Introduction
Liver cancer is one of the most frequently occurring
malignancies and the second leading cause of cancer-associated
mortality worldwide (1). Hepatitis B
virus (HBV) infection is a global health concern that increases the
risk of liver cancer 5-fold (2).
Epidemiological studies have reported that 240 million individuals
are infected with HBV and approximately 650,000 succumb to
HBV-associated complications each year (3). The HBV genome encodes four main
proteins, denoted as C, P, S and X. Accumulating evidence indicates
that HBV X protein (HBx) plays a crucial role in HBV-mediated
carcinogenesis and liver cancer progression through the direct or
indirect regulation of host gene transcription and protein activity
(4,5). Therefore, a better understanding of the
molecular mechanisms underlying HBV-mediated liver cancer
pathogenesis may improve the diagnosis and treatment of this
disease.
Nicotinamide phosphoribosyltransferase (NAMPT) is a
critical rate-limiting enzyme involved in NAD synthesis that
catalyzes the conversion of nicotinamide (NAM) to nicotinamide
mononucleotide (NMN). Since NAD acts as a co-enzyme in the
regulation of various metabolic signaling pathways, including
glycolysis, serine biosynthesis and fatty acid oxidation, the
continuous generation of NAD promotes the survival and
proliferation of cancer cells (6,7).
Previous studies have shown that NAMPT expression is upregulated in
different types of cancer, including breast, lung and prostate
cancer (8,9). Therefore, the inhibition of NAMPT
represents a potential therapeutic strategy for killing cancer
cells. The specific NAMPT inhibitor FK866 has been shown to exhibit
potent anticancer activity in several types of cancer, including
gastric, pancreatic cancer and chronic lymphocytic leukemia
(10–12). However, whether NAMPT is involved in
HBV replication and HBV-mediated liver cancer progression remains
unknown.
In the present study, the expression levels of NAMPT
were assessed in HBV-positive and HBV-negative liver cancer cells,
and whether HBV-induced NAMPT expression was HBx-dependent was
investigated. In addition, the role of NAMPT in HBV replication and
transcription, as well as HBV-mediated liver cancer cell growth was
explored. Moreover, the effects of NAMPT on the glycolytic pathway
of HBV-expressing liver cancer cells were evaluated.
Materials and methods
Cell culture and reagents
HBV negative liver cancer cells (HepG2 and Huh7)
were purchased from the Chinese Academy of Sciences Cell Bank
(Shanghai, China) and maintained in DMEM medium (HyClone; Cytiva)
at 37°C in a 5% CO2 incubator. The medium was
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific Inc.),
100 IU/ml penicillin and 100 IU/ml streptomycin. HBV positive liver
cancer cells (HepG2.2.15 and HepAD38) cells were cultured in DMEM
medium (HyClone; Cytiva) supplemented with 400 µg/ml G418 (Sangon
Biotech Co., Ltd) at 37°C in a 5% CO2 incubator as
described in our previous study (13). The HepG2 and HepG2.2.15 cells were
assessed by short tandem repeat (STR) analysis and no contamination
with other cell lines was present. Moreover, STR analysis verified
that the cell profile was consistent with that of the archived
cells.
The following reagents were used: NAMPT inhibitor
FK866 (S2799; Selleck Chemicals), NMN (S5259; Selleck Chemicals)
and trypan blue (T6146; Sigma-Aldrich; Merck KGaA). The cells were
treated with 20 nM FK866 alone or in combination with 500 µM NMN
for 24 h at 37°C.
Plasmid construction and cell
transfection
The HBV1.3 plasmid harboring the HBV genome (subtype
adw), the plasmid encoding HBx amplified from the HBV genome, and
the HBV expression plasmid with an HBx mutation (∆HBx) were
generated as described previously (14). HepG2 and Huh7 cells were transfected
with 4 µg of HBV1.3, HBx, or ∆HBx plasmid by using
Lipofectamine®3000 (cat. no. L3000015; Invitrogen;
Thermo Fisher Scientific, Inc.). HepG2.2.15 and HepAD38 cells were
transfected with 100 nmol/l of specific NAMPT siRNAs (cat. no.
sc-45843; Santa Cruz Biotechnology Inc.) or negative control siRNA
(cat. no. sc-37007; Santa Cruz Biotechnology Inc.) by using
Lipofectamine RNAiMAX (cat. no. 13778150; Thermo Fisher Scientific,
Inc.). After transfection at 37°C for 48 h, cells were harvested
for subsequent experiments.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted using from cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and reverse transcribed to cDNA using the PrimeScript™ RT
reagent Kit (Takara Bio, Inc.) according to the manufacturer's
protocol. The relative levels of the target gene mRNA transcripts
were determined by qPCR with SYBR® Premix Ex Taq™ II
(Takara Bio, Inc.) using an iCycler iQ™ Multicolor Real-Time PCR
Detection System (Bio-Rad Laboratories, Inc.) as previously
described (15). The thermocycling
conditions were as follows: Initial denaturation at 95°C for 5 min
followed by 40 cycles of 95°C for 15 sec, 60°C for 20 sec, and 72°C
for 30 sec followed by final elongation at 72°C for 2 min. The
following primers were used: Glucose transporter 1 (GLUT1),
5′-CGGGCCAAGAGTGTGCTAAA-3′ (forward) and 5′-TGACGATACCGGAGCCAATG-3′
(reverse); lactate dehydrogenase A (LDHA),
5′-GGCCTGTGCCATCAGTATCT-3′ (forward) and 5′-GGAGATCCATCATCTCTCCC-3′
(reverse); NAMPT, 5′-TACAAGTTGCTGCCACCTTATC-3′ (forward) and
5′-GCAAACCTCCACCAGAACC-3′ (reverse); pyruvate kinase M2 (PKM2),
5′-TGTCTGGAGAAACAGCCAAAGG-3′ (forward) and
5′-CGGAGTTCCTCAAATAATTGCAA-3′ (reverse); HBV,
5′-ACCGACCTTGAGGCATACTT-3′ (forward) and 5′-GCCTACAGCCTCCTAGTACA-3′
(reverse); HBV core antigen (HBcAg), 5′-CTGGGTGGGTGTTAATTTGG-3′
(forward) and 5′-TAAGCTGGAGGAGTGCGAAT-3′ (reverse); HBV surface
antigen (HBsAg), 5′-CTCCAATCACTCACCAACCT-3′ (forward) and
5′-TCCAGAAGAACCAACAAGAAGA-3′ (reverse); GAPDH,
5′-ACCACAGTCCATGCCATCAC-3′ (forward) and 5′-TCCACCACCCTGTTGCTGTA-3′
(reverse). The 2−ΔΔCq method was used in analyzing
relative gene expression data (16).
Western blot analysis
The cells were lysed with RIPA lysis buffer (50 mM
Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.5%
deoxycolic acid) containing protease inhibitors (Complete Protease
Inhibitor Cocktail Tablets; Roche Diagnostics) for 30 min on ice.
