Introduction
Screening modalities to determine candidate
downstream targets and signaling pathways after novel and
influential proteins/pathways identified are limited by
availability and utility. Small molecule inhibitor libraries in
cancer research introduce a unique modality to screen for various
biologic endpoints, including drug sensitivity, cell morphology,
cell proliferation, and survival (1). The Published Kinase Inhibitor Set
(PKIS) is an example of such a library; PKIS1 and PKIS2 are
collections of ATP-competitive kinase inhibitors representing
dozens of chemotypes (2). The
described inhibitors have a range of selectivity profiles against
various kinase targets (3) and can
thus be used in a screening approach to identify candidate kinases
targets or downstream signaling pathways to pursue in further
mechanistic studies for select genes of interest. Previously, we
demonstrated the successful application of the PKIS in a
morphology-based screen as a starting point to discover kinase
targets and signaling pathways that drove a specific phenotype in
TNBC cells (4).
In this report, we utilized genetically modified
breast cancer cell lines to demonstrate the application of this
phenotypic screening approach to identify candidate targets to
pursue for potential drug discovery applications. For these
experiments, we focused on studying the downstream effects of
over-activation of the CXC chemokine receptor 4 (CXCR4).
Chemokine-mediated signaling processes have integral roles in
cancer development and metastasis (5,6). While
chemokines can bind to various chemokine receptors, CXCR4 is unique
because it exclusively binds to the CXCL12 chemokine, also known as
stromal cell-derived factor-1 (7).
CXCR4 is a G protein receptor that subsequently activates
phospholipase C-β and phosphatidylinositol-3-kinase, or PI3K. These
signaling events cause downstream activation of protein kinase C
and mitogen-activated protein kinase, which leads to cell migration
(8). Small molecule therapies
targeting CXCR4 are currently being investigated as anti-cancer
therapeutics (9,10), providing evidence for CXCR4 as a
viable target in endocrine therapy-resistant breast cancer
(11,12). CXCR4 is expressed in many different
cancer types (7,13), and its expression is associated with
higher-grade cancers (14). CXCR4
has been implicated as a prognostic marker in breast cancer and is
associated with worse prognoses (15,16). In
triple-negative breast cancer (TNBC), a subtype lacking hormone
receptors or HER2/Neu amplification, activated CXCR4 is present in
75% of TNBC tumors, as was evaluated in microarray analysis
(17,18). CXCR4 expression drives breast cancer
cell invasion and metastasis (13,19–21).
Furthermore, the CXCR4-SDF-1 signaling axis regulates the activity
of circulating tumor cells in primary breast cancer (22). In metastasis, cells acquire
characteristics that drive an invasive and migratory phenotype in a
process known as epithelial-mesenchymal transition (EMT) (23). In EMT, cancer cells that are
epithelial-shaped and have epithelial molecular phenotypes acquire
mesenchymal molecular features which induce a change in cell
morphology to a more fibroblastic and stellate-appearing shape
(23,24). Acquisition of a mesenchymal phenotype
drives cell invasion and migration through the extracellular matrix
and intravasate into surrounding vasculature to disseminate to
distal tissue sites (25,26).
CXCR4 activates signaling pathways that drive tumor
growth and angiogenesis. CXCR4 is positively upregulated by the
hypoxia-inducible factor-1α and growth factors (FGF, VEGF, EGF)
(27). The COOH-terminal domain
(CTD) mediates receptor desensitization and downregulation
(27), and truncation of this domain
(ΔCTD) results in sensitization and upregulation of the receptor.
The CTD domain is necessary to drive a mesenchymal cell morphology
and cell motility through CXCR4 signaling (27–29):
ΔCTD, and not CXCR4 overexpressing cells, downregulated epithelial
protein expression (CDH1, ZO1), decreased cell-cell contact, and
increased cell migration (27).
CXCR4 activation is additionally associated with
endocrine therapy resistance through the downregulation of estrogen
receptor expression (30). CXCR4
signaling is implicated in other areas of drug resistance: In
breast cancer, CXCR4 silencing sensitizes TNBC cells to cisplatin
therapy (31), silencing of CXCR4
and SDF-1 sensitizes breast cancer cells to paclitaxel (32), and CXCR4 inhibition abrogates
trastuzumab resistance in HER2-positive breast cancer (33). We previously demonstrated that CXCR4
expression mediates estrogen-independent tumorigenesis, metastasis,
and resistance to endocrine therapies through increased MAPK
signaling (34,35). CXCR4 activates ER-mediated gene
transcription through phosphorylation of ERβ by MAPK family members
(36), inducing estrogen
independence in MCF-7-CXCR4 cells. Dubrovska et al found
that CXCR4 maintains a cancer stem cell-like progenitor population
in tamoxifen-resistant MCF-7 cells (37). Together, these findings support a
role for CXCR4 activation in endocrine therapy resistance in
addition to driving a mesenchymal and migratory phenotype.
Downstream signaling pathways of CXCR4 that are
responsible for these phenotypic changes remain widely unknown. To
address this knowledge gap, we employed the PKIS library in a
medium-throughput phenotypic screen using MCF-7 parental cells
(MCF-7), MCF-7 cells with constitutively active CXCR4 expression
(MCF-7-CXCR4-ΔCTD), fulvestrant resistant MCF-7 cells (MCF-7-FR),
and TNBC cell lines (BT-549, MDA-MB-231). We then compared relative
kinase activity of compounds within the same chemotype series that
were active or inactive in the screens. Our goal was to identify
candidate signaling pathways responsible for the observed
mesenchymal and fulvestrant-resistant phenotype of MCF-7-CXCR4-ΔCTD
cells. This aim of this study was to demonstrate the utility in
using a phenotypic screening approach with small molecule kinase
inhibitors to identify potential pathways and targets downstream to
pursue in CXCR4-activated breast cancer cells. Future experiments
will be required to validate and interrogate the kinase pathway
leads identified in this screen.
Materials and methods
Cell culture
Human MDA-MB-157, MDA-MB-231 and BT-549 cells were
acquired from the American Type Culture Collection (ATCC). Human
MCF-7 cells used for stable transfection of CXCR4 were generously
provided to our lab by Louisa Nutter (University of Minnesota,
Minneapolis, MN, USA). Cells were maintained in DMEM supplemented
with 10% fetal bovine serum, 1% non-essential amino acids (NEAA)
(Caisson Labs), MEM amino acids (Invitrogen; Thermo Fisher
Scientific, Inc.), antibiotic-antimycotic solution (100 U/ml;
Caissan Labs), sodium pyruvate (Invitrogen; Thermo Fisher
Scientific, Inc.) and insulin (1×10−10 mol/l;
Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C in humidified
5% CO2.
Generation of stably overexpressing
and resistant cell lines
MCF-7 cells were stably transfected with truncated
CXCR4 (ΔCTD), wild type CXCR4 (CXCR4), or empty vector as controls,
as previously described (27).
Fulvestrant resistant MCF-7N cells were generated by exposing the
cells to gradually increasing concentrations of fulvestrant, until
resistance was achieved, as described by Fan et al (38).
mRNA isolation
Cells were plated in 10% DMEM at 70% confluency
harvested after 24 h using a mix of phosphate-buffered saline and
EDTA. Total RNA was isolated using the RNeasy kit, according to
manufacturer's instructions (Qiagen, Inc.). Quantity and quality of
RNA were determined by absorbance at 260 and 280 nm using the
ND-1000 (NanoDrop).
Analysis of oligo-array data
Published oligo-array data by Ueda et al was
analyzed using GeneGo Metacore (Thomson Reuters) (27). The Enrichment Analysis Workflow was
performed using the gene list, fold-change, and P-value scores
generated by edgeR. A threshold P-value of <0.05, and threshold
fold-change <0.5 was set when performing the analysis in
GeneGo.
