Introduction
Sarcoma is a rare malignant tumor that frequently
occurs in, or originates from, the bone, cartilage or connective
tissue (1). Globally, almost 200,000
patients are affected by sarcoma each year (2). The prognosis of patients is poor, and
the choice of approved targeted drugs is somewhat limited (3,4). Surgery
is currently the primary treatment option for most sarcomas, but
local recurrence does occur (5).
Targeted molecular therapies have yielded improved clinical
outcomes (6–8). However, due to diagnostic difficulties,
bone and soft-tissue sarcomas are often only diagnosed at the
advanced stage, resulting in a 50–60% 5-year survival rate
(9,10). Therefore, a more accurate system for
sarcoma diagnosis and classification is urgently required.
Sarcoma includes soft-tissue sarcomas and primary
bone sarcomas (11). Soft-tissue
sarcomas comprise >50 subtypes (12), and the most frequently observed
subtypes include liposarcoma, leiomyosarcoma, undifferentiated
soft-tissue sarcoma, fibrosarcoma and synovial sarcoma (13). Primary bone sarcoma subtypes include
Ewing's sarcoma and osteosarcoma (14). Histopathological examination, such as
the analysis of histological sections and fluorescence in
situ hybridization (FISH), are still the only available methods
for the accurate diagnosis of sarcomas. However, due to their
rarity and diversity, the classification of sarcomas remains a
challenge. The identification and application of potential
biomarkers is a convenient, rapid and accurate strategy for
identifying sarcoma subtypes, and is conducive to improving
diagnosis and prognostic prediction (15–18).
With continuous developments in molecular biology,
next-generation sequencing (NGS) technology has enabled more
accurate and efficient molecular characterization. The Cancer
Genome Atlas and the International Cancer Genome Consortium have
characterized the genome and genomic alterations (GAs) of most
types of cancer (19,20), and a number of recent studies have
focused on the molecular profiling of sarcomas (21,22).
Sarcomas can be divided into the following subgroups according to
genetic heterogeneity: i) Gene fusions; ii) genomic amplifications;
and iii) extensive combinations of genomic imbalances and point
mutations (23–25). There is also evidence to suggest that
some sarcomas possess unique molecular characteristics, such as the
SYT-SSX fusion in synovial sarcoma, the EWS-ATF1
fusion in clear cell sarcoma, and the EWSR1-FLI1 fusion in
Ewing's sarcoma (26–28). Specific molecular characterizations
not only assistant in the classification of sarcoma, but can also
guide treatment programs. For example, imatinib has demonstrated
good efficacy in gastrointestinal stromal tumor (GIST) patients
with KIT, PDGFRA, CSF1 and ABL mutations (29–31);
since 2013, palbociclib has undergone phase II clinical trials in
liposarcoma patients with CDK4 mutations (32). Therefore, a clear classification
system and a precise molecular description of sarcoma subtypes are
necessary for subsequent diagnosis and treatment.
The present study aimed to identify GAs for the
mutational profiling of 199 patients with sarcoma (both soft-tissue
and primary bone sarcoma). By comparing molecular-based
classification with traditional immunohistochemical categorization,
the accuracy and necessity of NGS technology for sarcomas
classification was confirmed. The results provide comprehensive and
accurate information of GAs, which suggest novel biomarkers for
sarcoma diagnosis that may guide precise therapeutic strategies for
patients with bone and soft-tissue sarcomas.
Materials and methods
Patient enrollment and sample
collection
The present study was approved by the Ethics
Committees of National Cancer Center/Cancer Hospital, Chinese
Academy of Medical Sciences and Peking Union Medical College
(Beijing, China) and the First Affiliated Hospital of Sun Yat Sen
University (Guangzhou, China). A total of 199 patients with sarcoma
were enrolled between January 1, 2008 and December 31, 2018. Tumor
tissues were collected from patients, fixed in formalin and
embedded in paraffin. Matched blood samples were collected as
controls for GA detection.
Identification of GAs and measurement
of tumor mutational burden (TMB)
Total DNA was obtained from both formalin-fixed
paraffin-embedded (FFPE) tumor tissues and matched blood samples of
each patient using the QIAamp DNA FFPE Tissue Kit and QIAamp DNA
Blood Midi Kit (both Qiagen GmbH), respectively. The DNA samples
were sequenced using the next-generation sequencing-based
YuanSu450™ gene panel (OrigiMed®), in a laboratory
certified by the College of American Pathologists (CAP) and the
Clinical Laboratory Improvement Amendments (CLIA). The genes were
captured and sequenced with a mean depth of 800× using the NextSeq
500 system (Illumina, Inc.). Single nucleotide variants (SNVs) were
identified using MuTect (v1.7, www.broadinstitute.org/cancer/CGA), and
insertion-deletion polymorphisms (indels) were identified using
PINDEL (V0.2.5, http://www.pindel.com). The functional impact of these
mutations was annotated using SnpEff3.0 (http://snpeff.sourceforge.net). Copy number variation
(CNV) regions were identified by Control-FREEC (v9.7, http://boevalab.inf.ethz.ch/FREEC/index.html.) with
the following parameters: Window=50,000, and step=10,000. Gene
fusions/rearrangements were detected using the following in-house
pipeline: Paired-end reads with an abnormal insert size of
>2,000 bp (aligned to the same or different chromosomes) were
collected; discordant read pairs were clustered according to the
pairing relationship, and consistent breakpoints from the
paired-end discordant reads (within a single cluster) were
identified to establish potential fusion/rearrangement breakpoints.
Gene fusion/rearrangements were assessed using the Integrative
Genomics Viewer (v2.4, http://software.broadinstitute.org/software/igv/ReleaseNotes/2.4.x).
The TMB of each patient was calculated by counting the number of
somatic mutations (including SNVs and indels) per megabase (Mb) of
the sequence examined.
Statistical analysis
Statistical analyses were performed using SPSS
version 22.0 (IBM Corp) and significant differences were detected
using Fisher's exact test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Clinical characteristics of patients
with sarcoma
A total of 199 patients with soft-tissue or
osteogenic sarcomas were enrolled in the present study. This
included 105 male and 94 female patients, with a median age of 50
years (range, 1–86 years). The TMB values of all patients were
identified, from which 197 valid values were obtained with a median
of 1.5 muts/Mb (range, 0.7–24.5 muts/Mb) (Table I).
 | Table I.Clinicopathological features of 199
patients in the sarcoma cohort. |
Table I.
Clinicopathological features of 199
patients in the sarcoma cohort.
