Introduction
Metastasis is responsible for ~90% of cancer-related
deaths (1). Metastasis is a
multi-step process that begins with the acquisition of migratory
ability by tumor cells (1).
Therefore, the suppression of tumor cell migration may represent an
efficient strategy for the inhibition of cancer metastasis.
Inflammation is a critical factor for tumor malignancy, including
cancer cell migration and tumor metastasis (2–5). The
transcription factor nuclear factor-κB (NF-κB) serves an essential
role in inflammatory signaling (3).
Upon a stimulation by inflammatory cytokines, such as tumor
necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), transforming
growth factor β-activated kinase 1 (TAK1) is phosphorylated and
induces the activation of IκB kinases (IKKs) (4). IKKs subsequently phosphorylate p65, the
major component of NF-κB, and IκBα, the inhibitor of p65. At the
same time, phosphorylated IκBα is ubiquitinated and transferred to
the ubiquitin-proteasome system for degradation (4). Phosphorylated p65 then translocates
from the cytosol into the nucleus to activate the transcription of
genes that promote cell migration, such as matrix
metalloproteinases and epithelial-to-mesenchymal
transition-inducible genes (3–5).
Therefore, the suppression of p65 activation may be an effective
strategy for inhibiting cancer metastasis.
Erythropoietin-producing hepatocellular receptor A2
(EphA2) belongs to the receptor tyrosine kinase family and
maintains intercellular adhesion in normal cells (6,7).
However, it is upregulated in various types of malignant tumors,
such as lung and colorectal cancer, glioblastoma and melanoma,
particularly in metastatic tumors (6,7).
Previous studies have demonstrated that ligand- and tyrosine
kinase-independent EphA2 phosphorylation at S897, which is induced
by the activation of p90 ribosomal S6 kinase (RSK), controls cancer
cell migration (6,8). In addition, the RSK-EphA2 axis has been
implicated in the poor survival of patients with lung cancer,
suggesting its potential as a novel molecular target for
pharmacological interventions (8).
Ginseng is the rhizome of plants in the genus
Panax, such as South China ginseng (P. notoginseng),
Korean ginseng (P. ginseng) and American ginseng (P.
quinquefolius), and is traditionally used to treat various
diseases, such as inflammation, lung and colon cancer (9–11).
Steaming and heating processes have often been adopted to alter or
enhance the pharmacological activities of various natural
medicines; steamed or heat-processed ginseng has frequently been
used in the treatment of tumors (12), inflammation (13) and stress-related conditions (14,15).
Certain compounds in processed ginseng, including ginsenoside Rh2
and Rg5, appear to exert inhibitory effects against cancer cell
migration (12,16). Ginsenosides, also termed ginseng
saponins, are the main bioactive ingredients of ginseng (15). Various ginsenosides have been
reported to exert antimetastatic (17) and anti-inflammatory (16,18)
effects. However, sufficiently detailed research on ginsenosides,
particularly processed ginsenosides, at the molecular level has not
been performed to date. The present study screened ginsenosides
that exerted inhibitory effects on NF-κB activity and identified
ginsenoside Rg5. The aim of the present study was to elucidate the
molecular mechanisms of ginsenoside Rg5 and determine whether it
could block the migration of A549 cells.
Materials and methods
Antibodies and reagents
Antibodies against phospho-p65 (S536; cat. no.
3033), phospho-ERK (T202/Y204; cat. no. 4370), phospho-RSK (S380;
cat. no. 11989), phospho-TAK1 (T187; cat. no. 4536), phospho-IKKα/β
(S176/177; cat. no. 2697), phospho-EphA2 (S897; cat. no. 6347),
phospho-EphA2 (Y577; cat. no. 12677), TAK1 (D94D7; cat. no. 5206),
TAK1 binding protein 1 (TAB1) (C25E9; cat. no. 3226), TAB2 (C88H10;
cat. no. 3745), IKKβ (D30C6; cat. no. 8943), ERK1/2 (137F5; cat.
no. 4695), EphA2 (D4A2; cat. no. 6997) and β-actin (D6A8; cat. no.
