Introduction
Extraskeletal Ewing sarcoma (EES) is a rare entity
that belongs to the ES family of tumors (ESFT), which is a group of
small round tumor cells that share a common neural histology and
genetic mechanism (1–4). In addition to EES, ESFT includes the
classical ES of bone (ESB), which is the second most common primary
bone malignancy in the pediatric population, peripheral primitive
neuroectodermal tumor (pPNET) and Askin tumor of the chest wall,
which is a subtype of pPNET (5). EES
was first discovered in 1969 (6),
but it remains an elusive pathology in the literature.
Magnetic resonance (MR) and
fluorodeoxyglucose-positron emission tomography (FDG-PET) imaging
are used for initial diagnosis and detection of metastasis,
respectively (7–9). Definitive treatment for localized
disease include chemotherapy (10–12) and
surgery (13). Radiation therapy is
effective in unresectable diseases (14).
The present review discusses the imaging and
diagnostic modalities (histology and molecular genetics) used for
the initial diagnosis and detection of metastasis in EES. In
addition, the present review provides an update on the current
treatment protocols (chemotherapy, surgery and radiotherapy) at
different stages of the disease and their respective outcomes.
Epidemiology
The incidence of EES is 0.4 per million, which is 10
times less than that of ESB (1). Its
prevalence follows a bimodal distribution, peaking in those who are
<5 years and >35 years (15).
Patients with EES tend to be older than those with ESB, and EES is
not associated with sex or race unlike ESB (15–17).
EES is a rapidly growing mass that causes localized
pain (18). It develops within the
soft tissues of any anatomic region, but the most common sites
include the upper thigh, buttocks, upper arm and shoulders
(18). Conversely, metastases are
commonly observed in the lungs, bones and bone marrow (19). Thus, the symptoms of EES depend on
its primary site, as well as the site of metastases, which are
found in 25% of all cases at presentation (19).
Diagnosis
Diagnosis and local staging
Imaging plays a central role in the diagnosis,
staging, treatment monitoring and surveillance of EES (20). However, the imaging characteristics
of EES are non-specific (20–22). EES
can be diagnosed via ultrasonography (US), computed tomography (CT)
or MR imaging, with each modality having its own specific
indications (20).
With US, EES usually appears as a heterogenous mass
of low echogenicity with intratumor flow signals on a Doppler
study. With CT, EES presents as a large sharply demarcated mass