The concentration of extracted proteins were determined by Bradford
method. Cell lysates (40 µg/lane) were resolved by 12% SDS-PAGE and
transferred to PVDF membranes (EMD Millipore). After blocking with
5% skimmed milk at room temperature for 1 h, the membrane was
incubated overnight at 4°C with the following primary antibodies:
GLUT1 (1:1,000; cat. no. 12939; Cell Signaling Technology, Inc.),
LDHA (1:1,000; cat. no. 3582; Cell Signaling Technology, Inc.),
NAMPT (1:1,000; cat. no. ab45890; Abcam), PKM2 (1:1,000; cat. no.
4053; Cell Signaling Technology, Inc.), HBx (1:1,000; cat. no.
MA1-081; Thermo Fisher Scientific, Inc.) and β-actin (1:2,000; cat.
no. sc-47778; Santa Cruz Biotechnology, Inc.). After washing with
TBS with 0.1% Tween-20 (TBST) three times, the membranes were
subsequently incubated with HRP-conjugated secondary antibody
(1:3,000; cat. nos. sc-2004 and sc-2005; Santa Cruz Biotechnology,
Inc.) at room temperature for 1 h. Following three washes with
TBST, the membrane was visualized using an Immobilon™ Western
Chemiluminescent HRP substrate (EMD Millipore).
Determination of HBsAg, HBeAg and HBV
DNA
The quantities of HBsAg and HBeAg in the cell
supernatant were measured using ELISA kits (Kehua Bio-Engineering
Co., Ltd.) and the quantity of HBV DNA in the cell-free culture
medium was detected using the artus® HBV RG PCR Kit
(Qiagen AB) according to the manufacturer's instructions.
Measurement of cell viability
Cell viability analysis was performed using a Cell
Counting Kit-8 assay (CCK-8; KN867; Dojindo Molecular Technologies,
Inc.) as previously described (17).
Briefly, the cells were plated in 96-well plates (1×104
cells/well) and treated with 20 nM FK866 alone or in combination
with 500 µM NMN for 24 h at 37°C. After this, 10 µl CCK-8 solution
was added to each well and the plate was incubated for an
additional 4 h. The absorbance was then measured at 450 nm using a
microplate reader. For trypan blue exclusion assay,
2×105 cells were plated in 6-well plates and treated
with 20 nM FK866 alone or in combination with 500 µM NMN for 24 h
at 37°C. Then, the cells were digested with trypsin and resuspended
in PBS containing 0.04% trypan blue. The viable cells (clear
cytoplasm) and death cells (blue cytoplasm) were counted under a
light microscope.
Colony formation assay
Cells were plated into 24-well-plates (500
cells/well) and subsequently treated with 20 nM FK866 alone or in
combination with 500 µM NMN at 37°C. The colonies were allowed to
grow for ~10 days, and then fixed in 4% paraformaldehyde at room
temperature for 15 min and stained with crystal violet at room
temperature for 20 min. The number of colonies was counted using
ImagJ software (v.1.52a; National Institutes of Health).
Glycolysis examination
The extracellular acidification rate (ECAR) of the
cells was determined using a Seahorse XF Glycolysis Stress Test Kit
with an XF96 Extracellular Flux Analyzer (Seahorse Bioscience;
Agilent Technologies, Inc.) according to the manufacturer's
instructions. Briefly, 2×104 cells were plated into XF96
cell plates (Agilent Technologies, Inc.) and treated with 20 nM
FK866 alone or in combination with 500 µM NMN at 37°C for 24 h.
After washing with XF assay media, each well of the XF96 cartridge
was sequentially injected with glucose (detection of glycolysis),
oligomycin (an ATPase inhibitor, which restrains mitochondrial ATP
production) and 2-DG (a glucose analog, which inhibits glycolysis).
Glucose uptake, lactate production and ATP concentration were
measured in cell lysates using a Glucose Uptake Colorimetric Assay
Kit (BioVision, Inc.), Lactate Colorimetric/Fluorometric Assay Kit
