Introduction
Colorectal cancer (CRC) is the third most common
cancer and the fourth most common cause of cancer-associated death
worldwide (1). In China, the
incidence of CRC is increasing due to physical inactivity,
unhealthy diet, obesity and long-term smoking (2). Over the last decade, colonoscopy
screening has significantly improved the 5-year survival rates of
patients with CRC. For example, a previous study reported that
patients who did not take part in the screening program showed
significantly lower cancer-specific survival compared with those
who did in the Pudong New Area of Shanghai of China (hazard ratio
(HR)=1.46; 95% confidence interval (CI): 1.12–1.91) (3–5).
Nevertheless, ~40% of patients who undergo surgery, which is
applied in combination with chemotherapy or radiation therapy,
subsequently experience local and systemic recurrence or resistance
(6). Among primary risk factors,
complex genomic alterations, which are essential in the mechanism
of CRC pathophysiology, may induce resistance of CRC to drug
therapy, thus making CRC the fourth most common cause of
cancer-associated mortality (4,7).
Consequently, investigating the mechanisms underpinning CRC
chemosensitivity is important to improve treatment.
Under pathological conditions, the Janus kinase
(JAK)-STAT signaling pathway has been shown to regulate
proliferation, differentiation and angiogenesis of malignant tumor
cells and to promote the development of several malignant tumors,
such as breast cancer and esophageal cancer (8,9). This
pathway is constitutively activated in myeloproliferative diseases
and in various solid tumors, including hepatocellular carcinomas,
prostate, breast, head and neck, lung and CRC (10). Although the exact dysregulation
mechanism of JAK/STAT signaling in CRC remains unclear, the
JAK/STAT signaling pathway has been suggested as therapeutic target
for the treatment of CRC (11,12).
Myeloid cell leukemia-1 (Mcl-1) has sequence and
functional similarity to Bcl-2 (13). Mcl-1 has a short half-life and is a
highly regulated protein (14). The
upstream regulatory kinase cascades of Mcl-1 transcription include
JAK/STAT, PI3K and mitogen-activated protein kinases (15). Previous studies have suggested that
Mcl-1 may have a significant part in the survival or resistance of
a variety of cancer cells, such as liver cancer and multiple
myeloma (14,16).
Ruxolitinib (aslo named INCB018424, Jakari) is a
small molecule inhibitor of the JAK1/2 kinase, which has been
approved by the USA Food and Drug Administration for the treatment
of myelofibrosis and polycythemia vera (17,18).
Recently, new clinical trials have tested the efficacy of
ruxolitinib in the treatment of inflammatory-driven solid tumors
(19–21). It was reported that the therapeutic
effects observed in patients were not related to JAK mutational
capacity and that ruxolitinib initiates only restricted anticlonal
activity, thus poignantly altering the inflammatory
microenvironment (22). Therefore,
ruxolitinib may have potential for the treatment of
inflammatory-driven cancer types, such as pancreatic cancer and
CRC. The present study aimed to investigate the inhibitory
mechanisms of ruxolitinib on CRC cells both in vitro and
in vivo.
Materials and methods
Reagents
Primary antibodies against caspase-3 (cat. no.
9662S), cleaved caspase-3 (cat. no. 9664), caspase-9 (cat. no.
9502), cleaved caspase-9 (cat. no. 9501), caspase-8 (cat. no.
4790), PARP (cat. no. 9532), cleaved PARP (cat. no. 9541), Mcl-1
(cat. no. 5453), Bcl-2 (cat. no. 2872), Bcl-xL (cat. no. 2764), Bak
(cat. no. 12105), JAK1 (cat. no. 3344), JAK2 (cat. no. 3230), STAT1
(cat. no. 9172) and β-actin (cat. no. 4970) were acquired form Cell
Signaling Technology, Inc. Anti-Mcl-1 antibody (cat. no. S-19),
control siRNA-A (cat. no. sc-37007), and STAT1p84/p91 siRNA (h)
(cat. no. sc-44123) were from Santa Cruz Biotechnology, Inc.
Anti-STAT1 [phosphorylated (p)S727] (cat. no. ab109461) antibody
was acquired from Abcam. Annexin V-FITC was purchased from BD
Bioscience. Z-VAD-FMK was from Selleck Chemicals. Monoclonal
antibody anti-Bak (cat. no. TC-100) was from Merck KGaA. JC-1 dye
was purchased from AAT Bioquest, Inc.
Cell line and culture conditions
Human CRC cell lines LS411N, SW620, DLD-1, HCT116,
SW480 and mouse CRC cell line, CT-26, were obtained from the
American Type Culture Collection. Cells were cultured in Dulbecco's
Modified Eagle's Medium supplemented with 10% FBS (both purchased
from Gibco, Thermo Fisher Scientific), 100 U/ml of penicillin and
100 µg/ml of streptomycin (cat. no. 15070-063, Gibco; Thermo Fisher
Scientific, Inc.) in a humidified atmosphere containing 5%
CO2/95% air at 37°C.
Cell viability assay
An MTT assay was used for determining ruxolitinib
cytotoxicity against CRC cells. Briefly, 2,000 cells/well were
plated in 96-well plates and incubated for 24 h and then exposed to
gradually increasing concentrations (0, 2.5, 5, 10, 15, 20, 25, 30,
40, 50 µM) of ruxolitinib for 48 h. MTT solution (prepared in
serum-free growth medium) was added to each well and was incubated
for another 4 h at 37°C. DMSO was added and the absorbance at 570
nm was determined using a microplate reader. CCK-8 assay and
real-time monitoring of cell viability using an xCELLigence system
were performed to determine the cytotoxicity of ruxolitinib against
CRC cells. Briefly, LS411N and SW620 cells were seeded into 96-well
plates at a density of 2,000 cells/well, incubated for 24 h at 37°C
and then exposed to different concentrations (0, 2.5, 5, 10, 15,
20, 25, 30, 40, 50 µM) of ruxolitinib for 48 h at 37°C. CCK-8 was
added to the plates and incubated for 1 h at 37°C. The absorbance
was measured at a wavelength of 570 nm, using a microplate reader.
The in vitro cell growth assay was performed on an
xCELLigence system from ACEA Biosciences Inc., using E-plates.
Briefly, the E-plates with 50 µl of growth medium in each well were
placed into the RTCA-DP instrument for baseline test. The LS411N
and SW620 cells were seeded into the plates at a density of 5,000
cells/well in 100 µl of growth medium and treated with indicated
concentrations of ruxolitinib. The E-plates were incubated at room
temperature for 30 min and placed into the RTCA-DP instrument for
data collection at 15 min intervals.
Early/late apoptosis analyzed by flow
cytometry
After treatment with various concentrations of
ruxolitinib for 48, 72, 96 h, human CRC LS411N, SW620 cells were
collected and washed with ice-cold PBS before staining with Annexin
V-FITC for 15 min at room temperature in the dark followed by PI
staining for 5 min at room temperature. The samples were analyzed
with a BD FACSCanto flow cytometer (BD Biosciences). Data analysis
was performed by the software WinMDI 2.9 (BD Biosciences). Human
CRC LS411N and SW620 cells were treated with Z-VAD-FMK for 1 h
followed by ruxolitinib treatment for another 48 h, and were
collected and analyzed as aforementioned.
Western blotting
LS411N, SW620 cells were lysed in Laemmli lysis
buffer (Merck, cat. no. S3401). Proteins (25 µg/lane) were loaded
in denaturing 12% or 15% SDS-PAGE gels and transferred to
nitrocellulose membranes. The membranes were then incubated with 5%
non-fat milk for 2 h at room temperature, washed with PBS
containing 0.1% Tween-20 and incubated with primary antibodies
(1:1,000 dilution) overnight at 4°C. After washing with PBST the
membrane was incubated with secondary antibodies conjugated with
horseradish peroxidase (1:2,000 dilution) for 2 h at room
temperature. Signals were visualized using the Pierce ECL Plus kit
(cat. no. 32132) with a chemiluminescence system (Amersham;
Cyvita). β-actin was used as a loading control.
Mitochondrial membrane potential
assay
To examine the mitochondrial membrane potential,
JC-1 staining was used. Human CRC LS411N, SW620 cells were
incubated with various concentrations of ruxolitinib for 48 h as
aforementioned. Then, the cells were harvested, washed with PBS and
resuspended in JC-1 staining solution at room temperature for 10
min. Cells were detected by a BD FACSCanto flow cytometer (BD
Biosciences). Data analysis was performed using WinMDI 2.9 software
(BD Biosciences).
Plasmid construction and
transfection
The coding sequences of human Mcl-1 mRNA were
amplified by PCR using total cDNA as previously described (14). The PCR products were digested with
HindIII and EcoRI and subcloned into the pcDNA3.1
vector (obtained from co-author Shengjie Zhang's lab). The
integrity of the respective plasmid constructs was confirmed using
PCR. One microgram pcDNA3.1-negative control (0.2 µg/ml) and
pcDNA3.1/Mcl-1 (0.2 µg/ml) was transfected in SW620 and LS411N
cells using Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) at 37°C for 24 h. After 24 h, the LS411N
and SW620 cells were used for subsequent experiments.