with similar intensity to the surrounding muscles. Post-contrast
medium enhancement reveals areas of necrosis, while the surrounding
viable contour appears enhanced and heterogeneous (20). Calcification is present in only 10%
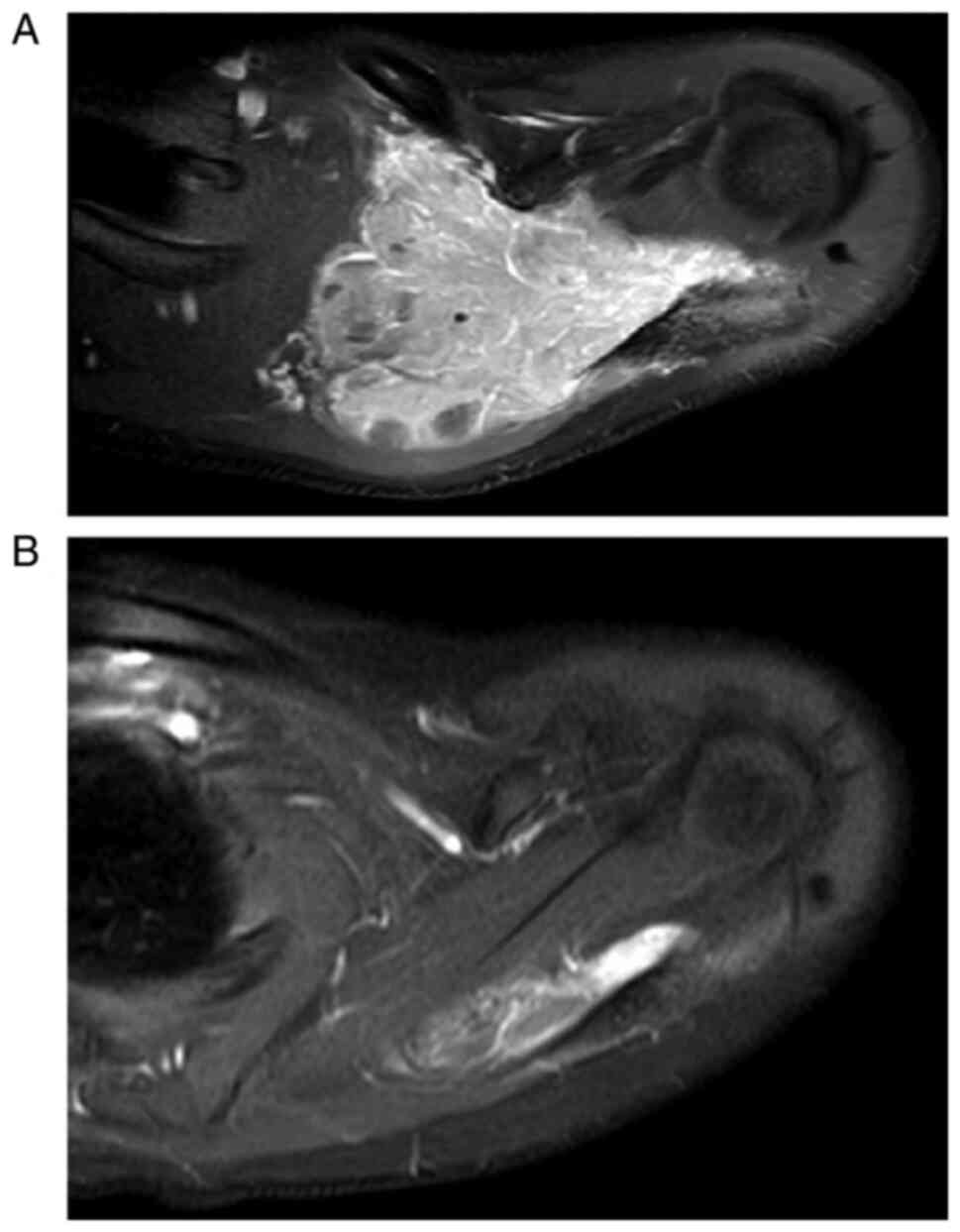
of cases, appearing faint and amorphous (20). With MR imaging, EES has
low-to-intermediate signal intensity on T1-weighted sequences and
displays high signal intensity on T2-weighted images, with variable
post-contrast enhancement (Fig. 1)
(20–23). MR imaging is performed prior to
biopsy to help determine the optimal biopsy site and to avoid the
distortion caused by post-biopsy changes (24). This is also recommended for restaging
purposes prior to local control, as the tumor may have receded or
progressed during neo-adjuvant chemotherapy (7).
MR imaging and CT scans are accurate for local
staging of malignant bone and soft-tissue tumors (7). However, MR imaging is used more
frequently due to its high detection sensitivity for soft tissue
contrast, as well as its ability to avoid radiation exposure from
CT scans (7).
Systemic staging and surveillance
In addition to local staging of the primary tumor,
imaging is also used to detect the presence of metastatic disease.
CT scans of the chest are superior to conventional radiographs in
detecting lung metastasis. Intravenous contrast is not required
unless there is hilar, mediastinal or chest wall involvement that
may require further delineation of these regions. With chest CT,
metastatic legions are typically round, ovoid, sharply demarcated
and located in the lung periphery (7). Notably, it is recommended to perform a
chest CT prior to biopsy to avoid the stimulation of metastasis due
to atelectasis secondary to general anesthesia (7).
Bone scans are also recommended for detection of
bone metastasis at presentation. If regions of increased uptake are
noted on the scan, radiographs are performed to further confirm the
presence of metastasis (7). This may
be followed by cross-sectional imaging modalities of these areas if
the diagnosis of metastasis is uncertain (7). FDG-PET appears to be superior to bone
scans for the detection of bone metastasis in ES (8,9). FDG-PET
is also used to assess chemotherapeutic response and detect
recurrent disease (7). Gerth et
al (25) reported that PET/CT is
sensitive (87%) and specific (97%) for detecting distant
metastasis, with an accuracy of 94%.
Overall, definitive diagnosis is made with a
CT-guided or ultrasound-guided core-needle biopsy, as well as
pathological examination of the resected surgical specimen
(26).