The Published Kinase Inhibitor Sets
(PKIS)
The PKIS1 and PKIS2 are first generation kinase
chemogenomic sets. They have now been supplanted by the KCGS
(Kinase Chemogenomic Set) which is openly available in screening
quantities from the SGC-UNC. Instructions for Requesting KCGS can
be found at www.sgc-unc.org. Chemical structures
and other pharmacologic activity for the PKIS compounds can be
found at https://www.ebi.ac.uk/chembldb/extra/PKIS/compounds.html.
The set is typically provided as 1 µl of a 10 mM solution in DMSO,
dispensed in 384-well plates. A material transfer agreement was
created to ensure that the screening results are made publicly
available. Larger aliquots of requested compounds were delivered as
solids, dissolved in DMSO to a 1 mM stock solution, and stored at
−20°C. The solutions were diluted in culture media and used at 1 µM
concentrations, as determined by dose-response studies.
Crystal violet staining
MDA-MB-157, MDA-MB-231, BT549, MCF-7-CXCR4-ΔCTD and
MCF-7-FR cells were plated in a 96-well plate format at 2,000 cells
per well. After 24 h, cells were exposed to 5% charcoal stripped
FBS media or phenol-free DMEM media (Invitrogen; Thermo Fisher
Scientific, Inc.) supplemented with charcoal-stripped FBS, NEAA,
MEM amino acids, Gluta-Max and penicillin (100 U/ml). After 48 h of
exposing the cells to CS DMEM media, cells were treated with the
vehicle or selected PKIS library compounds for 72 h and the plate
was incubated in 37°C, 5% CO2. The plate was then
harvested by adding glutaraldehyde (10 µl of 25% stock solution) to
each well for 20 min. After rinsing and drying the plate, the cells
were stained with 0.1% crystal violet in 90% methanol (50 µl) for
20 min. After another rinse, the cells were left overnight to dry,
and the following day morphological alterations of the cells were
visualized with an inverted microscope and images were recorded at
×200 magnification.
Results
Candidate kinases identified that are
responsible for promoting a mesenchymal phenotype in constitutive
CXCR4 activation
Cell morphology and cytoskeletal rearrangement have
important roles in suppressing metastasis, as epithelial-like cells
are not able to invade and migrate into the vasculature to spread
to distal tissue sites (25,26). Mesenchymal morphology characteristics
include bipolar cells often with protrusions that appear
fibroblast-like, with minimal cell-cell contacts. Epithelial
morphology cells have rounder shapes, increased circularity and
form closer cell-cell contacts that facilitate colony formation and
look more ‘cobblestone’ in appearance. Because some TNBC cell lines
have inherently mesenchymal cell morphologies due to these cells'
fibroblast-like characteristics, we chose to use three classic
mesenchymal lines as positive controls in our phenotypic screen:
MDA-MB-231, BT-549, MDA-MB-157. Only compounds were selected as
‘hits’ if they promoted an epithelial morphology in one or more