8457) were purchased from Cell Signaling Technology, Inc.
Antibodies against p65 (C-20; cat. no. sc-372), IKKα (H-744; cat.
no. sc-7218), IκBα (C-21; cat. no. sc-371), RSK1 (C-21; cat. no.
sc-231), RSK2 (C-19; cat. no. sc-1430) and Lamin B1 (8D1; cat. no.
sc-56144) were obtained from Santa Cruz Biotechnology, Inc.
Recombinant human TNF-α was obtained from R&D Systems, Inc.,
and recombinant human IL-1β from PeproTech, Inc. Ginsenoside Rg5
was purchased from Biopurify Phytochemicals Ltd. and dissolved in
dimethyl sulfoxide (Sigma-Aldrich; Merck KGaA). Cycloheximide,
MG-132 and bafilomycin A1 were purchased from Sigma-Aldrich; Merck
KGaA, and the cells were treated for 2–6 h at 37°C.
Cell culture and treatment
HeLa, A549 and 293T cells were purchased from
Shanghai Fan Shuo Biological Technology, and 293 cells were
obtained from ATCC. These cells were maintained in high-glucose
Dulbecco's modified Eagle's medium (Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin
and 100 µg/ml streptomycin at 37°C in 5% CO2. A549 cells
were cultured in RPMI-1640 medium (Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS, 100 U/ml penicillin and 100 µg/ml
streptomycin at 37°C in 5% CO2.
Cells were stimulated with recombinant human TNF-α
(10 ng/ml) or IL-1β (10 ng/ml) for 24 h at 37°C with 5%
CO2. Ginsenoside Rg5 or an equal volume of DMSO was
added for 30 min before the TNF-α or IL-1β stimulation.
Luciferase assay
HeLa cells were transferred with a luciferase
reporter plasmid provided by Professor Hiroaki Sakurai (University
of Toyama, Toyama, Japan) under the control of four sites
containing a neomycin resistance gene (19). A stable clone was isolated in DMEM
containing 500 µg/ml G418 (Thermo Fisher Scientific, Inc.). The
transfected cells (5×104) were seeded in a 96-well plate
and stimulated with TNF-α for 6 h at 37°C with 5% CO2.
Luciferase activity was measured using a Dual-Luciferase Reporter
Assay system (Promega Corporation). The experiments were conducted
in triplicate.
Immunoblotting
Whole-cell lysates of HeLa, A549, 293 and 293T cells
were prepared using a whole-cell lysate buffer [1.0 M HEPES-NaOH,
pH 7.7; 0.3 M NaCl; 0.1 M MgCl2; 0.5 M EDTA, pH 8.0; 10%
Triton X-100; and protease/phosphatase inhibitor cocktails (Roche
Diagnostics)]. The concentration of lysates was measured by BCA
assay (Thermo Fisher Scientific, Inc.), and each sample was
adjusted to the same concentration. Each sample was mixed with an
equal volume of SDS-PAGE sample buffer (195 mM Tris-HCl, pH 6.8; 3%
SDS; 15% DTT; 30% glycerol; and 0.10% bromophenol blue) and heated
at 95°C for 5 min. The samples (5–20 ng) were separated by 7.5 or
10% SDS-PAGE and transferred to Immobilon-P nylon membranes (EMD
Millipore). The membranes were treated with SuperBlock (Thermo
Fisher Scientific, Inc.) or BlockAce (KAC Co., Ltd.) overnight at
4°C and probed with primary antibodies (dilution, 1:1,000-2,000)
for 2 h at room temperature. The antibodies were detected using a
horseradish peroxidase-conjugated anti-rabbit, anti-mouse or
anti-goat secondary antibody (dilution, 1:5,000; cat. nos. 7074 and
7076; Cell Signaling Technology, Inc.; and cat. no. PA1-28664;
Thermo Fisher Scientific, Inc.) for 1 h at room temperature and
visualized by Immobilon Western Chemiluminescent HRP Substrate (EMD
Millipore) on Amersham Imager 600 (Cytiva) or X-ray film. Analyses
were performed using Adobe Photoshop CC (Ver. 14.2.1, Adobe, Inc.)