Histology
EES is a soft, multilobulated, gray-yellow tumor,
whose diameter rarely exceeds 10 cm (18). EES can contain cystic, hemorrhagic or
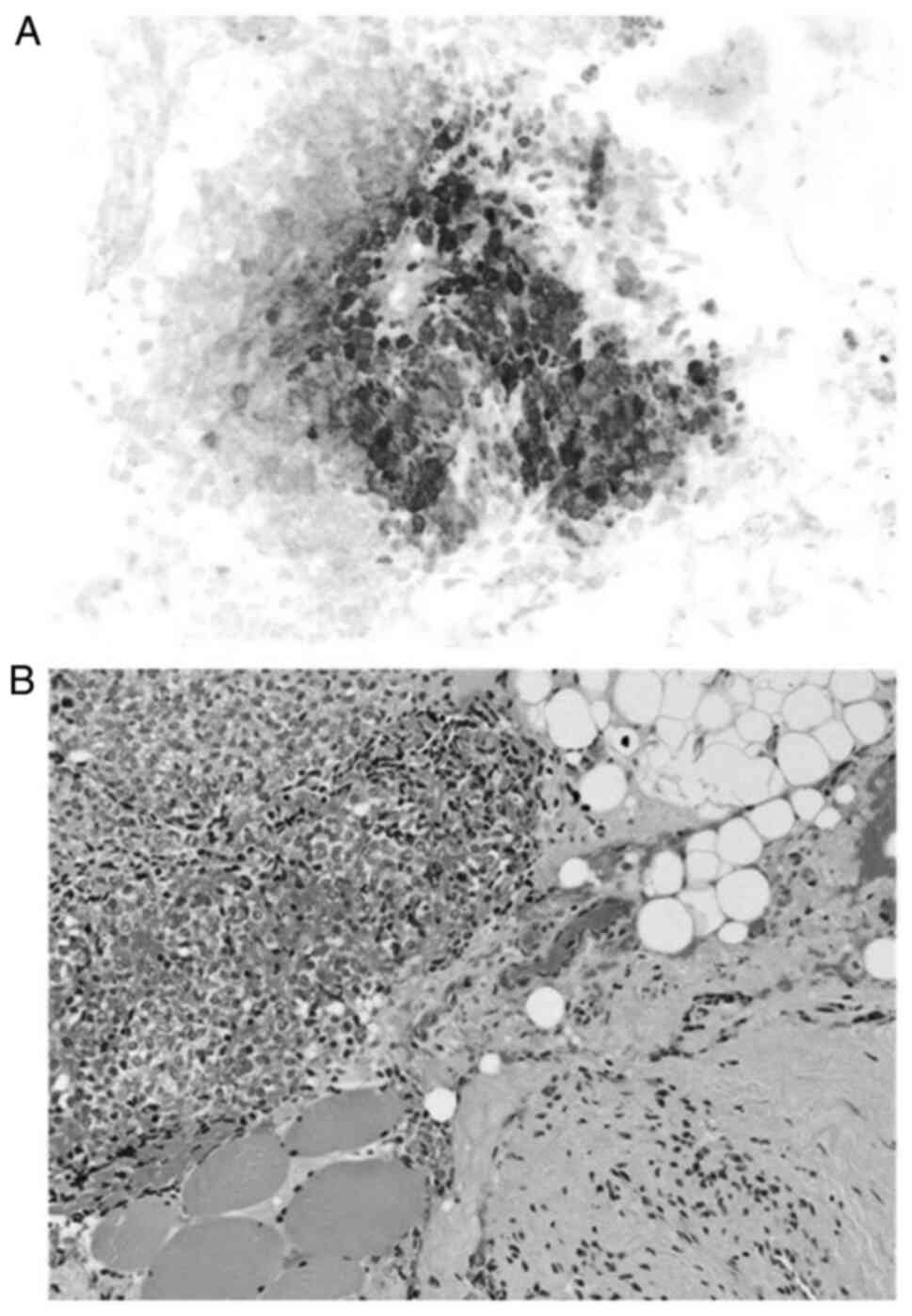
necrotic areas; however, calcifications are rare (18). Microscopically, EES appears as
monomorphic, small, round blue cells that contain large spherical
nuclei, inconspicuous nucleoli and indistinct cytoplasmic borders
(27). These cells lack
extracellular matrix and have low mitotic activity, albeit necrotic
areas are commonly observed (Figs. 2
and 3) (27).
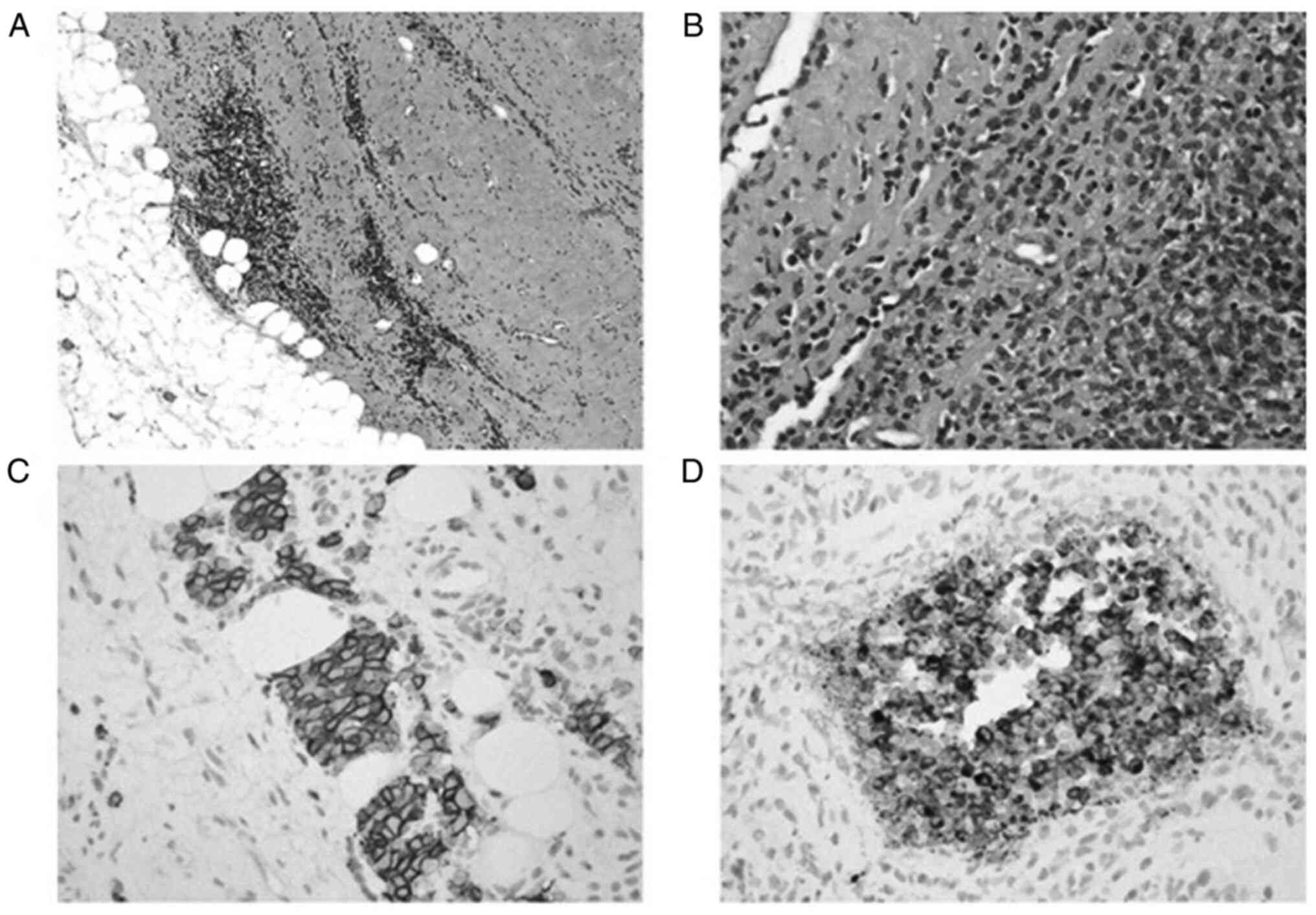
Immunohistochemistry is used in addition to light
microscopy to diagnosis EES (18). A
spectrum of immunohistochemical markers are used to study EES since
no marker is pathognomonic (18).
These markers include CD99 antigen, which is a single-chain type-1
glycoprotein that is highly sensitive but not specific, S-100
protein and synaptophysin, both of which are neural markers, and
FLI1, which was recently discovered as a DNA-binding transcription
factor that is involved in t(11;22) translocation and has higher
specificity than CD99 (18,28).
Molecular genetics
The use of pathology and immunohistochemistry for
the diagnosis of EES is sufficient in most cases. However,
molecular genetic analysis via reverse transcriptase (RT) PCR or
fluorescence in-situ hybridization (FISH) add an additional
diagnostic domain that is essential in unusual variants. The two
most common chromosomal translocations specific to EES and ESFT are
t(11;22)(q24;q12) and t(21;22)(q22;q12) (29). Translocation t(11;22)(q24;q12) is
present in 90% of all cases, which causes a fusion between the FLI
gene on 11q24 and the EWSR1 gene on 22q12, creating an EWS/FLI-1
transcript that has the DNA binding domain of FLI-1 instead of the
RNA-binding domain of EWS (29).
Conversely, translocation t(21;22)(q22;q12) fuses EWSR1 and ERG,
