Introduction
Lung cancer is the most common malignant tumor with
~1.6 million cases of lung cancer-associated mortality worldwide
per year (1). The most common
subtype is non-small cell lung cancer (NSCLC), which accounts for
~85% of all lung cancer cases (2).
The therapeutic strategies include surgery, traditional
chemotherapy, chemo- and radiotherapy, and molecular targets for
NSCLC therapy. However, treatment failure in patients with NSCLC
results from a lack of effective therapeutic strategies (2). Recent studies showed that patients
receiving epidermal growth factor receptor-tyrosine kinase
inhibitors (EGFR-TKIs), such as gefitinib or erlotinib, exhibited
higher positive response rates and longer progression-free survival
times compared with that in patients receiving traditional
chemotherapy, in the majority of non-smoking patients with
adenocarcinoma of Asian descent (3–5).
However, most patients eventually experience tumor progression due
to acquired resistance within ~12 months of gefitinib treatment
(6). Therefore, EGFR-TKI resistance
in patients with NSCLC is a critical problem associated with
EGFR-TKI treatment.
The mechanism of resistance to EGFR-TKIs has been
reported to include a secondary mutation (T790M), amplification of
MET, a Ras mutation, loss of PTEN and a change in
epithelial-to-mesenchymal transition (EMT) (7); however, the molecular mechanism of
primary resistance is still poorly understood. Increasing evidence
has suggested that EMT plays a vital role in EGFR-TKIs resistance.
The rate of EMT accounted for 14% of cases of acquired EGFR-TKIs
resistance in NSCLC (8). In
addition, reversal of EMT improved gefitinib sensitivity in HCC827
resistant cells or lung tumors (9–11).
However, a comprehensive understanding of the EMT regulatory
mechanism contributing to EGFR-TKIs resistance remains
necessary.
Contactin-1 (CNTN1), a neuronal cell adhesion
molecular, has been reported to be associated with carcinogenesis
and tumor progression. CNTN1 has alternative functions causing
invasion and metastasis of various types of cancer, including
prostate cancer (12), gastric
cancer (13), hepatocellular
carcinoma (14), breast cancer
(15), thyroid cancer (16) and lung adenocarcinoma (LUAD)
(17). Furthermore, overexpression
of CNTN1 has been associated with poor prognosis in patients with
oral squamous cell carcinoma (18).
Knockdown of CNTN1 expression decreased invasion and metastasis,
and inhibited tumor initiation in gastric and prostate cancers
(13,19). However, the function of CNTN1 in
EGFR-TKI resistance remains unclear. In the current study, the role
of CNTN1 in gefitinib resistance was evaluated and the molecular
mechanism associated with gefitinib resistance induced by CNTN1 was
investigated.
Materials and methods
Public databases
Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/gds) was used for the
analysis of CNTN1 mRNA expression levels between
gefitinib-sensitive and -insensitive NSCLC. The gene expression
datasets (GSE4342 and GSE38302) were downloaded from the GEO
database (20,21). The differential expression of genes
between gefitinib-sensitive cells (HCC827 and Calu3) and
intrinsically resistant cells (A549, H460 and H1975) in GSE4342,
and between acquired gefitinib resistant cells (PC9GR) and parental
cells (PC9) in GSE38302 was determined using GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r).
The Kaplan-Meier plot was used to determine overall survival (OS)
time using Kaplan-Meier Plotter database (https://kmplot.com) (22) and a log rank test. P<0.05 was
considered to indicate a statistically significant difference. The
total number of patients with LUAD was 719, which were divided into
the high and low expression level groups using an auto select best
cutoff, including 536 (74.5%) in the low CNTN1 expression group and
183 (25.5%) in the high CNTN1 expression group.
Cell line and plasmids
The NSCLC A549 cell line was purchased from the
American Type Culture Collection, and was cultured in high-glucose
DMEM (Hyclone; Cytiva), supplemented with 10% FBS
(Natocor-Industria Biológica) and 1% penicillin-streptomycin
(Sigma-Aldrich; Merck KGaA). The cell line was thawed every month.
Short hairpin (sh)RNA-CNTN1 and control shRNA [Luciferase shRNA
(shLuc)] was purchased from Sigma-Aldrich (cat. no. SHC007; Merck
KGaA). shRNA-CNTN1 was ligated into a lentiviral plasmid (Takara
Bio Inc.) and the shRNA had the following sequences: shCNTN1-1,
5′-CCGGGCCGTGGTTCAGACAATCATACTCGAGTATGATTGTCTGAACCACGGCTTTTTG-3′;
and shCNTN1-2,
5′-CCGGCCAAGGATCATCAGTTCAGTACTCGAGTACTGAACTGATGATCCTTGGTTTTTG-3′.
Virus packaging and transfection
Briefly, the 293T cells were purchased from the
American Type Culture Collection and were cultured in high-glucose
DMEM, supplemented with 10% FBS. The cells were cultured to 50–60%
confluency and co-transfected with either 10 µg control (shLuc) or
shRNA-CNTN1, with a packaging plasmid [7.5 µg psPAX2 (Invitrogen;
Thermo Fisher Scientific Inc.)] and an envelope plasmid [2.5 µg
pMD2.G (Invitrogen; Thermo Fisher Scientific Inc.)] using 40 µg PEI
reagent (Sigma-Aldrich; Merck KGaA). The lentiviral
particles-containing medium was harvested after transfection for 48
h at 37°C and filtered using a 0.22-µm filter. The A549 cells were
seeded into 6-well culture plates, at a density of 5×105
cells per well. When the confluency of the cells was 50–60%, the
culture medium was replaced with the 2 ml lentiviral particles
mixed with 4 µl polybrene (Sigma-Aldrich; Merck KGaA) for 24 h.
Next, the cells were selected for stable integration with 4 µg
puromycin dihydrochloride for 2 weeks (2 µg/ml; Sigma-Aldrich;
Merck KGaA). After this process, the stable cells were used for all
further experiments and untransfected cells were used as a negative
control.
In vitro drug sensitivity assay
For the MTT assay, the cells were seeded into a
96-well plate (5,000 cells/well) and incubated at 37°C in a
humidified incubator with 5% CO2. After being cultured
for 24 h, the cells were treated with increasing concentrations of
gefitinib (0, 1, 2, 5, 12.5, 25 and 50 µM) for 48 h. The culture
medium was replaced with fresh medium containing MTT solution (0.5
mg/ml; Sigma-Aldrich; Merck KGaA). After an additional 4-h
incubation, the blue MTT-formazan product was dissolved in 200 µl
DMSO for 30 min. The absorbance of the formazan solution was read
spectrophotometrically at 570 nm. The 50% inhibitory concentration
(IC50) was calculated using GraphPad Prism v7.0 software
(GraphPad Software, Inc.).
Migration assays
A wound healing assay was performed to evaluate the
migration ability of the A549 cells. Briefly, after the cells had
been seeded in a 12-well plate (2×105 cells per well)
and cultured overnight, the 100% confluent cell monolayer was
sequentially scratched in a straight line with a 200-µl pipette
tip, washed with sterile PBS twice and cultured at 37°C in fresh
FBS-free medium for 24 h. Images of the migrated cells were
captured at 0 and 24 h under a light microscope (magnification,
×10) (Zeiss AG). The cell migration ability in each group was
calculated using ImageJ software [version 1.52a; National
Institutes of Health (NIH)].
The migration ability assay was performed according
to the manufacturer's procedures (Corning Inc.). Briefly, a total
of 5×104 A549 cells in 0.5 ml serum-free DMEM were
seeded onto a Transwell plate, with an 8-µm pore size polycarbonate
membrane. Next, 0.5 ml DMEM with 10% FBS was placed in the lower
chamber. After 24 h, the cells that had migrated to the lower
surface of the membranes were fixed with 4% paraformaldehyde for 30
min at room temperature and stained with crystal violet (0.5%; cat.
no. C0775; Sigma-Aldrich; Merck KGaA) for 2 h at room temperature.