(BioVision, Inc.) and ATP Colorimetric/Fluorometric Assay Kit
(BioVision, Inc), respectively. The enzymatic activities of PKM2
and LDHA were determined using a Pyruvate Kinase Activity Assay Kit
(Beijing Solarbio Science & Technology Co., Ltd.) and Lactate
Dehydrogenase Activity Colorimetric Assay Kit (BioVision, Inc.),
respectively. All measurements were performed according to the
manufacturer's protocols and normalized to the cell protein
levels.
Statistical analysis
All quantitative data are expressed as the mean ±
SD. Comparisons between two groups were performed using the
Student's t-test. Differences in quantitative data among multiple
groups were analyzed by one-way ANOVA followed by Tukey's post hoc
test. Data were analyzed using SPSS statistical software (version
22.0; IBM Corp.). P<0.05 was considered to indicate a
statistically significant difference.
Results
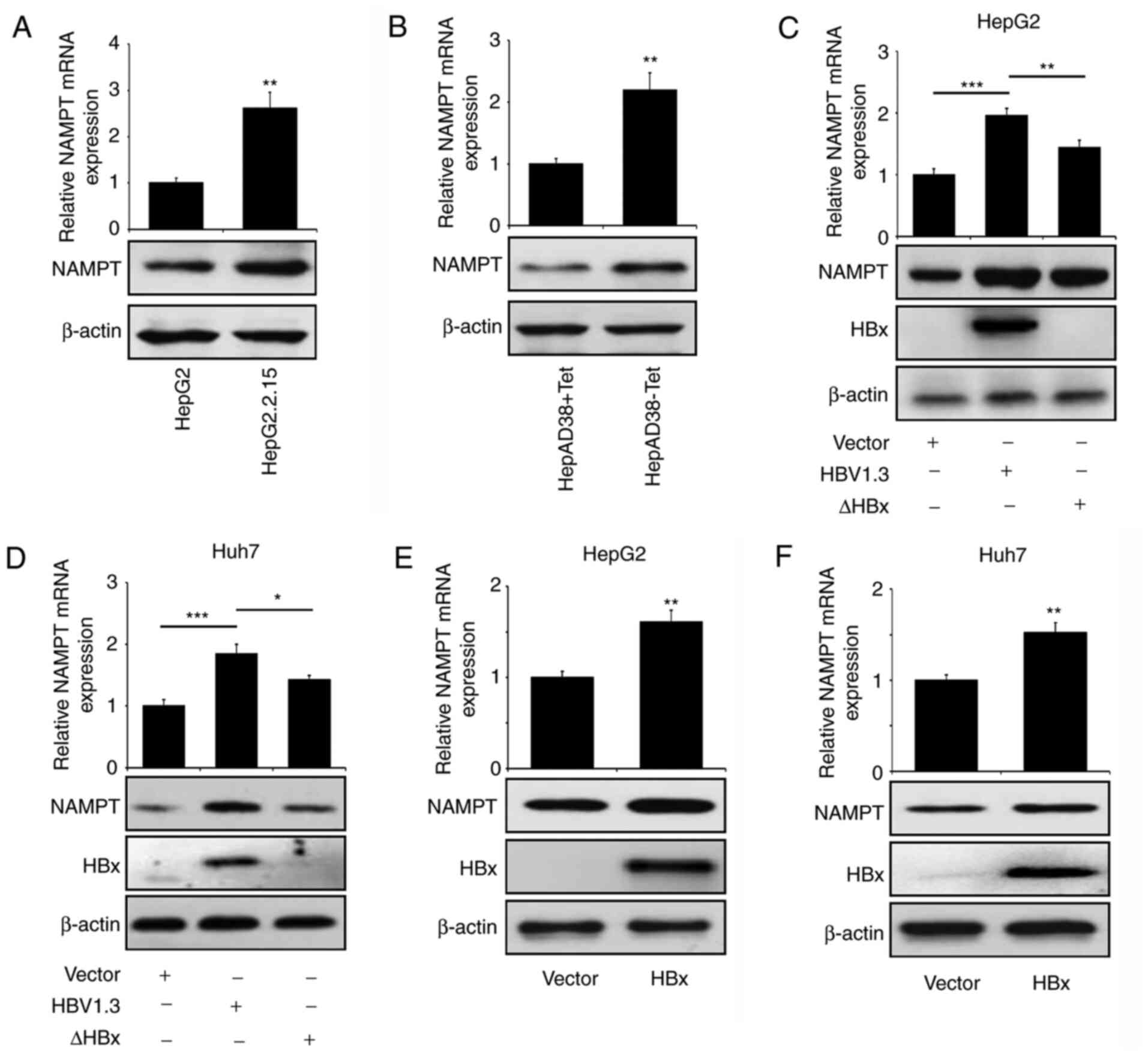
NAMPT expression levels are
upregulated by HBx
To explore the association between NAMPT and HBV
replication, the expression levels of NAMPT were assessed in
HBV-producing liver cancer cells. The mRNA and protein expression
levels of NAMPT were upregulated in HepG2.2.15 cells compared with
those in HepG2 cells (Fig. 1A). In
addition, elevated NAMPT levels were observed in untreated HepAD38
cells compared with tetracycline-treated HepAD38 cells (Fig. 1B). HepG2 and Huh7 cells transiently
transfected with HBV-expressing plasmid HBV1.3 (harboring a
1.3-fold overlength of the HBV genome) demonstrated increased
expression of NAMPT compared with that in cells transfected with
empty vector (Fig. 1C and D). These
data indicate that HBV replication promoted NAMPT expression in the
liver cancer cells. To further assess whether HBV-induced NAMPT
expression was HBx-dependent, the ∆HBx plasmid (HBV1.3 plasmid with
HBx mutation) was transiently transfected into HepG2 and Huh7 cells
and its effect on NAMPT expression was evaluated. As shown in
Fig. 1C, the ectopic introduction of
HBV1.3 plasmid but not that of ∆HBx plasmid, significantly
increased the expression levels of HBx in HepG2 cells. Notably, the
mutation of HBx significantly mitigated the elevation in the levels
of NAMPT induced by HBV replication in HepG2 cells. A similar
result was observed in Huh7 cells (Fig.
1D). When the HBx expression plasmid was successfully
introduced into HepG2 and Huh7 cells, the mRNA and protein
expression levels of NAMPT were increased in response to the HBx
stimulus (Fig. 1E and F). Therefore,
these results indicate that HBx enhances the expression levels of
NAMPT in liver cancer cells.
NAMPT activity is required for HBV
replication and transcription
To investigate the involvement of NAMPT in HBV
replication, the extracellular levels of HBV DNA were assessed in
HBV-producing liver cancer cells treated with the NAMPT inhibitor
FK866. Extracellular HBV DNA levels were significantly reduced in
HepG2.2.15 and HepAD38 cells following incubation with FK866
(Fig. 2A). Moreover, FK866 treatment
resulted in a 2-fold reduction in HBV transcription (Fig. 2B), suggesting that NAMPT activity
upregulates HBV production. To further verify the association
between NAMPT activation and HBV replication, the expression levels
of HBcAg and HBsAg, which are indicators of active HBV replication,
were assessed. The mRNA levels of HBcAg and HBsAg in FK866-treated
HepG2.2.15 and HepAD38 cells were lower than those observed in
vehicle-treated control cells (Fig. 2C
and D). In addition, ELISAs indicated that the inhibition of
NAMPT activity induced a significant reduction in the secretion of
HBeAg and HBsAg into the supernatant of HepG2.2.15 and HepAD38