which is another DNA-binding protein (30), creating an oncogenic transcription
factor that inhibits apoptosis (30). Less common translocations have also
been reported, all of which involve the EWSR1 gene on chromosome 22
(30).
Treatment
Only a few studies have investigated the optimal
treatment options and prognostic factors for EES (13,31–34).
Previously, EES was treated using the soft-tissue sarcomas protocol
(21); however, the current
treatment recommendation by the National Comprehensive Cancer
Network (NCCN) is local treatment (surgery and/or radiotherapy)
plus chemotherapy (31,32). According to the NCCN and the European
Society for Medical Oncology (31,32), all
members of the Ewing family can be treated with the same algorithm,
although the optimum treatment and natural history of EES remain
unknown (31,32). Previous studies have demonstrated the
role of surgery in EES compared with skeletal ES, suggesting that
wide surgical resection increases the survival rate of patients
with EES than those with ESB (33,34).
Systemic treatment: Chemotherapy
The use of systemic chemotherapy in the treatment of
localized ESFT has increased the 5-year survival rate from 5 to
10%, to >65%, which is primarily due to the elimination of
micrometastases (3). To the best of
our knowledge, Rud et al (34) was the first to report on the
importance of multiagent chemotherapy in the treatment of patients
with EES, demonstrating that the survival rate increased from 28%,
before 1970, to 48%, after 1970. In addition, Raney et al
(10) reported a 10-year survival
rate of 61–77% following multiagent chemotherapy. According to
Bacci et al (11),
neoadjuvant and adjuvant chemotherapies exhibited comparable
results in patients with localized disease. Overall, chemotherapy
improves the overall survival rates and decreases the likelihood of
recurrence following surgery (12).