TNBC cell lines and if they altered the morphology of
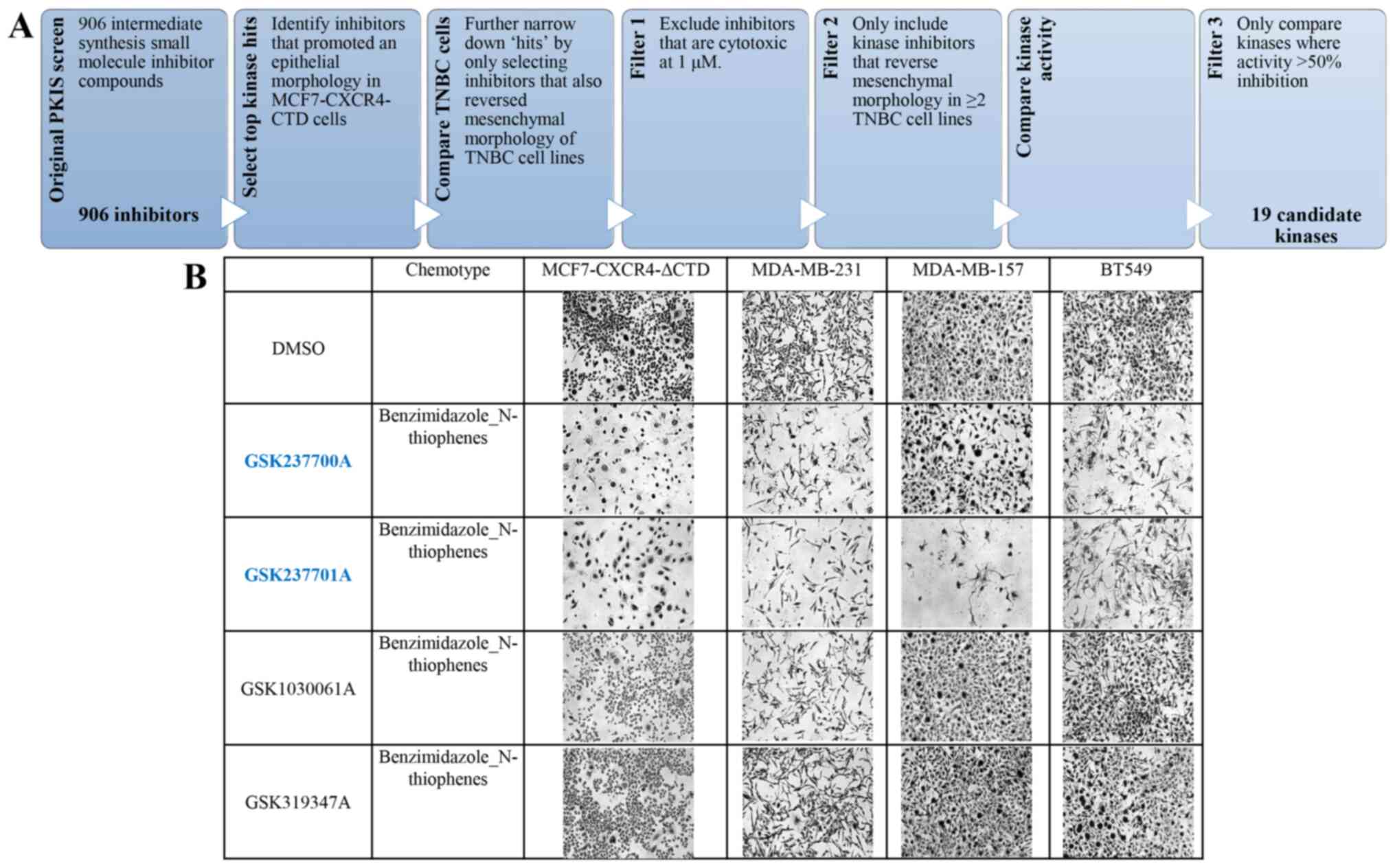
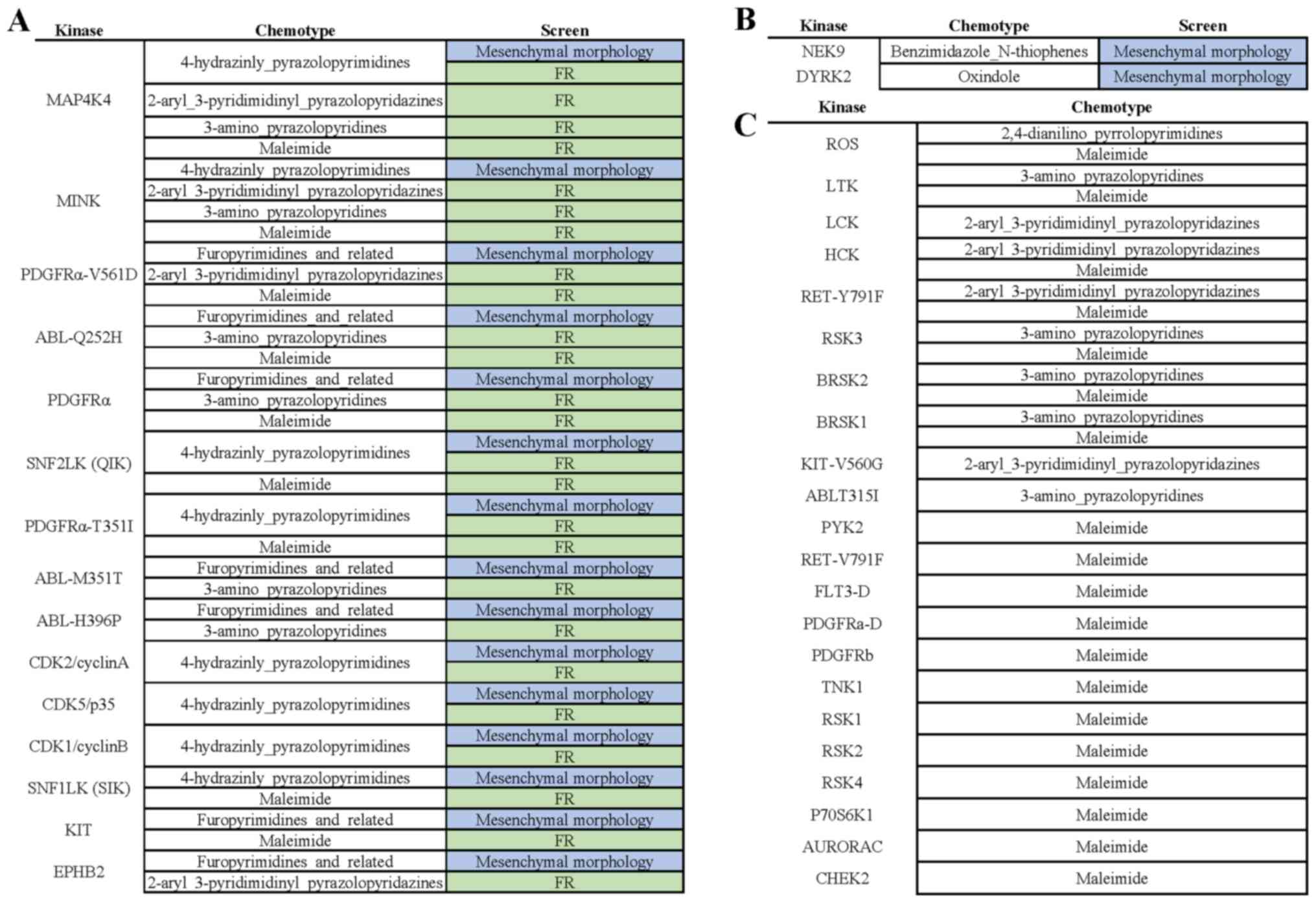
MCF-7-CXCR4-ΔCTD cells (Fig. 1A).
Overall, we observed four different chemotypes that contained
active and inactive compounds within the same chemotype, in which
we could compare kinase activity. The four chemotypes were:
Benzimidazole-N-thiophenes (Fig.
1B), oxindoles (Fig. S1),
4-hydrazinyl-pyrazolopyrimidines (Fig.
S2), and furopyrimidines (Fig.
S3).
Compounds in the PKIS library are non-selective
kinase inhibitors, and thus they have target various kinases in
addition to activity at the kinase for which they were originally
designed. Many of the compounds in the PKIS library have kinase
activity data described in a study by Elkins et al (2). Using these data sets we compared kinase
activity of compound ‘hits’, referred to as active compounds, to
activity of inactive compounds to find potential candidate kinases
responsible for the observed phenotypic changes. In this first
screen, active compounds altered cell morphologies and reversed the
mesenchymal phenotype in MCF-7-CXCR4-ΔCTD and TNBC cells, while
inactive compounds did not. For these analyses, we compared active
and inactive compounds that were within four chemotypes that had
available published kinase activity data sets. Within the oxindole
chemotype series, only one compound was active based on our initial
screen, out of the 19 tested inhibitors. When kinase activity was
compared in the active and inactive compounds, the only kinase
which the active compound exhibited anti-kinase activity was DYRK2
(52% anti-kinase activity). The inactive compounds in this series
exhibited less anti-kinase activity against DYRK2: GW305178A (36%),
GW300660A (33%), GR105659A (25%), GW429374A (16%), GW290597A (16%),
GW406108X (14%), GW284408A (11%), GW275616A (10%), GW300657A (8%),
GW301789A (7%), GW416469A (5%), GW442130A (5%), GW335962A (4%),
GW441756A (4%), GW282536A (3%), GW279320A (2%), GW300653A (0%),
GW352430A (−1%), GW278681A (−1%). Within the benzimidazole
N-thiophenes chemotype series, 7 of the 13compounds were active.
The only kinase which the active compounds exhibited anti-kinase
activity compared to inactive compounds was NEK9: GSK579289A (97%),
GSK237701A (92%), GSK317315A (84%), GW843682X (59%), GW852849X
(55%), GSK237700A (38%), GW853606X (29%). Anti-NEK9 kinase activity
of the inactive compounds includes: GW853609X (10%), GSK1030061A
(9%), GSK1030058A (7%), GSK1030059A (6%), GSK1030062A (3%),
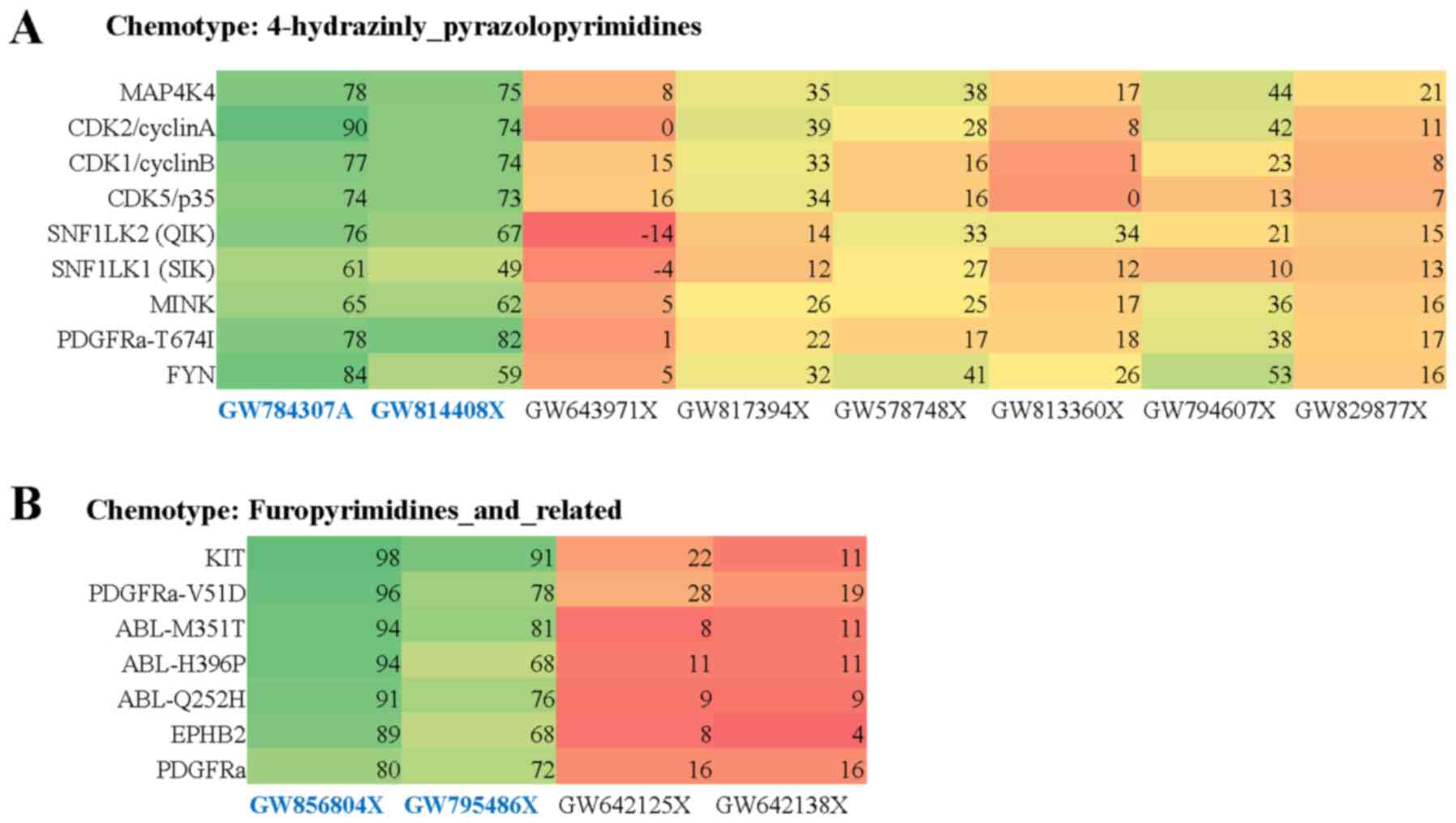
GSK319347A (2%). Within the 4-hydrazinly-pyrazolopyrimidines
chemotype series, 2 out of 8 compounds tested were active based on
the initial screen. Kinases specific for the active compounds
compared to inactive compounds included: MAP4K4, CDK2/cyclinA,
CDK1/cyclinB, CDK5/p35, SNF1LK2(QIK), SNF1LK1(SIK), MINK,
PDGFRα-T674I, FYN, KIT (Fig. 2A).