cells (Fig. 2E and F). Collectively,
these data imply that the activation of NAMPT is essential for HBV
replication and transcription.
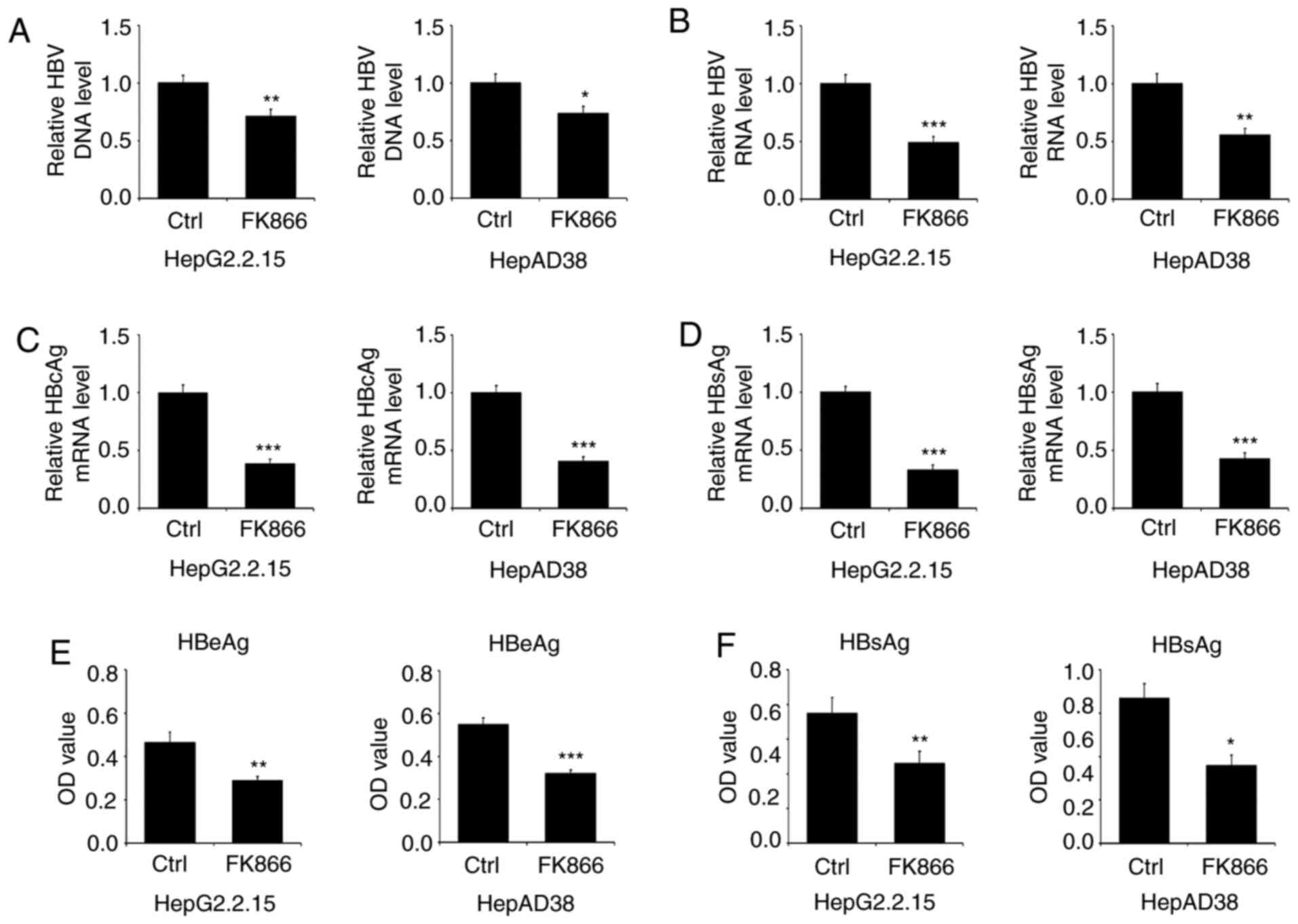 | Figure 2.Inhibition of NAMPT by FK866
represses HBV replication and transcription. HepG2.2.15 and HepAD38
cells were treated with 20 nM FK866 for 24 h. (A) The number of HBV
DNA copies relative to those of untreated cells in the cell-free
culture media were measured by qPCR analysis. (B) The relative
levels of HBV RNA, (C) HBcAg mRNA and (D) HBsAg mRNA were detected
by RT-qPCR analysis. The levels of secreted (E) HBeAg and (F) HBsAg
in the culture media were measured by ELISA. *P<0.05,
**P<0.01,***P<0.001. NAMPT, nicotinamide
phosphoribosyltransferase; HBV, hepatitis B virus; HBcAg, HBV core
antigen; HBsAg, HBV surface antigen; RT-qPCR, reverse
transcriptase-quantitative PCR; Ctrl, control; OD, optical
density. |
NAMPT inhibition by FK866 represses
HBV-mediated cell survival
To further investigate the effect of NAMPT on
HBV-mediated liver cancer cell growth, liver cancer cells were
treated with FK866 and cell proliferation was assessed using a
CCK-8 assay. Treatment with FK866 resulted in a significant
reduction in the viability of HBV-producing liver cancer cells,
whereas the exogenous addition of NMN attenuated the FK866-induced
cytotoxicity. Consistent with the CCK-8 assay results, the number
of colonies formed by FK866-treated HepG2.2.15 and HepAD38 cells
was significantly lower compared with that of vehicle-treated
cells, while the co-administration of NMN rescued the colony
formation of HepG2.2.15 and HepAD38 cells following FK866
supplementation (Fig. 3B).
Furthermore, the inhibition of NAMPT by FK866 induced an increase
in the death rate of HBV-expressing liver cancer cells that was
reversed by NMN treatment (Fig. 3C).
In addition, FK866 reduced markedly the protein expression level of
Bcl2 and increased the levels of cleaved caspase-3, cleaved PARP
and Bax (Fig. 3D). Co-treatment of
the cells with NMN alleviated the FK866-induced effects on the
expression of apoptosis-associated proteins (Fig. 3D). Therefore, these findings indicate
that NAMPT contributes to the HBV-mediated malignant transformation
of liver cancer.
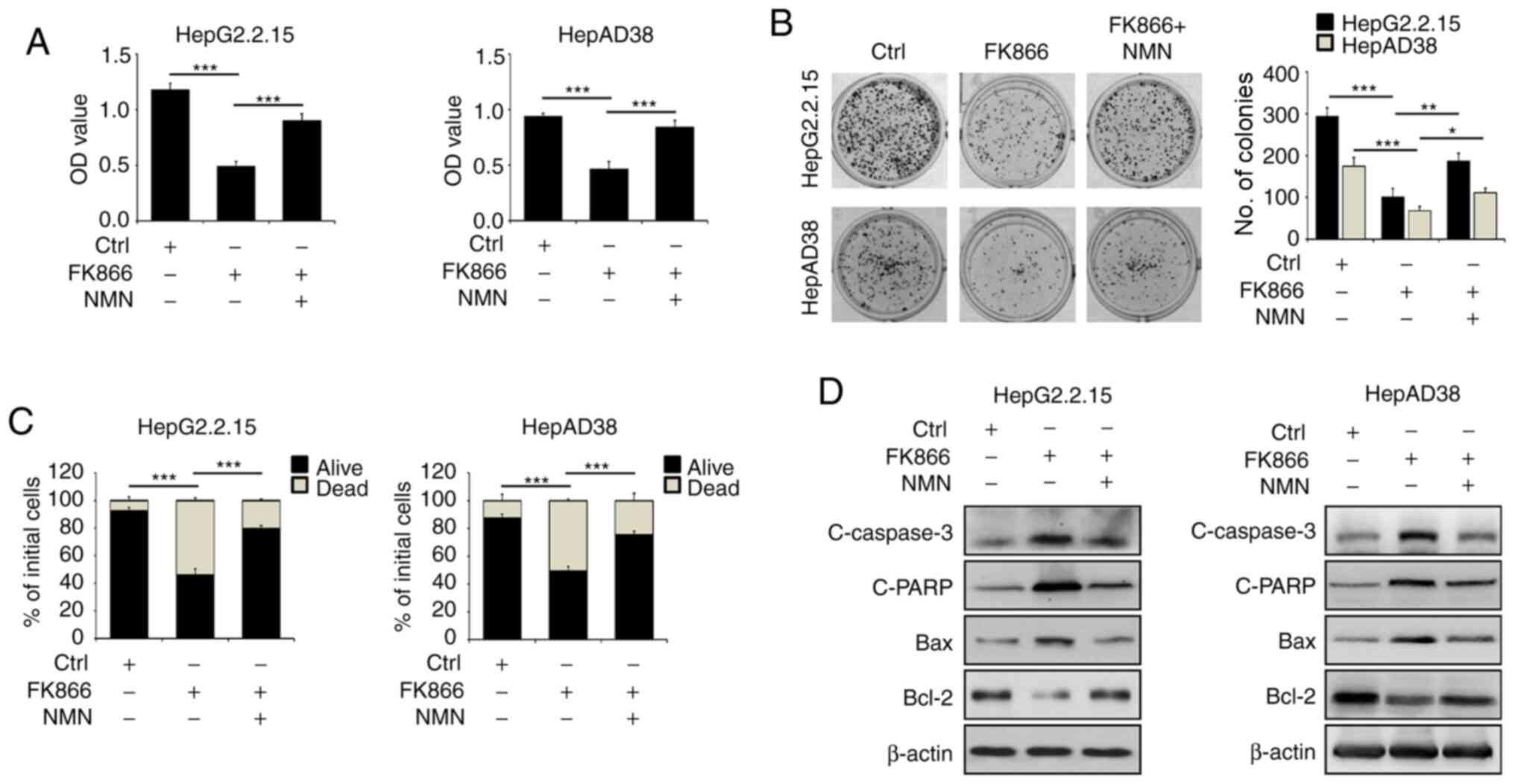 | Figure 3.FK866-induced NAMPT inhibition
decreases cell survival and promotes cell death in HBV-expressing
liver cancer cells. HepG2.2.15 and HepAD38 cells were treated with
FK866 alone or in combination with NMN for 24 h. (A) Cell viability
was measured by CCK-8 assay, (B) a colony formation assay was
performed and (C) cell death was determined by the trypan blue
exclusion assay. (D) The treated HepG2.2.15 and HepAD38 cells were
also subjected to immunoblot analysis for the detection of
apoptosis-associated proteins. *P<0.05, **P<0.01,
***P<0.001. NAMPT, nicotinamide phosphoribosyltransferase; HBV,
hepatitis B virus; NMN, nicotinamide mononucleotide; CCK-8, Cell
Counting Kit-8; C, cleaved; PARP, poly (ADP-ribose) polymerase;
Ctrl, control; OD, optical density. |
NAMPT regulates aerobic glycolysis in
HBV-expressing liver cancer cells
Altered energy metabolism is a hallmark of cancer
and has been shown to play a critical role in tumor initiation and
development (18). The effects of
NAMPT on glycolysis in HBV-expressing liver cancer cells were
assessed in the present study. The inactivation of NAMPT by FK866
resulted in a markedly decreased ECAR during the stages of
glycolysis following glucose and oligomycin injection, whereas
co-treatment with NMN partially rescued the glycolytic capacity of
HepG2.2.15 and HepAD38 cells (Fig.