The current regimens include alternating
vincristine-doxorubicin, cyclophosphamide and ifosfamide-etoposide
cycles every 3 weeks (Tables I and
II) (35). Womer et al (35) demonstrated that patients who received
the same chemotherapy regimen every 2 weeks instead of 3 weeks,
known as interval compression, exhibited a better event-free
survival rate (73 vs. 65%) (36).
Although chemotherapy programs are essential and have proven
effective, chemotherapy alone without surgery and/or radiotherapy
is insufficient as a treatment option (36).
 | Table I.Interval compressed chemotherapy for
Ewing sarcoma (35). Induction
chemotherapy. |
Table I.
Interval compressed chemotherapy for
Ewing sarcoma (35). Induction
chemotherapy.
| Regimen | Drug | Timing |
|---|
| A | Vincristine | Weeks 1, 5 and
6 |
|
| Doxorubicin |
|
|
|
Cyclophosphamide |
|
|
| Filgrastim |
|
| B | Ifosfamide | Weeks 3, 7 and
11 |
|
| Etoposide |
|
|
| Filgrastim |
|
| Table II.Interval compressed chemotherapy for
Ewing sarcoma (35). Consolidation
therapy. |
Table II.
Interval compressed chemotherapy for
Ewing sarcoma (35). Consolidation
therapy.
| Regimen | Drug | Surgery alone | Radiotherapy
alone | Surgery and
radiotherapy |
|---|
| A | Vincristine | Weeks 15 and
19 | Weeks 13 (with the
start of RT) and 25 | Weeks 15 (with the
start of RT) and 27 |
|
| Doxorubicin |
|
|
|
|
|
Cyclophosphamide |
|
|
|
|
| Filgrastim |
|
|
|
| B | Ifosfamide | Weeks 17, 21, 25
and 29 | Weeks 15, 19, 23
and 27 | Weeks 17, 21, 25
and 29 |
|
| Etoposide |
|
|
|
|
| Filgrastim |
|
|
|
| C | Vincristine | Weeks 23 and
27 | Weeks 17 and
21 | Weeks 19 and
23 |
|
|
Cyclophosphamide |
|
|
|
|
| Filgrastim |
|
|
|
Localized treatment: Surgery and/or
radiotherapy
EES is radiosensitive; however, the use of
radiotherapy alone for local control has proved less effective over
the years. This is due to advancements in surgical techniques for
limb salvage, as well as the adverse effects of radiotherapy and
the high incidence of local recurrence (>30–35%). Nonetheless,
studies comparing surgery with radiotherapy may have overlooked the
importance of selection factors dictating local therapy decisions
(4,32). Surgery is performed in cases where
marginal or wide resection is possible (32,37).
Resectable lesions are usually small, peripheral in location and
have a good response to induction chemotherapy (36). Conversely, irradiated lesions are
often large, central in location and have a poor response to
induction chemotherapy (36). The
ability to obtain adequate negative margins has the strongest
influence on local control of malignancy. Wider margins are
required when the response to chemotherapy is not adequate
(4,32). When it is not possible to obtain wide
margins due to the presence of fixed structures, such as vessels
and/or nerves, postoperative radiotherapy can be implemented for
better local control (4,32).
The overall 5-year survival rate is better in
patients who undergo complete resection, with wide surgical margins
compared with suboptimal margins (13). However, if the tumor is not
resectable with clear margins (spine or skull) or if surgery would
involve vital fixed structures, definitive radiotherapy can be
implemented (14,37).
According to the European Cooperative Ewing Sarcoma
Studies (CESS) and European Intergroup Cooperative Ewing's Sarcoma
Study (EICESS) trials (38),
intralesional resection with radiotherapy did not result in a
superior local control rate compared with radiotherapy alone. In
such cases, surgery can be avoided in favor of radiotherapy
(38). Currently, definitive
radiotherapy is only indicated for inoperable lesions, with a
recommended dose of 54–55 Gy depending on the involved site
(10,39). However, larger tumors may require
higher doses (10,39).
Metastatic disease
Metastatic disease or unresectable recurrent disease
is treated with the same approach as localized disease but carries
a worse prognosis (32). In such
cases, chemotherapy is an option, with the limited benefit of
extending progression-free survival (23). In patients with lung metastasis,
whole lung irradiation has been demonstrated to offer survival
benefit (40). Chemotherapy regimens
similar to localized disease can be used in such cases but are less
effective (32).
Prognosis
The prognosis of EES is more favorable compared with
the skeletal subtype, although factors affecting prognosis seem to
be similar in both subtypes (16,17,41).
Notably, the 5-year overall survival rate is superior for localized
EES compared with localized skeletal ES (15).