The number of cells that had migrated to the lower side of the
membrane were counted using ImageJ software (version 1.52a;
NIH).
Colony formation
A549-shLuc and A549-shCNTN1-1 cells were seeded in a
6-well plate (1,000 cells/well) and treated with DMSO or 12.5 µM
gefitinib for 2 weeks. The cells were subsequently stained with
0.5% crystal violet for 2 h at room temperature. Quantification was
performed using ImageJ software (version 1.52a; NIH).
Cell cycle and apoptosis assay
For the cell cycle analysis, 2×105
A549-shLuc and A549-shCNTN1-1 cells were seeded in a 6-well plate.
The cells were subsequently harvested and washed with PBS, then
fixed with 70% ethanol for 30 min at room temperature. The fixed
cells were then stained with fresh propidium iodide (PI) containing
RNase A for 30 min at 37°C. Finally, a total of 1×104
cells were analyzed from each sample using a BD Accuri C6 flow
cytometer (BD Biosciences).
For apoptosis analysis, the transfected cells were
seeded in a 6-well plate (2×105 cells/well) and treated
with 12.5 µM gefitinib. After incubation for 48 h, the cells were
digested with trypsin, washed with cold PBS twice, resuspended in
500 µl binding buffer, then incubated with 5 µl FITC-Annexin V and
5 µl PI for 15 min at room temperature, in the dark. Flow cytometry
was performed using a BD Accuri C6 flow cytometer (BD Biosciences)
following the manufacturer's instructions, to detect cell apoptosis
by determining the relative amount of Annexin V-FITC-positive,
PI-negative cells or FITC-Annexin V-positive and PI-positive cells.
The cell cycle and apoptosis data analysis was performed using BD
Accuri C6 Software v.1.0.264.21 (BD Biosciences).
Western blot analysis
Total protein from A549-shLuc and A549-shCNTN1-1
cells was extracted using RIPA lysis buffer (Beyotime Institute of
Biotechnology) with a protease cocktail inhibitor (Roche). For
detecting Akt phosphorylation (Thr308 and Ser473), the cells were
harvested within 30 min after gefitinib or DMSO treatment to
monitor the rapid Akt pathway activation. Protein concentration
were measured using a BCA protein assay kit (Beyotime Institute of
Biotechnology). Total protein (20 µg/lane) was loaded and separated
by 10% SDS-PAGE and transferred to 0.45-µm PVDF membranes (EMD
Millipore). Membranes were blocked with 5% skimmed milk for 2 h at
room temperature. The membranes were then incubated with the
following specific monoclonal or polyclonal primary antibodies
overnight at 4°C: Anti-CNTN1 (1:200; cat. no. sc-136133; Santa Cruz
Biotechnology, Inc.), anti-Akt Ser473 phosphorylation (1:1,000;
cat. no. 4060; Cell Signaling Technology, Inc.), anti-Akt Thr308
phosphorylation (1:1,000; cat. no. 9275; Cell Signaling Technology,
Inc.), anti-Akt (1:1,000; cat. no. 9275; Cell Signaling Technology,
Inc.), anti-E-cadherin (1:200; cat. no. sc-8426; Santa Cruz
Biotechnology, Inc.), anti-N-cadherin (1:200; cat. no. sc-7939;
Santa Cruz Biotechnology, Inc.), anti-vimentin (1:200; cat. no.
sc-6260; Santa Cruz Biotechnology, Inc.) and anti-Actin (1:5,000;
cat. no. A5441; Sigma-Aldrich; Merck KGaA). Subsequently, the
membranes were incubated with the corresponding horseradish
peroxidase-conjugated anti-mouse IgG (1:10,000; cat. no. 7076; Cell
Signaling Technology, Inc.) or anti-rabbit IgG antibodies
(1:10,000; cat. no. 7074; Cell Signaling Technology, Inc.) for 1 h
at room temperature. Signal detection was performed using an
enhanced chemiluminescence kit (GE Healthcare) and photographed
using an imaging system (Epson). Finally, data were analyzed using
ImageJ software v.1.52a (National Institutes of Health).
Immunofluorescence staining
The cells were seeded on 8-well cell culture slides,
fixed with 4% paraformaldehyde for 30 min at room temperature and
blocked with 3% BSA (Beyotime Institute of Biotechnology) in PBS
for 2 h at room temperature. An anti-α-tubulin (cat. no. ab24610;
Abcam) antibody was added to the slides at a dilution of 1:200 and
incubated overnight at 4°C. The samples were then treated with an
Alexa Fluor-conjugated anti-mouse antibody (1:1,000; cat. no.
R37115; Thermo Fisher Scientific, Inc.) for 1 h protected from
light at room temperature. The nuclei were stained with DAPI
(Thermo Fisher Scientific, Inc.) for 15 min at room temperature.
Images were captured using a Zeiss fluorescence microscope
(magnification, ×20) (Zeiss AG).
Statistical analysis
All the data were obtained from three independent
experiments and presented as the mean ± standard deviation, with
between-group differences analyzed by multiple t-tests followed by
the Holm-Sidak correction test. P<0.05 was considered to
indicate a statistically significant difference. All statistical
analyses were performed using GraphPad Prism v7.0 (GraphPad
Software, Inc.).
Results
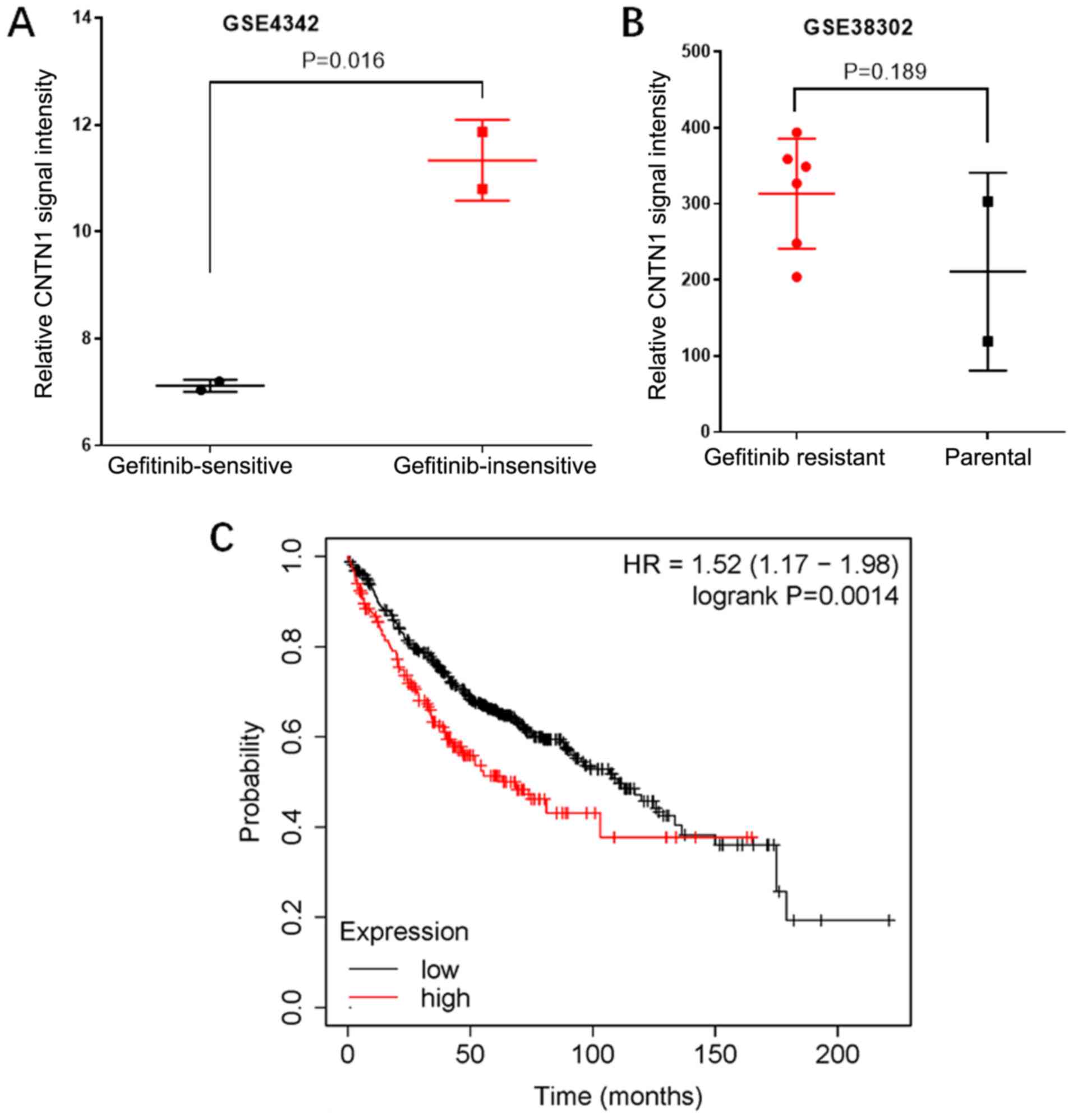
CNTN1 is abnormally upregulated in
gefitinib-insensitive and -resistant NSCLC
To investigate the role of CNTN1 in the gefitinib
resistance of NSCLC, the mRNA expression level of CNTN1 in
gefitinib-sensitive and -insensitive NSCLC was analyzed using data
from the GEO database. In the GSE4342 dataset, the mRNA expression
level of CNTN1 in gefitinib-insensitive lung cancer cells was
significantly higher compared with that in gefitinib-sensitive lung
cancer cells (P<0.05; Fig. 1A).