| Variable | Value |
|---|
| Sex, n (%) |
|
|
Male | 105 (52.76) |
|
Female | 94 (47.24) |
| Median age (range),
years | 50 (1–86) |
| Median TMB (range),
muts/Mb | 1.5 (0.7–42.5) |
GAs in 199 patients with sarcoma
Based on NGS targeting of 450 cancer-associated
genes, a total of 1,077 clinically relevant GAs were identified in
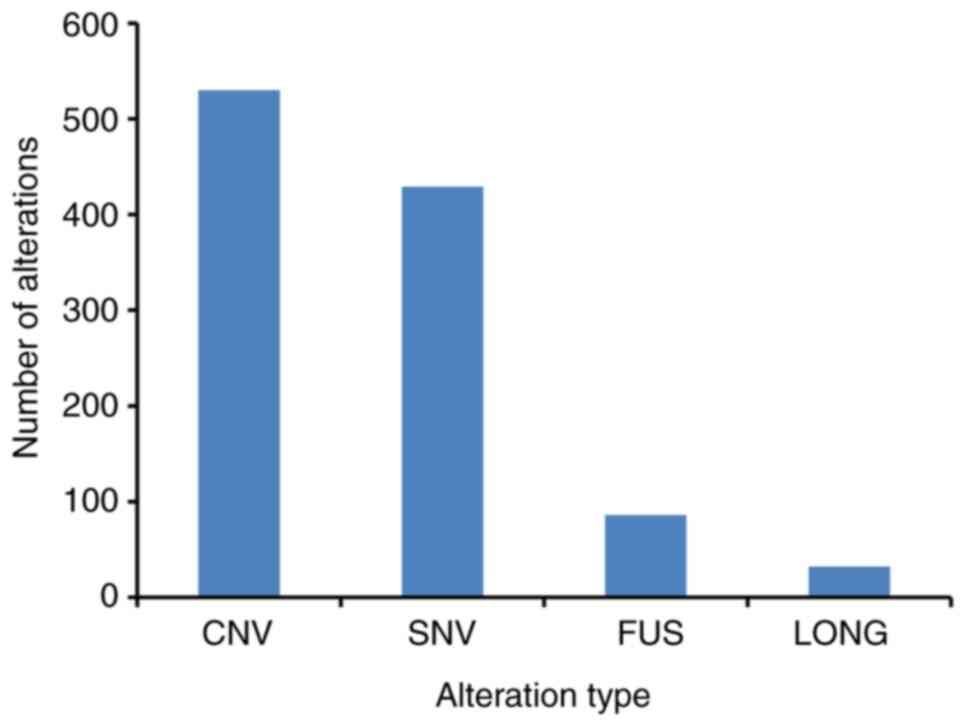
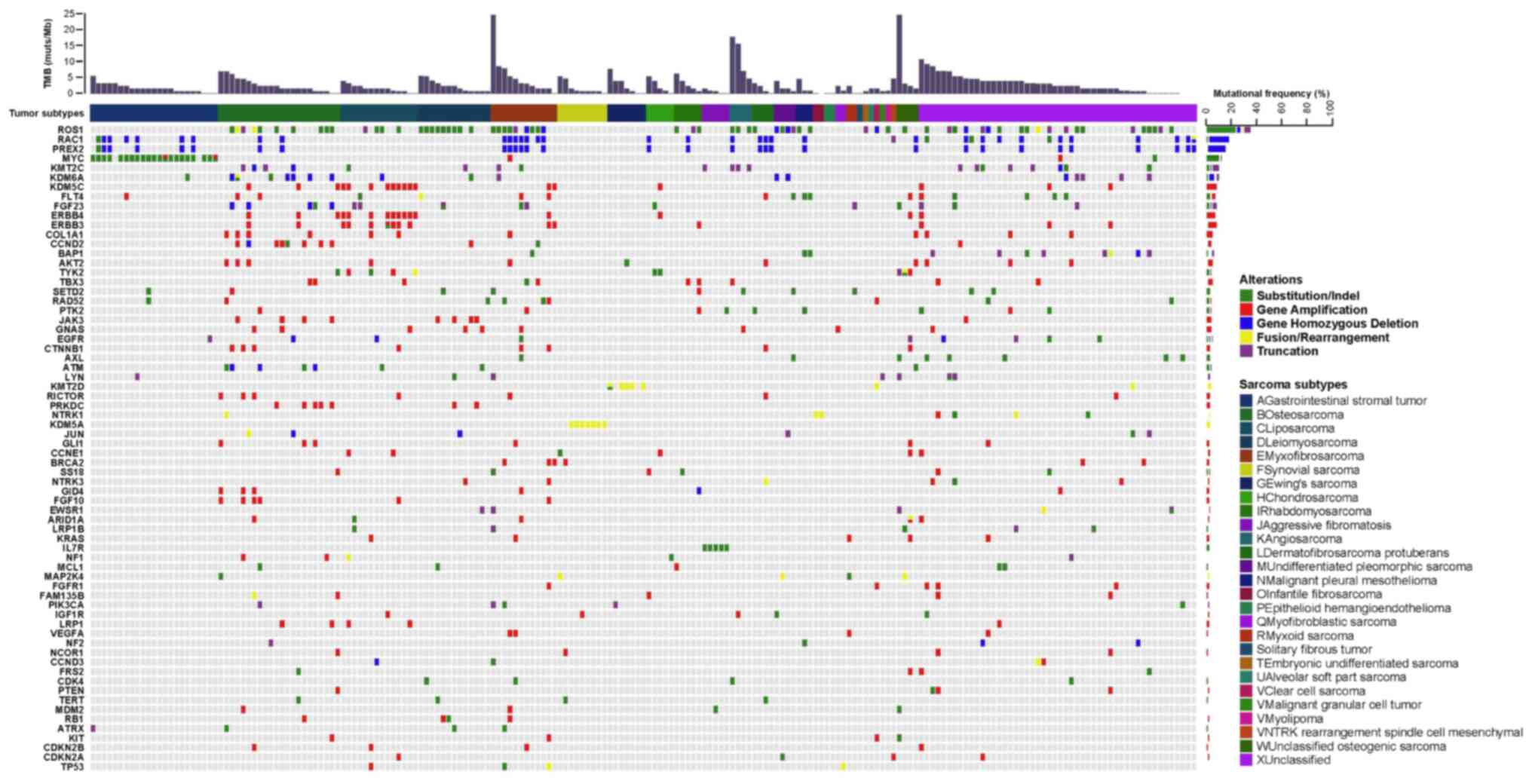
288 genes (Fig. 1), with an average
of 5.41 alterations per sample (range, 0–21). Among these GAs, CNV
was the most frequent mutation type (49.21%, 530/1,077), followed
by SNV/short indel (39.83%, 429/1077), gene fusion (7.99%,
86/1,077) and long indel (2.97%, 32/1,077) (Fig. 1 and Table
SI). The most commonly mutated genes with a mutation frequency
of >10% were TP53 (39.70%, 79/199), CDKN2A
(19.10%, 38/199), CDKN2B (15.08%, 30/199), KIT
(14.07%, 28/199), ATRX (10.05%, 20/199) and RB1
(10.05%, 20/199). Notably, most mutations in TP53, KIT and
ATRX were SNVs, while those in CDKN2A, CDKN2B and
RB1 were CNVs (Fig. 2).
NGS aids the diagnosis of sarcoma
All sarcomas were pathologically diagnosed before
sample collection, after which an experienced pathologist was
invited to make a second diagnosis. Only those with the same
diagnostic results were considered to be classified, while those
with inconsistent or undetermined diagnostic results were
considered as unclassified. As a result, 23 GISTs, 22
osteosarcomas, 12 myxofibrosarcoma, 11 liposarcomas, 11
leiomyosarcomas, 9 synovial sarcomas, 5 chondrosarcomas, 5
aggressive fibromatosis, 5 rhabdomyosarcomas, 4 Ewing's sarcomas, 4
angiosarcomas, 4 undefferentiated pleomorphic sarcomas, 3
mesotheliomas, 2 epithelioid hemangioendotheliomas, 2
myofibroblastic sarcomas, 2 myxoid sarcomas, 1 dermatofibrosarcoma
protuberan, 1 solitary fibrous tumor, 1 embryonic undifferentiated
sarcoma, 1 alveolar soft part sarcoma, 1 clear cell sarcoma, 1
malignant granular cell tumor, 1 myolipoma, 64 unclassified
soft-tissue sarcomas and 4 unclassified osteogenic sarcomas were
identified (Table II). Therefore,
further classification was carried out according to the results of
NGS.
| Table II.Comparison of sarcoma subtypes
identified by histochemistry- and NGS-based methods. |
Table II.
Comparison of sarcoma subtypes
identified by histochemistry- and NGS-based methods.
| Sarcoma
subtype |
Histochemistry-based, n | NGS-based, n |
|---|
| Gastrointestinal
stromal tumor | 23 | 23 |
| Osteosarcoma | 22 | 22 |
| Liposarcoma | 11 | 14 |
| Leiomyosarcoma | 11 | 13 |
|
Myxofibrosarcoma | 12 | 12 |
| Synovial
sarcoma | 9 | 9 |
| Ewing's
sarcoma | 4 | 7 |
| Chondrosarcoma | 5 | 5 |
|
Rhabdomyosarcoma | 5 | 5 |
| Aggressive
fibromatosis | 5 | 5 |
| Angiosarcoma | 4 | 4 |
| Dermatofibrosarcoma
protuberans | 1 | 4 |
| Undifferentiated
pleomorphic sarcoma | 4 | 4 |
| Malignant pleural
mesothelioma | 3 | 3 |
| Infantile
fibrosarcoma | 0 | 2 |
| Epithelioid
hemangioendothelioma | 2 | 2 |
| Myofibroblastic
sarcoma | 2 | 2 |
| Myxoid sarcoma | 2 | 2 |
| Solitary fibrous
tumor | 1 | 1 |
| Embryonic
undifferentiated sarcoma | 1 | 1 |
| Alveolar soft part
sarcoma | 1 | 1 |
| Clear cell
sarcoma | 1 | 1 |
| Malignant granular
cell tumor | 1 | 1 |
| Myolipoma | 1 | 1 |
| NTRK rearrangement
spindle cell mesenchymal tumors | 0 | 1 |
| Unclassified
osteogenic sarcoma | 4 | 4 |
| Unclassified | 64 | 50 |
According to the NGS detection results, 15
additional sarcoma cases were identified and classified, including
3 liposarcomas with amplifications in MDM2 and CDK4,
3 Ewing's sarcomas with EWSR1 fusions, 3 dermatofibrosarcoma
protuberans (DFSP) cases with fusions of PDGFB
(COL1A1-PDGFB), 2 leiomyosarcomas with mutations of
RB1 and TP53, 2 infantile fibrosarcomas with the
fusion of ETV6-NTRK3, 1 GIST with a mutation in
PDGFRA, and 1 NTRK rearranged spindle cell
mesenchymal tumor. Among them, 1 leiomyosarcoma was misdiagnosed as
a GIST before NGS auxiliary diagnosis. However, 54 cases remained
unclassifiable. The primary characteristic mutations of these 15
sarcomas are listed in Table
III.
| Table III.List of cases diagnosed by
next-generation sequencing. |
Table III.