or ImageJ (Ver. 1.52; National Institutes of Health) and GraphPad
Prism 7 (Ver. 7.0.0.159; GraphPad Software, Inc.) at least three
times, and representative images are presented.
Thermal shift assay
Samples of HeLa, A549 or 293T cells were prepared as
described previously (20) and
analyzed by immunoblotting as aforementioned.
Transfection of plasmid DNA
293T and 293 cells at 50% confluency were
transfected using Effectene Transfection Reagent (Qiagen, Inc.) and
Lipofectamine® 2000 reagent (Thermo Fisher Scientific,
Inc.), respectively, according to the manufacturer's instructions.
Expression vectors for human TAK1, TAB1, TAB2, EphA2, RSK1 and p65
were provided by Professor Hiroaki Sakurai (University of Toyama,
Toyama, Japan). The amount of plasmid DNA was 0.5 ng. After 24-h
incubation at 37°C with 5% CO2, the transfected cells
were used in subsequent experiments. Successful transfection was
determined by immunoblotting.
Reverse transcription-quantitative
(RT-q)PCR analysis
Total RNAs were extracted from HeLa and A549 cells
using RNA Faster200 reagent (Shanghai Fastagen Biotechnology Co.,
Ltd.) according to the manufacturer's instructions. RNA was
reverse-transcribed to generate first-strand cDNAs using the
PrimeScript™ RT Master Mix Kit (Takara Bio, Inc.); the reaction
conditions were 37°C for 15 min and 85°C for 5 sec. The qPCR
analysis was performed using the SYBR® Premix Ex Taq™
(Tli RNaseH Plus) Kit (Takara Bio, Inc.) according to the
manufacturer's instructions on a 6000 Real-Time PCR System (Thermo
Fisher Scientific, Inc.), and the relative expression levels were
quantified using the 2−ΔΔCq method (21). The cycle threshold values of the
target genes were normalized to those of GAPDH from the same
sample. The primer sequences were as follows: EPHA2 forward,
5′-CCATCCATCCTGTGTCA-3′ and reverse, 5′-TCGCTGCTTCTCTGTGT-3′; and
GAPDH forward, 5′-GGGAAGGTGAAGGTCGGAGT-3′ and reverse,
5′-GGGGTCATTGATGGCAACA-3′.
Wound healing assay
A549 or 293 cells were cultured in a 3.5 cm dish
until they formed a monolayer, which was scratched using a 200-µl
pipette tip. Cells were washed to remove cellular debris, fresh
medium with 10% FBS was added, and the cells were allowed to
migrate for 24 h at 37°C with 5% CO2. Due to the high
toxicity of ginsenoside Rg5 and the low cell viability in presence
of lower amounts of FBS, the assays were performed using complete
medium. Images were captured by an Olympus CKX53 (magnification,
×10; Olympus Corporation) or a Nikon Diaphot (magnification, ×4;
Nikon Instruments, Inc.) phase contrast microscope at the same
position within the wound region at 0 and 24 h, and the distance of
cell migration was measured using ImageJ. Analyses were performed
at least three times and representative results are shown.
Migration assay
A migration assay was performed using Transwell
chambers (Costar; Corning, Inc.), and the lower surface was
pre-coated with 1.25 µg fibronectin for 2 at room temperature
(Sigma-Aldrich; Merck KGaA). A549 cells (2×105
cells/well) pre-treated with ginsenoside Rg5 and TNF-α or IL-1β
were added to the upper compartment of the chamber. Following 8-h
incubation at 37°C with 5% CO2, the cells were fixed
with 100% methanol for 1 min at room temperature and stained with
hematoxylin and eosin (3 min at room temperature). The non-migrated
cells were removed by a cotton swab. Images of the migrated cells
were captured in five fields by an Olympus CKX53 microscope
(magnification, ×10) and counted manually.