In addition, the mRNA expression level of CNTN1 in
gefitinib-acquired resistant cells was significantly higher
compared with that in the parental cells according to the GSE38302
data (P<0.05; Fig. 1B).
Furthermore, the prognostic value of CNTN1 was analyzed using the
Kaplan-Meier plotter database. The results showed that patients
with LUAD and high mRNA expression levels of CNTN1 showed reduced
overall survival times (Fig. 1C),
which indicated that CNTN1 was upregulated in gefitinib-resistant
and -insensitive NSCLC, and was associated with worse prognosis.
Collectively, the results from publicly available data suggested
that CNTN1 might play an important role in EGFR-TKI resistance.
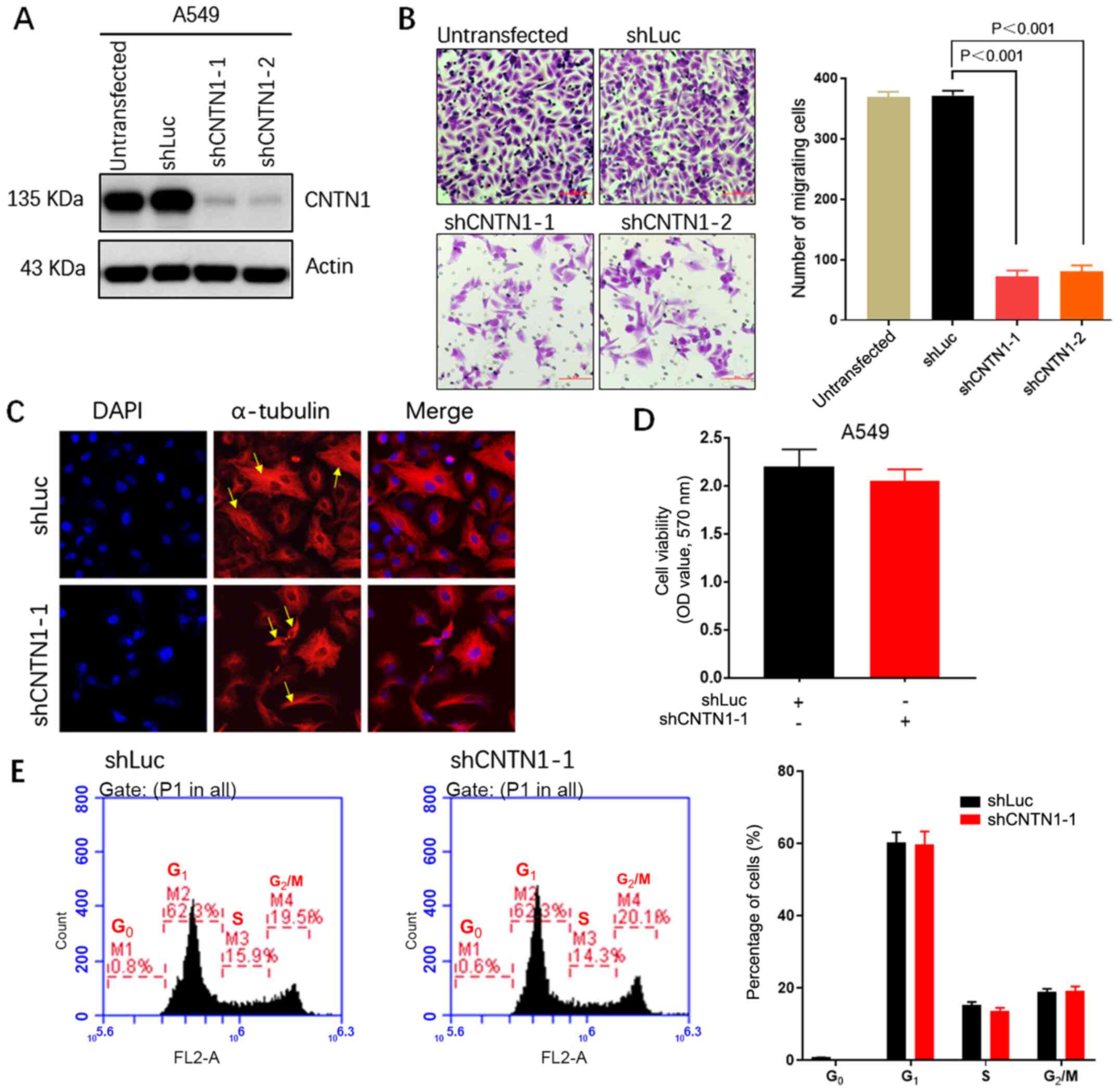
CNTN1 knockdown inhibits the migration
but not the proliferation ability of A549 cells
To further understand the function of CNTN1 in cell
migration, CNTN1 was silenced in the high-metastatic LUAD A549 cell
line using CNTN1-targeted shRNA. The western blot analysis showed
that shCNTN1-1 and shCNTN1-2 markedly inhibited CNTN1 protein
expression level (Fig. 2A).
shCNTN1-1 was selected for subsequent experimentation due shCNTN1-1
and shCNTN1-2 having comparable effects. Transwell analysis
confirmed that CNTN1 knockdown significantly decreased the
migration ability of the A549 cells compared with untransfected
A549 cells or A549 cells transfected with shLuc (Fig. 2B). To further understand the role of
CNTN1 in the arrangement of the cytoskeleton and in adhesion
structures which can affect cell motility, the A549-shLuc and
A549-shCNTN1-1 cells were stained with α-tubulin and characterized
using a fluorescent microscope. The A549-shLuc cells displayed
relaxing-formed α-tubulin-containing microtubules within the
cytoplasm and below the plasma membrane. However, the
A549-shCNTN1-1 cells displayed firm-formed microtubules (Fig. 2C). Accordingly, these findings
strongly suggested that CNTN1 could regulate the migration ability
of LUAD cells.
Next, the proliferation role of CNTN1 in the A549
cells transfected with shLuc or shCNTN1-1 was evaluated. MTT
analyses showed that knockdown of CNTN1 was not associated with
cell proliferation of A549 cells (Fig.
2D). Analysis of cell cycle distribution using flow cytometry
showed that CNTN1 knockdown had no significant effect on the cell
cycle in the A549 cells (Fig.
2E).