List of cases diagnosed by
next-generation sequencing.
| Case | Sarcoma
subtype | Mutated genes | Mutation type |
|---|
| 1 | DFSP |
COL1A1-PDGFB | FUS |
| 2 | DFSP |
COL1A1-PDGFB | FUS |
| 3 | DFSP |
COL1A1-PDGFB | FUS |
| 4 | Ewing's
sarcoma | EWSR1
(EWSR1-FLI1) | FUS |
| 5 | Ewing's
sarcoma | EWSR1
(EWSR1-ERG) | FUS |
| 6 | Ewing's
sarcoma | EWSR1
(EWSR1-Intergenic) | FUS |
| 7 | Liposarcoma | CDK4 | CNV |
|
|
| MDM2 | CNV |
| 8 | Liposarcoma | CDK4 | CNV |
|
|
| MDM2 | CNV |
| 9 | Liposarcoma | CDK4 | CNV |
|
|
| MDM2 | CNV |
| 10 |
Leiomyosarcomas | RB1 | SNV |
|
|
| TP53 | SNV |
| 11 |
Leiomyosarcomas | TP53 | SNV |
| 12 | Infantile
fibrosarcoma |
ETV6-NTRK3 | FUS |
| 13 | Infantile
fibrosarcoma |
ETV6-NTRK3 | FUS |
| 14 | GIST | PDGFRA | SNV |
| 15 | Spindle cell
mesenchymal tumor | NTRK1
(LMNA-NTRK1) | FUS |
Association of GAs with sarcoma
subtypes
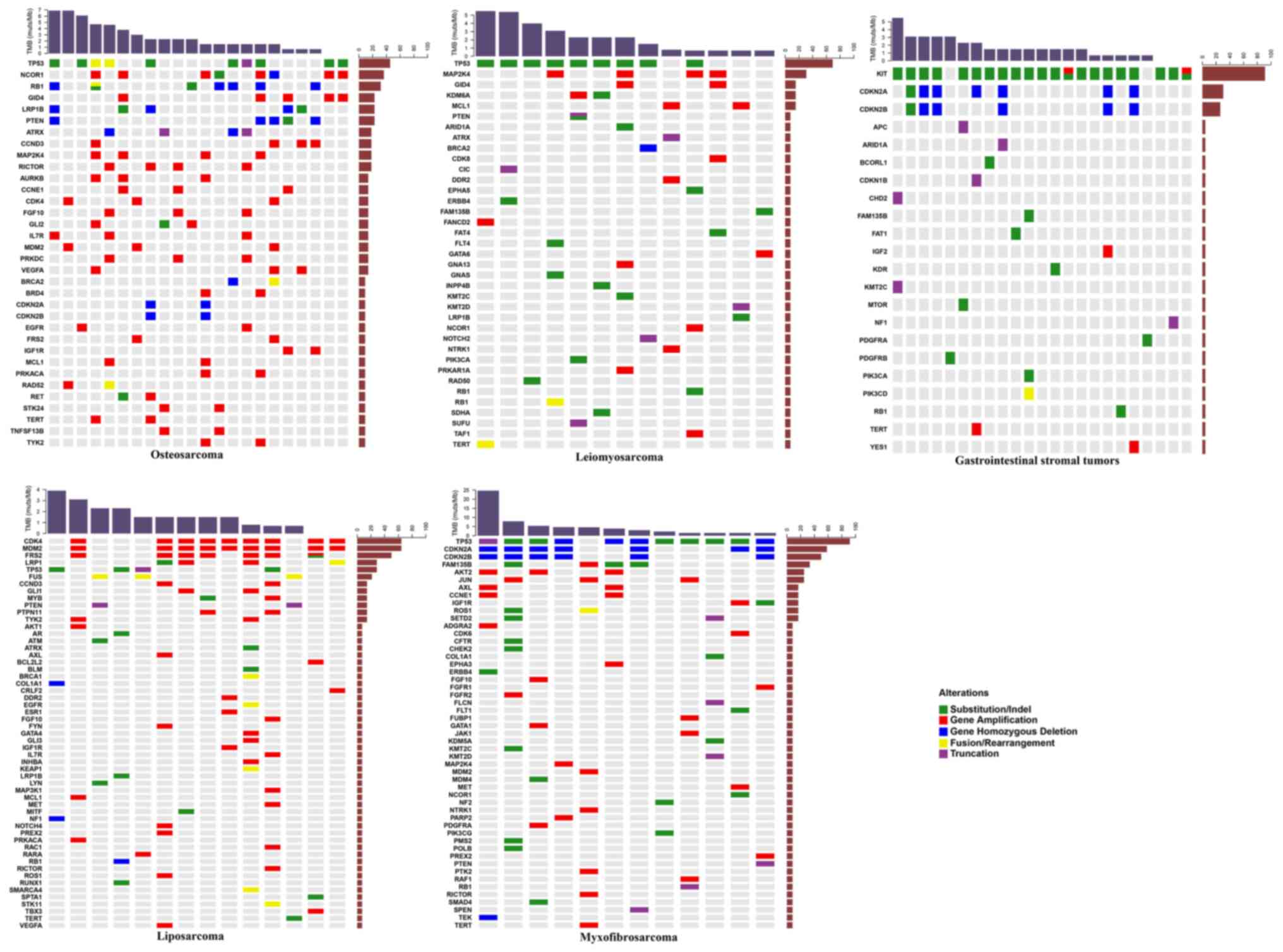
The mutational landscapes of sarcoma subtypes
including GIST, osteosarcoma, liposarcoma, leiomyosarcoma and
myxofibrosarcoma, were subsequently analyzed. The most common
mutated genes in GISTs were KIT, CDKN2A and CDKN2B,
and the most commonly mutated genes in osteosarcoma were TP53,
NCOR1, RB1, GID4, LRP1B, PTEN, ATRX, CCND3, MAP2K4 and
RICTOR. In liposarcoma, CDK4, MDM2, FRS2, LRP1 and
TP53 were the most frequently mutated, as were TP53,
MAP2K4, GID4, KDM6A and MCL1 in leiomyosarcoma. The most
commonly mutated genes in myxofibrosarcoma were TP53, CDKN2A,
CDKN2B, FAM135B, AKT2 and JUN (Fig. 3).
Statistical analysis revealed that mutations in
TP53, AKT2, FAM135B, CDKN2A, JUN, CDKN2B, ROS1, AXL, SETD2
and CCNE1 were significantly associated with
myxofibrosarcoma (Table IV). The
primary mutation type in GISTs was SNV, and mutations in KIT
and TP53 were significantly associated with GISTs. Gene
amplifications were the most common mutations in osteosarcoma and
liposarcoma (Fig. 2). Mutations in
NCOR1, GID4, LRP1B, RB1, AURKB, GLI2, RICTOR, MAP2K4, STK24,
TNFSF13B, CCNE1, PRKDC, PTEN, CCND3, FGF10, BRD4, PRKACA, RET
and IL7R were significantly associated with osteosarcoma,
while those in CDK4, MDM2, FRS2, FUS, LRP1, MYB, PTPN11 and
TYK2 were significantly associated with liposarcoma
(Table IV). Furthermore, mutations
in MAP2K4, TP53 and KDM6A were significantly
associated with leiomyosarcoma (Table
IV). Except for the negative association between TP53
mutations and GISTs, the majority of these frequently-mutated genes
significantly occurred in corresponding sarcomas. Although only 9
synovial sarcomas and 7 Ewing's sarcomas were identified in the
present study, the mutations in SS18 and EWSR1
significantly occurred in synovial sarcoma and Ewing's sarcoma,
respectively. Notably, the mutation of TP53 was also
significantly negatively associated with synovial sarcoma (Table IV).
| Table IV.Association between mutated genes and
sarcoma subtypes. |
Table IV.
Association between mutated genes and
sarcoma subtypes.