Within the furopyrimidines and related chemotype series, 2 out of 4
inhibitors tested in the initial screen were active. Kinases
specific for active compounds compared to inactive compounds
included: PDGFRα-V561D, ABL-M351T, ABL-H396P, ABL-Q252H, EPHB2,
PDGFRα (Fig. 2B). Together, these
data demonstrate the utility of this small molecule inhibitor
phenotypic screen approach and comparing kinase activity of active
and inactive compounds to identify candidate kinases downstream of
CXCR4 activation.
Use of phenotypically mesenchymal
MCF-7 cells to identify candidate kinases that promote a
mesenchymal and fulvestrant resistant phenotype in the setting of
constitutive CXCR4 activation
MCF-7-FR cells have acquired resistance to
fulvestrant and exhibit a mesenchymal cell phenotype (38). Here, we utilized these cells as
another positive control in our screen to find candidate kinase
targets that reversed the mesenchymal phenotype in addition to
targets responsible for acquisition of an endocrine resistant
phenotype (using cell viability as an endpoint) in CXCR4 activated
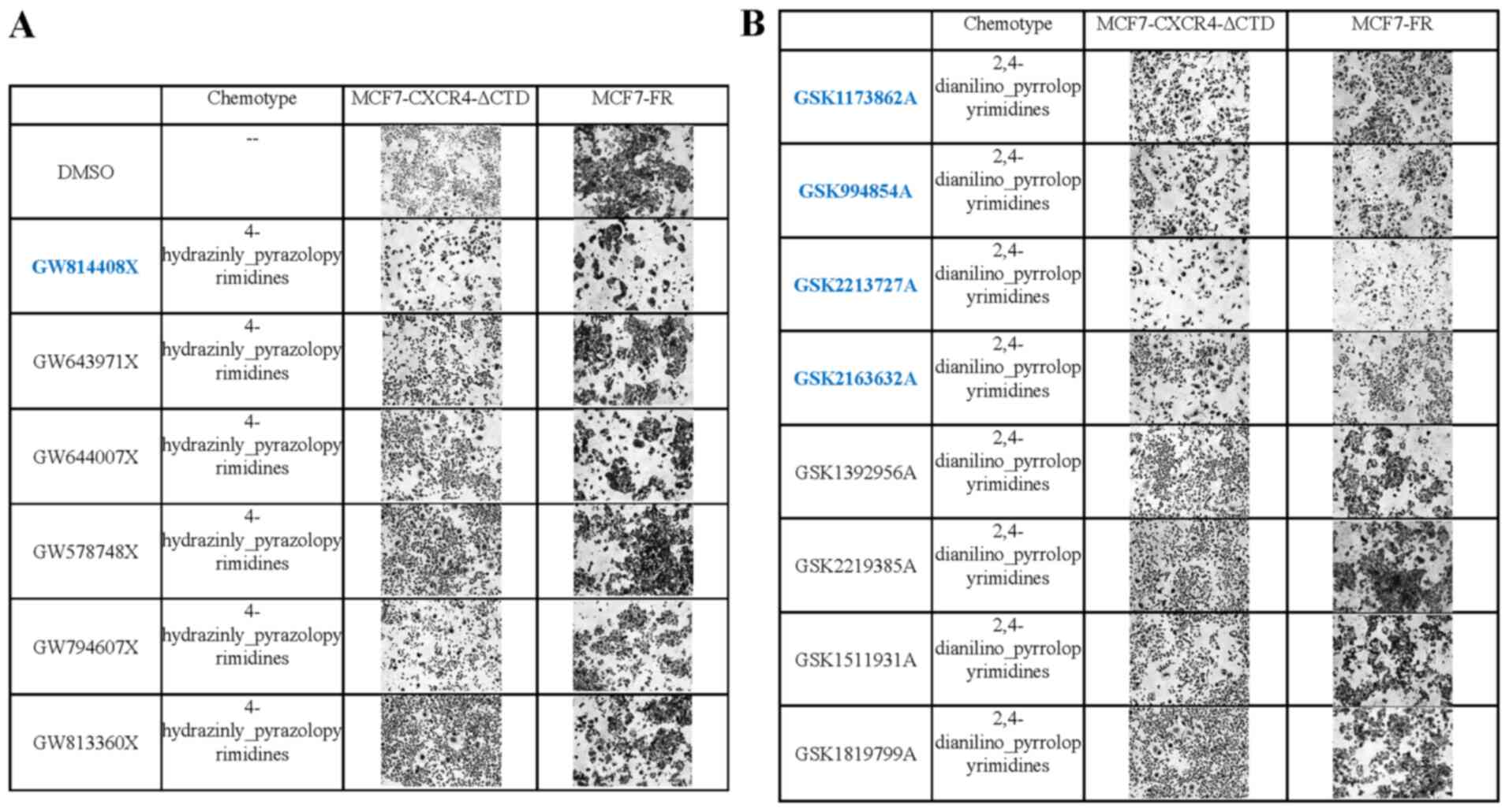
MCF-7 cells. We discovered active inhibitors within the PKIS set
that promoted an epithelial morphology and/or reduced cell
viability in MCF-7-CXCR-ΔCTD cells and MCF-7-FR cells (Fig. 3A and B). Then we compared kinase
activity of active and inactive compounds within the same chemotype
series. Within the 2,4-dianilino pyrrolopyrimidines chemotype
series 4 out of 8 tested inhibitors were active and ROS was the
only kinase which active compounds targeted compared to inactive
compounds (Fig. S4). Active
compound kinase activity includes GSK2213727A (86%), GSK2163632A
(83%), GSK1173862A (81%), GSK994854A (61%). Inactive compounds ROS
kinase activity includes GSK1392956A (60%), GSK1819799A (55%),
GSK2219385A (50%), GSK1511931A (45%). Within the
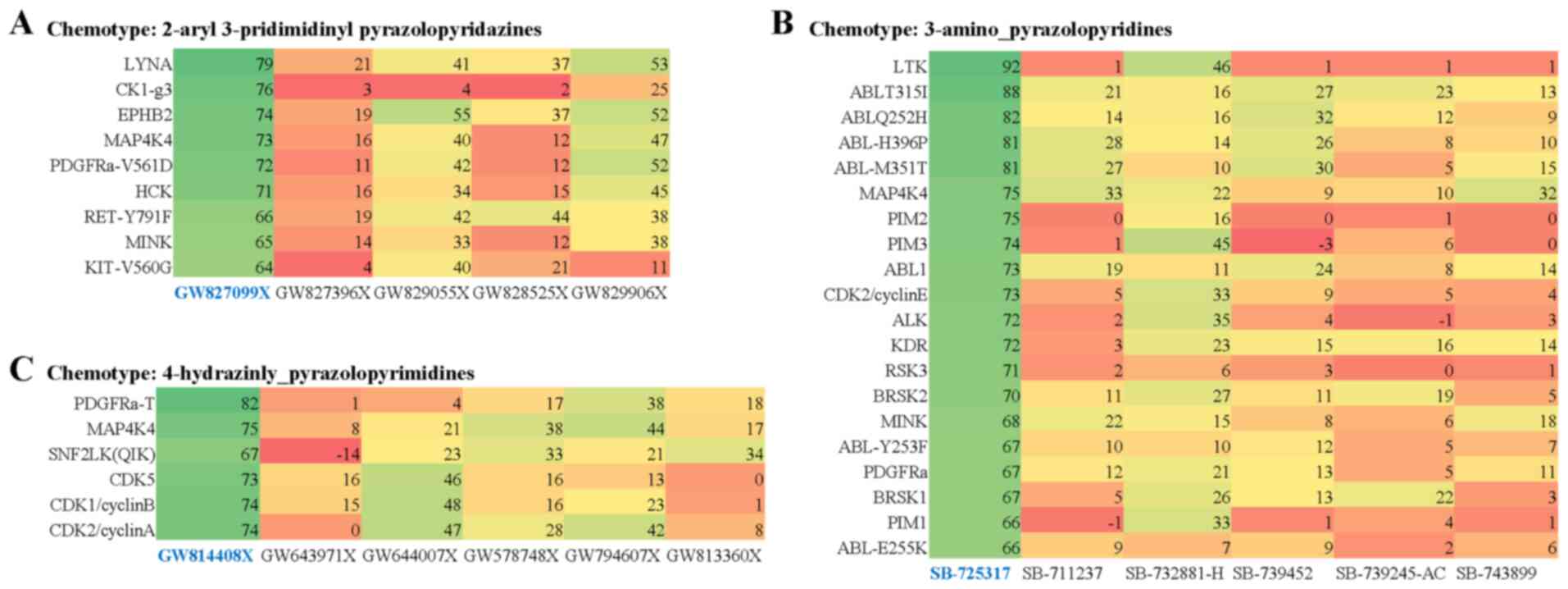
2-aryl-3-pridimidinyl pyrazolopyridazines chemotype series one out
of 5 screened inhibitors were active (Fig. S5), within the 3-amino