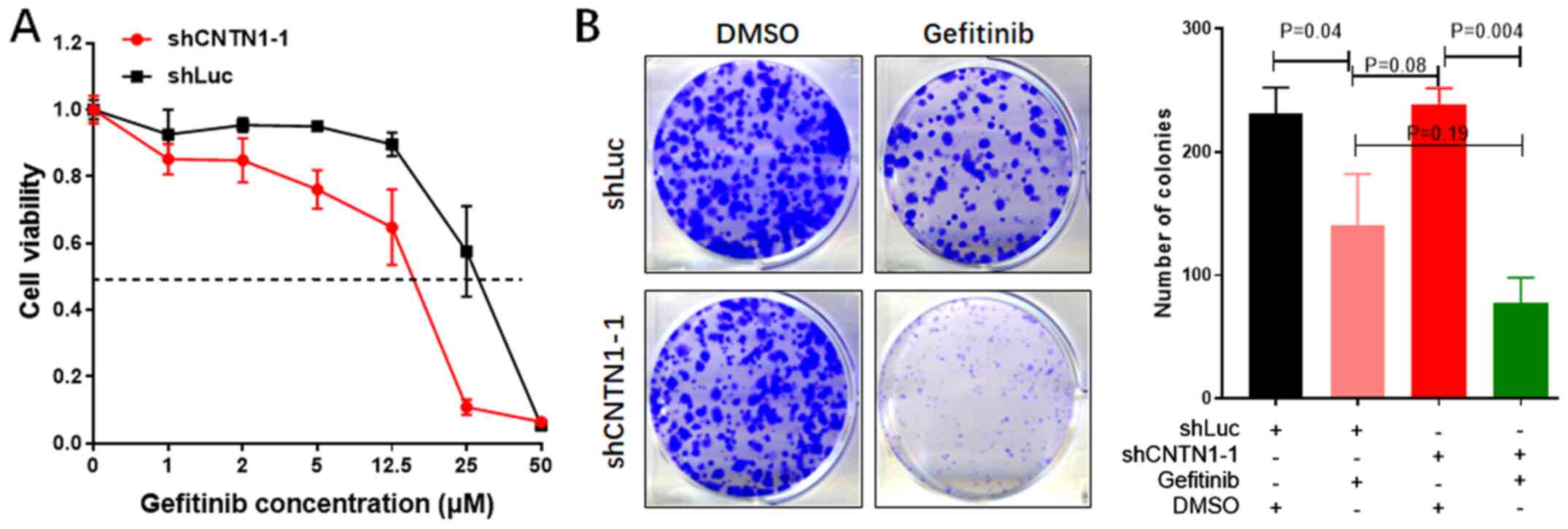
Knockdown of CNTN1 enhances gefitinib
sensitivity in the A549 cells
To further understand the association between CNTN1
expression level and gefitinib sensitivity, the IC50 of
the A549 cells transfected with shLuc or shCNTN1-1 was determined.
As shown in Fig. 3A, the
IC50 values of gefitinib in the A549-shCNTN1-1 and
A549-shLuc cells were 14.45 and 28.33 µM, respectively, confirming
that CNTN1 mRNA expression level could induce gefitinib resistance
in the A549 cells.
In addition, the colony formation assay showed that
CNTN1 knockdown of A549 treated with DMSO had no impact on colony
formation capacity compared with the shLuc group treated with DMSO.
However, the A549 cells transfected with shCNTN1-1 or shLuc were
treated with 12.5 µM gefitinib for 2 weeks and the numbers of
colonies derived from the CNTN1-knockdown cells were significantly
decreased compared with that in cells transfected with shLuc
(P<0.05; Fig. 3B). These results
indicated that knockdown of CNTN1 enhanced gefitinib sensitivity in
the A549 cells.
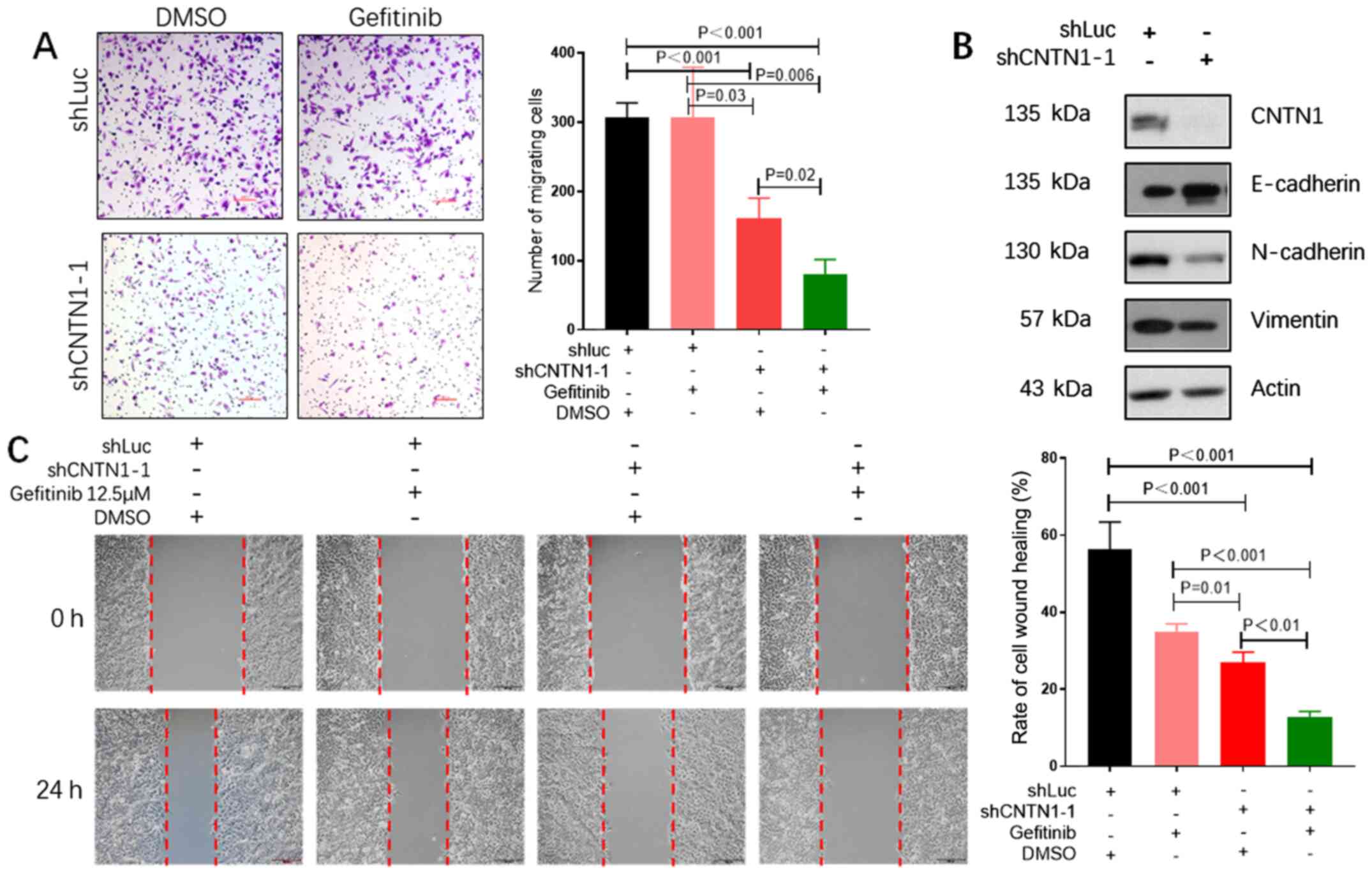
CNTN1 knockdown enhances the effect of
gefitinib on the migration of the A549 cells
As CNTN1 could promote cell migration in previous
studies (15,23), it was hypothesized that knockdown of
CNTN1 could enhance the effect of gefitinib on inhibition of the
cell migration abilities of A549 cells. Transwell and wound healing
assays were used to investigate the role of CNTN1 and gefitinib in
combination on A549 cell migration. The number of migrated cells
was significantly decreased in both knockdown of CNTN1 and
gefitinib-treated group compared with that in the shLuc group
treated with gefitinib (Fig. 4A).