| Sarcoma
subtype | Mutated gene | Mutation frequency
within subtype, % | Mutation frequency
outside of subtype, % | P-value |
|---|
| Osteosarcoma | NCOR1 | 36.36 | 1.69 |
8.14×10−7 |
|
| GID4 | 22.73 | 1.13 |
1.92×10−4 |
|
| LRP1B | 22.73 | 1.69 |
4.75×10−4 |
|
| RB1 | 31.82 | 6.21 |
1.12×10−3 |
|
| AURKB | 13.64 | 0.00 |
1.19×10−3 |
|
| GLI2 | 13.64 | 0.00 |
1.19×10−3 |
|
| RICTOR | 18.18 | 1.13 |
1.15×10−3 |
|
| MAP2K4 | 18.18 | 2.82 |
9.90×10−3 |
|
| STK24 | 9.09 | 0.00 | 0.012 |
|
|
TNFSF13B | 9.09 | 0.00 | 0.012 |
|
| CCNE1 | 13.64 | 1.69 | 0.019 |
|
| PRKDC | 13.64 | 1.69 | 0.019 |
|
| PTEN | 22.73 | 6.21 | 0.020 |
|
| CCND3 | 18.18 | 3.95 | 0.022 |
|
| FGF10 | 13.64 | 2.26 | 0.031 |
|
| BRD4 | 9.09 | 0.56 | 0.033 |
|
| PRKACA | 9.09 | 0.56 | 0.033 |
|
| RET | 9.09 | 0.56 | 0.033 |
|
| IL7R | 13.64 | 2.82 | 0.046 |
|
Myxofibrosarcoma | TP53 | 91.67 | 31.02 |
4.28×10−5 |
|
| AKT2 | 25.00 | 0.53 |
6.57×10−4 |
|
| FAM135B | 33.33 | 2.67 |
8.32×10−4 |
|
| CDKN2A | 58.33 | 16.04 |
1.77×10−3 |
|
| JUN | 25.00 | 1.60 |
3.06×10−3 |
|
| CDKN2B | 50.00 | 12.83 |
3.48×10−3 |
|
| ROS1 | 16.67 | 1.07 | 0.019 |
|
| AXL | 16.67 | 1.60 | 0.030 |
|
| SETD2 | 16.67 | 1.60 | 0.030 |
|
| CCNE1 | 16.67 | 2.14 | 0.044 |
| Leiomyosarcoma | MAP2K4 | 30.77 | 2.69 |
1.17×10−3 |
|
| TP53 | 69.23 | 32.26 | 0.013 |
|
| KDM6A | 15.38 | 1.08 | 0.022 |
| GIST | KIT | 91.30 | 1.70 |
4.00×10−23 |
|
| TP53 | 0.00 | 39.20 |
3.36×10−5 |
| Liposarcoma | CDK4 | 64.29 | 3.78 |
1.72×10−8 |
|
| MDM2 | 64.29 | 4.86 |
6.96×10−8 |
|
| FRS2 | 50.00 | 4.32 |
7.70×10−6 |
|
| FUS | 21.43 | 0.00 |
2.81×10−4 |
|
| LRP1 | 28.57 | 3.24 |
2.58×10−3 |
|
| MYB | 14.29 | 0.00 |
4.62×10−3 |
|
| PTPN11 | 14.29 | 0.54 | 0.013 |
|
| TYK2 | 14.29 | 1.62 | 0.041 |
| Synovial
sarcoma | SS18 | 77.78 | 0.00 |
1.63×10−11 |
|
| TP53 | 0.00 | 36.32 | 0.029 |
|
| CREB3L1 | 11.11 | 0.00 | 0.045 |
|
| PDK1 | 11.11 | 0.00 | 0.045 |
|
| TET1 | 11.11 | 0.00 | 0.045 |
| Ewing's
sarcoma | EWSR1 | 71.43 | 1.04 |
1.75×10−7 |
|
| EPHB1 | 14.29 | 0.00 | 0.035 |
|
| FEV | 14.29 | 0.00 | 0.035 |
|
| VGLL3 | 14.29 | 0.00 | 0.035 |
Association between TMB value, sarcoma
subtype and patient demographics
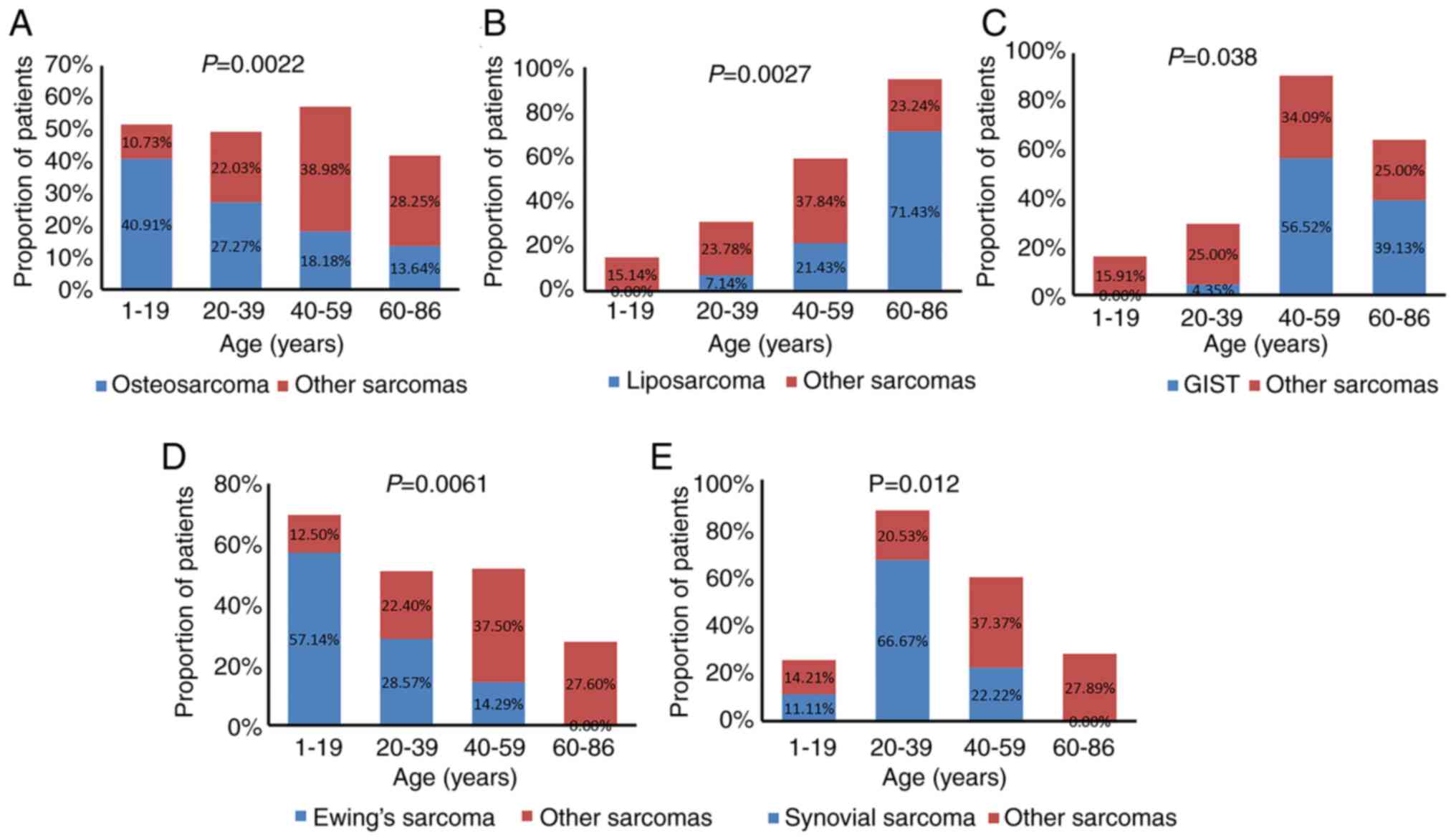
The associations between sarcoma subtype, TMB value
and patient sex and age were further analyzed. Based on age
distribution, the patients we categorized into 4 groups: i) 1–19
years; ii) 20–39 years; iii) 40–59 years; iv) and 60–86 years of
age. Osteosarcoma and Ewing's sarcoma commonly occurred in younger
patients (1–19 years old), accounting for 40.91% (9/22) and 57.14%
(4/7), respectively; GISTs and liposarcomas were more common in
elderly patients (60–86 years old), accounting for 39.13% (9/23)
and 71.43% (10/14), respectively; and synovial sarcoma commonly
occurred in young patients (20–39 years old), accounting for 66.67%
(6/9). Statistical analysis showed that osteosarcomas, Ewing's
sarcomas and synovial sarcomas significantly occurred in younger
patients, while liposarcomas and GISTs significantly occurred in
older patients (Fig. 4).
Most of the sarcoma subtypes had comparable
frequencies in male and female patients, while 11 of the 13
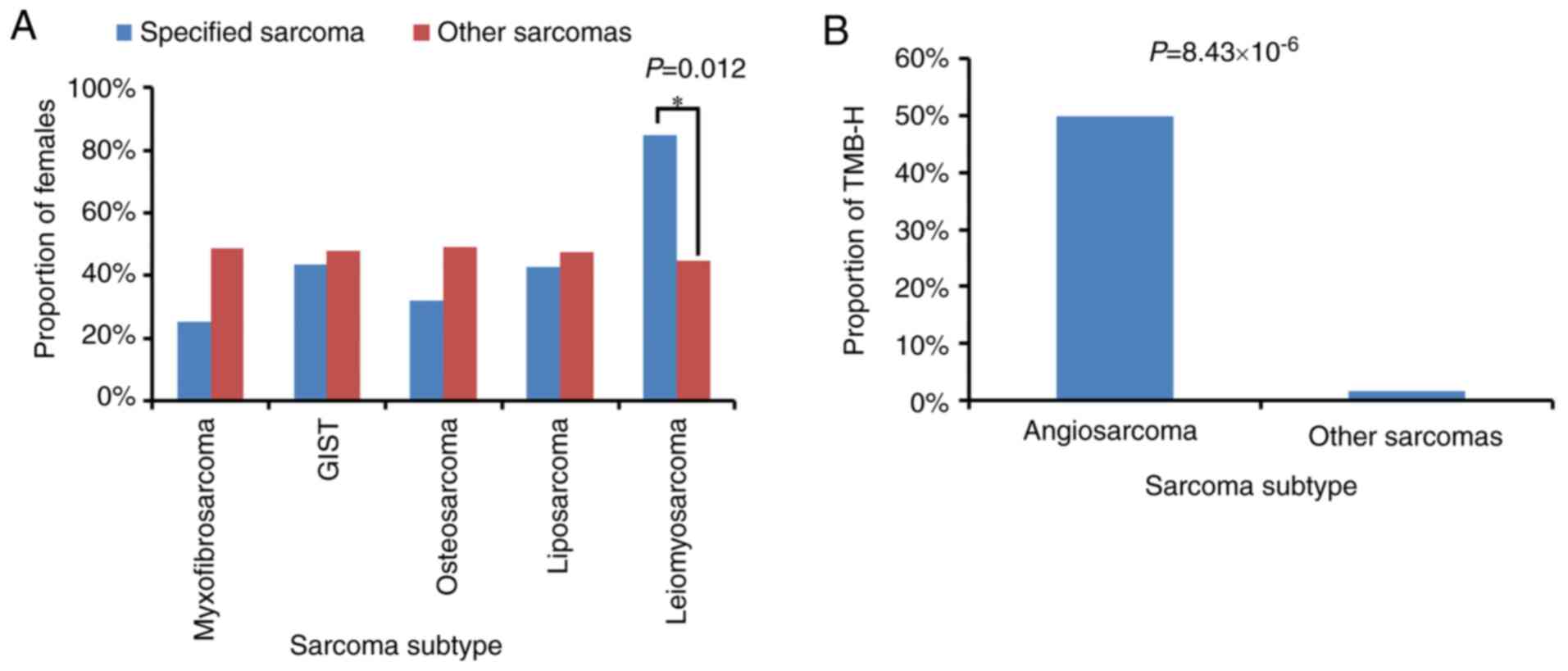
leiomyosarcomas occurred in females. Statistical analysis showed
that leiomyosarcomas occurred significantly more often in women
than in men (Fig. 5A). TMB values
were obtained from 194 of the 199 enrolled patients. High TMB
(TMB-H), which was defined as a TMB value >10 muts/Mb, was
observed in 5 patients, including 1 with fibrosarcoma, 2 with
angiosarcoma and 2 patients with unclassified soft-tissue sarcoma.