4A). In addition, FK866 suppressed glucose uptake, lactate
production and ATP levels, and these effects were also reversed by
NMN (Fig. 4B-D). Since PKM2 and LDHA
are required for lactate production and GLUT1 is directly
associated with glucose uptake, whether FK866 affected the
expression of these glycolysis-associated genes was investigated.
The results indicated that the mRNA and protein expression levels
of GLUT1, PKM2 and LDHA were decreased following FK866 treatment in
HepG2.2.15 and HepAD38 cells (Fig. 4E
and F), and co-treatment with NMN markedly attenuated the
FK866-induced effects on the levels of GLUT1, PKM2 and LDHA
(Fig. 4E and F). The enzymatic
activity of PKM2 in HepG2.2.15 and HepAD38 cells was observed to be
reduced in response to FK866 treatment, and this reduction in
activity was diminished by NMN (Fig.
4G). A similar result was noted in the enzymatic activity of
LDHA (Fig. 4H).
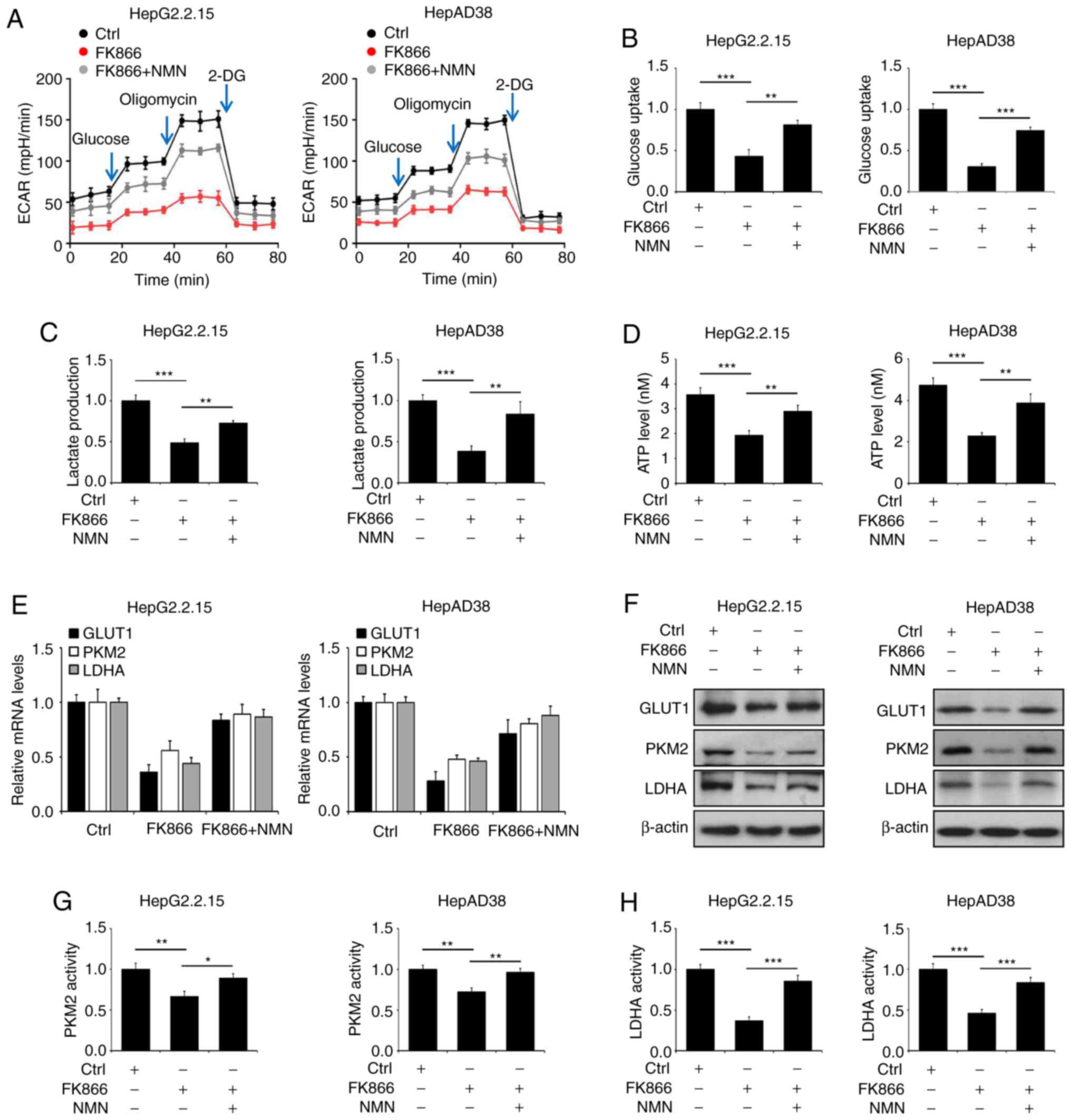 | Figure 4.NAMPT inactivation impairs aerobic
glycolysis in HBV-expressing liver cancer cells. HepG2.2.15 and
HepAD38 cells were treated with FK866 alone or in combination with
NMN for 24 h. (A) The ECAR, (B) glucose uptake, (C) lactate
production and (D) ATP levels of the cells were evaluated. (E) The
levels of GLUT1, PKM2 and LDHA mRNA relative to those in untreated
controls were detected by RT-qPCR analysis. (F) Immunoblot analysis
was performed to detect the expression of GLUT1, PKM2 and LDHA, and
the enzymatic activity of (G) PKM2 and (H) LDHA was determined.