Reverse transcription-quantitative
(RT-q)PCR analysis
Total RNA was isolated from LS411N and SW620 cells
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocols
(23). Complementary DNA synthesis
was performed using the PrimeScript RT Reagent kit (Takara Bio,
Inc.). Real-time PCR amplification was performed using the ABI 7300
Fast Real-Time PCR system (Applied Bioscience; Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions.
The amplification reactions were performed using 1X Power SYBR
Green PCR Master mix (Applied Biosystems; Thermo Fisher Scientific,
Inc.). PCR primers for Mcl-1 (forward, 5′-GCTTCGGAAACTGGACAT-3′;
reverse, 5′-CACAAACCCATCCCAGCC-3′), β-actin (forward,
5′-ACACCCCAGCCATGTACGTT-3′; reverse, 5′-TCACCGGAGTCCATCACGAT-3′)
were designed using Primer Premier 5 software (Premier Biosoft
International). The standard temperature profile, including initial
denaturing, annealing and extension, was used as previously
described (14).
RNA interference of STAT1
Short interfering (si)RNAs for STAT1 (cat. no.
sc-44123) and non-targeting siRNA negative control (cat. no.
sc-37007) were obtained from Santa Cruz Biotechnology, Inc. LS411N
and SW620 cells were transfected with 1 µg siRNA (0.2 µg/ml) using
Lipofectamine 2000 reagent at 37°C for 24 h, according to the
manufacturer's instructions. Before treatment, cells were incubated
for 24 h and the silencing efficiency of the siRNA was determined
using western blotting as aforementioned. Subsequent experiments
were performed 24 h post-transfection.
Measurement of Bak conformational
change
LS411N and SW620 cells were fixed in 4%
paraformaldehyde for 10 min at room temperature, washed three times
with PBS and permeabilized with 0.1% PBS-Tween-20 for 20 min. Cells
were then incubated with anti-active Bak for 30 min at 4°C,
followed by incubation with 1:200 dilution of FITC-conjugated
goat-anti-mouse IgG (Santa Cruz Biotechnology, Inc. cat. no.
sc-2010) for 30 min at 4°C and analyzed using a BD FACSCanto flow
cytometer (BD Biosciences). Data analysis was performed using
WinMDI 2.9 software (BD Biosciences).
Immunoprecipitation
Protein-protein interactions were determined by
co-immunoprecipitation (Co-IP) analysis. LS411N and SW620 cells
were lysed in co-immunoprecipitation lyse-bind-wash buffer [50 mM
Tris-HCl, 150 mM NaCl, 1% NP-40 (V/V), 2 mM EDTA] containing 1%
protease inhibitor cocktail (Pierce; Thermo Fisher Scientific,
Inc.) at 4°C with rotation for 60 min. The lysate was then
centrifuged at 12,000 × g for 10 min at 4°C. After that, samples
(800 µg protein for control or indicated concentrations of
ruxolitinib treatment were incubated with anti-Bak antibody at 4°C
overnight. A total of 50 µl Protein A/G agarose was added to each
sample and incubated for another 4 h at 4°C. Samples were washed
three times and subjected to immunoblot analysis using anti-Mcl-1
antibody as aforementioned.
Animals and antitumor activity in
vivo
In total, 14 Balb/c nude mice, male, 6 weeks old,
weighing 20–25 g, were obtained from Vital River Laboratories. All
the animals were housed in an environment with temperature of
22±1°C, relative humidity of 50±1% and a light/dark cycle of 12/12
h and allowed access ad libitum to water and diet. All
animal studies (including the mice euthanasia procedure) were
approved and performed in compliance with the regulations and
guidelines of Zhejiang University Institutional Animal Care
Committee (Hangzhou, China; approval no. ZJU20180108020) and
conducted according to the Association for Assessment and
Accreditation of Laboratory Animal Care International and the
Institutional Animal Care and Use Committee guidelines.
Human CRC LS411N xenografts were established by
subcutaneously injecting 1×107 cells into the right
flank of the nude mice. When the tumors were palpable, the
tumor-bearing mice were randomized into two groups: Treatment group
and control group with seven mice/group. The treatment group
received ruxolitinib (150 mg/kg body weight) by oral gavage every 2
days for 14 days, while the control group received the same amount
of saline. At the end of experiment, mice were anesthetized with an
intraperitoneal injection of sodium pentobarbital (50 mg/kg body
weight) and euthanized by cervical dislocation.
Tumor size was measured using a micrometer caliper.
Tumor volume (mm3) was calculated using the following
formula: Length (mm) × width (mm) × width (mm)/2. Tumor volume data
are expressed as the mean ± SEM, while the rest of the data are
presented as mean ± SD. In addition, body weight for each mouse was
measured to evaluate the toxicity of the treatment. Moreover, blood
was collected at end of time point. The blood samples were
centrifuged at 500 × g for 5 min at 4°C, and then the serum was
collected. Serum creatine kinase and alanine aminotransferase
levels were detected using ELISA kits (creatine kinase, Biovision,
cat. no. E4607) and alanine aminotransferase (Biovision, cat. no.
K752), according to the manufacturer's instructions.
Immunohistochemistry
In total, 44 pairs of human CRC biopsy and adjacent
normal tissue specimens (5/20/2009-08/05/2015) were collected from
patients at Zhejiang Cancer Hospital (Hangzhou, China). This
included 32 men and 12 women, with an age range of 27–76 years and
median age of 57.5 years. The normal tissue specimens were
collected from the far end of the surgical tissues (≥5 cm from
tumor tissues) All patients were provided written informed consent
for the project, which was approved by the Medical Ethics Committee
of Zhejiang Cancer Hospital (Hangzhou, China). The tissue chip was
made and the hematoxylin and eosin was staining by Servicebio
Company, Hangzhou (https://www.servicebio.cn).
For hematoxylin and eosin staining, the paraffinized
sections were dewaxed using dimethylbenzene, 100% ethanol and 75%
ethanol followed by tap water rinse. Sections were subsequently
stained with hematoxylin solution for 3–5 min at room temperature
followed by tap water rinse. The samples were treated with
Hematoxylin Differentiation solution and Hematoxylin Scott Tap
Bluing (cat. no. G1003; Wuhan Servicebio Technology Co., Ltd.),
according to the manufacturer's protocol, and then rinsed with tap
water, 85% ethanol and 95% ethanol. The sections were stained with
eosin dye for 5 min at room temperature. The sections were
dehydrated with 100% ethanol (three times) and dimethylbenzene (two
times). The sections were cover slipped and sealed with neutral
gum.
For immunohistochemistry analysis, tissues were
deparaffinized, rehydrated and incubated in 3% hydrogen peroxide in
H2O. Heat-induced antigen retrieval was carried out for
all sections in 0.01 M citrate buffer, pH 6.0, using a steamer at
95°C. All aforementioned primary antibodies were diluted with PBST
to a concentration of 1:50 and were applied to the sections.
Incubation lasted for 30 min at room temperature followed by
incubation with a DakoEnVision + System-HRP Labelled Polymer (Dako;
Agilent Technologies) for 10 min at room temperature.
Diaminobenzidine was then applied for 10 min at room temperature.
The sections were counterstained for 5 min with hematoxylin,
dehydrated (including 3 min with 75% ethanol, 3 min with 95%
ethanol, 3 min with absolute ethanol, 3 min with dimethylbenzene,
and another 3 min with dimethylbenzene at room temperature), cover
slipped and visualized at room temperature. Slides were blindly
scored by two independent gastrointestinal pathologists. Samples
were evaluated for staining using the following scale: Scarce
positive cells=0, low abundance of positive cells=1, moderate
abundance of positive cells=2 and high abundance of positive
cells=3+. The two sets of scores were combined for the highest
possible score of 6.
Statistical analysis
Data are expressed as the mean ± SD or SEM from at
least three independent experiments. Immunohistochemistry data were
statistically analyzed using the Wilcoxon matched-pairs test. All
statistical analyses were performed using GraphPad Prism 5.0
(GraphPad Software, Inc.). Statistical significance was determined
using an ANOVA followed by Tukey's post hoc test, and P<0.05 was
considered to indicate a statistically significant difference.