pyrazolopyridines series one out of 6 screened inhibitors were
active (Fig. S6), within the
4-hydrazinly pyrazolopyrimidines group one out of 6 was an active
inhibitor, within the maleimide chemotype series, one out of 10
screened inhibitors was active (Fig.
S7) and in the furopyrimidines and related series two out of
four tested inhibitors were active (Fig. S8).
In this phenotypic screen comparing the CXCR4
activated MCF-7 cells to fulvestrant resistant cells, kinases that
were targeted by the active compounds and not in the inactive
compounds included LYNA, CK1-g3, EPHB2, PDGFRα-V561D, HCK,
RET-Y791F, MINK, KIT-V560G (Fig.
4A), LTK, ABL variants, PIM1, PIM2, PIM3, CDK2, ALK, KDR, RSK3,
BRSK1, BRSK2, MINK, PDGFRα (Fig.
4B), PDGFRα-T, QIK, CDK1, CDK2, CDK5 (Fig. 4C) and other kinases (Fig. S9). Notably MAP4K4, HCK and ABL,
PDGFR, PIM, and CDK family members were commonly targeted by active
compounds amongst the various chemotype series (Figs. 4A-C and S9).
Then we compared results from the morphology screen
(MCF7-CXCR4-ΔCTD compared to phenotypically mesenchymal TNBC cells)
and viability (comparing MCF7-CXCR4-ΔCTD and MCF7-FR) screens.
Eleven kinases were commonly targeted by active compounds in both
screens compared to inactive compounds, suggesting these kinases
were possibly responsible for both affecting the mesenchymal cell
morphologies and sensitivity to endocrine-targeted therapies. These
kinases included: MAP4K4, MINK, PDGFRα-V561D, ABL-Q252H, PDGFRα,
SNF2LK(QIK), PDGFRα-T351I, ABL-M351T, ABL-H386P, CDK2/cyclinA,
CDK5/p35, CDK1/cyclin B, SNF1LK(SIK), KIT and EPHB2 (Fig. 5A). Kinases that were unique hits in
the morphology screen were NEK9 and DYRK2 (Fig. 5B). Kinase targets that were unique
hits in the fulvestrant resistance screen that had over 70% kinase
activity in the active compounds were ROS, LYK, LCK, HCK,
RET-Y791F, RSK3, BRSK2, BRSK1, KIT-V560G, ABL-T351I, PYK2,
RET-V791F, FLT3-D, PDGFRα-D, PDGFRb, TNK1, RSK1, RSK2, RSK4,
P70S6K1, AURORAC and CHEK2 (Fig.
5C).