Notably, only 4 angiosarcomas were identified in the cohort, and
statistical analysis showed that angiosarcoma was significantly
associated with TMB-H (Fig. 5B).
New neurotrophic tyrosine kinase
(NTRK)1/3 fusions in the current cohort
NTRK1/3 mutations were detected in 11 of the
199 patients with sarcoma. Among these patients, 4 NTRK1 and
1 NTRK3 mutations were gene amplifications, and 2
NTRK1 and 4 NTRK3 mutations were gene fusions,
including 1 LMNA-NTRK1, 1 IRF2BP2-NTRK1, 1
MEF2A-NTRK3, 1 ITFG1-NTRK3 and 2 ETV6-NTRK3
fusions (Table V). Similar to
previous reports (33,34), gene fusions of ETV6-NTRK3 and
LMNA-NTRK1 were detected in 2 infantile fibrosarcoma cases
and 1 unclassified sarcoma, respectively. To the best of our
knowledge, the present study is the first to describe the fusion of
IRF2BP2-NTRK1, MEF2A-NTRK3 and ITFG1-NTRK3 in
sarcoma. Therefore, sarcoma patients with NTRK fusions or
amplifications may potentially benefit from NTRK inhibitor
therapy.
| Table V.List of NTRK fusions in the present
cohort. |
Table V.
List of NTRK fusions in the present
cohort.
| Case | Gene | Mutation type | DNA change |
|---|
| 1 | NTRK1 | CNV | Gene
amplification |
| 2 | NTRK1 | CNV | Gene
amplification |
| 3 | NTRK1 | CNV | Gene
amplification |
| 4 | NTRK1 | CNV | Gene
amplification |
| 5 | NTRK3 | CNV | Gene
amplification |
| 6 | NTRK1 | FUSION | LMNA-NTRK1 |
| 7 | NTRK1 | FUSION | IRF2BP2-NTRK1 |
| 8 | NTRK3 | FUSION | MEF2A-NTRK3 |
| 9 | NTRK3 | FUSION | ITFG1-NTRK3 |
| 10 | NTRK3 | FUSION | ETV6-NTRK3 |
| 11 | NTRK3 | FUSION | ETV6-NTRK3 |
Discussion
The tumorigenesis of sarcoma is characterized by
genomic abnormalities, manifested as multiple phenotypic changes
and divided into various subtypes (35). To date, histological examination
remains the primary method of sarcoma diagnosis (36). The histological and molecular
heterogeneity of sarcoma make it particularly difficult to
diagnose, though with the rapid development of NGS technology,
increasing numbers of sarcoma genome sequencing studies have
emerged (37–40). In the present study, the most
commonly mutated genes were identified in 199 patients with
sarcoma, and included TP53, CDKN2A, CDKN2B, KIT, ATRX and
RB1. TP53 encodes the p53 protein and functions in
the p53 pathway, while the CDKN2B and CDKN2A genes
are associated with the regulation of p53 pathways (41). These findings suggest that p53
pathway mutations frequently occurred in the present cohort, which
is consistent with a previous report (42). Somatic mutations of TP53 are
associated with poor prognosis and low chemotherapy response rates
in various tumor types (43,44). Poor patient prognosis is associated
with TP53 mutations in various sarcoma subtypes, such as
gliosarcoma (45), osteosarcoma
(46), Ewing's sarcoma (47), chondrosarcoma (48) and liposarcoma (49). In the present study, the highly
frequent TP53 mutations were identified in osteosarcoma,
fibrosarcoma, liposarcoma and leiomyosarcoma, suggesting an
association with poor prognosis in these subtypes.
Molecular diagnosis based on NGS detection can
accurately characterize sarcomas according to molecular
characteristics, which is a powerful complement to histological
identification (50) since
pathologists often provide descriptions such as ‘probable’ or
‘possible’ during sarcoma diagnosis. The molecular features of
different sarcoma subtypes have been extensively studied. For
example, PDGFB rearrangement in DFSP, MDM2 and
CDK4 amplification in liposarcoma, EWSR1
translocation in Ewing's sarcoma, and SS18 translocation in
synovial sarcoma (51–54). With the additional assistance of NGS
detection, 15 sarcomas that were difficult diagnose by histological
examination were further classified. Notably, one misclassified
sarcoma subtype was also successfully corrected. These results
suggest that NGS technology can effectively assist in the diagnosis
and classification of sarcoma subtypes. However, 54 cases were
still not well classified, which may be due to the fact that the
corresponding molecular characteristics or biomarkers of sarcoma
are still not clearly understood. Therefore, the identification of
sarcoma biomarkers is important for further diagnostic
advancements.
A number of specific mutations have been used for
the classification of sarcoma. For example, Pierron et al
(55) defined a novel type of bone
sarcoma by identifying the BCOR-CCNB3 gene fusion. Yoshida
et al (56) identified that
CIC-rearranged sarcomas were distinctly different from Ewing's
sarcomas, clinically, morphologically and immunohistochemically.
Furthermore, Michal et al (57) reported a
EWSR1-SMAD3-rearranged fibroblastic tumor that represented a
novel subtype, and Chiang et al (58) identified a novel tumor type with the
features of fibrosarcoma by NTRK fusion. However, few
reports have focused on the association between gene mutations and
different sarcoma subtypes. In the present study, the associations
between GAs and tumor subtypes, patient demographics and TMB values
were analyzed, which may provide potential biomarkers for the
future diagnosis of sarcoma.
KIT mutations are a significant phenotypic
feature of GISTs (59). As
predicted, the association between KIT mutations and GISTs
was also identified in the present study. In addition, a
significant negative association was observed between TP53
mutations and GISTs. These results suggest that mutations in both
KIT and TP53 may be used as biomarkers for GIST
diagnosis.
In osteosarcoma, the frequent mutation of RB1
was highly prevalent, and was thus proposed as a potential
prognostic biomarker (60). With the
exception RB1, the association between NCOR1 mutation
and osteosarcoma was also identified in the present study.
NCOR1 is a transcription factor that regulates various
biological functions (61). As a
tumor suppressor gene, mutation in NCOR1 was confirmed to be
associated with the prognostic prediction of numerous cancers, such
as breast cancer, lung adenocarcinoma and GISTs (62,63).
These findings suggest that NCOR1 mutations are a potential
biomarker for the molecular diagnosis and prognosis of
osteosarcoma. In the present study, genes such as GID4,
LRP1B and PTEN were found to be significantly associated
with osteosarcoma. These results have important relevance for
guiding the diagnosis of osteosarcoma.
The amplification of MDM2 and CDK4 has
been reported to occur in liposarcoma, and may therefore be
considered as therapeutic targets (64), as well as used to assist the
diagnosis of well-differentiated and dedifferentiated liposarcomas
(65). The significant association
between CDK4 and MDM2 amplification and liposarcoma
was detected in the present study, and was able to successfully
classify 2 cases of liposarcoma from soft-tissue sarcomas. These
results support the significance of NGS detection in the diagnosis
of liposarcoma. In addition to CDK4 and MDM2, 6
additional mutated genes (including FRS2, FUS, LRP1, MYB,
PTPN11 and TYK2) were also associated with the
liposarcoma subtype, indicating the potential diagnostic value of
these genes in liposarcoma.