Results
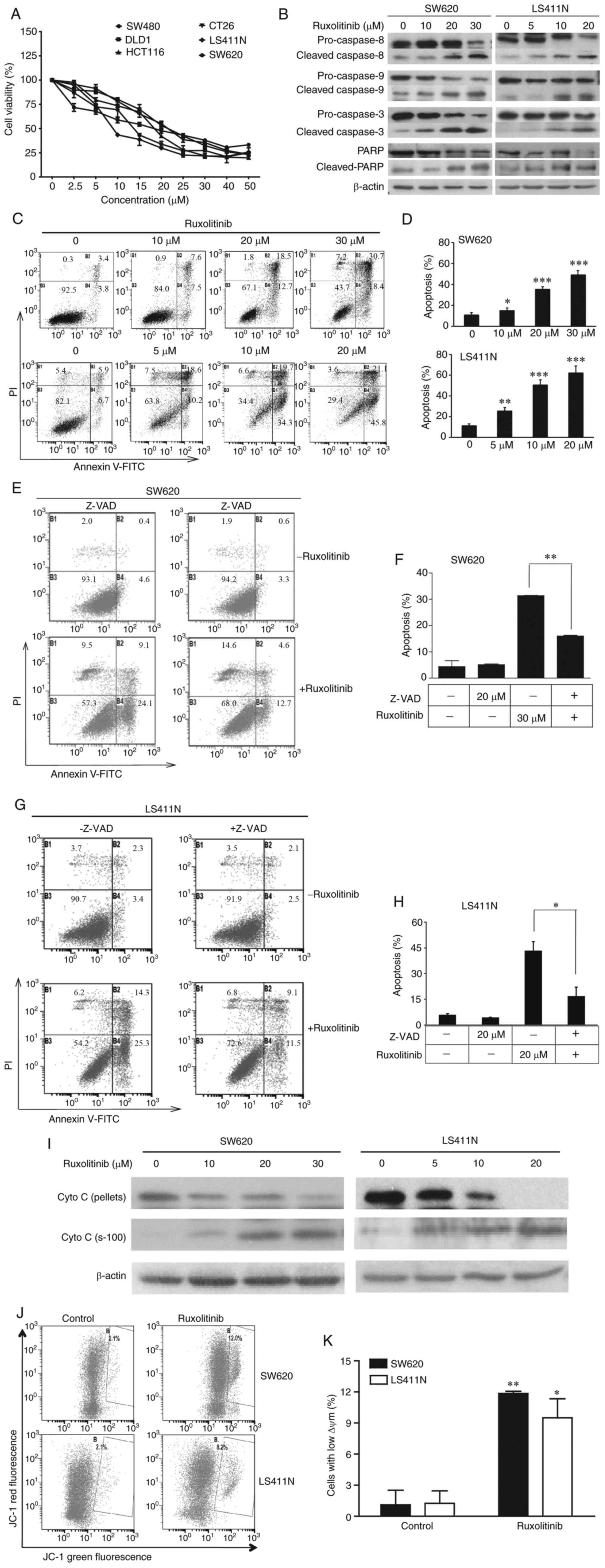
Ruxolitinib induces CRC cell
apoptosis
The cytotoxicity of ruxolitinib on LS411N, CT-26,
SW620, DLD-1, HCT116 and SW480 cells was examined using MTT assays.
As shown in Fig. 1A and Table I, all cancer cell lines were
sensitive to ruxolitinib with the IC50 ranging from 8 to
25 µM. To confirm the cytotoxicity of ruxolitinib on LS411N and
SW620 cells, CCK-8 assays and real-time monitoring of cell
viability using an xCELLigence system were performed (24). As presented in Fig. S1, ruxolitinib significantly
inhibited LS411N and SW620 cell proliferation in a dose-dependent
manner.
 | Table I.Cytotoxicity of ruxolitinib against
human colorectal cancer cells. |
Table I.
Cytotoxicity of ruxolitinib against
human colorectal cancer cells.
| Cell line | IC50
(µM) |
|---|
| LS411N | 8.44±0.42 |
| SW620 | 13.42±0.47 |
| SW480 | 19.70±1.71 |
| HCT116 | 18.41±0.20 |
| DLD1 | 20.90±1.51 |
| CT26 | 21.50±0.80 |
To elucidate the mechanism underlying
ruxolitinib-mediated suppression of CRC cell proliferation, two
sensitive cell lines were selected, LS411N and SW620. Ruxolitinib
induced discernible apoptosis in both cell lines in a
dose-dependent manner, with an increased percentage of Annexin
V-positive cells (Fig. 1C and D).
The results of long-term apoptotic assay are shown in Fig. S2. A time-dependent manner was
observed between 72 and 96 h treatments in both two cell lines. In
addition, increased expression of cleaved caspase 3, 8, 9 and PARP
(Fig. 1B) was observed in the LS411N
and SW620 cells 48 h after incubation with increasing concentration
of ruxolitinib. To further confirm ruxolitinib-induced apoptosis,
LS411N and SW620 cells were cultured with pan-caspase inhibitor
Z-VAD-FMK. As expected, Z-VAD-FMK blocked ruxolitinib-induced
apoptosis (Fig. 1E-H).
Furthermore, another biochemical marker of apoptosis
pathways, cytochrome c was investigated. The western blot analysis
indicated that ruxolitinib treatment induced the release of
cytochrome c in a dose-dependent manner (Fig. 1I). JC-1 staining together with flow
cytometry analysis was used to determine the effect of ruxolitinib
on mitochondrial outer membrane permeabilization (MOMP). As shown
in Fig. 1J and K, the disruption of
MOMP induced by ruxolitinib was upregulated compared with the
control. After treatment with 20 and 30 µM ruxolitinib, the
percentage of cells with depolarized MOMP increased to 9.50±1.84,
and 11.85±0.21% in LS411N and SW620 cells, respectively. These data
suggested that ruxolitinib inhibits human CRC cell proliferation
through an apoptosis-dependent mechanism.
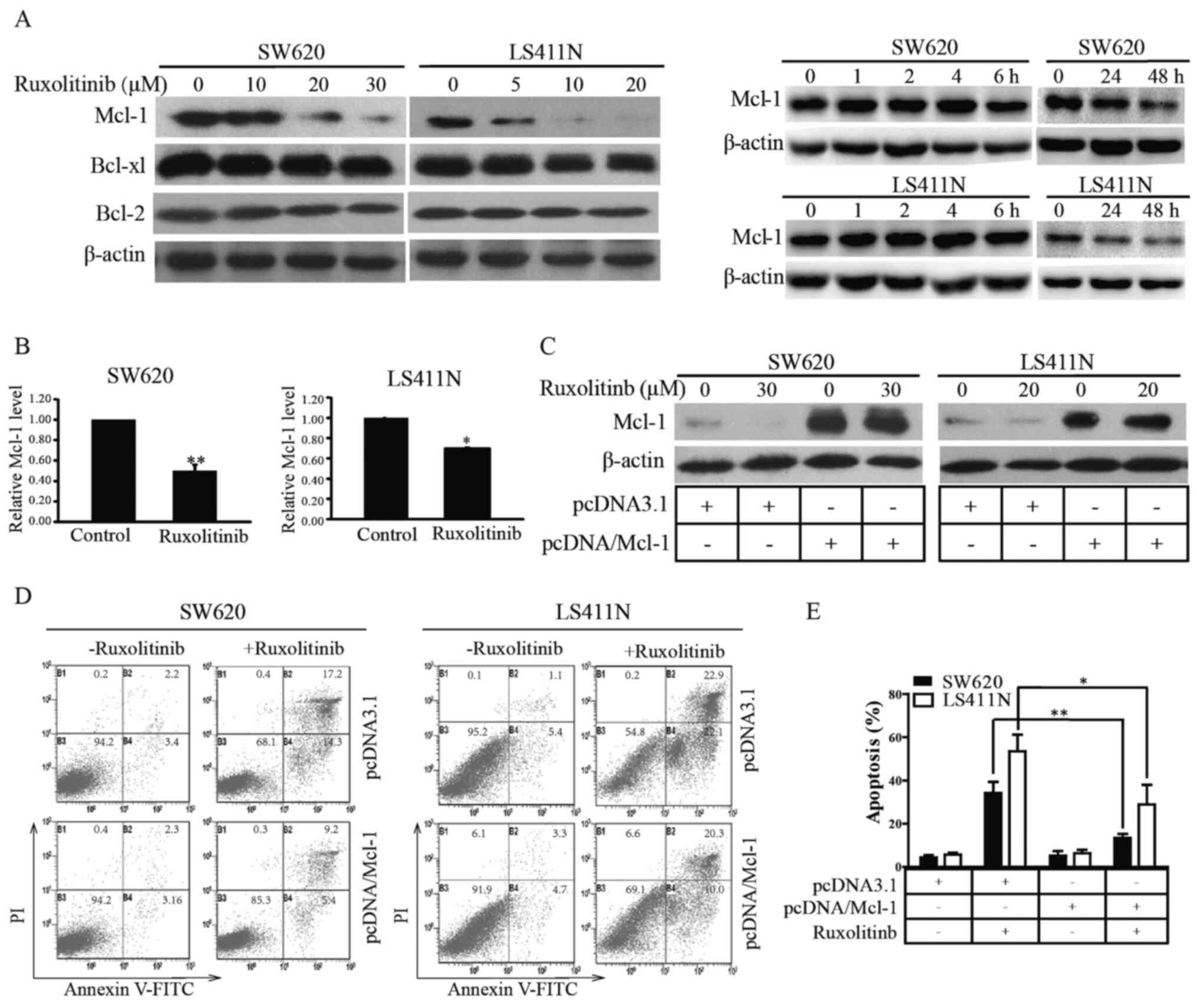
Mcl-1 is the molecular target of
ruxolitinib
It was demonstrated that ruxolitinib partly
suppressed CRC cell proliferation by inducing apoptosis through
activating the intrinsic pathway. Bcl-2 family proteins that are
key mediators of the intrinsic apoptosis pathway were screened out
(25,26). As shown in Fig. 2A, ruxolitinib selectively decreased
the Mcl-1 protein level in LS411N and SW620 cells in dose- and long
time-dependent manner, rather than short time-dependent within 6 h,
suggesting that Mcl-1 was a molecular target of ruxolitinib.