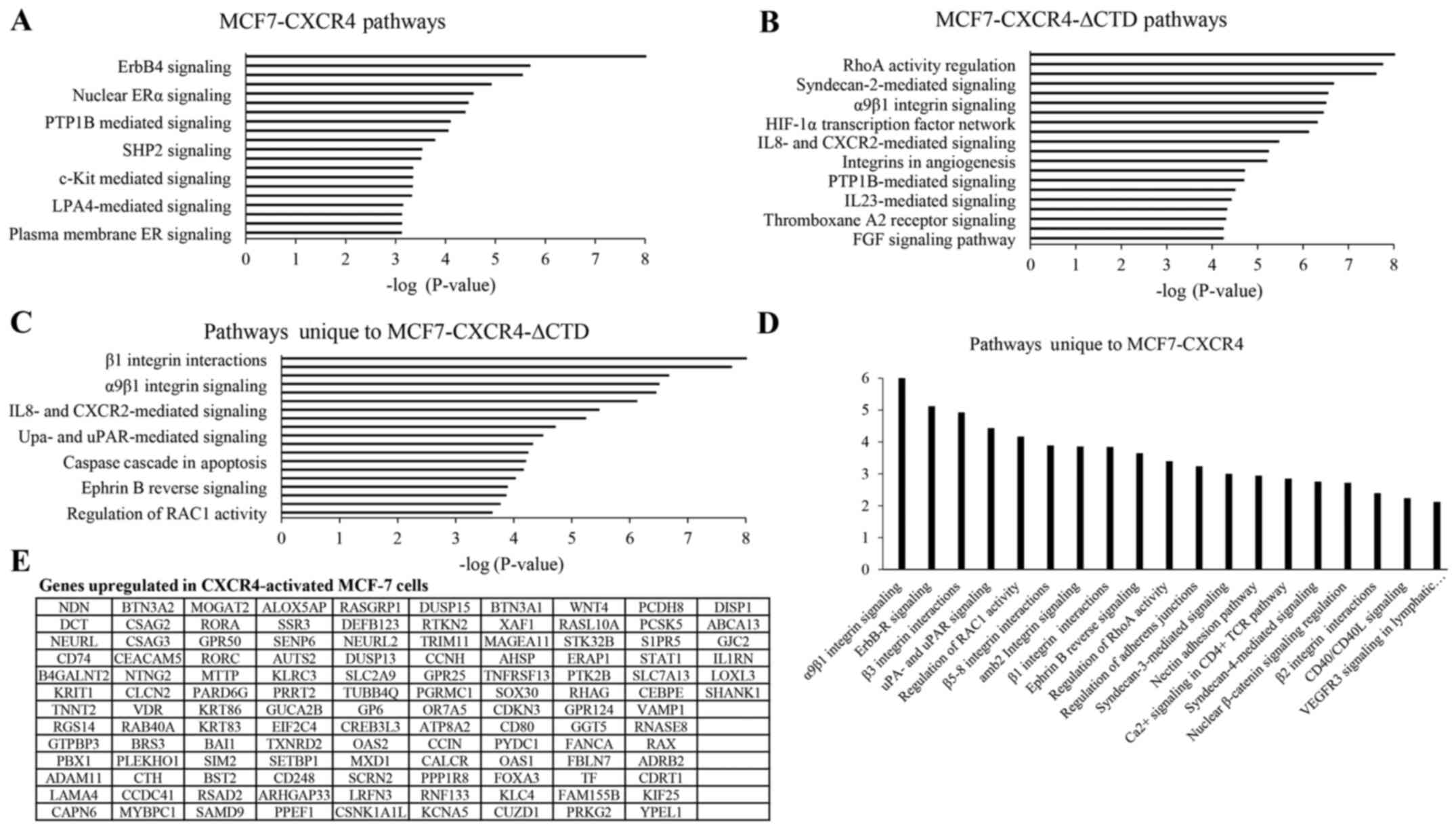
Upregulated pathways regulated by
CXCR4 identified through the screening approach were similar to
those identified through oligoarrays
To compare potential mechanisms identified through
our screen to other testing modalities, we performed a pathway
analysis using oligo-array data provided by Ueda et al
(27). For these analyses, we
evaluated gene expression changes in MCF-7-CXCR4 and
MCF-7-CXCR4-ΔCTD cells compared to the empty vector controls
(MCF-7-VEC). We found that in CXCR4 overexpressing cells the most
upregulated pathways included HDAC Class III, ERBB4, Endothelin,
FOXA1, Nuclear ERα, IL-2, and p75-mediated signaling pathways
(Fig. 6A). In MCF-7-CXCR4-ΔCTD
cells, there was an upregulation of β1 integrin, RhoA,
CXCR4-mediated, syndecan 2, LPA receptor and other integrin
mediated signaling pathways (Fig.
6B). When pathways that were unique to MCF7-CXCR4-ΔCTD were
examined, integrin-mediated signaling and RhoA signaling were
within the top 10 most upregulated signaling pathways. Other
signaling pathways included the uPA/uPAR, FAK, glypican 1,
IL-8/CXCR2 and ephrin B-mediated signaling pathways (Fig. 6C). Pathways were then analyzed in
genes that had over 3-fold difference in expression in MCF7-CXCR4
cells compared to vector control cells. In addition to integrin
pathways, there was upregulation in ErbB receptor, ephrin B,
adherent junctions and RhoA signaling pathways (Fig. 6D). Specific genes upregulated in
CXCR4 activated cells were also shown in Fig. 6E. These data demonstrate potential
downstream signaling mechanisms that CXCR4 employs to drive a
mesenchymal phenotype.
Discussion
Estrogen independent hormone receptor positive
breast cancers have acquired drug resistance, inhibiting response
to endocrine-targeted therapies. Mesenchymal features of cancer
cells that drive metastatic cancers are difficult to treat with
currently available therapeutic regimens. In this
proof-of-principle study, we introduce a new application of an
available small molecule inhibitor chemogenomic library in a
phenotypic screen approach to identify candidate kinases to pursue
as potential therapeutic targets for the mesenchymal/metastatic
phenotype and endocrine therapy resistance. Future investigations
are required to interrogate these kinases and associated signaling
pathways as potential downstream mechanisms of CXCR4.
Kinase profiling of the PKIS data sets using two
endpoints (cell morphology and sensitivity to fulvestrant resistant
cell lines) was used to identify known signaling pathways and novel
candidate kinases responsible for the observed phenotypic and
proliferative changes induced by constitutive CXCR4 activation.
Kinases that reversed mesenchymal morphology compared to TNBC cell
lines, were MINK, FYN, NEK9, and DYRK2. Kinases that were unique to
the endocrine sensitivity screen were ROS, LCK, LYNA, p38β, CK1-g3,
LYNB, HCK, RET-Y791F, KIT-V560G, LTK, PIM2, PIM3, ABL1,
CDK2/cyclinE, ALK, KDR, RSK3, BRSK2, BRSK1, PIM1, and ABL-E255K.
The data suggests these kinases specifically affect the processes
in which the CXCR4-activated cells either acquire a mesenchymal
phenotype or acquire an endocrine therapy-resistant phenotype. We
found another subset of kinases that were hits in both the
morphology and fulvestrant resistant screens: MAP4K4, PDGFRα-V561D,
ABL-Q252H, PDGFRα, SNF2LK(QIK), PDGFRα-T351I, ABL-M351T, ABL-H386P,
CDK2/cyclinA, CDK1/cyclinB, CDK5/p35, SNF1LK(SIK), EPHB2, and KIT.
Interestingly, three members of the CDK family were identified in
our screen: CDK2/cyclinA, CDK1/cyclinB, CDK5/p35. While the role of
CXCR4 in breast cancer metastasis is well established, Yi et
al were the first to attempt to thoroughly interrogate possible
downstream mechanisms of CXCR4 activity using
phosphoproteomic-based methods (21). In their studies, CXCR4 substantially
increased phosphorylation of CDK1, CDK3, and CDK7, which was novel
because of the role for SDF-1/CXCR4 signaling in cell cycle
regulation was not characterized (20). Here, our findings confirm a possible
role for SDF-1/CXCR4 in CDK-mediated cell cycle processes.
Interestingly, variants in both the ABL and PDGFRα
signaling pathways were within these kinase ‘hits’, indicating ABL
and PDGFRα pathways have roles in CXCR4 downstream mechanisms.
Crosstalk between ABL and CXCR4 signaling pathways exist through
the Src kinase LYN (39,40). Furthermore, other studies have shown
ABL kinases are activated downstream of the CXCR4 receptor,
facilitating ABL-mediated cell invasion and matrix degradation and,
ultimately, metastasis (41). Our
identification of ABL as a possible downstream kinase of CXCR4
activity in our screen in combination with these published studies,
validates our approach.
Some of the candidate kinases identified in this
screen had no previous associations to CXCR4 signaling. Examples of
such kinases included MAP4K4, SNF2LK, SNF1LK, EPBH2, and KIT. Yi
et al revealed a potential relationship between CXCR4
signaling and phosphorylation of MAP4K4 (21), but the mechanism behind this
association has not yet been described. Here we further validate
MAP4K4 as a candidate downstream kinase of CXCR4 activity to
interrogate in future studies. SNF1LK and SNF2LK are
serine/threonine kinases; SFK1LK, or SIK1, is downstream of LKB1, a
well-described tumor suppressor protein. The association between
SNF1LK and CXCR4 has not yet been reported. Ephrin B signaling was
one of the pathways upregulated in MCF7-CXCR4-CTD cells, as
identified in the oligoarray analyses. We further validate this
finding in our small molecule inhibitor screen when EPHB2 was found
to be a novel candidate downstream target of CXCR4 activity. EPHB2
is estrogen-independent, while other ephrin family members are
estrogen dependent (42). Reverse
signaling of ephrin B2 in endothelial cells is required for
angiogenesis and is integral in metastasis (42,43). Our
data suggest that ephrin B signaling should be interrogated further
as downstream regulators of CXCR4 function, and EPHB2 is the
specific ephrin B family member on which to focus future
mechanistic studies.