Risk factors associated with worse prognosis in EES
include older age (26), pelvic
involvement (42), high WBC,
elevated LDH and low Hb at the time of diagnosis (43,44).
Intitial tumor size is also a risk factor and is considered a
strong prognostic factor in localized disease (26). However, for those treated with
neoadjuvant chemotherapy, histological response is regarded as the
strongest independent prognostic factor (37). Metastatic disease is a bad prognostic
factor, with a 5-year overall survival rate <30% and <20% in
the case of extrapulmonary involvement (45). The favorable prognositc factors
include lesions at the extremity and surgery (46). Notably, recurrent ES, whether
localized or metastatic, is almost always fatal (10).
Conclusions
In the evaluation and diagnosis of EES, while MR
imaging is used for local staging, FDG-PET is used to detect
metastatic disease. Immunohistochemical analysis, as well as RT PCR
and FISH are performed to detect genetic translocations to confirm
the diagnosis. Definitive treatment for localized disease include
neoadjuvant chemotherapy followed by surgery. Radiation therapy
plays a role in unresectable disease or when negative margins are
not possible.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
AA, KM, MS, RH, RS, NK, AT and SS contributed
equally to the preparation of the present study, including the
literature review and drafting the initial manuscript. AA and KM
confirmed the authenticity of all the raw data. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
EES
|
extraskeletal Ewing sarcoma
|
|
ESFT
|
Ewing sarcoma family of tumors
|
|
ESB
|
Ewing sarcoma of bone
|
|
pPNET
|
peripheral primitive neuroectodermal
tumor
|
|
US
|
ultrasonography
|
|
CT
|
computed tomography
|
|
MR
|
magnetic resonance
|
|
FDG-PET
|
fluorodeoxyglucose-positron emission
tomography
|
|
NCCN
|
National Comprehensive Cancer
Network
|
|
CESS
|
European Cooperative Ewing Sarcoma
Studies
|
|
EICESS
|
European Intergroup Cooperative
Ewing's Sarcoma Study
|
References
|
1
|
Van den Berg H, Heinen RC, van der Pal HJ
and Merks JH: Extra-osseous Ewing sarcoma. Pediatr Hematol Oncol.
26:175–185. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Peabody TD and Attar S: SpringerLink:
Orthopaedic Oncology: Primary and metastatic tumors of the skeletal
system. Cancer Treatment and Research, Cham. Springer International
Publishing; Imprint, Springer, Berlin, Germay: pp. 203–223.
2014
|
|
3
|
El Weshi A, Allam A, Ajarim D, Al Dayel F,
Pant R, Bazarbashi S and Memon M: Extraskeletal Ewing's sarcoma
family of tumours in adults: Analysis of 57 patients from a single
institution. Clin Oncol (R Coll Radiol). 22:374–381. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carpentieri DF, Qualman SJ, Bowen J,
Krausz T, Marchevsky A and Dickman PS; Cancer Committee, College of
American Pathologists, : Protocol for the examination of specimens
from pediatric and adult patients with osseous and extraosseous
Ewing sarcoma family of tumors, including peripheral primitive
neuroectodermal tumor and Ewing sarcoma. Arch Pathol Lab Med.
129:866–873. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Iwamoto Y: Diagnosis and treatment of
Ewing's sarcoma. Jpn J Clin Oncol. 37:79–89. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tefft M, Vawter GF and Mitus A:
Paravertebral ‘round cell’ tumors in children. Radiology.
92:1501–1509. 1969. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Meyer JS, Nadel HR, Marina N, Womer RB,
Brown KL, Eary JF, Gorlick R, Grier HE, Randall RL, Lawlor ER, et
al: Imaging guidelines for children with Ewing sarcoma and
osteosarcoma: A report from the children's oncology group bone
tumor committee. Pediatr Blood Cancer. 51:163–170. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Franzius C, Sciuk J, Daldrup-Link HE,
Jürgens H and Schober O: FDG-PET for detection of osseous
metastases from malignant primary bone tumours: Comparison with
bone scintigraphy. Eur J Nucl Med. 27:1305–1311. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Völker T, Denecke T, Steffen I, Misch D,
Schönberger S, Plotkin M, Ruf J, Furth C, Stöver B, Hautzel H, et
al: Positron emission tomography for staging of pediatric sarcoma
patients: Results of a prospective multicenter trial. J Clin Oncol.
25:5435–5441. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Raney RB, Asmar L, Newton WA Jr, Bagwell
C, Breneman JC, Crist W, Gehan EA, Webber B, Wharam M, Wiener ES,
et al: Ewing's sarcoma of soft tissues in childhood: A report from
the intergroup rhabdomyosarcoma study, 1972 to 1991. J Clin Oncol.
15:574–582. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bacci G, Balladelli A, Forni C, Ferrari S,
Longhi A, Bacchini P, Alberghini M, Fabbri N, Benassi M, Briccoli A
and Picci P: Adjuvant and neoadjuvant chemotherapy for Ewing
sarcoma family tumors in patients aged between 40 and 60: Report of
35 cases and comparison of results with 586 younger patients
treated with the same protocols in the same years. Cancer.