To further explore whether the ruxolitinib-mediated
decrease of Mcl-1 occurred at the transcriptional level, RT-PCR was
used to identify the effects of ruxolitinib on Mcl-1 mRNA. The
results showed that the Mcl-1 mRNA level was markedly reduced in
LS411N and SW620 cells compared with the control (Fig. 2B). To determine whether decreased
Mcl-1 mRNA was due to reduced Mcl-1 mRNA stability, ruxolitinib was
incubated with or without actinomycin D, a transcription inhibitor
(27), in LS411N and SW620 cells. As
shown in Fig. S3, there was no
significant difference between Mcl-1 mRNA level in the actinomycin
D alone and combination group. These results suggested that
ruxolitinib-mediated Mcl-1 decrease was more likely to occur at the
transcriptional level without affecting stability of Mcl-1
mRNA.
To confirm the effects on Mcl-1 in
ruxolitinib-mediated apoptosis in CRC cells, Mcl-1 was
overexpressed in LS411N and SW620 cells through cell transfection
(Fig. 2C). Overexpression of Mcl-1
significantly decreased LS411N and SW620 sensitivity to ruxolitinib
by inducing apoptosis (Fig. 2D and
E). These investigations suggested that ruxolitinib targeted
Mcl-1 to trigger CRC cell apoptosis.
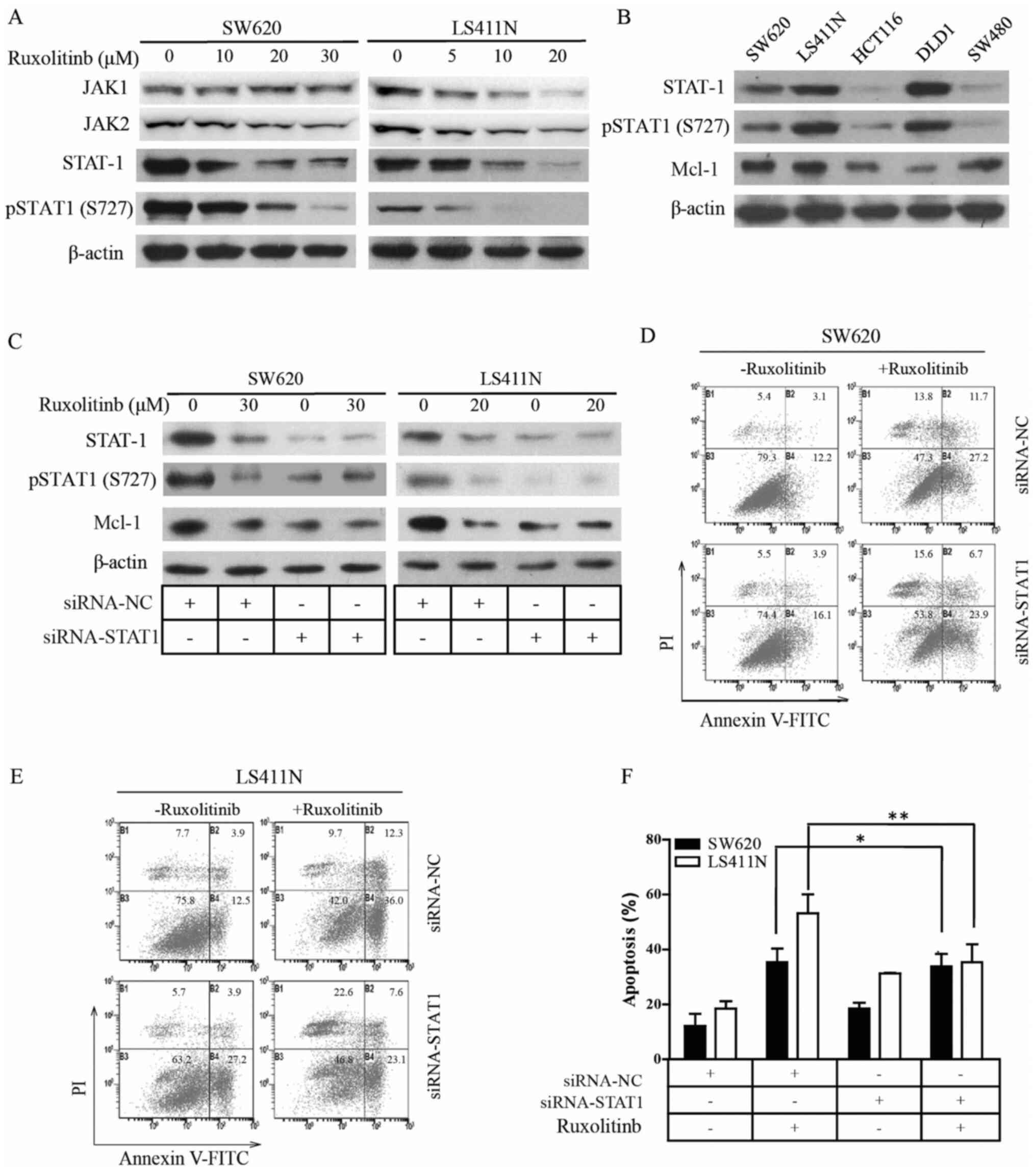
Involvement of pSTAT1 (S727) in the
ruxolitinib-mediated decrease of Mcl-1
According to Timofeeva's report,
serine-phosphorylated STAT1 is a pro-survival factor in Wilms'
tumor pathogenesis (28). To study
the inhibitory effects of ruxolitinib on STAT1 activity in CRC, the
endogenous phosphorylation status of STAT1 in various CRC cell
lines was examined using western blotting. As shown in Fig. 3A, ruxolitinib decreased JAK1, JAK2,
STAT1 and pSTAT1 (S727) protein levels in a dose-dependent manner
in LS411N and SW620 cells. Furthermore, different Mcl-1, STAT1 and
pSTAT1 (S727) protein levels were found in the CRC cell lines
(Fig. 3B); very low STAT1, pSTAT1
(S727) and high Mcl-1 were observed in HCT116 and SW480 cells, high
STAT1, pSTAT1 (S727) and low Mcl-1 in DLD-1 cells and high STAT1,
pSTAT1 (S727) and Mcl-1 were detected in LS411N and SW620
cells.
To validate the function of pSTAT1 (S727) in
ruxolitinib-induced apoptosis in CRC cells, STAT1 was knocked down
in LS411N and SW620 cells using transfection. As expected, western
blot analysis indicated that the transfection of LS411N and SW620
cells with siRNA-STAT1 notably decreased STAT1 and pSTAT1 (S727)
protein levels in the tumor cells (Fig.
3C). These data suggested that the ruxolitinib decreased the
transcriptional level of Mcl-1. If ruxolitinib indeed induced
apoptosis by decreasing transcriptional levels of Mcl-1, knockdown
of STAT1 and pSTAT1 (S727) would diminish ruxolitinib-induced
apoptosis in the tumor cells. Knockdown of STAT1 significantly
decreased LS411N and SW620 cell sensitivity to ruxolitinib by
neither inducing Mcl-1 protein levels or increasing apoptosis
(Fig. 3C-F). These observations
suggested that ruxolitinib may decrease Mcl-1 protein levels by
inhibiting transcription factor pSTAT1 (S727), and, in turn,
inducing CRC cell apoptosis.