Cell viability assay
A549 cells (2×104 cells/well) treated
with ginsenoside Rg5, and subsequently incubated at 37°C with 5%
CO2. After 24 h, cells were subjected to Cell Counting
Kit-8 (Dojindo Molecular Technologies, Inc.) according to the
manufacturer's instructions (incubated at 37°C for 30 min).
Absorbance at 450 nm was assessed using a microplate reader.
Statistical analysis
Data are presented as the mean ± SD. Statistical
analyses were performed using GraphPad Prism 7 software (GraphPad
Software, Inc.). The differences between two groups were analyzed
by Student's t-test. Differences among multiple groups were
analyzed by one-way ANOVA with Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Ginsenoside Rg5 suppresses
TNF-α-induced p65 activation
Ginsenosides were screened to identify compounds
that exert inhibitory effects on p65 activation using HeLa cells
stably transfected with a p65-dependent reporter plasmid (data not
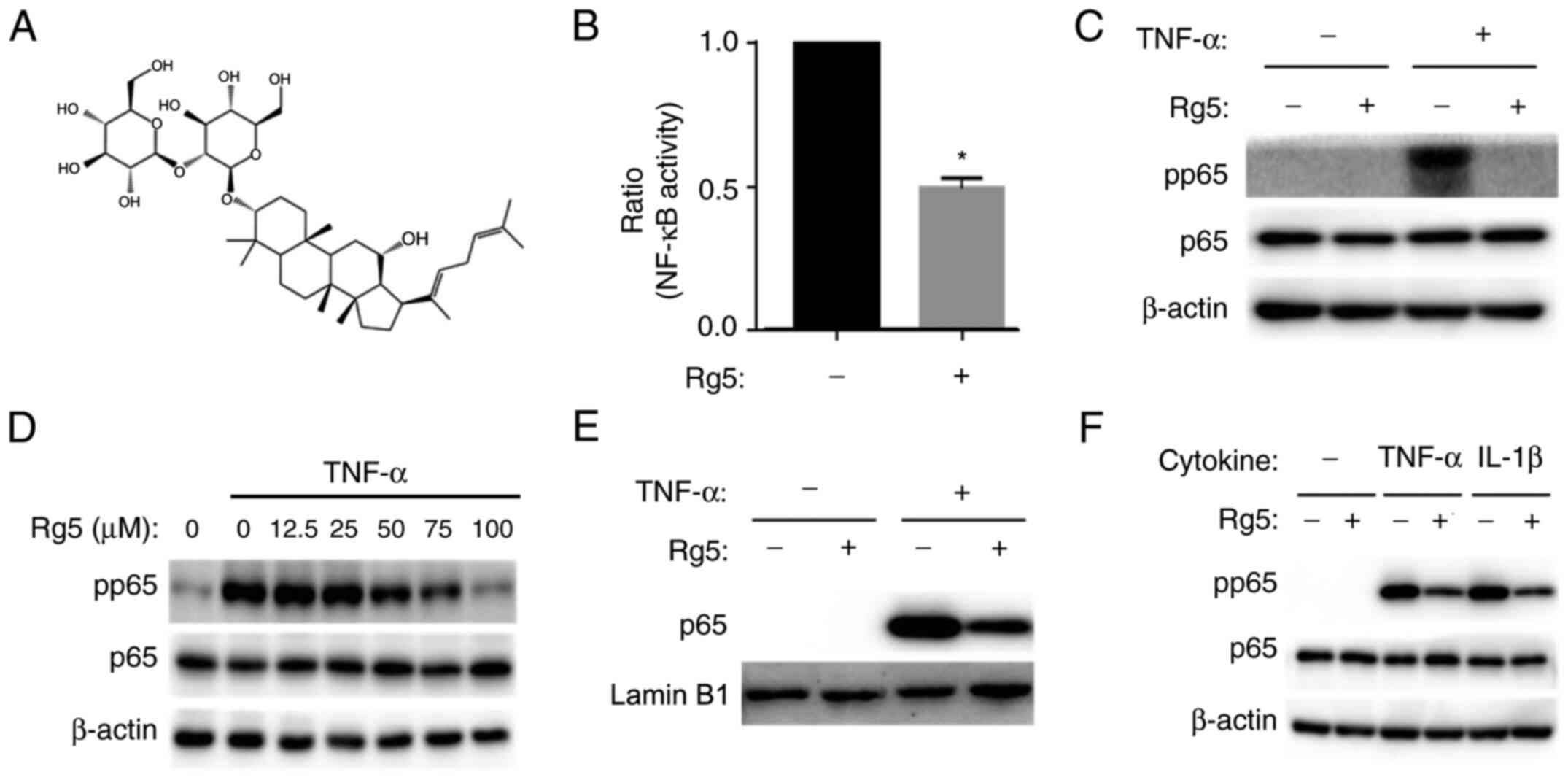
shown), and the results demonstrated that ginsenoside Rg5 (Fig. 1A) inhibited TNF-α-induced p65
activation compared with that in untreated cells (Fig. 1B). Following TNF-α stimulation, p65
is phosphorylated and transferred to the nucleus (3,5). As
presented in Fig. 1C-E, the
phosphorylation of p65 (Figs. 1C and
D, and S1A and B) and its
nuclear translocation (Figs. 1E and
S1C) were inhibited by the
ginsenoside Rg5 pretreatment. Ginsenoside Rg5 inhibited not only