109:780–786. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Castex MP, Rubie H, Stevens MC, Escribano
CC, de Gauzy JS, Gomez-Brouchet A, Rey A, Delattre O and Oberlin O:
Extraosseous localized Ewing tumors: Improved outcome with
anthracyclines-the French society of pediatric oncology and
international society of pediatric oncology. J Clin Oncol.
25:1176–1182. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ahmad R, Mayol BR, Davis M and Rougraff
BT: Extraskeletal Ewing's sarcoma. Cancer. 85:725–731. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dunst J and Schuck A: Role of radiotherapy
in Ewing tumors. Pediatr Blood Cancer. 42:465–470. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Applebaum MA, Worch J, Matthay KK, Goldsby
R, Neuhaus J, West DC and Dubois SG: Clinical features and outcomes
in patients with extraskeletal Ewing sarcoma. Cancer.
117:3027–3032. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lynch AD, Gani F, Meyer CF, Morris CD,
Ahuja N and Johnston FM: Extraskeletal versus skeletal Ewing
sarcoma in the adult population: Controversies in care. Surg Oncol.
27:373–379. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cash T, McIlvaine E, Krailo MD, Lessnick
SL, Lawlor ER, Laack N, Sorger J, Marina N, Grier HE, Granowetter
L, et al: Comparison of clinical features and outcomes in patients
with extraskeletal versus skeletal localized Ewing sarcoma: A
report from the children's oncology group. Pediatr Blood Cancer.
63:1771–1779. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Goldblum J, Folpe A and Weiss S: Enzinger
and weiss's soft tissue tumors. 6th edition. Elsevier Health
Sciences; 2014
|
|
19
|
Grier HE: The Ewing family of tumors.
Ewing's sarcoma and primitive neuroectodermal tumors. Pediatr Clin
North Am. 44:991–1004. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Javery O, Krajewski K, O'Regan K, Kis B,
Giardino A, Jagannathan J and Ramaiya NH: A to Z of extraskeletal
Ewing sarcoma family of tumors in adults: Imaging features of
primary disease, metastatic patterns, and treatment responses. AJR
Am J Roentgenol. 197:W1015–W1022. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huh J, Kim KW, Park SJ, Kim HJ, Lee JS, Ha
HK, Tirumani SH and Ramaiya NH: Imaging features of primary tumors
and metastatic patterns of the extraskeletal Ewing sarcoma family
of tumors in adults: A 17-year experience at a single institution.
Korean J Radiol. 16:783–790. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Somarouthu BS, Shinagare AB, Rosenthal MH,
Tirumani H, Hornick JL, Ramaiya NH and Tirumani SH: Multimodality
imaging features, metastatic pattern and clinical outcome in adult
extraskeletal Ewing sarcoma: Experience in 26 patients. Br J
Radiol. 87:201401232014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Galyfos G, Karantzikos GA, Kavouras N,
Sianou A, Palogos K and Filis K: Extraosseous Ewing sarcoma:
Diagnosis, prognosis and optimal management. Indian J Surg.
78:49–53. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brisse H, Ollivier L, Edeline V,
Pacquement H, Michon J, Glorion C and Neuenschwander S: Imaging of
malignant tumours of the long bones in children: Monitoring
response to neoadjuvant chemotherapy and preoperative assessment.
Pediatr Radiol. 34:595–605. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gerth HU, Juergens KU, Dirksen U, Gerss J,
Schober O and Franzius C: Significant benefit of multimodal
imaging: PET/CT compared with PET alone in staging and follow-up of
patients with Ewing tumors. J Nucl Med. 48:1932–1939. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brinkhuis M, Wijnaendts LC, van der Linden
JC, van Unnik AJ, Voûte PA, Baak JP and Meijer CJ: Peripheral
primitive neuroectodermal tumour and extra-osseous Ewing's sarcoma;
a histological, immunohistochemical and DNA flow cytometric study.
Virchows Arch. 425:611–616. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Folpe AL; Inwards CY: Bone and Soft Tissue
Pathology, Saunders. Elsevier; Philadelphia, PA, USA: pp. 367–379.
2010
|
|
28
|
Hornick JL: Practical soft tissue
pathology: A diagnostic approach. Elsevier; Philadelphia, PA, USA:
pp. 269–297. 2018
|
|
29
|
Downing JR, Head DR, Parham DM, Douglass
EC, Hulshof MG, Link MP, Motroni TA, Grier HE, Curcio-Brint AM and
Shapiro DN: Detection of the (11;22)(q24;q12) translocation of
Ewing's sarcoma and peripheral neuroectodermal tumor by reverse
transcription polymerase chain reaction. Am J Pathol.