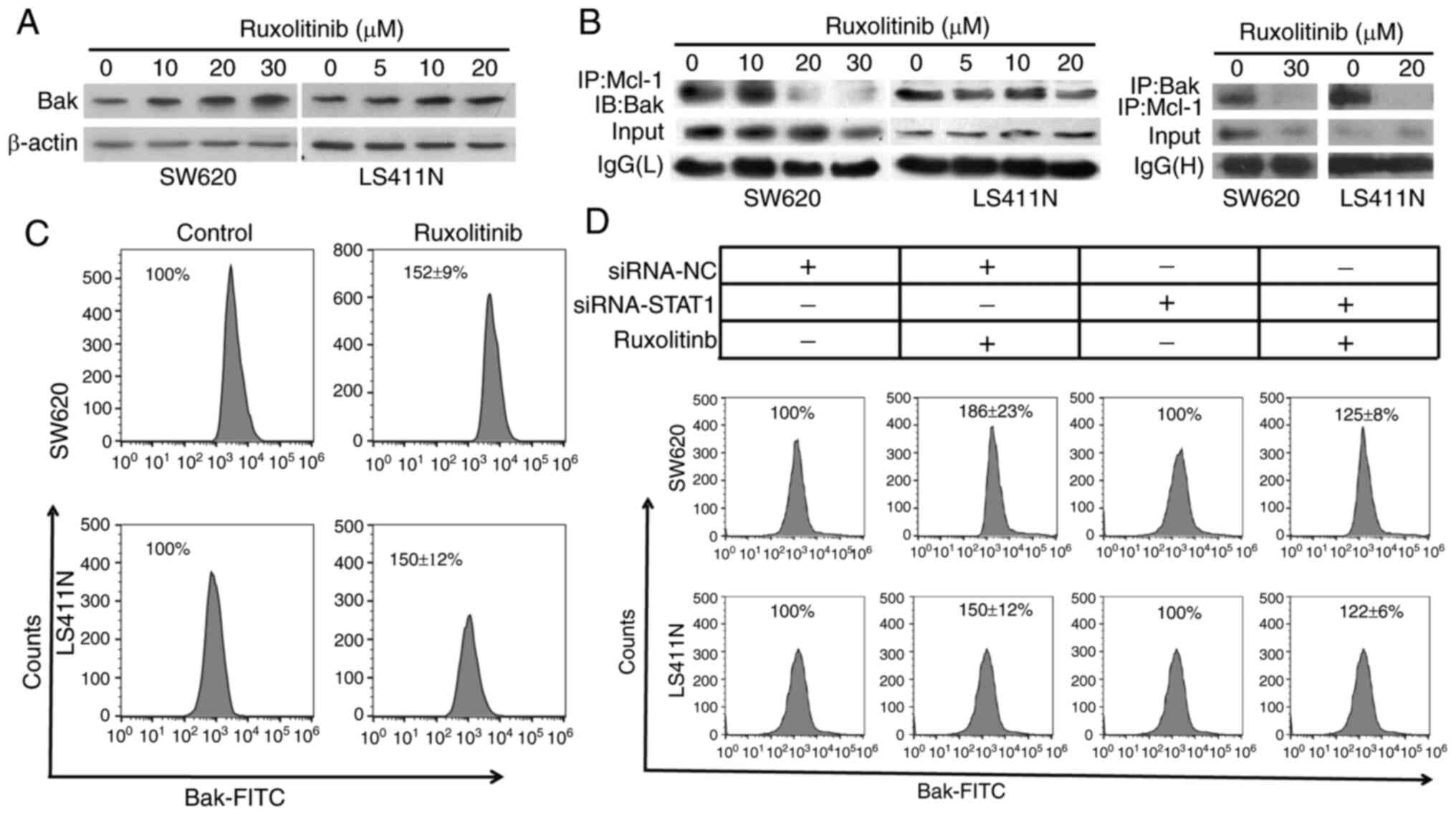
Ruxolitinib disrupts the association
of Bak with Mcl-1 and induces Bak activation
Bak has an important role in the intrinsic pathway
of apoptosis (29). Mcl-1 can bind
and thereby inactivate pro-apoptotic Bak (30). Based on this connection between Mcl-1
and Bak, it was hypothesized that the inhibition of Mcl-1 by
ruxolitinib might release Bak, which, in turn, may induce
apoptosis. To verify this hypothesis, the effect of ruxolitinib on
Bak activation and combination of Bak with Mcl-1 was examined.
Western blot analysis revealed that ruxolitinib treatment increased
total Bak protein levels (Fig. 4A).
In addition, the Co-IP analysis indicated that Mcl-1 bound Bak in
LS411N and SW620 cells, and ruxolitinib treatment decreased this
binding (Fig. 4B). Furthermore, an
increase in Bak conformational change was also observed after
ruxolitinib treatment (Fig. 4C).
To validate the aforementioned findings, STAT1 was
knocked down in LS411N and SW620 cells, and Bak activation was
analyzed using flow cytometry. Bak activation is related with a
conformational change, which can be detected by antibodies only
recognizing the active protein conformer (31). Knockdown of STAT1 blocked
ruxolitinib-induced Bak activation (Fig.
4D). In short, these data suggested that ruxolitinib decreased
Mcl-1 to release Bak, whose activation could trigger apoptosis.
Oral administration of ruxolitinib
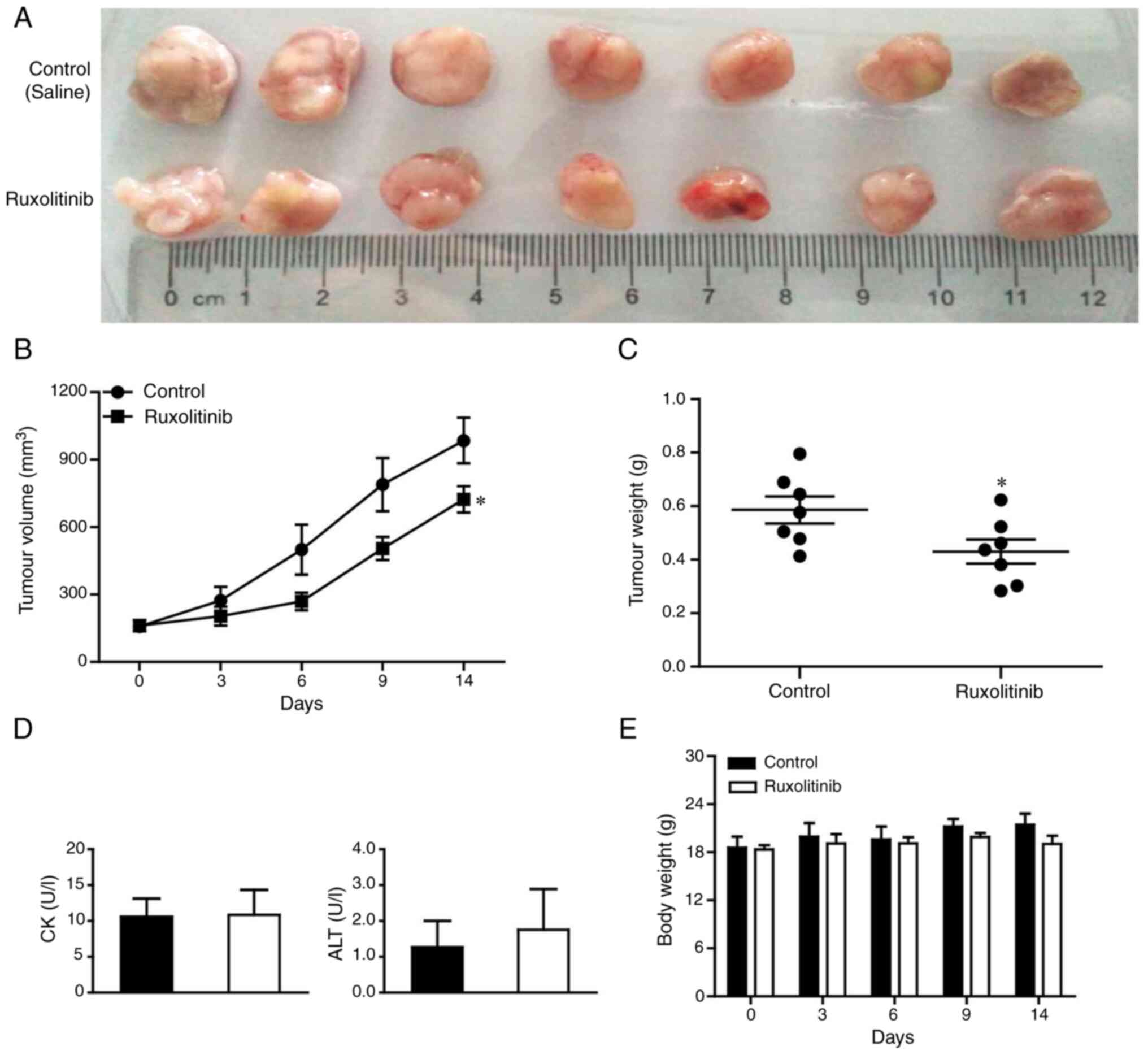
significantly suppresses CRC tumor growth in vivo
To further determine the anticancer efficacy of
ruxolitinib treatment in vivo, a xenograft model was
constructed using LS411N cells. As shown in Fig. 5A-C, significantly lower tumor volume
and weight were observed in mice treated with ruxolitinib compared
with the control group (both P<0.05).
Furthermore, serum creatine kinase and alanine
aminotransferase levels were analyzed to determine ruxolitinib
liver toxicity. The two enzymes were unchanged by ruxolitinib
(Fig. 5D). Meanwhile, no significant
differences in animal body weight were found between the treatment
and control group (Fig. 5E), which
further suggested that ruxolitinib was not toxic to mice. Taken
together, these data showed that ruxolitinib could effectively
suppress LS411N xenograft growth in vivo without inducing
liver toxicity.