The results were also confirmed by the wound healing assay, in
which the wound closure rate was significantly inhibited in A549
cells with knockdown of CNTN1 and gefitinib combination compared
with that in cells treated with shLuc and gefitinib combination
(Fig. 4C). Furthermore, western blot
analysis revealed that the protein expression level of the
epithelial marker, E-cadherin was increased, whereas the protein
expression level of the mesenchymal markers (N-cadherin and
vimentin) was notably decreased in the CNTN1-knockdown A549 cells
(Fig. 4B). Collectively, these
findings suggested that the EMT process was associated with
malignant behavior in A549 cells via CNTN1.
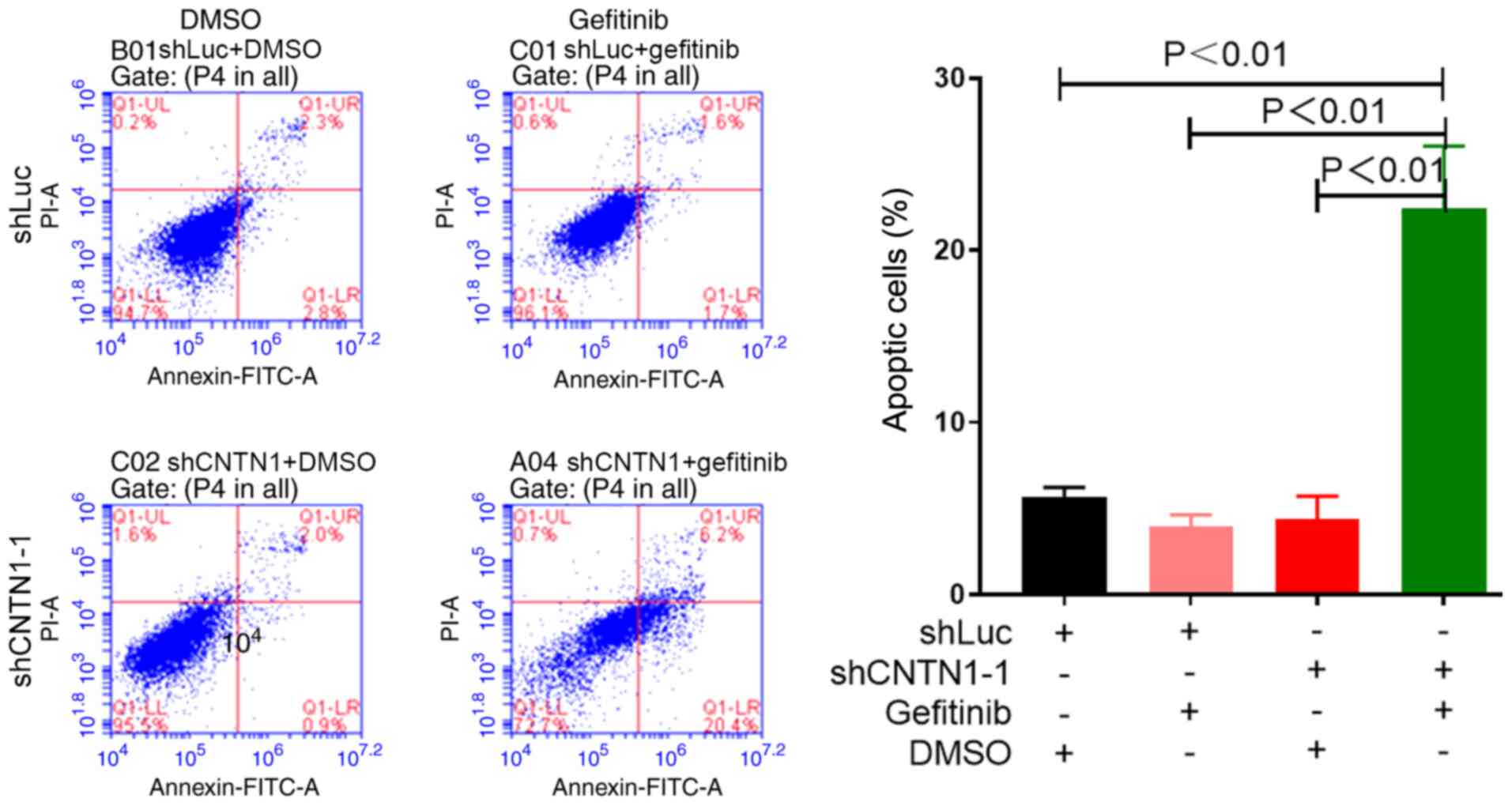
CNTN1 knockdown induces A549 cell
apoptosis in the presence of gefitinib
As shown in Fig. 3A,
MTT assay showed that knockdown of CNTN1 significantly improved
gefitinib sensitivity in the A549 cells. Therefore, to confirm that
CNTN1 knockdown inhibited the proliferation of the A549 cells in
the presence of gefitinib, cell apoptosis was analyzed using
Annexin V-FITC and PI double staining, followed by flow cytometry.
The apoptosis rate of A549-shLuc/DMSO, A549-shLuc/gefitinib,
A549-shCNTN1-1/DMSO and A549-shCNTN1-1/gefitinib cells was
5.29±1.22, 3.94±2.28, 4.32±1.82 and 22.47±3.03%, respectively. The
results showed that cell apoptosis increased by ~18% in A549 cells
transfected with shCNTN1-1 compared with that of cells transfected
with shLuc in the presence of 12.5 µM gefitinib (Fig. 5), indicating that cell apoptosis was
induced by CNTN1 knockdown.
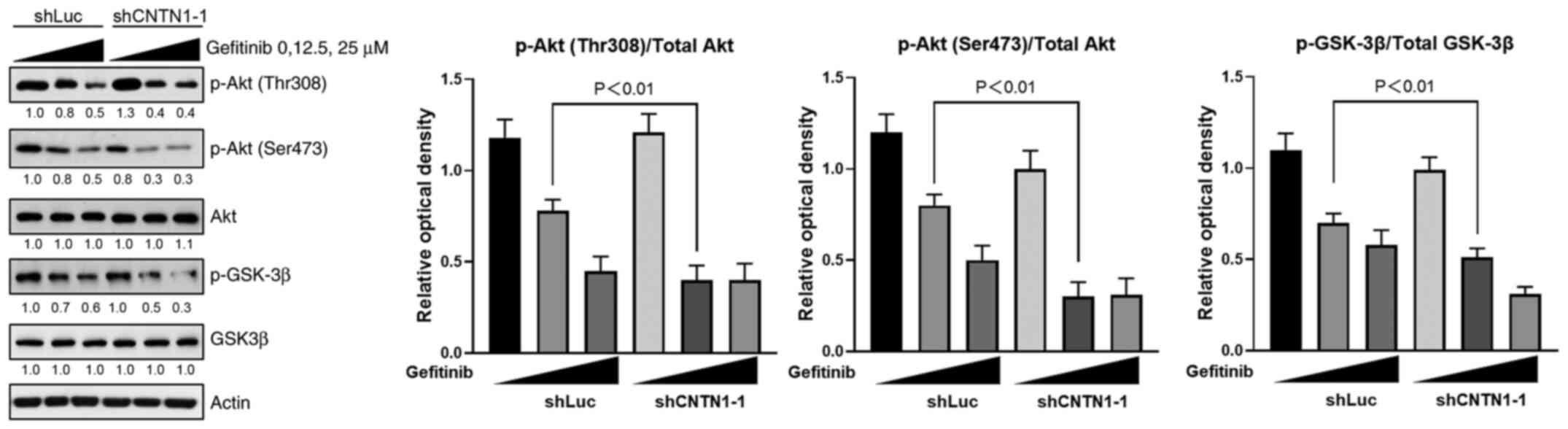
CNTN1 knockdown reverses gefitinib
resistance by inhibiting the PI3K/Akt signaling pathway
The PI3K/Akt pathway is not only a vital signaling
pathway in the regulation of cell survival and growth, but it is
also an efficient target to promote drug sensitivity in various
types of cancer cells (24). To
further investigate the mechanism underlying CNTN1-mediated
gefitinib resistance and the EMT phenotype, the expression level of
proteins in the PI3K/Akt signaling pathway was analyzed. As shown
in Fig. 6, western blot analysis
confirmed that knockdown of CNTN1 in the A549 cells did not affect
the protein expression levels of total Akt or GSK-3β. However,
there was a greater decrease in the protein expression level of
phosphorylated (p)Akt (T308 and S473) and p-GSK-3β in the
CNTN1-knockdown A549 cells treated with 12.5 µM gefitinib for 30
min compared with A549 transfected with shLuc and treated with 12.5
µM gefitinib for 30 min (Fig. 6).