*P<0.05, **P<0.01, ***P<0.001. NAMPT, nicotinamide
phosphoribosyltransferase; HBV, hepatitis B virus; NMN,
nicotinamide mononucleotide; ECAR, extracellular acidification
rate; GLUT1, glucose transporter 1; PKM2, pyruvate kinase M2; LDHA,
lactate dehydrogenase A; RT-qPCR, reverse
transcriptase-quantitative PCR; Ctrl, control; 2-DG,
2-deoxy-D-glucose. |
Further experiments were conducted using HepG2.2.15
and HepAD38 cells transfected with specific siRNA targeting NAMPT
(Fig. 5A). The cells transfected
with siNAMPT displayed decreased cell viability and increased cell
death compared with those transfected with siNC (Fig. 5B and C). In addition, the knockdown
of NAMPT inhibited glycolytic capacity, glucose uptake and lactate
production in HepG2.2.15 and HepAD38 cells (Fig. 5D-F). These data suggest that NAMPT
regulates energy metabolism by enhancing aerobic glycolysis in
HBV-expressing liver cancer cells.
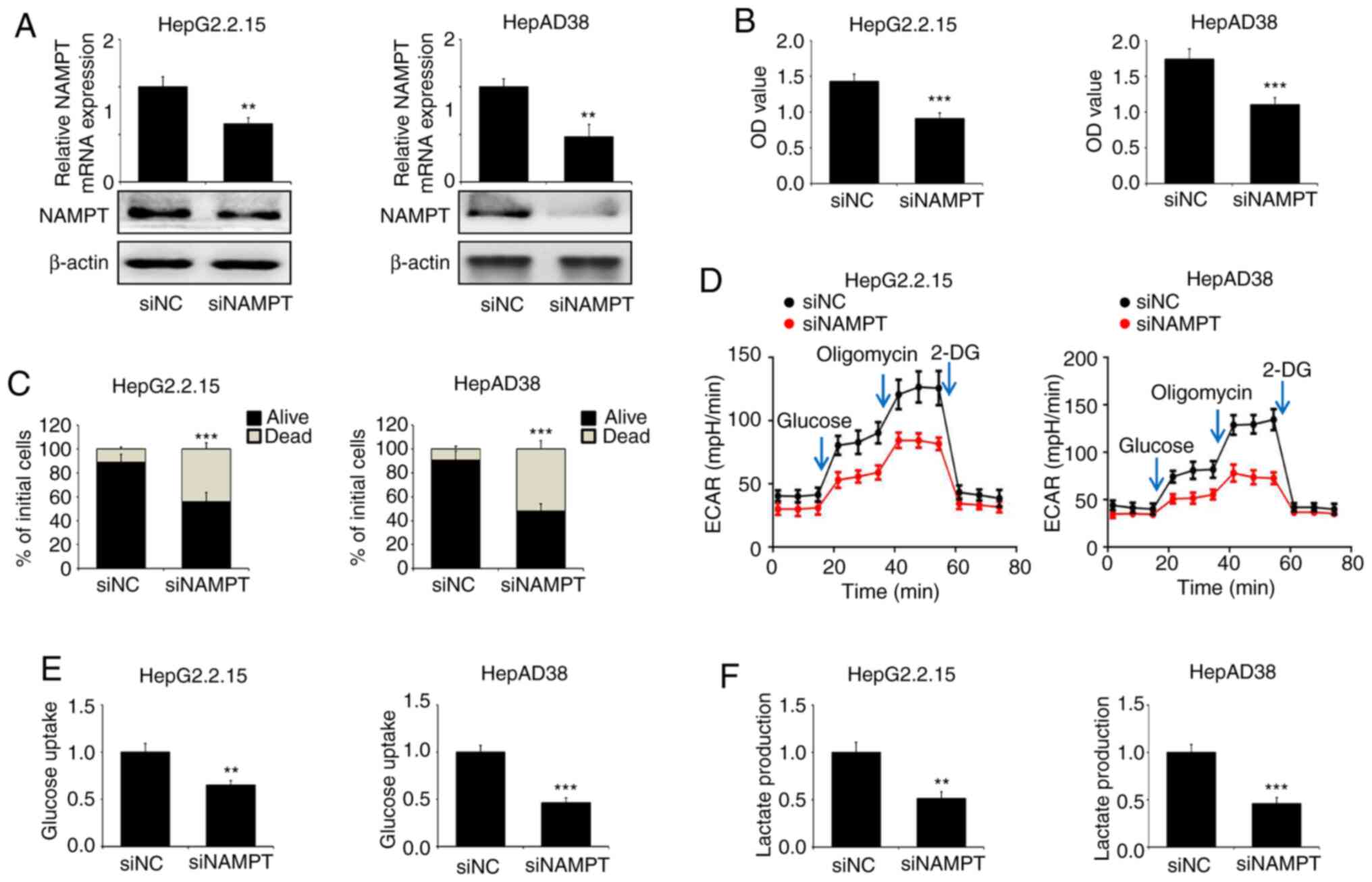 | Figure 5.NAMPT suppression diminishes cell
growth and aerobic glycolysis in HBV-expressing liver cancer cells.
HepG2 and HepG2.2.15 cells were transfected with siRNA targeting
NAMPT or siNC. (A) RT-qPCR and western blot analyses of NAMPT
expression in the cells. (B) Cell viability was measured using the
CCK-8 assay and (C) cell death was determined by the trypan blue
exclusion assay. (D) The ECAR was determined, and (E) glucose
uptake and (F) lactate production assays were performed.
**P<0.01, ***P<0.001. NAMPT, nicotinamide
phosphoribosyltransferase; HBV, hepatitis B virus; RT-qPCR, reverse
transcriptase-quantitative PCR; siRNA, small interfering RNA;
siNAMPT, siRNA targeting NAMPT; siNC, negative control siRNA;
CCK-8, Cell Counting Kit-8; ECAR, extracellular acidification rate;
OD, optical density. |
Discussion
HBV is an enveloped DNA virus with four overlapping
genes and can cause acute and chronic infections. Chronic infection
with HBV contributes to chronic inflammation of the liver, leading
to liver impairment, liver cirrhosis and eventually the development
of liver cancer (19). Therefore,
chronic HBV infection is a major risk factor for liver cancer. HBV
utilizes the cellular machinery of the host for viral DNA
replication, protein production and packaging. HBx is a
non-structural protein of the HBV genome, which contributes to the
development of liver cancer by modulating various cellular
signaling pathways to enhance HBV replication and transcription
(20). A recent study indicated that
a C-terminal-truncated HBx protein promoted hepatocarcinogenesis
via activation of the caveolin-1/low-density lipoprotein
receptor-related protein 6/FERM domain containing 5 axis (21). HBx has also be shown to maintain the
proliferation of liver cancer cells via the microRNA-155-mediated
zinc fingers and homeoboxes 2 gene silencing (22). In addition, HBx has been found to be
localized in the lumen of the endoplasmic reticulum (ER) where it
interacts with 78-kDa glucose-regulated protein, leading to a
reduction in ER stress and DNA repair that prevents liver cancer
cells from undergoing apoptosis (23). However, the role of NAMPT in
HBV-associated liver cancer progression is currently unclear.