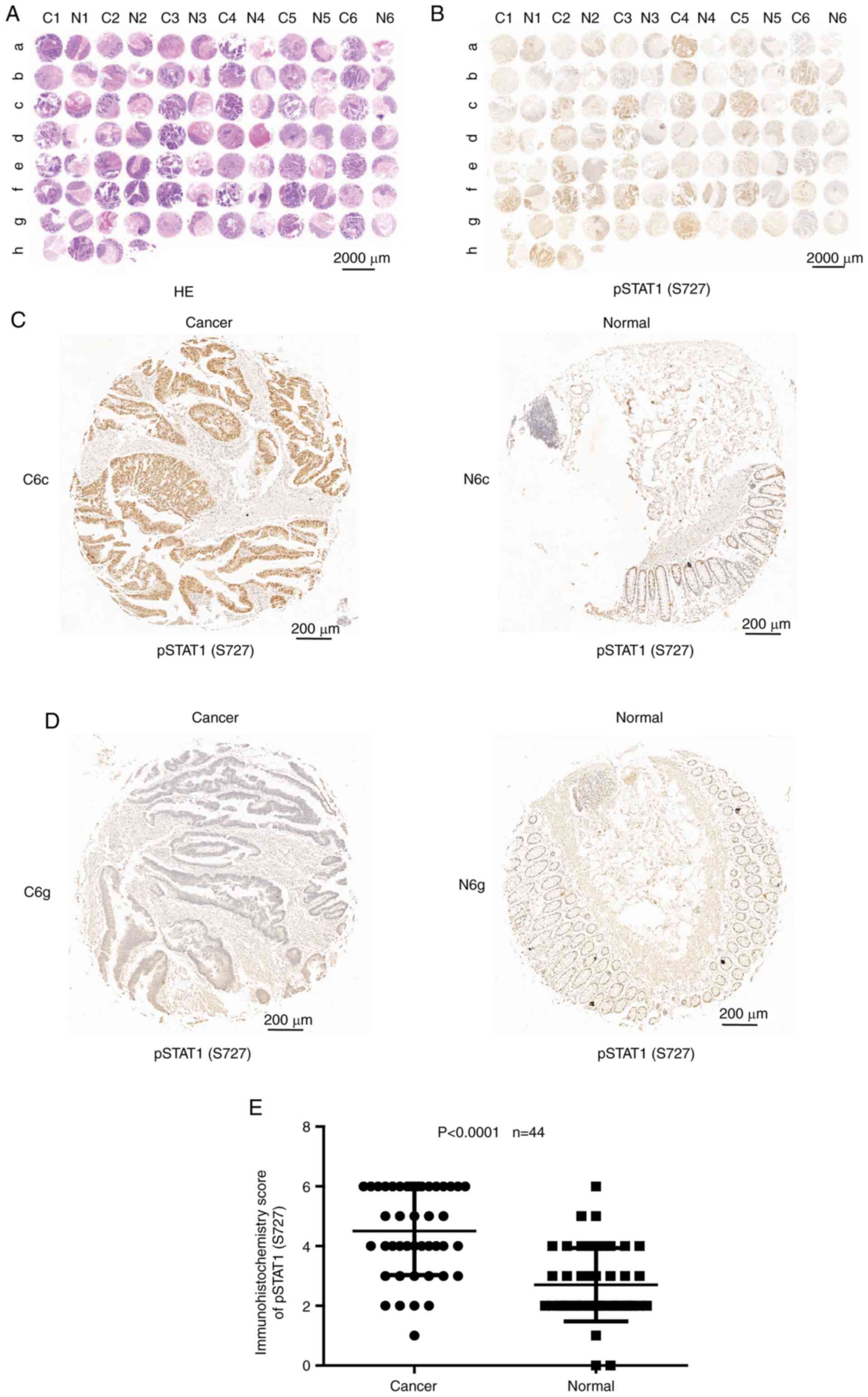
Expression of pSTAT1 (S727) protein
level in patients with CRC
To determine the expression levels of pSTAT1 (S727)
in human colon carcinoma, 44 pairs of human colon carcinoma and
adjacent normal tissue specimens were examined using
immunohistochemical staining. The results showed that hematoxylin
and eosin staining (Servicebio Company, Hangzhou) (Fig. 6A) and pSTAT1 (S727) staining
(Fig. 6B) images from tumor samples
and paired normal tissues from patients with CRC. Furthermore,
Fig. 6C and D shows the
representative pSTAT1 (S727) high and low staining images of tumors
and paired normal tissues. In total, 36.4% (16/44) of patients
showed the positive expression of pSTAT1 (S727) in normal tissues;
however, 81.8% (36/44) of patients showed positive pSTAT1 (S727) in
paired tumor tissues. The staining scores indicated that the
expression levels of pSTAT1 (S727) were significantly higher in
tumor tissues compared with those in matched normal tissues
(P<0.0001; Fig. 6E). STAT1
phosphorylation on S727 in most CRC specimens illustrated that this
modification might be involved in the pathogenesis of this
tumor.
Discussion
JAK, a kind of tyrosine kinase, serves key roles in
the differentiation, proliferation and cell death of both normal
and cancer cells (32). JAKs are
activated by growth factor receptors, cytokines, such as IL-6 and
IFN-γ, and downstream signaling proteins, such as STATs (33). STAT1 is part of the JAK/STAT
signaling cascade. STAT1 modulates various cellular processes, such
as antimicrobial activities, cell proliferation and cell death
(34). STAT1 is activated by
different cytokines, including type I–III interferons, interleukin
(IL)-21 and IL-27, and is transiently and tightly regulated
(35). Tyrosine 701 phosphorylation
on STAT1 through JAKs leads to its activation and nuclear
translocation. Meanwhile, serine 727 phosphorylation is required
for transcriptional activation in response to cellular stress
(36). Yet, the role of STAT1 in
carcinogenesis is still not well defined. Some studies have
suggested that STAT1 may suppress tumorigenesis and/or metastasis
in various types of cancer, including hepatocellular carcinoma,
esophageal cancer, CRC, pancreatic cancer and metastatic melanoma
(37–41). In addition, other studies have
suggested that STAT1 promotes tumor growth by inhibiting tumor
immune surveillance and by promoting tumor resistance against
chemotherapy and irradiation. For example, activation of STAT1 by
the cancer-up-regulated gene 2 enhances metastasis and drug
resistance in colon cancer cells (42–44).
Taken together, these data suggest that JAK-STAT activity is likely
cancer type-dependent, especially in CRC.
The present study examined the inhibitory effects of
ruxolitinib on human CRC cells. It was demonstrated that
ruxolitinib inhibited CRC cell viability through the induction of
apoptosis and by activating both intrinsic and extrinsic pathways.
Furthermore, Mcl-1 was identified as the downstream molecular
target of ruxolitinib, which inhibited JAK1 and/or JAK2. It was
also reported that Mcl-1 is regulated by transcription factor
pSTAT1 (S727). Ruxolitinib decreased Mcl-1 mRNA without affecting
mRNA stability, which in turn enabled Bak to trigger CRC apoptosis.
In addition, the in vivo experiment suggested that
ruxolitinib was effective in the suppression of CRC tumor growth
without inducing liver toxicity. Moreover, an analysis of 44
matched pairs of normal human colorectal tissues and CRC tissues
revealed that pSTAT1 (S727) expression was significantly higher in
the tumor tissues compared with in the adjacent normal colorectal
tissues.
Mcl-1 is a member of the antiapoptotic Bcl-2 family
of proteins that represses apoptosis through binding with
pro-apoptotic proteins, such as Bak, that inhibit its activation
and subsequently prevent cytochrome c release from mitochondria
(45). Mcl-1 is a highly regulated
protein with a short half-life; it is induced by a broad range of
survival signals, such as AKT and ERK signals, and is quickly
downregulated during apoptosis (14). Mcl-1 can be regulated at both the
transcriptional and translational levels. Transcriptionally, Mcl-1
expression can be induced by various cytokines and signaling
pathways, including PI3K/AKT and JAK/STAT pathways (46,47).
Previous studies have highlighted the importance of microRNAs in
Mcl-1 regulation at the translational level (48–50). In
addition, Mcl-1 is highly expressed in human cancer cell lines,
including breast, colon, lung, ovarian, prostate and renal cancer,
and melanoma (51). Aberrant
expression of Mcl-1 is an important genomic change present in
multiple cancer types, a number of which become dependent upon this
protein for cell survival and resistance to chemotherapy. For
example, our previous study revealed that Mcl-1, which induces
proteasome-dependent degradation by imperatorin, is an important
factor in the mechanism of liver cancer pathophysiology that can
also cause resistance to doxorubicin-based therapy (14). It is known that, transcriptionally,
Mcl-1 expression can be regulated by STAT-3 (46). Downregulation of STAT-3 induces a
decrease in Mcl-1 transcription, indicating that STAT-3 inhibition
could play a significant role in the induction of apoptosis
(52). Timofeeva et al
reported that serine-phosphorylated STAT1 promotes cancer cell
survival through downregulation of Mcl-1 expression (28). Some reports have reported a
correlation between STAT-1 or STAT-3 serine phosphorylation with
increased DNA-binding ability; however, sufficient evidence for
this observation has not been provided (36,53,54). In
the present study, Mcl-1 was downregulated in SW620 and LS411N
cells under ruxolitinib treatment when transcription factor
serine-phosphorylated STAT1 was decreased. Moreover, an
overexpression of Mcl-1 effectively reduced LS411N and SW620 cells
sensitivity to ruxolitinib by inducing apoptosis. Notably,
knockdown of STAT1 induced apoptosis in LS411N and SW620 cells and
also significantly decreased the sensitivity of these cells to
ruxolitinib-induced apoptosis. Therefore, these observations
suggested that serine-phosphorylated STAT1 can maintain the
expression of Mcl-1 in LS411N and SW620 cells, thus enabling the
survival of tumor cells under the same conditions.