Considering these results, it was concluded that the regulatory
effects of CNTN1 on gefitinib sensitivity and the EMT phenotype
could be partially attributed to the PI3K/Akt/GSK-3β signaling
pathway.
Discussion
Gefitinib, an EGFR-TKI, inhibits the intracellular
tyrosine kinase domain of EGFR by competitively binding with ATP;
thus, leading to the apoptosis of LUAD cells, and has been used as
a first-line drug for NSCLC (25).
However, the majority of patients develop acquired resistance to
gefitinib within 12 months, which is still a main obstacle for the
successful treatment of NSCLC (26).
Further investigation of the potential mechanisms of gefitinib
resistance may provide potential therapeutic strategies for
addressing this challenge (7,27,28).
Several studies have reported that CNTN1 not only facilitated the
migration and metastasis of cancer cells via EMT, but also promoted
drug resistance (12,15,19,23).
Therefore, identifying and confirming CNTN1 as a novel therapeutic
target associated with EGFR-TKIs resistance is important. The
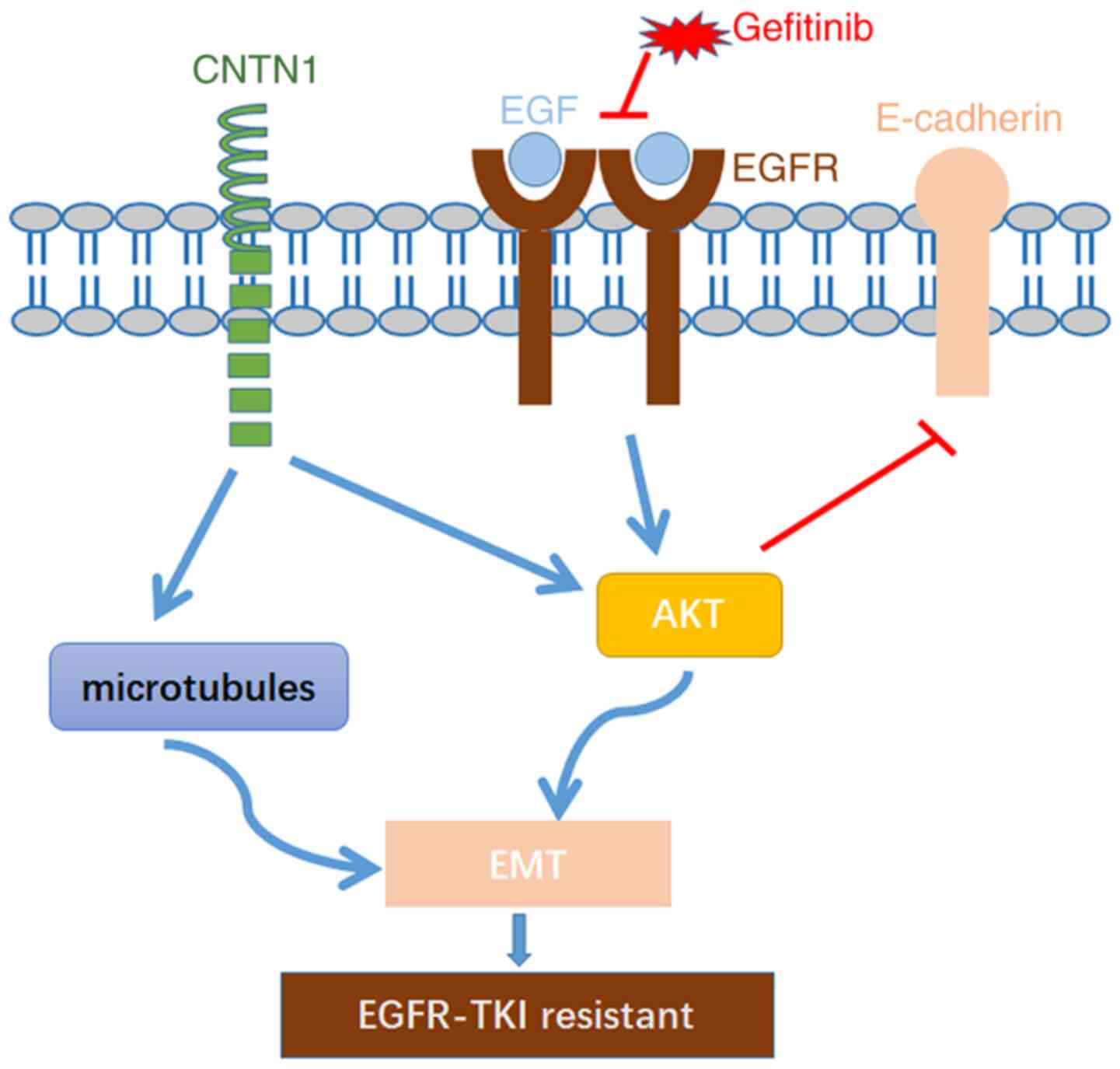
present study aimed to investigate silencing of CNTN1 enhance
gefitinib sensitivity by reversing EMT in A549 cell and the
mechanism of CNTN1 is described in Fig.
7. The current study found that knockdown of CNTN1 reversed
gefitinib resistance and EMT by inactivating the PI3K/Akt signaling
pathway and rearranging the cytoskeleton, suggesting that CNTN1 may
be a potential target to reverse gefitinib resistance in lung
adenocarcinoma A549 cell.
CNTN1, which is located on chromosome 12q11-q12, is
a novel member of the contactin sub-group of the immunoglobulin
superfamily (29). The expression
level and function of CNTN1 has been characterized in the nervous
system, and has been associated with the regulation of neurite
outgrowth, anchor neural cell adhesion and myelin organization
(30,31). Previous studies have demonstrated
that CNTN1 played a key role in cancer progression in various types
of cancer, including prostate cancer (19), esophageal squamous cell carcinoma
(32), oral squamous cell carcinoma
(33), hepatocellular carcinoma
(14), gastric cancer (13) and LUAD (34). Chen et al (13) reported that CNTN1 protein expression
level was increased in patients with primary gastric carcinoma
compared with that in adjacent normal gastric mucosa. Similarly, Su
et al (35) demonstrated that
the mRNA expression level of CNTN1 in the specimens from patients
with metastatic LUAD was significantly higher compared with that in
samples derived from patients with LUAD without metastasis. In
previous studies, CNTN1 promoted cisplatin resistance in
cisplatin-resistant lung cancer cells, and knockdown of CNTN1
reversed cisplatin resistance in LUAD (23,36).
Consistent with these prior findings, the results from the present
study showed that CNTN1 was increased in gefitinib-insensitive and
-resistant NSCLC, and knockdown of CNTN1 reversed gefitinib
resistance in the A549 cell line. Based on these observations, the
present study described the role of CNTN1 in gefitinib resistance
and partially investigated the mechanism of CNTN1-mediated EMT.