Fibrosarcoma can also be divided into multiple
subtypes, such as myxofibrosarcoma, DFSP, solitary fibrous tumor
and infantile fibrosarcoma. COL1A1-PDGFB fusion is a
prominent molecular feature of DFSP (66). Mutations within the telomerase
reverse transcriptase promoter were reported to be associated with
the histologically malignant features of solitary fibrous tumors,
and to some extent, to play an auxiliary role in their diagnosis
and treatment (67). Although there
are high mutational frequencies of TP53, RB1, CDKN2A, CDKN2B,
NF1 and NTRK1, few molecular predictors of
myxofibrosarcoma have been identified (68). Due to the limited number of subtypes
across the samples, only the association between the mutated genes
and myxofibrosarcoma was analyzed in the present study, and the
results showed that mutations in TP53, AKT2, FAM135B, CDKN2A,
JUN, CDKN2B, ROS1, AXL, SETD2 and CCNE1 were
significantly associated with myxofibrosarcoma. These results may
be helpful for the diagnosis of myxofibrosarcoma. Although the
number of cases was not large, the association between mutated
genes and sarcoma subtype may still be used to guide molecular
diagnoses. For example, based on 9 cases, a positive association
was detected between the SS18 mutation and synovial sarcoma,
and based on 7 cases, a significant association was also detected
between the EWSR1 mutation and Ewing's sarcoma. However,
studies with larger cohorts are required to identify potential
biomarkers for the auxiliary diagnosis of sarcomas.
The incidence rate of different sarcoma subtypes
varies with sex and age. Classical osteosarcoma and
rhabdomyosarcoma frequently occur in children and adolescents,
while myxofibrosarcoma, synovial sarcoma, angiosarcoma, DFSP and
clear cell sarcoma are more common in patients >20 years of age
(69,70). Also, myxofibrosarcoma,
rhabdomyosarcoma and synovial sarcoma may be more likely to occur
in men, while the occurrence of leiomyosarcoma was notably more
common in female participants (70).
The results of the present study support previous studies
suggesting that osteosarcomas, Ewing's sarcomas, GISTs and
liposarcomas are associated with patient age, and that
leiomyosarcoma is associated with patient sex.
TMB is a novel biomarker for the prognosis of cancer
patients treated with immune checkpoint inhibitors (ICPIs)
(71,72). The majority of sarcomas (such as
osteosarcomas, GISTs and Ewing's sarcomas) are reported to have a
low TMB (73,74), while Trabucco (75) reported TMB-H in skin atypical
fibroxanthoma and skin sarcoma. However, the results of the present
study support that with the exception of 2 angiosarcoma cases, the
TMB value of most sarcomas is low. Though only 4 cases were
included in the current cohort, a significant association was
detected between angiosarcomas and TMB-H, indicating that patients
with angiosarcomas may benefit from ICPI therapy.
NTRK functions in the development, differentiation
and metabolism of nerves and other tissues. NTRK inhibitors can be
used as targeted agents for tumor therapy, thus the detection of
NTRK fusions has important clinical significance (76–78).
ETV6-NTRK3 fusion is common in infantile fibrosarcoma, and
the tropomyosin-related kinase inhibitor LOXO-101 was reported to
benefit infantile fibrosarcoma patients harboring ETV6-NTRK3
fusions (78). A metastatic
infantile fibrosarcoma patient harboring LMNA-NTRK1 showed a
complete and durable response to crizotinib (79). Furthermore, Wong et al
(77) presented a case of a reverse
transcription PCR ETV6-NTRK3-negative congenital infantile
fibrosarcoma harboring a LMNA-NTRK1 gene fusion with a
near-complete response to crizotinib. The data support the
assumption that NTRK fusions are the drug target of LOXO-101
or crizotinib in sarcomas. In the present study, ETV6-NTRK3
and LMNA-NTRK1 fusions were successfully detected,
indicating a potential treatment target for these patients.
Follow-up information on the targeted treatment of patients with
new NTRK fusions of IRF2BP2-NTRK1, MEF2A-NTRK3 and
ITFG1-NTRK3 may also guide and expand the use of NTRK fusion
therapy in patients with sarcoma.
In conclusion, the present study investigated the
genomic mutation profiles of pan-sarcomas, identified potential
biomarkers, and accurately classified sarcoma subtypes with the
assistance of NGS. The identification of NTRK fusions in
sarcoma provides important value for NTRK inhibitor therapy. The
absence of FISH confirmation is a limitation of the present study.
However, the results support that NGS targeting may effectively
promote the accurate classification and diagnosis of sarcomas, and
provide guidance for precise therapeutic strategies for bone and
soft-tissue sarcomas.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed in the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
LX, XX and JW recruited the patients, collected the
samples, analyzed the data and wrote the manuscript; XS, PZ and AL
processed samples, conducted experiments, analyzed data and
reviewed the manuscript; XS and PZ are responsible for the
authenticity of the raw data. BZ designed and supervised the study.
All authors read and approved the final manuscript.
Ethical approval and consent to
participate
The present study was approved by the Ethics
Committees of the National Cancer Center/Cancer Hospital, Chinese
Academy of Medical Sciences and Peking Union Medical College and
the First Affiliated Hospital of Sun Yat Sen University. Written
informed consent for participation was obtained from all
subjects.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
NGS
|
next-generation sequencing
|
|
FISH
|
fluorescence in situ
hybridization
|
|
GA
|
genomic alteration
|
|
GIST
|
gastrointestinal stromal tumor
|
|
SNV
|
single nucleotide variant
|
|
Indel
|
insertion-deletion polymorphism
|
|
CNV
|
copy number variation
|
|
TMB
|
tumor mutational burden
|
|
DFSP
|
dermatofibrosarcoma protuberans
|
|
TMB-H
|
high tumor mutational burden
|
|
ICPI
|
immune checkpoint inhibitor
|
References
|
1
|
Taylor BS, Barretina J, Maki RG, Antonescu
CR, Singer S and Ladanyi M: Advances in sarcoma genomics and new
therapeutic targets. Nat Rev Cancer. 11:541–557. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Szkandera J, Absenger G, Liegl-Atzwanger
B, Pichler M, Stotz M, Samonigg H, Glehr M, Zacherl M, Stojakovic
T, Gerger A and Leithner A: Elevated preoperative
neutrophil/lymphocyte ratio is associated with poor prognosis in
soft-tissue sarcoma patients. Br J Cancer. 108:1677–1683. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chan JY, Zhang Z, Chew W, Tan GF, Lim CL,
Zhou L, Goh WL, Poon E, Somasundaram N, Selvarajan S, et al:
Biological significance and prognostic relevance of peripheral
blood neutrophil-to-lymphocyte ratio in soft tissue sarcoma. Sci
Rep. 8:119592018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Somasundaram N, Chan SH, Quek R and Ngeow
J: Chapter 43-Advances in Sarcoma Genomics and Therapeutic
Management. In: Oncogenomics. Dammacco F and Silvestris F: Academic
Press; pp. 609–621. 2019
|
|
6
|
Penel N, Ray-Coquard I, Bal-Mahieu C,
Chevreau C, Le Cesne A, Italiano A, Bompas E, Clisant S, Baldeyrou
B, Lansiaux A, et al: Low level of baseline circulating VEGF-A is
associated with better outcome in patients with vascular sarcomas
receiving sorafenib: An ancillary study from a phase II trial.
Target Oncol. 9:273–277. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Serranogarcia C, Heinrich MC, Zhu M, Raut
CP, Eilers G, Ravegnini G, Demetri GD, Bauer S, Fletcher JA and
George S: In vitro and in vivo activity of regorafenib (REGO) in
drug-resistant gastrointestinal stromal tumors (GIST). J Clin
Oncol. 31 (Suppl 15):10510. 2013. View Article : Google Scholar
|
|
8
|
Rickel K, Fang F and Tao J: Molecular
genetics of osteosarcoma. Bone. 102:69–79. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Maguire M: Comment on: Early symptoms of
bone and soft tissue sarcomas: Could they be diagnosed earlier? Ann
R Coll Surg Engl. 94:4512012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Scurr M and Judson I: Sarcoma. Cancer
Chemother Biol Response Modif. 21:637–653. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hastings CA, Torkildson JC and Agrawal AK:
Sarcomas of the Soft Tissues and Bone. Handbook of Pediatric
Hematology and Oncology: Children's Hospital & Research Center
Oakland: Second edition. pp. 183–192. 2012, View Article : Google Scholar
|
|
13
|
Sinha S and Peach AH: Diagnosis and
management of soft tissue sarcoma. BMJ. 341:c71702010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Whelan J, McTiernan A, Cooper N, Wong YK,
Francis M, Vernon S and Strauss SJ: Incidence and survival of
malignant bone sarcomas in England 1979–2007. Int J Cancer.
131:E508–E517. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang L, Chen Y, Cui T, Knösel T, Zhang Q,
Geier C, Katenkamp D and Petersen I: Identification of biomarkers
to distinguish clear cell sarcoma from malignant melanoma. Hum
Pathol. 43:1463–1470. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
van Brummelen EMJ, de Boer A, Beijnen JH
and Schellens JHM: BRAF mutations as predictive biomarker for
response to Anti-EGFR monoclonal antibodies. Oncologist.
22:864–872. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Evola FR, Costarella L, Pavone V, Caff G,
Cannavò L, Sessa A, Avondo S and Sessa G: Biomarkers of
osteosarcoma, chondrosarcoma, and ewing sarcoma. Front Pharmacol.