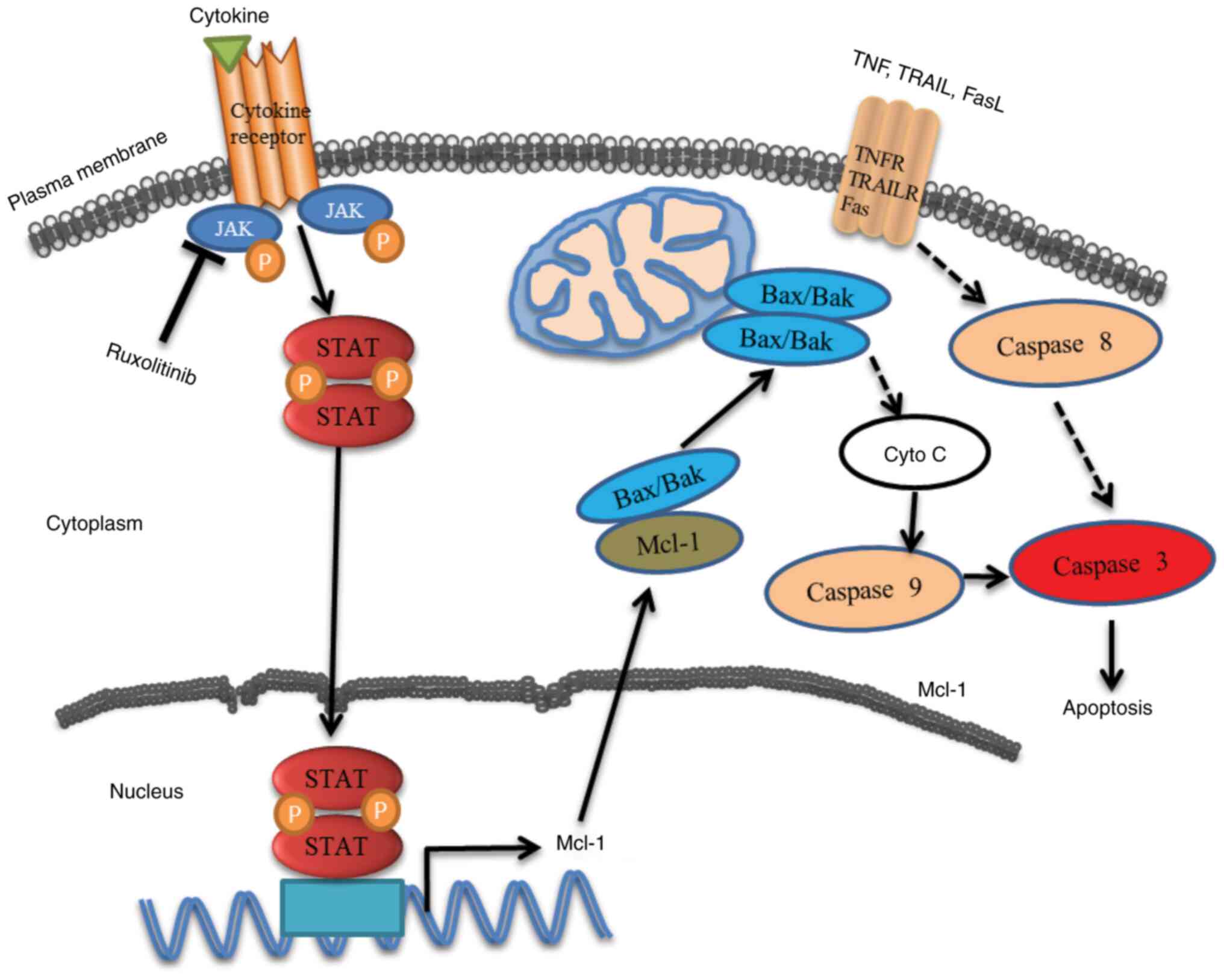
In summary, the results suggested ruxolitinib that
inhibited JAK1 and/or JAK2 and could decrease STAT1 S727
phosphorylation. In addition, it was demonstrated that Mcl-1 is
regulated by the transcription factor pSTAT1 (S727). Ruxolitinib
decreased Mcl-1 mRNA levels without affecting mRNA stability, which
in turn enabled Bak to trigger caspase-dependent apoptosis in human
CRC cells (Fig. 7). The present
study identified JAK1/2-STAT1-Mcl-1 as important factors and
potential molecular therapeutic targets in human CRC. However,
based on the current results, the JAK1/2-STAT1-Mcl-1 axis is
insufficient to function as a biomarker for CRC prognosis.
Prospective studies will aim to investigate the mechanisms of
pSTAT1 (S727) in CRC and the interaction of relevant factors.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant nos. 31570811, 81673813,
81502603 and 81903641) the Zhejiang Provincial Natural Science
Foundation of China (grant nos. LQ18H160016, LGF18H160016 and
LY20H160039), the International Cooperation Fund from Science
Technology Department of Zhejiang Province (grant no. LGJ19H160001)
and the Medicine & Health Science Fund of Zhejiang Province
(grant no. 2018RC021).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
XL, WC and FL drafted the initial manuscript and
confirmed the authenticity of all the raw data. XL, ZW, WC and FL
participated in the collection, analysis and interpretation of
data. XL, ZW, SZ, QY, WC and FL performed the experiments, and
collected and analyzed the data. WC and FL conceived of the study
and participated in its design and coordination and provided
important inputs for the manuscript. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Medical Ethics
Committee of Zhejiang Cancer Hospital (Hangzhou, China; approval
no. ZJU20180108020). All patients provided written informed consent
prior to the study start.
Patient consent for publication
Not applicable.
Competing interests
They authors declare that they have no competing
interests.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li X, Zhou Y, Luo Z, Gu Y, Chen Y, Yang C,
Wang J, Xiao S, Sun Q, Qian M and Zhao G: The impact of screening
on the survival of colorectal cancer in Shanghai, China: A
population based study. BMC Public Health. 19:10162019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Meads MB, Gatenby RA and Dalton WS:
Environment-mediated drug resistance: A major contributor to
minimal residual disease. Nat Rev Cancer. 9:665–674. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhao H, Zhang N, Ho V, Ding M, He W, Niu
J, Yang M, Du XL, Zorzi D, Chavez-MacGregor M and Giordano SH:
Adherence to treatment guidelines and survival for older patients
with stage II or III colon cancer in Texas from 2001 through 2011.
Cancer. 124:679–687. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lieberman DA, Rex DK, Winawer SJ,
Giardiello FM, Johnson DA and Levin TR: Guidelines for colonoscopy
surveillance after screening and polypectomy: A consensus update by
the US multi-society task force on colorectal cancer.
Gastroenterology. 143:844–857. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Harada T, Yamamoto E, Yamano HO, Aoki H,
Matsushita HO, Yoshikawa K, Takagi R, Harada E, Tanaka Y, Yoshida
Y, et al: Surface microstructures are associated with mutational
intratumoral heterogeneity in colorectal tumors. J Gastroenterol.
53:1241–1252. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rawlings JS, Rosler KM and Harrison DA:
The JAK/STAT signaling pathway. J Cell Sci. 117:1281–1283. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
O'Shea JJ, Schwartz DM, Villarino AV,
Gadina M, McInnes IB and Laurence A: The JAK-STAT pathway: Impact
on human disease and therapeutic intervention. Annu Rev Med.
66:311–328. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sansone P and Bromberg J: Targeting the
interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol.
30:1005–1014. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Spano JP, Milano G, Rixe C and Fagard R:
JAK/STAT signalling pathway in colorectal cancer: A new biological
target with therapeutic implications. Eur J Cancer. 42:2668–2670.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang SW and Sun YM: The IL-6/JAK/STAT3
pathway: Potential therapeutic strategies in treating colorectal
cancer (Review). Int J Oncol. 44:1032–1040. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yamaguchi R, Lartigue L and Perkins G:
Targeting Mcl-1 and other Bcl-2 family member proteins in cancer
therapy. Pharmacol Ther. 195:13–20. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li X, Zeng X, Sun J, Li H, Wu P, Fung KP
and Liu F: Imperatorin induces Mcl-1 degradation to cooperatively
trigger Bax translocation and Bak activation to suppress
drug-resistant human hepatoma. Cancer Lett. 348:146–155. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Craig RW: MCL1 provides a window on the
role of the BCL2 family in cell proliferation, differentiation and
tumorigenesis. Leukemia. 16:444–454. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gomez-Bougie P, Halliez M, Moreau P,
Pellat-Deceunynck C and Amiot M: Repression of Mcl-1 and disruption
of the Mcl-1/Bak interaction in myeloma cells couple ER stress to
mitochondrial apoptosis. Cancer Lett. 383:204–211. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Verstovsek S, Kantarjian H, Mesa RA,
Pardanani AD, Cortes-Franco J, Thomas DA, Estrov Z, Fridman JS,
Bradley EC, Erickson-Viitanen S, et al: Safety and efficacy of
INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J
Med. 363:1117–1127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shilling AD, Nedza FM, Emm T, Diamond S,
McKeever E, Punwani N, Williams W, Arvanitis A, Galya LG, Li M, et
al: Metabolism, excretion, and pharmacokinetics of [14C]INCB018424,
a selective Janus tyrosine kinase 1/2 inhibitor, in humans. Drug
Metab Dispos. 38:2023–2031. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stover DG, Gil Del Alcazar CR, Brock J,
Guo H, Overmoyer B, Balko J, Xu Q, Bardia A, Tolaney SM, Gelman R,
et al: Phase II study of ruxolitinib, a selective JAK1/2 inhibitor,
in patients with metastatic triple-negative breast cancer. NPJ
Breast Cancer. 4:102018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Galvez Acosta S and Javalera Rincon M:
Ruxolitinib as first-line therapy in secondary hemophagocytic
lymphohistiocytosis and HIV infection. Int J Hematol. 112:418–421.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Harrison C, Kiladjian JJ, Al-Ali HK,
Gisslinger H, Waltzman R, Stalbovskaya V, McQuitty M, Hunter DS,
Levy R, Knoops L, et al: JAK inhibition with ruxolitinib versus
best available therapy for myelofibrosis. N Engl J Med.