The EMT process has been associated with migration,
invasion and EGFR-TKI resistance in lung cancer. In the clinic, the
detection rate of the EMT phenotype was ~14% in patients with NSCLC
and EGFR-TKI resistance (8). Weng
et al (37) reported that
gefitinib-resistant cells exhibited an EMT phenotype, with a
decrease in E-cadherin protein expression and an increase in
vimentin protein expression, which led to resistant cells with
enhanced migration and invasion abilities. Furthermore, previous
studies showed that CNTN1 rearranged the actin cytoskeleton and
focal adhesion structures, and promoted cancer invasion and
metastasis (15,35). Overexpression of CNTN1 in the A549
cells promoted EMT progression, and enhanced migration, invasion
and cisplatin resistance compared with that in the control cells
(23). In the present study,
knockdown of CNTN1 reduced cell migration. Further investigation of
the mechanism involved indicated that CNTN1 rearranged the
cytoskeleton and adhesion structures via the microtubules to affect
cell motility. Furthermore, knockdown of CNTN1 upregulated
E-cadherin protein expression levels, and downregulated N-cadherin
and vimentin protein expression levels. Collectively, CNTN1
overexpression in the A549 cells may contribute to gefitinib
resistance and a more aggressive EMT phenotype.
The PI3K/Akt signaling pathway is a vital pathway,
that regulates cell proliferation, migration and invasion, as well
as drug resistance (24). Activation
of the Akt signaling pathway not only inhibited E-cadherin protein
expression, for facilitating EMT development, but also promoted
gefitinib resistance in NSCLC (23).
A previous study showed that Akt phosphorylation (Ser473) was
elevated within 5 min after serum stimulation in serum-starved
cells and that this was maintained for up to 30 min, which suggests
a very fast activation of Akt, which lasts for some time (38). Wang et al (39) showed that Akt phosphorylation (Thr308
and Ser473) increased in serum-starved cells who were subsequently
treated with EGF for 5 min or IGF-1 for 30 min. Han et al
(40) also demonstrated that Akt
phosphorylation (Thr308 and Ser473) was upregulated within 5 min,
after being induced by EGF. Therefore, Akt phosphorylation (Thr308
and Ser473) was detected within 30 min after gefitinib treatment to
monitor the rapid Akt pathway activation. However, previous studies
reported that Akt phosphorylation was also detectable 6, 24 and 48
h after gefitinib treatment (41,42).
There is a limitation on how long inhibition of to the present
study Akt activation after gefitinib treatment, as Akt
phosphorylation in cells treated with gefitinib for ≥1 h was not
investigated.
In the present study, CNTN1 knockdown together with
gefitinib treatment significantly inhibited the activation of the
PI3K/Akt signaling pathway in the A549 cells. Similar findings have
also been reported in lung cancer cell lines. For example, Zhang
et al (23) reported that
CNTN1 promoted cisplatin resistance in LUAD by inducing EMT
progression following activation of the PI3K/Akt signaling pathway.
Furthermore, overexpression of CNTN reduced E-cadherin protein
expression level to promote lung cancer metastasis, and was
associated with the activation of the PI3K/Akt signaling pathway
(17). Therefore, CNTN1 could
promote gefitinib resistance by activating the PI3K/Akt signaling
pathway.
In summary, the present study demonstrated that
CNTN1 plays an important role in EGFR-TKI resistance. Knockdown of
CNTN1 reversed the EMT phenotype and enhanced gefitinib sensitivity
in the A549 cells by inhibiting the activation of the PI3K/Akt
signaling pathway. Taken together, these results suggested that
CNTN1 may represent a potential therapeutic target for preventing
EGFR-TKI resistance in NSCLC.
Acknowledgements
Not applicable.
Funding
This study was financially supported by the National
Natural Science Foundation of China (grant no. 81902357), and the
Science and Technology Research Program of Chongqing University of
Arts and Sciences (grant no. 2017RBX10 and R2016BX09).
Availability of data and materials
The datasets used and/or analyzed in the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
CSH performed experimental work, data analysis,
manuscript preparation and editing. JHH assisted with data analysis
and acquired funding. DLY assisted with western blotting
experiments. CX assisted with bioinformatics analysis. ZGX assisted
with data analysis and manuscript preparation. HBT designed the
experiments, assisted with data analysis and edited the manuscript.
ZZC designed and supervised the experiments, edited the manuscript
and acquired funding. CSH and ZZC confirmed the authenticity of all
raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Xu Z, Yang Q, Chen X, Zheng L, Zhang L, Yu
Y, Chen M, You Q and Sun J: Clinical associations and prognostic
value of site-specific metastases in non-small cell lung cancer: A
population-based study. Oncol Lett. 17:5590–5600. 2019.PubMed/NCBI
|
|
2
|
Herbst RS, Morgensztern D and Boshoff C:
The biology and management of non-small cell lung cancer. Nature.
553:446–454. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cho BC, Chewaskulyong B, Lee KH,
Dechaphunkul A, Sriuranpong V, Imamura F, Nogami N, Kurata T,
Okamoto I, Zhou C, et al: Osimertinib versus standard of care EGFR
TKI as first-line treatment in patients with EGFRm advanced NSCLC:
FLAURA Asian subset. J Thorac Oncol. 14:99–106. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Han X, Luo R, Wang L, Zhang L, Wang T,
Zhao Y, Xiao S, Qiao N, Xu C, Ding L, et al: Potential predictive
value of serum targeted metabolites and concurrently mutated genes
for EGFR-TKI therapeutic efficacy in lung adenocarcinoma patients
with EGFR sensitizing mutations. Am J Cancer Res. 10:4266–4286.
2020.PubMed/NCBI
|
|
5
|
Nagasaka M, Zhu VW, Lim SM, Greco M, Wu F
and Ignatius Ou SH: Beyond osimertinib: The development of
third-generation EGFR tyrosine kinase inhibitors for advanced EGFR+
NSCLC. J Thorac Oncol. Dec 15–2020.(Epub ahead of print).
View Article : Google Scholar
|
|
6
|
Hirsch FR, Scagliotti GV, Mulshine JL,
Kwon R, Curran WJ Jr, Wu YL and Paz-Ares L: Lung cancer: Current
therapies and new targeted treatments. Lancet. 389:299–311. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hu C and Tan H: The E3 ubiquitin ligase
NEDD4 mediates EGFR-TKI acquired resistance in non-small cell lung
cancer. Int J Clin Exp Med. 12:12013–12019. 2019.
|
|
8
|
Sequist LV, Waltman BA, Dias-Santagata D,
Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger
S, Cosper AK, et al: Genotypic and histological evolution of lung
cancers acquiring resistance to EGFR inhibitors. Sci Transl Med.
3:75ra262011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yue J, Lv D, Wang C, Li L, Zhao Q, Chen H
and Xu L: Epigenetic silencing of miR-483-3p promotes acquired
gefitinib resistance and EMT in EGFR-mutant NSCLC by targeting
integrin β3. Oncogene. 37:4300–4312. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang X, Peng Y, Jiang X, Lu X, Duan W,
Zhang S, Dai N, Shan J, Feng Y, Li X, et al: The regulatory role of
APE1 in epithelial-to-mesenchymal transition and in determining
EGFR-TKI responsiveness in non-small-cell lung cancer. Cancer Med.
7:4406–4419. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lou W, Chen Y, Zhu KY, Deng H, Wu T and
Wang J: Polyphyllin I overcomes EMT-associated resistance to
erlotinib in lung cancer cells via IL-6/STAT3 pathway inhibition.
Biol Pharm Bull. 40:1306–1313. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang B, Yang X, Zhao T, Du H, Wang T,
Zhong S, Yang B and Li H: Upregulation of contactin-1 expression
promotes prostate cancer progression. Oncol Lett. 19:1611–1618.
2020.PubMed/NCBI
|
|
13
|
Chen DH, Yu JW, Wu JG, Wang SL and Jiang
BJ: Significances of contactin-1 expression in human gastric cancer
and knockdown of contactin-1 expression inhibits invasion and
metastasis of MKN45 gastric cancer cells. J Cancer Res Clin Oncol.