the TNF-α-, but also the IL-1β-induced phosphorylation of p65 in
A549 cells (Figs. 1F, S1D, S2 and
S3). These results suggested that
ginsenoside Rg5 suppressed the activation of p65 by inhibiting the
phosphorylation of p65 and nuclear translocation of p65
independently of cell and cytokine types.
Ginsenoside Rg5 inhibits TAK1
phosphorylation
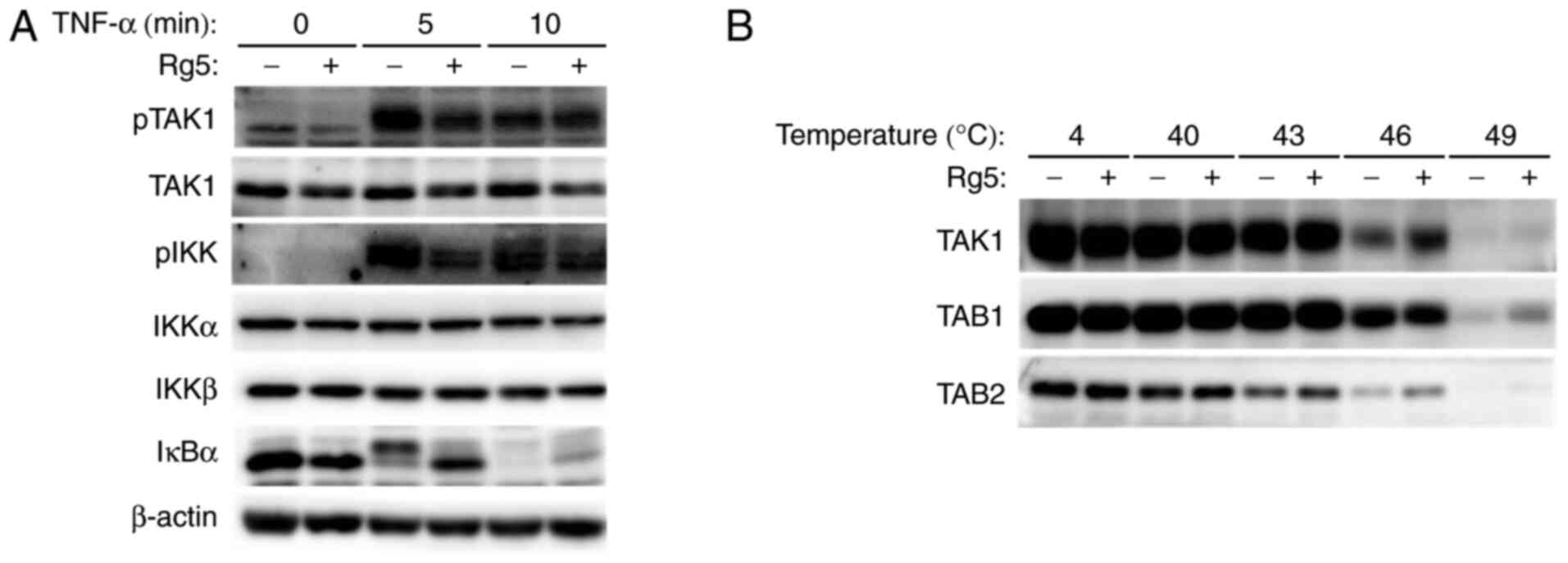
To elucidate the molecular mechanism by which
ginsenoside Rg5 inhibits the activation of p65, the phosphorylation
levels of the upstream molecules of p65, namely TAK1, IKK and IκBα,
were evaluated (Figs. 2A and
S4A-C). TAK1 and IKK were
phosphorylated 5 min post-TNF-α stimulation, whereas ginsenoside
Rg5 moderately inhibited the phosphorylation of these kinases. A
band shift in IκBα, which indicated the phosphorylation of IκBα,
occurred after 5 min and completely degraded after 10 min;
ginsenoside Rg5 inhibited the phosphorylation and degradation of
IκBα (Figs. 2A and S4A-C). Similar results were obtained in
the TNF-α- or IL-1β-treated A549 cells (Figs. S5A and B, and S6A and B). These results demonstrated that
ginsenoside Rg5 inhibited the phosphorylation of TAK1 to suppress
the activation of p65. TAK1 is stabilized by binding with its
adaptor proteins TAB1 and TAB2; to investigate the potential
interactions between ginsenoside Rg5 and the TAK1 complex, the
present study used a cellular thermal shift assay, which identifies