143:1294–1300. 1993.PubMed/NCBI
|
|
30
|
Sorensen PH, Lessnick SL, Lopez-Terrada D,
Liu XF, Triche TJ and Denny CT: A second Ewing's sarcoma
translocation, t(21;22), fuses the EWS gene to another ETS-family
transcription factor, ERG. Nat Genet. 6:146–151. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Biermann JS: Updates in the treatment of
bone cancer. J Natl Compr Canc Netw. 11 (Suppl 5):S681–S683. 2013.
View Article : Google Scholar
|
|
32
|
Casali PG, Bielack S, Abecassis N, Aro HT,
Bauer S, Biagini R, Bonvalot S, Boukovinas I, Bovee JVMG, Brennan
B, et al: Bone sarcomas: ESMO-PaedCan-EURACAN clinical practice
guidelines for diagnosis, treatment and follow-up. Ann Oncol. 29
(Suppl 4):iv79–iv95. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Covelli HD, Beekman JF and Kingry RL:
Extraskeletal Ewing's sarcoma: Prolonged survival with recurrence
after operation. South Med J. 73:1294–1295. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rud NP, Reiman HM, Pritchard DJ, Frassica
FJ and Smithson WA: Extraosseous Ewing's sarcoma. A study of 42
cases. Cancer. 64:1548–1553. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Womer RB, West DC, Krailo MD, Dickman PS,
Pawel BR, Grier HE, Marcus K, Sailer S, Healey JH, Dormans JP and
Weiss AR: Randomized controlled trial of interval-compressed
chemotherapy for the treatment of localized Ewing sarcoma: A report
from the children's oncology group. J Clin Oncol. 30:4148–4154.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Donaldson SS: Ewing sarcoma: Radiation
dose and target volume. Pediatr Blood Cancer. 42:471–476. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gaspar N, Hawkins DS, Dirksen U, Lewis IJ,
Ferrari S, Le Deley MC, Kovar H, Grimer R, Whelan J, Claude L, et
al: Ewing sarcoma: Current management and future approaches through
collaboration. J Clin Oncol. 33:3036–3046. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Schuck A, Ahrens S, Paulussen M, Kuhlen M,
Könemann S, Rübe C, Winkelmann W, Kotz R, Dunst J, Willich N and
Jürgens H: Local therapy in localized Ewing tumors: Results of 1058
patients treated in the CESS 81, CESS 86, and EICESS 92 trials. Int
J Radiat Oncol Biol Phys. 55:168–177. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Donaldson SS, Torrey M, Link MP, Glicksman
A, Gilula L, Laurie F, Manning J, Neff J, Reinus W, Thompson E and
Shuster JJ: A multidisciplinary study investigating radiotherapy in
Ewing's sarcoma: End results of POG #8346. Pediatric oncology
group. Int J Radiat Oncol Biol Phys. 42:125–135. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bolling T, Schuck A, Paulussen M, Dirksen
U, Ranft A, Könemann S, Dunst J, Willich N and Jürgens H: Whole
lung irradiation in patients with exclusively pulmonary metastases
of Ewing tumors. Toxicity analysis and treatment results of the
EICESS-92 trial. Strahlenther Onkol. 184:193–197. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Takenaka S, Naka N, Obata H, Joyama S,
Hamada K, Imura Y, Kakunaga S, Aoki Y, Ueda T, Araki N and
Yoshikawa H: Treatment outcomes of Japanese patients with Ewing
sarcoma: Differences between skeletal and extraskeletal Ewing
sarcoma. Jpn J Clin Oncol. 46:522–528. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee JA, Kim DH, Lim JS, Koh JS, Kim MS,
Kong CB, Song WS, Cho WH, Lee SY and Jeon DG: Soft-tissue Ewing
sarcoma in a low-incidence population: Comparison to skeletal Ewing
sarcoma for clinical characteristics and treatment outcome. Jpn J
Clin Oncol. 40:1060–1067. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Orr WS, Denbo JW, Billups CA, Wu J, Navid
F, Rao BN, Davidoff AM and Krasin MJ: Analysis of prognostic
factors in extraosseous Ewing sarcoma family of tumors: review of
St. Jude children's research hospital experience. Ann Surg Oncol.
19:3816–3822. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Biswas B, Shukla NK, Deo SV, Agarwala S,
Sharma DN, Vishnubhatla S and Bakhshi S: Evaluation of outcome and
prognostic factors in extraosseous Ewing sarcoma. Pediatr Blood
Cancer. 61:1925–1931. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Haeusler J, Ranft A, Boelling T, Gosheger
G, Braun-Munzinger G, Vieth V, Burdach S, van den Berg H, Juergens
H and Dirksen U: The value of local treatment in patients with
primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer.
116:443–450. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kinsella TJ, Triche TJ, Dickman PS, Costa
J, Tepper JE and Glaubiger D: Extraskeletal Ewing's sarcoma:
Results of combined modality treatment. J Clin Oncol. 1:489–495.
1983. View Article : Google Scholar : PubMed/NCBI
|