141:2109–2120. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li GY, Huang M, Pan TT and Jia WD:
Expression and prognostic significance of contactin 1 in human
hepatocellular carcinoma. Onco Targets Ther. 9:387–394. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen N, He S, Geng J, Song ZJ, Han PH, Qin
J, Zhao Z, Song YC, Wang HX and Dang CX: Overexpression of
contactin 1 promotes growth, migration and invasion in Hs578T
breast cancer cells. BMC Cell Biol. 19:52018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shi K, Xu D, Yang C, Wang L, Pan W, Zheng
C and Fan L: Contactin 1 as a potential biomarker promotes cell
proliferation and invasion in thyroid cancer. Int J Clin Exp
Pathol. 8:12473–12481. 2015.PubMed/NCBI
|
|
17
|
Yan J, Wong N, Hung C, Chen WX and Tang D:
Contactin-1 reduces E-cadherin expression via activating AKT in
lung cancer. PLoS One. 8:e654632013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wu HM, Cao W, Ye D, Ren GX, Wu YN and Guo
W: Contactin 1 (CNTN1) expression associates with regional lymph
node metastasis and is a novel predictor of prognosis in patients
with oral squamous cell carcinoma. Mol Med Rep. 6:265–270.
2012.PubMed/NCBI
|
|
19
|
Yan J, Ojo D, Kapoor A, Lin X, Pinthus JH,
Aziz T, Bismar TA, Wei F, Wong N, De Melo J, et al: Neural cell
adhesion protein CNTN1 promotes the metastatic progression of
prostate cancer. Cancer Res. 76:1603–1614. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Coldren CD, Helfrich BA, Witta SE, Sugita
M, Lapadat R, Zeng C, Barón A, Franklin WA, Hirsch FR, Geraci MW
and Bunn PA Jr: Baseline gene expression predicts sensitivity to
gefitinib in non-small cell lung cancer cell lines. Mol Cancer Res.
4:521–528. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Terai H, Soejima K, Yasuda H, Nakayama S,
Hamamoto J, Arai D, Ishioka K, Ohgino K, Ikemura S, Sato T, et al:
Activation of the FGF2-FGFR1 autocrine pathway: A novel mechanism
of acquired resistance to gefitinib in NSCLC. Mol Cancer Res.
11:759–767. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gyorffy B, Surowiak P, Budczies J and
Lanczky A: Online survival analysis software to assess the
prognostic value of biomarkers using transcriptomic data in
non-small-cell lung cancer. PLoS One. 8:e822412013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang R, Sun S, Ji F, Liu C, Lin H, Xie L,
Yang H, Tang W, Zhou Y, Xu J and Li P: CNTN-1 enhances
chemoresistance in human lung adenocarcinoma through induction of
epithelial-mesenchymal transition by targeting the PI3K/Akt
pathway. Cell Physiol Biochem. 43:465–480. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Song M, Bode AM, Dong Z and Lee MH: AKT as
a therapeutic target for cancer. Cancer Res. 79:1019–1031. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu Q, Yu S, Zhao W, Qin S, Chu Q and Wu
K: EGFR-TKIs resistance via EGFR-independent signaling pathways.
Mol Cancer. 17:532018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Maemondo M, Inoue A, Kobayashi K, Sugawara
S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I,
et al: Gefitinib or chemotherapy for non-small-cell lung cancer
with mutated EGFR. N Engl J Med. 362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lei Y, Guo W, Chen B, Chen L, Gong J and
Li W: Tumorreleased lncRNA H19 promotes gefitinib resistance via
packaging into exosomes in nonsmall cell lung cancer. Oncol Rep.
40:3438–3446. 2018.PubMed/NCBI
|
|
28
|
Cho JH, You YM, Yeom YI, Lee DC, Kim BK,
Won M, Cho BC, Kang M, Park S, Yang SJ, et al: RNF25 promotes
gefitinib resistance in EGFR-mutant NSCLC cells by inducing
NF-kB-mediated ERK reactivation. Cell Death Dis. 9:5872018.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Berglund EO and Ranscht B: Molecular
cloning and in situ localization of the human contactin gene
(CNTN1) on chromosome 12q11-q12. Genomics. 21:571–582. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Haenisch C, Diekmann H, Klinger M,
Gennarini G, Kuwada JY and Stuermer CA: The neuronal growth and
regeneration associated Cntn1 (F3/F11/Contactin) gene is duplicated
in fish: Expression during development and retinal axon
regeneration. Mol Cell Neurosci. 28:361–374. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mohebiany AN, Harroch S and Bouyain S: New
insights into the roles of the contactin cell adhesion molecules in
neural development. Adv Neurobiol. 8:165–194. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu P, Chen S, Wu W, Liu B, Shen W, Wang
F, He X and Zhang S: Contactin-1 (CNTN-1) overexpression is
correlated with advanced clinical stage and lymph node metastasis
in oesophageal squamous cell carcinomas. Jap J Clin Oncol.
42:612–618. 2012. View Article : Google Scholar
|
|
33
|
Shigetomi S, Imanishi Y, Shibata K, Sakai
N, Sakamoto K, Fujii R, Habu N, Otsuka K, Sato Y, Watanabe Y, et
al: VEGF-C/Flt-4 axis in tumor cells contributes to the progression
of oral squamous cell carcinoma via upregulating VEGF-C itself and
contactin-1 in an autocrine manner. Am J Cancer Res. 8:2046–2063.
2018.PubMed/NCBI
|
|
34
|
Su JL, Yang PC, Shih JY, Yang CY, Wei LH,
Hsieh CY, Chou CH, Jeng YM, Wang MY, Chang KJ, et al: The
VEGF-C/Flt-4 axis promotes invasion and metastasis of cancer cells.
Cancer Cell. 9:209–223. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Su JL, Yang CY, Shih JY, Wei LH, Hsieh CY,
Jeng YM, Wang MY, Yang PC and Kuo ML: Knockdown of contactin-1
expression suppresses invasion and metastasis of lung
adenocarcinoma. Cancer Res. 66:2553–2561. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang R, Yao W, Qian P, Li Y, Jiang C, Ao
Z, Qian G, Wang C, Wu G and Li J: Increased sensitivity of human
lung adenocarcinoma cells to cisplatin associated with
downregulated contactin-1. Biomed Pharmacother. 71:172–184. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Weng CH, Chen LY, Lin YC, Shih JY, Lin YC,
Tseng RY, Chiu AC, Yeh YH, Liu C, Lin YT, et al:
Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per
se is a common mechanism for acquired resistance to EGFR TKI.
Oncogene. 38:455–468. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Abkhezr M, Keramati AR, Ostad SN, Davoodi
J and Ghahremani MH: The time course of Akt and ERK activation on
XIAP expression in HEK 293 cell line. Mol Biol Rep. 37:2037–2042.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang G, Long J, Gao Y, Zhang W, Han F, Xu
C, Sun L, Yang SC, Lan J, Hou Z, et al: SETDB1-mediated methylation
of Akt promotes its K63-linked ubiquitination and activation
leading to tumorigenesis. Nat Cell Biol. 21:214–225. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Han F, Li CF, Cai Z, Zhang X, Jin G, Zhang
WN, Xu C, Wang CY, Morrow J, Zhang S, et al: The critical role of
AMPK in driving Akt activation under stress, tumorigenesis and drug
resistance. Nat Commun. 9:47282018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang Y, Zhang W, Wen L, Yang H, Wen M, Yun
Y, Zhao L, Zhu X, Tian L, Luo E, et al: FOXM1 confers resistance to
gefitinib in lung adenocarcinoma via a MET/AKT-dependent positive
feedback loop. Oncotarget. 7:59245–59259. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang J, Qu Z, Yao H, Sun L, Harata-Lee Y,
Cui J, Aung TN, Liu X, You R, Wang W, et al: An effective drug
sensitizing agent increases gefitinib treatment by down regulating
PI3K/Akt/mTOR pathway and up regulating autophagy in non-small cell
lung cancer. Biomed Pharmacother. 118:1091692019. View Article : Google Scholar : PubMed/NCBI
|