366:787–798. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schonberg K, Rudolph J, Vonnahme M,
Parampalli Yajnanarayana S, Cornez I, Hejazi M, Manser AR, Uhrberg
M, Verbeek W, Koschmieder S, et al: JAK inhibition impairs NK cell
function in myeloproliferative neoplasms. Cancer Res. 75:2187–2199.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Klatte M and Bauer P: Accurate real-time
reverse transcription quantitative PCR. Methods Mol Biol.
479:61–77. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li X, Wu J, Zhang X and Chen W:
Glutathione reductase-mediated thiol oxidative stress suppresses
metastasis of murine melanoma cells. Free Radic Biol Med.
129:256–267. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Du H, Chen L, Luo F, Chen X, Li Y and
Cheng Q: Beclin-1 expression is associated with prognosis in a
Bcl-2-dependent manner in non-small cell lung cancer. Oncol Lett.
20:92020.PubMed/NCBI
|
|
26
|
Yuan B, Hao J, Zhang Q, Wang Y and Zhu Y:
Role of Bcl-2 on drug resistance in breast cancer
polyploidy-induced spindle poisons. Oncol Lett. 19:1701–1710.
2020.PubMed/NCBI
|
|
27
|
Zhou Y, Zhou Y, Yang M, Wang K, Liu Y,
Zhang M, Yang Y, Jin C, Wang R and Hu R: Digoxin sensitizes
gemcitabine-resistant pancreatic cancer cells to gemcitabine via
inhibiting Nrf2 signaling pathway. Redox Biol. 22:1011312019.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Timofeeva OA, Plisov S, Evseev AA, Peng S,
Jose-Kampfner M, Lovvorn HN, Dome JS and Perantoni AO:
Serine-phosphorylated STAT1 is a prosurvival factor in Wilms' tumor
pathogenesis. Oncogene. 25:7555–7564. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Reed JC: Proapoptotic multidomain
Bcl-2/Bax-family proteins: Mechanisms, physiological roles, and
therapeutic opportunities. Cell Death Differ. 13:1378–1386. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cuconati A, Mukherjee C, Perez D and White
E: DNA damage response and MCL-1 destruction initiate apoptosis in
adenovirus-infected cells. Genes Dev. 17:2922–2932. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen S, Dai Y, Harada H, Dent P and Grant
S: Mcl-1 down-regulation potentiates ABT-737 lethality by
cooperatively inducing Bak activation and Bax translocation. Cancer
Res. 67:782–791. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rane SG and Reddy EP: Janus kinases:
Components of multiple signaling pathways. Oncogene. 19:5662–5679.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Turkson J and Jove R: STAT proteins: Novel
molecular targets for cancer drug discovery. Oncogene.
19:6613–6626. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Meissl K, Macho-Maschler S, Muller M and
Strobl B: The good and the bad faces of STAT1 in solid tumours.
Cytokine. 89:12–20. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Boisson-Dupuis S, Kong XF, Okada S,
Cypowyj S, Puel A, Abel L and Casanova JL: Inborn errors of human
STAT1: Allelic heterogeneity governs the diversity of immunological
and infectious phenotypes. Curr Opin Immunol. 24:364–378. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Decker T and Kovarik P: Serine
phosphorylation of STATs. Oncogene. 19:2628–2637. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen J, Wang H, Wang J, Huang S and Zhang
W: STAT1 inhibits human hepatocellular carcinoma cell growth
through induction of p53 and Fbxw7. Cancer Cell Int. 15:1112015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang Y, Molavi O, Su M and Lai R: The
clinical and biological significance of STAT1 in esophageal
squamous cell carcinoma. BMC Cancer. 14:7912014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang X, Li X, Tan F, Yu N and Pei H:
STAT1 inhibits MiR-181a expression to suppress colorectal cancer
cell proliferation through PTEN/Akt. J Cell Biochem. 118:3435–3443.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sun Y, Yang S, Sun N and Chen J:
Differential expression of STAT1 and p21 proteins predicts
pancreatic cancer progression and prognosis. Pancreas. 43:619–623.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Osborn JL and Greer SF: Metastatic
melanoma cells evade immune detection by silencing STAT1. Int J Mol
Sci. 16:4343–4361. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang N, Li F, Gao J, Zhang S and Wang Q:
Osteopontin accelerates the development and metastasis of bladder
cancer via activating JAK1/STAT1 pathway. Genes Genomics.
42:467–475. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wu J, Gao F, Xu T, Li J, Hu Z, Wang C,
Long Y, He X, Deng X, Ren D, et al: CLDN1 induces autophagy to
promote proliferation and metastasis of esophageal squamous
carcinoma through AMPK/STAT1/ULK1 signaling. J Cell Physiol.
235:2245–2259. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Malilas W, Koh SS, Kim S, Srisuttee R, Cho
IR, Moon J, Yoo HS, Oh S, Johnston RN and Chung YH: Cancer
upregulated gene 2, a novel oncogene, enhances migration and drug
resistance of colon cancer cells via STAT1 activation. Int J Oncol.
43:1111–1116. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Inoue-Yamauchi A, Jeng PS, Kim K, Chen HC,
Han S, Ganesan YT, Ishizawa K, Jebiwott S, Dong Y, Pietanza MC, et
al: Targeting the differential addiction to anti-apoptotic BCL-2
family for cancer therapy. Nat Commun. 8:160782017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Quinn BA, Dash R, Azab B, Sarkar S, Das
SK, Kumar S, Oyesanya RA, Dasgupta S, Dent P, Grant S, et al:
Targeting Mcl-1 for the therapy of cancer. Expert Opin Investig
Drugs. 20:1397–1411. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Radhakrishnan H, Ilm K, Walther W,
Shirasawa S, Sasazuki T, Daniel PT, Gillissen B and Stein U: MACC1
regulates Fas mediated apoptosis through STAT1/3 - Mcl-1 signaling
in solid cancers. Cancer Lett. 403:231–245. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jin X, Yu Y, Zou Q, Wang M, Cui Y, Xie J
and Wang Z: MicroRNA-105 promotes epithelial-mesenchymal transition
of nonsmall lung cancer cells through upregulating Mcl-1. J Cell
Biochem. 120:5880–5888. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhao C, Wang Y, Jin H and Yu T: Knockdown
of microRNA-203 alleviates LPS-induced injury by targeting MCL-1 in
C28/I2 chondrocytes. Exp Cell Res. 359:171–178. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gong J, Zhang JP, Li B, Zeng C, You K,
Chen MX, Yuan Y and Zhuang SM: MicroRNA-125b promotes apoptosis by
regulating the expression of Mcl-1, Bcl-w and IL-6R. Oncogene.
32:3071–3079. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Placzek WJ, Wei J, Kitada S, Zhai D, Reed
JC and Pellecchia M: A survey of the anti-apoptotic Bcl-2 subfamily
expression in cancer types provides a platform to predict the
efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis.
1:e402010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang X, Zhang J, Wei H and Tian Z:
STAT3-decoy oligodeoxynucleotide inhibits the growth of human lung
cancer via down-regulating its target genes. Oncol Rep.
17:1377–1382. 2007.PubMed/NCBI
|
|
53
|
Salazar-Montes A, Ruiz-Corro L,
Sandoval-Rodriguez A, Lopez-Reyes A and Armendariz-Borunda J:
Increased DNA binding activity of NF-kappaB, STAT-3, SMAD3 and AP-1
in acutely damaged liver. World J Gastroenterol. 12:5995–6001.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Townsend PA, Scarabelli TM, Davidson SM,
Knight RA, Latchman DS and Stephanou A: STAT-1 interacts with p53
to enhance DNA damage-induced apoptosis. J Biol Chem.
279:5811–5820. 2004. View Article : Google Scholar : PubMed/NCBI
|