, Vahteristo P,
Aaltonen LA and Nevanlinna H: CHEK2 I157T associates with familial
and sporadic colorectal cancer. J Med Genet. 43:e342006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Skierucha M, Milne AN, Offerhaus GJA,
Polkowski WP, Maciejewski R and Sitarz R: Molecular alterations in
gastric cancer with special reference to the early-onset subtype.
World J Gastroenterol. 22:2460–2474. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Teodorczyk U, Cybulski C, Wokołorczyk D,
Jakubowska A, Starzyńska T, Lawniczak M, Domagała P, Ferenc K,
Marlicz K, Banaszkiewicz Z, et al: The risk of gastric cancer in
carriers of CHEK2 mutations. Fam Cancer. 12:473–478. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Buscemi G, Carlessi L, Zannini L, Lisanti
S, Fontanella E, Canevari S and Delia D: DNA damage-induced cell
cycle regulation and function of novel Chk2 phosphoresidues. Mol
Cell Biol. 26:7832–7845, 20060. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McBride OW, Merry D and Givol D: The gene
for human p53 cellular tumor antigen is located on chromosome 17
short arm (17p13). Proc Natl Acad Sci USA. 83:130–134. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Barnoud T, Parris JLD and Murphy ME:
Common genetic variants in the TP53 pathway and their impact on
cancer. J Mol Cell Biol. 11:578–585. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fenoglio-Preiser CM, Wang J, Stemmermann
GN and Noffsinger A: TP53 and gastric carcinoma: A review. Hum
Mutat. 21:258–270. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Blondal JA and Benchimol S: The role of
p53 in tumor progression. Semin Cancer Biol. 5:177–186.
1994.PubMed/NCBI
|
|
22
|
Lakin ND and Jackson SP: Regulation of p53
in response to DNA damage. Oncogene. 18:7644–7655. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Laurén P: The two histological main types
of gastric carcinoma: Diffuse and so-called intestinal-type
carcinoma. An attempt at a histo-clinical classification. Acta
Pathol Microbiol Scand. 64:31–49. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Milne AN, Carvalho R, Morsink FM, Musler
AR, de Leng WW, Ristimäki A and Offerhaus GJ: Early-onset gastric
cancers have a different molecular expression profile than
conventional gastric cancers. Mod Pathol. 19:564–572. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sitarz R, Leguit RJ, de Leng WW, Polak M,
Morsink FM, Bakker O, Maciejewski R, Offerhaus GJ and Milne AN: The
COX-2 promoter polymorphism-765 G>C is associated with
early-onset, conventional and stump gastric cancers. Mod Pathol.
21:685–690. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R; 1000 Genome
Project Data Processing Subgroup, : The sequence Alignment/Map
format and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Narzisi G, O'Rawe JA, Iossifov I, Fang H,
Lee YH, Wang Z, Wu Y, Lyon GJ, Wigler M and Schatz MC: Accurate de
novo and transmitted indel detection in exome-capture data using
microassembly. Nat Methods. 11:1033–1036. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cingolani P, Platts A, Wang le L, Coon M,
Nguyen T, Wang L, Land SJ, Lu X and Ruden DM: A program for
annotating and predicting the effects of single nucleotide
polymorphisms, SnpEff: SNPs in the genome of Drosophila
melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 6:80–92.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rugge M, Shiao YH, Busatto G, Cassaro M,
Strobbe C, Russo VM, Leo G, Parenti AR, Scapinello A, Arslan P and
Egarter-Vigl E: The p53 gene in patients under the age of 40 with
gastric cancer: Mutation rates are low but are associated with a
cardiac location. Mol Pathol. 53:207–210. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu JH, Chen LR, Wang HJ and Yao LF:
Application of immunohistochemistry-Laser microdissection-PCR
technique in detecting p53 gene mutation in paraffin sections of
gastric cancer. Zhonghua Bing Li Xue Za Zhi. 33:342–345. 2004.(In
Chinese). PubMed/NCBI
|
|
32
|
Gleeson CM, Sloan JM, McManus DT, Maxwell
P, Arthur K, McGuigan JA, Ritchie AJ and Russell SE: Comparison of
p53 and DNA content abnormalities in adenocarcinoma of the
oesophagus and gastric cardia. Br J Cancer. 77:277–286. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim SS, Bhang CS, Min KO, Chae HS, Choi
SW, Lee CD, Lim KW, Chung IS and Park DH: p53 mutations and
microsatellite instabilities in the subtype of intestinal
metaplasia of the stomach. J Korean Med Sci. 17:490–496. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bartek J and Lukas J: Chk1 and Chk2
kinases in checkpoint control and cancer. Cancer Cell. 3:421–429.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cybulski C, Wokołorczyk D, Jakubowska A,
Huzarski T, Byrski T, Gronwald J, Masojć B, Deebniak T, Górski B,
Blecharz P, et al: Risk of breast cancer in women with a CHEK2
mutation with and without a family history of breast cancer. J Clin
Oncol. 29:3747–3752. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Siołek M, Cybulski C, Gąsior-Perczak D,
Kowalik A, Kozak-Klonowska B, Kowalska A, Chłopek M, Kluźniak W,
Wokołorczyk D, Pałyga I, et al: CHEK2 mutations and the risk of
papillary thyroid cancer. Int J Cancer. 137:548–552. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zoppoli G, Solier S, Reinhold WC, Liu H,
Connelly JW Jr, Monks A, Shoemaker RH, Abaan OD, Davis SR, Meltzer
PS, et al: CHEK2 genomic and proteomic analyses reveal genetic
inactivation or endogenous activation across the 60 cell lines of
the US National Cancer Institute. Oncogene. 31:403–418. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li J and Stern DF: DNA damage regulates
Chk2 association with chromatin. J Biol Chem. 280:37948–37956.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ćmielová J, Lesná M and Řezáčová M:
Subcellular localization of proteins responding to
mitoxantrone-induced DNA damage in leukaemic cells. Folia Biol
(Praha). 61:60–65. 2015.PubMed/NCBI
|
|
40
|
Li WQ, Hu N, Hyland PL, Gao Y, Wang ZM, Yu
K, Su H, Wang CY, Wang LM, Chanock SJ, et al: Genetic variants in
DNA repair pathway genes and risk of esophageal squamous cell
carcinoma and gastric adenocarcinoma in a Chinese population.
Carcinogenesis. 34:1536–1542. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lane DP: Cancer. p53, guardian of the
genome. Nature. 358:15–16. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Vousden KH and Prives C: P53 and
prognosis: New insights and further complexity. Cell. 120:7–10.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Feng Z, Hu W, Teresky AK, Hernando E,
Cordon-Cardo C and Levine AJ: Declining p53 function in the aging
process: A possible mechanism for the increased tumor incidence in
older populations. Proc Natl Acad Sci USA. 104:16633–16638. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
DiTullio RA Jr, Mochan TA, Venere M,
Bartkova J, Sehested M, Bartek J and Halazonetis TD: 53BP1
functions in an ATM-dependent checkpoint pathway that is
constitutively activated in human cancer. Nat Cell Biol.
4:998–1002. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
46
|
Steels E, Paesmans M, Berghmans T, Branle
F, Lemaitre F, Mascaux C, Meert AP, Vallot F, Lafitte JJ and
Sculier JP: Role of p53 as a prognostic factor for survival in lung
cancer: A systematic review of the literature with a meta-analysis.
Eur Respir J. 18:705–719. 2001. View Article : Google Scholar : PubMed/NCBI
|