In the present study, the data indicated that the
mRNA and protein expression levels of NAMPT were higher in
HBV-positive liver cancer cells than in HBV-negative liver cancer
cells. Moreover, the HBx-expressing HepG2 and Huh-7 cells had
higher NAMPT levels than the corresponding untransfected liver
cancer cells. These data imply that HBx promotes the expression of
NAMPT. The inhibition of NAMPT by FK866 significantly diminished
the secretion of HBV DNA and the mRNA expression levels of HBV RNA,
HBcAg and HBsAg, suggesting a critical role of NAMPT in HBV
replication. Furthermore, FK866 also reduced the secretion of HBeAg
and HBsAg by HBV-producing liver cancer cells. Therefore, the data
suggest the existence of a positive feedback loop between NAMPT and
HBV replication. HBV infection has been shown to cause metabolic
reprogramming in host cells to generate the energy and molecular
components required for their replication and activation (24,25). For
example, a study reported that the levels of metabolic
intermediates, including phenylalanine, L-glutamic acid and serine,
were significantly elevated in the serum of patients with HBV
compared with healthy controls (26). In addition, another study
demonstrated that HBV replication accelerated the biosynthesis of
hexosamine and phosphatidylcholine, leading to the dysregulated
expression of glycolytic enzymes and increased secretion of lactate
(27). In the present study, the
data indicated that the FK866-induced inactivation of NAMPT
inhibited glycolysis and the glycolytic capacity of HBV-expressing
liver cancer cells. In addition, the expression levels and
enzymatic activities of PKM2 and LDHA, which are key regulators of
glucose metabolism, were decreased in response to FK866. These
results indicate an important role of NAMPT in the regulation of
glucose metabolic reprogramming, whereas the disruption of NAD
metabolism may be considered a potential strategy for cancer
therapy. The inhibition of NAMPT by FK866 resulted in a substantial
reduction of cell viability and increased cell death in
HBV-expressing liver cancer cells, whereas the supplementation of
NMN partially reversed the effect of FK866. The present study
further supports the essential function of NAMPT in HBV-mediated
liver malignancy.
Accumulating evidence suggests that oxidative stress
is necessary for HBV-mediated liver cancer development and
progression. For example, HBV infection promotes an intracellular
accumulation of reactive oxygen species that is necessary for HBV
replication in host cells (28,29).
Increased oxidative stress leads to elevated levels of DNA damage,
which activates poly (ADP-ribose) polymerase (PARP)-mediated
repair. As PARP-1 consumes NAD, which is the substrate for its
activation (30), HBV-positive liver
cancer cells may become sensitive to NAD deficiency triggered by
NAMPT inhibition. Furthermore, sirtuins are a family of
NAD-dependent deacetylases that have been shown to regulate
numerous biological functions, including lipid and energy
metabolism, DNA damage, oxidative stress and cell growth, survival
and death (31–33). Sirtuin 6 promotes the transcription
and replication of HBV by upregulating the expression of peroxisome
proliferator-activated receptor α (34). In addition, sirtuin 1 has been shown
to be essential for HBV-associated liver cancer tumorigenesis
(35–37). Therefore, the reduction of NAD
synthesis by NAMPT inhibition may be an effective and promising
method for the treatment of patients with HBV-associated liver
cancer.
In conclusion, the findings of the present study
provide the first evidence that HBV promotes NAMPT expression, and
indicate that the activation of NAMPT is positively associated with
HBV replication and transcription in liver cancer cells. The
knockdown of NAMPT repressed cell growth and promoted cell death in
HBV-positive liver cancer cells. Moreover, the inactivation of
NAMPT suppressed glucose uptake, lactate production and ATP levels,
suggesting a critical role of NAMPT in the regulation of aerobic
glycolysis. Based on this evidence, the present study suggests an
important molecular mechanism by which NAMPT promotes HBV
replication and HBV-mediated cell growth through the manipulation
of glucose metabolic reprogramming. In addition, it highlights
NAMPT as a promising target for the therapy of patients with
HBV-associated liver cancer.
Acknowledgements
Not applicable.
Funding
The study was supported by the Development and
Regeneration Key Laboratory of Sichuan Province, Chengdu Medical
College (grant no. SYS20-05), the National Undergraduate Training
Program for Innovation and Entrepreneurship (grant no.
S202013705006) and the Research Fund of Chengdu Medical College
(grant no. CYZ19-12).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
CFM and ZS conceived the study. HJG, HYL and ZHC
performed the experiments, and WJZ, JJL, JYZ and JW helped to
conduct the experiments. CFM and HJG wrote the manuscript. ZS, XYL
and TZ interpreted the data and revised the manuscript. CFM and ZS
supervised the study. CFM and ZS confirm the authenticity of all
the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
World Health Organization, . Guidelines
for the Prevention, Care and Treatment of Persons with Chronic
Hepatitis B Infection. 2015.
|
|
4
|
Levrero M and Zucman-Rossi J: Mechanisms
of HBV-induced hepatocellular carcinoma. J Hepatol. 64 (1
Suppl):S84–S101. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xu QG, Yuan SX, Tao QF, Yu J, Cai J, Yang
Y, Guo XG, Lin KY, Ma JZ, Dai DS, et al: A novel HBx genotype
serves as a preoperative predictor and fails to activate the
JAK1/STATs pathway in hepatocellular carcinoma. J Hepatol.
70:904–917. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xiao W, Wang RS, Handy DE and Loscalzo J:
NAD(H) and NADP(H) redox couples and cellular energy metabolism.
Antioxid Redox Signal. 28:251–272. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yaku K, Okabe K and Nakagawa T: NAD
metabolism: Implications in aging and longevity. Ageing Res Rev.