Employing another modality, such as an oligoarray,
to assess potential mechanisms downstream of CXCR4 signaling
validated the utility of our phenotypic screen approach. CXCR4
drives breast cancer metastasis by activating CXCR2 and MEK/PI3K
pathways (28). Using oligoarrays,
we found that CXCR4 overexpression increased expression of ERBB4
signaling as well as Rho/RAC signaling pathways. Similarly, in the
screen within the top ten inhibitors that reversed the mesenchymal
phenotype in TNBC cells in addition to altering the phenotypes of
constitutively active CXCR4 cells, targets of these compounds
included ERBB family members, GSK-3β and AKT.
The primary objective of this study was to
demonstrate the utility of a comprehensive medium-throughput
phenotypic screen using a readily available non-selective inhibitor
set in a proof-of-concept study. A limitation of our study was that
we only used one endocrine targeting therapy in the resistance
screen, MCF-7 cells that were resistant to fulvestrant. We expect
acquired resistance to other endocrine-targeting drugs to affect
different kinase signaling pathways (44,45).
Another limitation was that there was only available off-target
kinase comparison data for the PKIS1 library set. Because we
screened PKIS1 and PKIS2 libraries, we hypothesized that comparing
relative kinase activity in the PKIS2 set could lead to discovery
of more targets, or validate the targets identified in the
screen.
The interaction between SDF-1 and CXCR4 promotes a
mesenchymal and migratory breast cancer cell phenotype, ultimately
resulting in metastasis. SDF-1/CXCR4 signaling also facilities the
acquisition of a resistant phenotype to endocrine targeting
therapies. However, the mechanisms through which CXCR4 functions to
promote this phenotype are not well characterized. In this study,
using a phenotypic screen approach using the PKIS small molecule
inhibitor set, we discovered candidate kinases and signaling
pathways downstream of CXCR4 to be interrogated in future
validation studies. This project provides valuable insight into
novel mechanisms of CXCR4 activity and identifies potential
pathways and targets to pursue using a comprehensive phenotypic
screen approach. While our screening tool has promising preliminary
findings, future projects are required to validate and interrogate
the kinase pathway leads identified in this screen.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to acknowledge the Structural
Genomics Consortium (SGC), which provided the PKIS1 and PKIS2. The
SGC is a registered charity (no. 1097737) that receives funds from
AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation
for Innovation, Eshelman Institute for Innovation, Genome Canada,
Innovative Medicines Initiative (EU/EFPIA), Janssen, Merck KGaA
Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of
Economic Development and Innovation, Pfizer, Takeda and Wellcome
(106169/ZZ14/Z). The authors would like to acknowledge Mr. Brandon
Burow for his contributions in editing the manuscript.
Funding
The present study was funded by the National
Institutes of Health (grant no. 1R01CA174785-01A1).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
MDM wrote the first draft of the manuscript. MDM,
SE, VTH and HEB performed data acquisition and analysis. LVR, ECM,
WJZ, DHD, BMCB and MEB made substantial contributions to conception
and design of the study, and interpretation of the data. MDM, WJZ,
DHD and MEB were responsible for confirming the authenticity of the
data. All authors provided revisions for the manuscript, read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Eggert US: The why and how of phenotypic
small-molecule screens. Nat Chem Biol. 9:206–209. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Elkins JM, Fedele V, Szklarz M, Abdul
Azeez KR, Salah E, Mikolajczyk J, Romanov S, Sepetov N, Huang XP,
Roth BL, et al: Comprehensive characterization of the published
kinase inhibitor set. Nat Biotechnol. 34:95–103. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Drewry DH, Wells CI, Andrews DM, Angell R,
Al-Ali H, Axtman AD, Capuzzi SJ, Elkins JM, Ettmayer P, Frederiksen
M, et al: Progress towards a public chemogenomic set for protein
kinases and a call for contributions. PLoS One. 12:e01815852017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Matossian MD, Elliott S, Hoang VT, Burks
HE, Phamduy TB, Chrisey DB, Zuercher WJ, Drewry DH, Wells C,
Collins-Burrow B and Burrow ME: Novel application of the published
kinase inhibitor set to identify therapeutic targets and pathways
in triple negative breast cancer subtypes. PLoS One.
12:e01778022017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rubin JB: Chemokine signaling in cancer:
One hump or two? Semin Cancer Biol. 19:116–122. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Balkwill FR: The chemokine system and
cancer. J Pathol. 226:148–157. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chatterjee S, Behnam Azad B and Nimmagadda
S: The intricate role of CXCR4 in cancer. Adv Cancer Res.
124:31–82. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bendall LJ, Baraz R, Juarez J, Shen W and
Bradstock KF: Defective p38 mitogen-activated protein kinase
signaling impairs chemotaxic but not proliferative responses to
stromal-derived factor-1alpha in acute lymphoblastic leukemia.
Cancer Res. 65:3290–3298. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Grande F, Giancotti G, Ioele G, Occhiuzzi
MA and Garofalo A: An update on small molecules targeting CXCR4 as
starting points for the development of anti-cancer therapeutics.
Eur J Med Chem. 139:519–530. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Walenkamp AME, Lapa C, Herrmann K and
Wester HJ: CXCR4 Ligands: The next big hit? J Nuc Med. 58 (Suppl
2):77S–82S. 2017. View Article : Google Scholar
|
|
11
|
Xu C, Zhao H, Chen H and Yao Q: CXCR4 in
breast cancer: Oncogenic role and therapeutic targeting. Drug Des
Devel Ther. 9:4953–4964. 2015.PubMed/NCBI
|
|
12
|
Eckert F, Schilbach K, Klumpp L, Bardoscia
L, Sezgin EC, Schwab M, Zips D and Huber SM: Potential role of
CXCR4 targeting in the context of radiotherapy and immunotherapy of
cancer. Front Immunol. 9:30182018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mukherjee D and Zhao J: The role of
chemokine receptor CXCR4 in breast cancer metastasis. Am J Cancer
Res. 3:46–57. 2013.PubMed/NCBI
|
|
14
|
Salvucci O, Bouchard A, Baccarelli A,
Deschênes J, Sauter G, Simon R, Bianchi R and Basik M: The role of
CXCR4 receptor expression in breast cancer: A large tissue
microarray study. Breast Cancer Res Treat. 97:275–283. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Z, Ni C, Chen W, Wu P, Wang Z, Yin
J, Huang J and Qiu F: Expression of CXCR4 and breast cancer
prognosis: A systematic review and meta-analysis. BMC Cancer.
14:492014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Okuyama Kishima M, de Oliveira CE,
Banin-Hirata BK, Losi-Guembarovski R, Brajão de Oliveira K,
Amarante MK and Watanabe MA: Immunohistochemical expression of
CXCR4 on breast cancer and its clinical significance. Anal Cell
Pathol (Amst). 2015:8910202015.PubMed/NCBI
|
|
17
|
Chu QD, Panu L, Holm NT, Li BD, Johnson LW
and Zhang S: High chemokine receptor CXCR4 level in triple negative
breast cancer specimens predicts poor clinical outcome. J Surg Res.