8:1502017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hutanu D, Popescu R, Stefanescu H, Pirtea
L, Candea A, Sarau C, Boruga O, Mehdi L, Ciuca I and Tanasescu S:
The molecular genetic expression as a novel biomarker in the
evaluation and monitoring of patients with osteosarcoma-subtype
bone cancer disease. Biochem Genet. 55:291–299. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cancer Genome Atlas Research Network, .
Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA,
Ellrott K, Shmulevich I, Sander C and Stuart JM: The cancer genome
atlas Pan-cancer analysis project. Nat Genet. 45:1113–1120. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cancer Genome Atlas Research Network, .
Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H,
Robertson AG, Pashtan I, Shen R, et al: Integrated genomic
characterization of endometrial carcinoma. Nature. 497:67–73. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cancer Genome Atlas Research Network, .
Electronic address simpleelizabeth.demicco@sinaihealthsystem.ca.
et al Comprehensive and integrated genomic characterization of
adult soft tissue sarcomas. Cell. 171:950–965.e28. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chudasama P, Mughal SS, Sanders MA,
Hübschmann D, Chung I, Deeg KI, Wong SH, Rabe S, Hlevnjak M,
Zapatka M, et al: Integrative genomic and transcriptomic analysis
of leiomyosarcoma. Nat Commun. 9:1442018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mertens F, Antonescu CR and Mitelman F:
Gene fusions in soft tissue tumors: Recurrent and overlapping
pathogenetic themes. Genes Chromosomes Cancer. 55:291–310. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Italiano A, Bianchini L, Gjernes E,
Keslair F, Ranchere-Vince D, Dumollard JM, Haudebourg J, Leroux A,
Mainguené C, Terrier P, et al: Clinical and biological significance
of CDK4 amplification in well-differentiated and dedifferentiated
liposarcomas. Clin Cancer Res. 15:5696–5703. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chibon F, Lagarde P, Salas S, Pérot G,
Brouste V, Tirode F, Lucchesi C, de Reynies A, Kauffmann A, Bui B,
et al: Validated prediction of clinical outcome in sarcomas and
multiple types of cancer on the basis of a gene expression
signature related to genome complexity. Nat Med. 16:781–787. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sankar S and Lessnick SL: Promiscuous
partnerships in Ewing's sarcoma. Cancer Genet. 204:351–365. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fligman I, Lonardo F, Jhanwar SC, Gerald
WL, Woodruff J and Ladanyi M: Molecular diagnosis of synovial
sarcoma and characterization of a variant SYT-SSX2 fusion
transcript. Am J Pathol. 147:1592–1599. 1995.PubMed/NCBI
|
|
28
|
Antonescu CR, Tschernyavsky SJ, Woodruff
JM, Jungbluth AA, Brennan MF and Ladanyi M: Molecular diagnosis of
clear cell sarcoma: Detection of EWS-ATF1 and MITF-M transcripts
and histopathological and ultrastructural analysis of 12 cases. J
Mol Diagn. 4:44–52. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kubota T: Gastrointestinal stromal tumor
(GIST) and imatinib. Int J Clin Oncol. 11:184–189. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stacchiotti S, Crippa F, Messina A,
Pilotti S, Gronchi A, Blay JY and Casali PG: Response to imatinib
in villonodular pigmented synovitis (PVNS) resistant to nilotinib.
Clin Sarcoma Res. 3:82013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rausch JL, Boichuk S, Ali AA, Patil SS,
Liu L, Lee DM, Brown MF, Makielski KR, Liu Y, Taguchi T, et al:
Opposing roles of KIT and ABL1 in the therapeutic response of
gastrointestinal stromal tumor (GIST) cells to imatinib mesylate.
Oncotarget. 8:4471–4483. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dickson MA, Tap WD, Keohan ML, D'Angelo
SP, Gounder MM, Antonescu CR, Landa J, Qin LX, Rathbone DD, Condy
MM, et al: Phase II trial of the CDK4 inhibitor PD0332991 in
patients with advanced CDK4-amplified well-differentiated or
dedifferentiated liposarcoma. J Clin Oncol. 31:2024–2028. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Knezevich SR, McFadden DE, Tao W, Lim JF
and Sorensen PH: A novel ETV6-NTRK3 gene fusion in congenital
fibrosarcoma. Nat Genet. 18:184–187. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Haller F, Knopf J, Ackermann A, Bieg M,
Kleinheinz K, Schlesner M, Moskalev EA, Will R, Satir AA,
Abdelmagid IE, et al: Paediatric and adult soft tissue sarcomas
with NTRK1 gene fusions: A subset of spindle cell sarcomas unified
by a prominent myopericytic/haemangiopericytic pattern. J Pathol.
238:700–710. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gibault L, Pérot G, Chibon F, Bonnin S,
Lagarde P, Terrier P, Coindre JM and Aurias A: New insights in
sarcoma oncogenesis: A comprehensive analysis of a large series of
160 soft tissue sarcomas with complex genomics. J Pathol.
223:64–71. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schaefer IM, Cote GM and Hornick JL:
Contemporary sarcoma diagnosis, genetics, and genomics. J Clin
Oncol. 36:101–110. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Krystel-Whittemore M, Taylor MS, Rivera M,
Lennerz JK, Le LP, Dias-Santagata D, Iafrate AJ, Deshpande V,
Chebib I, Nielsen GP, et al: Novel and established EWSR1 gene
fusions and associations identified by next generation sequencing
and fluorescence in-situ hybridization. Hum Pathol. 93:65–73. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liang X, Li Q, Xu B, Hu S, Wang Q, Li Y,
Zong Y, Zhang S and Li C: Mutation landscape and tumor mutation
burden analysis of Chinese patients with pulmonary sarcomatoid
carcinomas. Int J Clin Oncol. 24:1061–1068. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zheng B, Qu Y, Wang J, Shi Y and Yan W:
Pathogenic and targetable genetic alterations in resected recurrent
undifferentiated pleomorphic sarcomas identified by targeted
next-generation sequencing. Cancer Genomics Proteomics. 16:221–228.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nagy A, Bhaduri A, Shahmarvand N,
Shahryari J, Zehnder JL, Warnke RA, Mughal T, Ali S and Ohgami RS:
Next-generation sequencing of idiopathic multicentric and
unicentric Castleman disease and follicular dendritic cell
sarcomas. Blood Adv. 2:481–491. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nakashima R, Fujita M, Enomoto T, Haba T,
Yoshino K, Wada H, Kurachi H, Sasaki M, Wakasa K, Inoue M, et al:
Alteration of p16 and p15 genes in human uterine tumours. Br J
Cancer. 80:458–467. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Toguchida J, Yamaguchi T, Ritchie B,
Beauchamp RL, Dayton SH, Herrera GE, Yamamuro T, Kotoura Y, Sasaki
MS, Little JB, et al: Mutation spectrum of the p53 gene in bone and
soft tissue sarcomas. Cancer Res. 52:6194–6199. 1992.PubMed/NCBI
|
|
43
|
Quinlan DC, Davidson AG, Summers CL,
Warden HE and Doshi HM: Accumulation of p53 protein correlates with
a poor prognosis in human lung cancer. Cancer Res. 52:4828–4831.
1992.PubMed/NCBI
|
|
44
|
Li JP, Zhang XM, Zhang Z, Zheng LH, Jindal
S and Liu YJ: Association of p53 expression with poor prognosis in
patients with triple-negative breast invasive ductal carcinoma.