47:1–17. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Garten A, Schuster S, Penke M, Gorski T,
de Giorgis T and Kiess W: Physiological and pathophysiological
roles of NAMPT and NAD metabolism. Nat Rev Endocrinol. 11:535–546.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sampath D, Zabka TS, Misner DL, O'Brien T
and Dragovich PS: Inhibition of nicotinamide
phosphoribosyltransferase (NAMPT) as a therapeutic strategy in
cancer. Pharmacol Ther. 151:16–31. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee J, Kim H, Lee JE, Shin SJ, Oh S, Kwon
G, Kim H, Choi YY, White MA, Paik S, et al: Selective cytotoxicity
of the NAMPT inhibitor FK866 toward gastric cancer cells with
markers of the epithelial-mesenchymal transition, due to loss of
NAPRT. Gastroenterology. 155:799–814 e13. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Espindola-Netto JM, Chini CCS, Tarrago M,
Wang E, Dutta S, Pal K, Mukhopadhyay D, Sola-Penna M and Chini EN:
Preclinical efficacy of the novel competitive NAMPT inhibitor
STF-118804 in pancreatic cancer. Oncotarget. 8:85054–85067. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gehrke I, Bouchard ED, Beiggi S, Poeppl
AG, Johnston JB, Gibson SB and Banerji V: On-target effect of
FK866, a nicotinamide phosphoribosyl transferase inhibitor, by
apoptosis-mediated death in chronic lymphocytic leukemia cells.
Clin Cancer Res. 20:4861–4872. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mo CF, Li J, Yang SX, Guo HJ, Liu Y, Luo
XY, Wang YT, Li MH, Li JY and Zou Q: IQGAP1 promotes anoikis
resistance and metastasis through Rac1-dependent ROS accumulation
and activation of Src/FAK signalling in hepatocellular carcinoma.
Br J Cancer. 123:1154–1163. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu J, Liu H, Chen L, Wang S, Zhou L, Yun
X, Sun L, Wen Y and Gu J: Hepatitis B virus X protein confers
resistance of hepatoma cells to anoikis by up-regulating and
activating p21-activated kinase 1. Gastroenterology.
143:199–212.e4. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li K, Mo C, Gong D, Chen Y, Huang Z, Li Y,
Zhang J, Huang L, Li Y, Fuller-Pace FV, et al: DDX17
nucleocytoplasmic shuttling promotes acquired gefitinib resistance
in non-small cell lung cancer cells via activation of β-catenin.
Cancer Lett. 400:194–202. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak JK and Schmittgen Td: Analysis of
relative gene expression data using quantitative Pcr and the
2(-delta delta c(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li K, Gao B, Li J, Chen H, Li Y, Wei Y,
Gong D, Gao J, Zhang J, Tan W, et al: ZNF32 protects against
oxidative stress-induced apoptosis by modulating C1QBP
transcription. Oncotarget. 6:38107–38126. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ringelhan M, O'Connor T, Protzer U and
Heikenwalder M: The direct and indirect roles of HBV in liver
cancer: Prospective markers for HCC screening and potential
therapeutic targets. J Pathol. 235:355–367. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Buendia MA and Neuveut C: Hepatocellular
carcinoma. Cold Spring Harb Perspect Med. 5:a0214442015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mao X, Tey SK, Ko FCF, Kwong EML, Gao Y,
Ng IO, Cheung ST, Guan XY and Yam JWP: C-terminal truncated HBx
protein activates caveolin-1/LRP6/β-catenin/FRMD5 axis in promoting
hepatocarcinogenesis. Cancer Lett. 444:60–69. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Song X, Tan S, Wu Z, Xu L, Wang Z, Xu Y,
Wang T, Gao C, Gong Y, Liang X, et al: HBV suppresses ZHX2
expression to promote proliferation of HCC through miR-155
activation. Int J Cancer. 143:3120–3130. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li J, He J, Fu Y, Hu X, Sun LQ, Huang Y
and Fan X: Hepatitis B virus X protein inhibits apoptosis by
modulating endoplasmic reticulum stress response. Oncotarget.
8:96027–96034. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pallett LJ, Gill US, Quaglia A, Sinclair
LV, Jover-Cobos M, Schurich A, Singh KP, Thomas N, Das A, Chen A,
et al: Metabolic regulation of hepatitis B immunopathology by
myeloid-derived suppressor cells. Nat Med. 21:591–600. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fisicaro P, Boni C, Barili V, Laccabue D
and Ferrari C: Strategies to overcome HBV-specific T cell
exhaustion: Checkpoint inhibitors and metabolic re-programming.
Curr Opin Virol. 30:1–8. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao R, Cheng J, Fan C, Shi X, Cao Y, Sun
B, Ding H, Hu C, Dong F and Yan X: Serum metabolomics to identify
the liver disease-specific biomarkers for the progression of
hepatitis to hepatocellular carcinoma. Sci Rep. 5:181752015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li H, Zhu W, Zhang L, Lei H, Wu X, Guo L,
Chen X, Wang Y and Tang H: The metabolic responses to hepatitis B
virus infection shed new light on pathogenesis and targets for
treatment. Sci Rep. 5:84212015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schinzari V, Barnaba V and Piconese S:
Chronic hepatitis B virus and hepatitis C virus infections and
cancer: Synergy between viral and host factors. Clin Microbiol
Infect. 21:969–974. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ganesan M, Eikenberry A, Poluektova LY,
Kharbanda KK and Osna NA: Role of alcohol in pathogenesis of
hepatitis B virus infection. World J Gastroenterol. 26:883–903.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ying W, Alano CC, Garnier P and Swanson
RA: NAD+ as a metabolic link between DNA damage and cell death. J
Neurosci Res. 79:216–223. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Roulston A and Shore GC: New strategies to
maximize therapeutic opportunities for NAMPT inhibitors in
oncology. Mol Cell Oncol. 3:e10521802016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Griffiths HBS, Williams C, King SJ and
Allison SJ: Nicotinamide adenine dinucleotide (NAD+):
Essential redox metabolite, co-substrate and an anti-cancer and
anti-ageing therapeutic target. Biochem Soc Trans. 48:733–744.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yaku K, Okabe K, Hikosaka K and Nakagawa
T: NAD metabolism in cancer therapeutics. Front Oncol. 8:6222018.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jiang H, Cheng ST, Ren JH, Ren F, Yu HB,
Wang Q, Huang AL and Chen J: SIRT6 inhibitor, OSS_128167 restricts
hepatitis B virus transcription and replication through targeting
transcription factor peroxisome proliferator-activated receptors α.
Front Pharmacol. 10:12702019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ren JH, Tao Y, Zhang ZZ, Chen WX, Cai XF,
Chen K, Ko BC, Song CL, Ran LK, Li WY, et al: Sirtuin 1 regulates
hepatitis B virus transcription and replication by targeting
transcription factor AP-1. J Virol. 88:2442–2451. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pant K, Mishra AK, Pradhan SM, Nayak B,
Das P, Shalimar D, Saraya A and Venugopal SK: Butyrate inhibits HBV
replication and HBV-induced hepatoma cell proliferation via
modulating SIRT-1/Ac-p53 regulatory axis. Mol Carcinog. 58:524–532.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang Q, Cheng ST and Chen J: HBx mediated
increase of SIRT1 contributes to HBV-related hepatocellular
carcinoma tumorigenesis. Int J Med Sci. 17:1783–1794. 2020.
View Article : Google Scholar : PubMed/NCBI
|