159:689–695. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hassan S, Ferrario C, Saragovi U,
Quenneville L, Gaboury L, Baccarelli A, Salvucci O and Basik M: The
influence of tumor-host interactions in the stromal cell-derived
factor-1/CXCR4 ligand/receptor axis in determining metastatic risk
in breast cancer. Am J Pathol. 175:66–73. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Krohn A, Song YH, Muehlberg F, Dross L,
Bechmann C and Alt E: CXCR4 receptor positive spheroid forming
cells are responsible for tumor invasion in vitro. Cancer Lett.
280:65–71. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Graham NA and Graeber TG: Complexity of
metastasis-associated SDF-1 ligand signaling in breast cancer stem
cells. Proc Natl Acad Sci USA. 111:7503–7504. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yi T, Zhai B, Yu Y, Kiyotsugu Y, Raschle
T, Etzkorn M, Seo HC, Nagiec M, Luna RE, Reinherz EL, et al:
Quantitative phosphoproteomic analysis reveals system-wide
signaling pathways downstream of SDF-1/CXCR4 in breast cancer stem
cells. Proc Natl Acad Sci USA. 111:E2182–E2190. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mego M, Cholujova D, Minarik G, Sedlackova
T, Gronesova P, Karaba M, Benca J, Cingelova S, Cierna Z, Manasova
D, et al: CXCR4-SDF-1 interaction potentially mediates trafficking
of circulating tumor cells in primary breast cancer. BMC Cancer.
16:1272016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brabletz T, Kalluri R, Nieto MA and
Weinberg RA: EMT in cancer. Nat Rev Cancer. 18:128–134. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Felipe Lima J, Nofech-Mozes S, Bayani J
and Bartlett JM: EMT in breast carcinoma-A review. J Clin Med.
5:652016. View Article : Google Scholar
|
|
25
|
Leggett SE, Sim JY, Rubins JE, Neronha ZJ,
Williams EK and Wong IY: Morphological single cell profiling of the
epithelial-mesenchymal transition. Integr Biol (Camb). 8:1133–1144.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nelson CM, Khauv D, Bissell MJ and Radisky
DC: Change in cell shape is required for matrix
metalloproteinase-induced epithelial-mesenchymal transition of
mammary epithelial cells. J Cell Biochem. 105:25–33. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ueda Y, Neel NF, Schutyser E, Raman D and
Richmond A: Deletion of the COOH-terminal domain of CXC chemokine
receptor 4 leads to the down-regulation of cell-to-cell contact,
enhanced motility and proliferation in breast carcinoma cells.
Cancer Res. 66:5665–5675. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sobolik T, Su YJ, Wells S, Ayers GD, Cook
RS and Richmond A: CXCR4 drives the metastatic phenotype in breast
cancer through induction of CXCR2 and activation of MEK and PI3K
pathways. Mol Biol Cell. 25:566–582. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun X, Cheng G, Hao M, Zheng J, Zhou X,
Zhang J, Taichman RS, Pienta KJ and Wang J: CXCL12/CXCR4/CXCR7
chemokine axis and cancer progression. Cancer Metastasis Rev.
29:709–722. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rhodes LR, Short SP, Neel NF, Salvo VA,
Zhu Y, Elliott S, Wei Y, Yu D, Sun M, Muir SE, et al: Cytokine
receptor CXCR4 mediates estrogen-independent tumorigenesis,
metastasis, and resistance to endocrine therapy in human breast
cancer. Cancer Res. 71:603–613. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liang S, Peng X, Li X, Yang P, Xie L, Li
Y, Du C and Zhang F: Silencing of CXCR4 sensitizes triple-negative
breast cancer cells to cisplatin. Oncotarget. 6:1020–1030. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang Y, Yan L, Zhang L, Xu H, Chen T, Li
Y, Wang H, Chen S, Wang W, Chen C and Yang Q: NT21MP negatively
regulates paclitaxel-resistant cells by targeting miR-155-3p and
miR-155-5p via the CXCR4 pathway in breast cancer. Int J Oncol.
53:1043–1054. 2018.PubMed/NCBI
|
|
33
|
Liu S, Xie SM, Yang-Kolodji G and Tripathy
D: Targeting the tumor microenviroment by CXCR4 inhibition to
abrogate trastuzumab resistance in HER2-positive breast cancer.
Cancer Res. 79 (Suppl 4):P5-03-04. 2019.
|
|
34
|
Rhodes LV, Antoon JW, Muir SE, Elliott S,
Beckman BS and Burrow ME: Effects of human mesenchymal stem cells
on ER-positive human breast carcinoma cells mediated through
ER-SDF-1/CXCR4 crosstalk. Mol Cancer. 9:2952010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rhodes LV, Bratton MR, Zhu Y, Tilghman SL,
Muir SE, Salvo VA, Tate CR, Elliott S, Nephew KP, Collins-Burow BM
and Burow ME: Effects of SDF-1-CXCR4 signaling on microRNA
expression and tumorigenesis in estrogen receptor-alpha
(ER-α)-positive breast cancer cells. Exp Cell Res. 317:2573–2581.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sauvé K, Lepage J, Sanchez M, Heveker N
and Tremblay A: Positive feedback activation of estrogen receptors
by the CXCL12-CXCR4 pathway. Cancer Res. 69:5793–5800. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dubrovska A, Hartung A, Bouchez LC, Walker
JR, Reddy VA, Cho CY and Schultz PG: CXCR4 activation maintains a
stem cell population in tamoxifen-resistant breast cancer cells
through AhR signalling. British J Cancer. 107:43–52. 2012.
View Article : Google Scholar
|
|
38
|
Fan M, Yan PS, Hartman-Frey C, Chen L,
Paik H, Oyer SL, Salisbury JD, Cheng AS, Li L, Abbosh PH, et al:
Diverse gene expression and DNA methylation profiles correlate with
differential adaptation of breast cancer cells to the antiestrogens
tamoxifen and fulvestrant. Cancer Res. 66:11954–11966. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ptasznik A, Urbanowska E, Chinta S, Costa
MA, Katz BA, Stanislaus MA, Demir G, Linnekin D, Pan ZK and Gewirtz
AM: Crosstalk between BCR/ABL oncoprotein and CXCR4 signaling
through a Src family kinase in human leukemia cells. J Exp Med.
196:667–678. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dillmann F, Veldwijk MR, Laufs S,
Sperandio M, Calandra G, Wenz F, Zeller J and Fruehauf S:
Plerixafor inhibits chemotaxis toward SDF-1 and CXCR4-mediated
stroma contact in a dose-dependent manner resulting in increased
susceptibility of BCR-ABL+ cell to Imatinib and Nilotinib. Leuk
Lymphoma. 50:1676–1686. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Smith-Pearson PS, Greuber EK, Yogalingam G
and Pendergast AM: Abl kinases are required for invadopodia
formation and chemokine-induced invasion. J Biol Chem.
285:40201–40211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kaenel P, Mosimann M and Andres AC: The
multifaceted roles of Eph/ephrin signaling in breast cancer. Cell
Adh Migr. 6:138–147. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Haldimann M, Custer D, Munarini N,
Stirnimann C, Zürcher G, Rohrbach V, Djonov V, Ziemiecki A and
Andres AC: Deregulated ephrin-B2 expression in the mammary gland
interferes with the development of both the glandular epithelium
and vasculature and promotes metastasis formation. Int J Oncol.
35:525–536. 2009.PubMed/NCBI
|
|
44
|
Lamb CA, Helguero LA, Fabris V, Lucas C,
Molinolo AA and Lanari C: Differential effects of raloxifene,
tamoxifen and fulvestrant on a murine mammary carcinoma. Breast
Cancer Res Treat. 79:25–35. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Giuliano M, Schiff R, Osborne CK and
Trivedi MV: Biological mechanisms and clinical implications of
endocrine resistance in breast cancer. Breast. 20 (Suppl
3):S42–S49. 2011. View Article : Google Scholar : PubMed/NCBI
|