Medicine (Baltimore). 98:e154492019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cho SY, Park C, Na D, Han JY, Lee J, Park
OK, Zhang C, Sung CO, Moon HE, Kim Y, et al: High prevalence of
TP53 mutations is associated with poor survival and an EMT
signature in gliosarcoma patients. Exp Mol Med. 49:e3172017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tsuchiya T, Sekine K, Hinohara S, Namiki
T, Nobori T and Kaneko Y: Analysis of the p16INK4, p14ARF, p15,
TP53, and MDM2 genes and their prognostic implications in
osteosarcoma and Ewing sarcoma. Cancer Genet Cytogenet. 120:91–98.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Huang HY, Illei PB, Zhao Z, Mazumdar M,
Huvos AG, Healey JH, Wexler LH, Gorlick R, Meyers P and Ladanyi M:
Ewing sarcomas with p53 mutation or p16/p14ARF homozygous deletion:
A highly lethal subset associated with poor chemoresponse. J Clin
Oncol. 23:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Oshiro Y, Chaturvedi V, Hayden D, Nazeer
T, Johnson M, Johnston DA, Ordóñez NG, Ayala AG and Czerniak B:
Altered p53 is associated with aggressive behavior of
chondrosarcoma: A long term follow-up study. Cancer. 83:2324–2334.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Antonescu CR, Tschernyavsky SJ, Decuseara
R, Leung DH, Woodruff JM, Brennan MF, Bridge JA, Neff JR, Goldblum
JR and Ladanyi M: Prognostic impact of P53 status, TLS-CHOP fusion
transcript structure, and histological grade in myxoid liposarcoma:
A molecular and clinicopathologic study of 82 cases. Clin Cancer
Res. 7:3977–3987. 2001.PubMed/NCBI
|
|
50
|
Italiano A, Di Mauro I, Rapp J, Pierron G,
Auger N, Alberti L, Chibon F, Escande F, Voegeli AC, Ghnassia JP,
et al: Clinical effect of molecular methods in sarcoma diagnosis
(GENSARC): A prospective, multicentre, observational study. Lancet
Oncol. 17:532–538. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Llombart B, Monteagudo C, Sanmartin O,
López-Guerrero JA, Serra-Guillén C, Poveda A, Jorda E,
Fernandez-Serra A, Pellín A, Guillén C and Llombart-Bosch A:
Dermatofibrosarcoma protuberans: A clinicopathological,
immunohistochemical, genetic (COL1A1-PDGFB), and therapeutic study
of low-grade versus high-grade (fibrosarcomatous) tumors. J Am Acad
Dermatol. 65:564–575. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kammerer-Jacquet SF, Thierry S, Cabillic
F, Lannes M, Burtin F, Henno S, Dugay F, Bouzillé G, Rioux-Leclercq
N, Belaud-Rotureau MA and Stock N: Differential diagnosis of
atypical lipomatous tumor/well-differentiated liposarcoma and
dedifferentiated liposarcoma: Utility of p16 in combination with
MDM2 and CDK4 immunohistochemistry. Hum Pathol. 59:34–40. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kim SK and Park YK: Ewing sarcoma: A
chronicle of molecular pathogenesis. Hum Pathol. 55:91–100. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
El Beaino M, Araujo DM, Lazar AJ and Lin
PP: Synovial sarcoma: Advances in diagnosis and treatment
identification of new biologic targets to improve multimodal
therapy. Ann Surg Oncol. 24:2145–2154. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Pierron G, Tirode F, Lucchesi C, Reynaud
S, Ballet S, Cohen-Gogo S, Perrin V, Coindre JM and Delattre O: A
new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat
Genet. 44:461–466. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yoshida A, Goto K, Kodaira M, Kobayashi E,
Kawamoto H, Mori T, Yoshimoto S, Endo O, Kodama N, Kushima R, et
al: CIC-rearranged Sarcomas: A study of 20 cases and comparisons
with Ewing sarcomas. Am J Surg Pathol. 40:313–323. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Michal M, Berry RS, Rubin BP, Kilpatrick
SE, Agaimy A, Kazakov DV, Steiner P, Ptakova N, Martinek P,
Hadravsky L, et al: EWSR1-SMAD3-rearranged Fibroblastic Tumor: An
Emerging Entity in an Increasingly More Complex Group of
Fibroblastic/Myofibroblastic Neoplasms. Am J Surg Pathol.
42:1325–1333. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chiang S, Cotzia P, Hyman DM, Drilon A,
Tap WD, Zhang L, Hechtman JF, Frosina D, Jungbluth AA, Murali R, et
al: NTRK fusions define a novel uterine sarcoma subtype with
features of fibrosarcoma. Am J Surg Pathol. 42:791–798. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Fletcher JA and Rubin BP: KIT mutations in
GIST. Curr Opin Genet Dev. 17:3–7. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ren W and Gu G: Prognostic implications of
RB1 tumour suppressor gene alterations in the clinical outcome of
human osteosarcoma: A meta-analysis. Eur J Cancer Care (Engl).
262017.doi: 10.1111/ecc.12401.
|
|
61
|
Müller L, Hainberger D, Stolz V, Hamminger
P, Hassan H, Preglej T, Boucheron N, Sakaguchi S, Wiegers GJ,
Villunger A, et al: The corepressor NCOR1 regulates the survival of
single-positive thymocytes. Sci Rep. 7:159282017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Noblejas-Lopez MDM, Morcillo-Garcia S,
Nieto-Jimenez C, Nuncia-Cantarero M, Győrffy B, Galan-Moya EM,
Pandiella A and Ocaña A: Evaluation of transcriptionally regulated
genes identifies NCOR1 in hormone receptor negative breast tumors
and lung adenocarcinomas as a potential tumor suppressor gene. PLoS
One. 13:e02077762018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wang W, Song XW, Bu XM, Zhang N and Zhao
CH: PDCD2 and NCoR1 as putative tumor suppressors in gastric
gastrointestinal stromal tumors. Cell Oncol (Dordr). 39:129–137.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kanojia D, Nagata Y, Garg M, Lee DH, Sato
A, Yoshida K, Sato Y, Sanada M, Mayakonda A, Bartenhagen C, et al:
Genomic landscape of liposarcoma. Oncotarget. 6:42429–42444. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Binh MB, Sastre-Garau X, Guillou L, de
Pinieux G, Terrier P, Lagacé R, Aurias A, Hostein I and Coindre JM:
MDM2 and CDK4 immunostainings are useful adjuncts in diagnosing
well-differentiated and dedifferentiated liposarcoma subtypes: A
comparative analysis of 559 soft tissue neoplasms with genetic
data. Am J Surg Pathol. 29:1340–1347. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhang Z, Chen H, Chen M, He X, Wang Y and
Zhang H: Application of COL1A1-PDGFB fusion gene detection by
fluorescence in situ hybridization in biopsy tissue of
dermatofibrosarcoma protuberans. J Dermatol. 44:798–802. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bianchi G, Sambri A, Pedrini E, Pazzaglia
L, Sangiorgi L, Ruengwanichayakun P, Donati D, Benassi MS and Righi
A: Histological and molecular features of solitary fibrous tumor of
the extremities: Clinical correlation. Virchows Arch. 476:445–454.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ogura K, Hosoda F, Arai Y, Nakamura H,
Hama N, Totoki Y, Yoshida A, Nagai M, Kato M, Arakawa E, et al:
Integrated genetic and epigenetic analysis of myxofibrosarcoma. Nat
Commun. 9:27652018. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Rogozhin DV, Bulycheva IV, Konovalov DM,
Talalaev AG, Roshchin VY, Ektova AP, Bogoroditsky YS, Strykov VA,
Kazakova AN, Olshanskaya YV, et al: Classical osteosarcoma in
children and adolescent. Arkh Patol. 77:68–74. 2015.(In Russian).
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Mans DR, Budhu Lall AE, Macnack VL, van
Tholl JA, Zandveld EB and Vrede MA: Incidence, and gender, age and
ethnic distribution of sarcomas in the republic of Suriname from
1980 to 2008. West Indian Med J. 63:121–127. 2014.PubMed/NCBI
|
|
71
|
Knepper TC, Montesion M, Russell JS, Sokol
ES, Frampton GM, Miller VA, Albacker LA, McLeod HL, Eroglu Z,
Khushalani NI, et al: The genomic landscape of Merkel cell
carcinoma and clinicogenomic biomarkers of response to immune
checkpoint inhibitor therapy. Clin Cancer Res. 25:5961–5971. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Chalmers ZR, Connelly CF, Fabrizio D, Gay
L, Ali SM, Ennis R, Schrock A, Campbell B, Shlien A, Chmielecki J,
et al: Analysis of 100,000 human cancer genomes reveals the
landscape of tumor mutational burden. Genome Med. 9:342017.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Cote GM, He J and Choy E: Next-generation
sequencing for patients with sarcoma: A single center experience.
Oncologist. 23:234–242. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Chen X, Bahrami A, Pappo A, Easton J,
Dalton J, Hedlund E, Ellison D, Shurtleff S, Wu G, Wei L, et al:
Recurrent somatic structural variations contribute to tumorigenesis
in pediatric osteosarcoma. Cell Rep. 7:104–112. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Trabucco SE, Ali SM, Sokol E, Schrock AB,
Albacker LA, Chung J, Xavier MF, Disel U, Miller VA, Ross JS, et
al: Frequency of genomic biomarkers of response to immunotherapy in
sarcoma. J Clin Oncol. 36:11579. 2018. View Article : Google Scholar
|
|
76
|
Lange AM and Lo HW: Inhibiting TRK
proteins in clinical cancer therapy. Cancers (Basel). 10:1052018.
View Article : Google Scholar
|
|
77
|
Wong V, Pavlick D, Brennan T, Yelensky R,
Crawford J, Ross JS, Miller VA, Malicki D, Stephens PJ, Ali SM and
Ahn H: Evaluation of a congenital infantile fibrosarcoma by
comprehensive genomic profiling reveals an LMNA-NTRK1 gene fusion
responsive to Crizotinib. J Natl Cancer Inst. 108:djv3072016.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Nagasubramanian R, Wei J, Gordon P,
Rastatter JC, Cox MC and Pappo A: Infantile fibrosarcoma with
NTRK3-ETV6 fusion successfully treated with the tropomyosin-related
kinase inhibitor LOXO-101. Pediatric Blood Cancer. 63:1468–1470.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Bender J, Anderson B, Bloom DA, Rabah R,
McDougall R, Vats P and Mody R: Refractory and metastatic infantile
fibrosarcoma harboring LMNA-NTRK1 fusion shows complete and durable
response to crizotinib. Cold Spring Harb Mol Case Stud.
5:a0033762019. View Article : Google Scholar : PubMed/NCBI
|