compound-protein interactions based on increased stability of the
complex compared with that of a single protein at the expected
denaturation temperature, resulting in higher protein expression
levels in the complex-forming sample compared with those in the
non-forming sample at a specific temperature (20). As presented in Figs. 2B and S4D-F, denaturation of TAK1 was observed at
46°C, and TAK1 was mostly denatured at 49°C. At 46–49°C, the
expression levels of TAK1 were higher in ginsenoside Rg5-treated
cells compared with those in untreated cells, suggesting that
ginsenoside Rg5 bound to the TAK1 complex. The expression levels of
TAB1 and TAB2 were also higher in ginsenoside Rg5-treated cells
compared with those in untreated cells at 49°C and 43–46°C,
respectively. Similar results were obtained for A549 cells
(Figs. S5C and S6C-E) or TAK1-, TAB1- or
TAB2-overexpressing 293T cells (Figs.
S5D and E and S6F-H).
Therefore, these results suggested that ginsenoside Rg5 directly
interacted with the TAK1 complex to inhibit TAK1 phosphorylation
and suppress its downstream NF-κB signaling.
Ginsenoside Rg5 inhibits the
phosphorylation of EphA2 at S897
The inhibition of NF-κB signaling represents one
strategy for suppressing cancer metastasis; however, our previous
study demonstrated that TNF-α-induced activation of the RSK-EphA2
pathway promoted cell motility (8).
Therefore, the effects of ginsenoside Rg5 on the RSK-EphA2 pathway
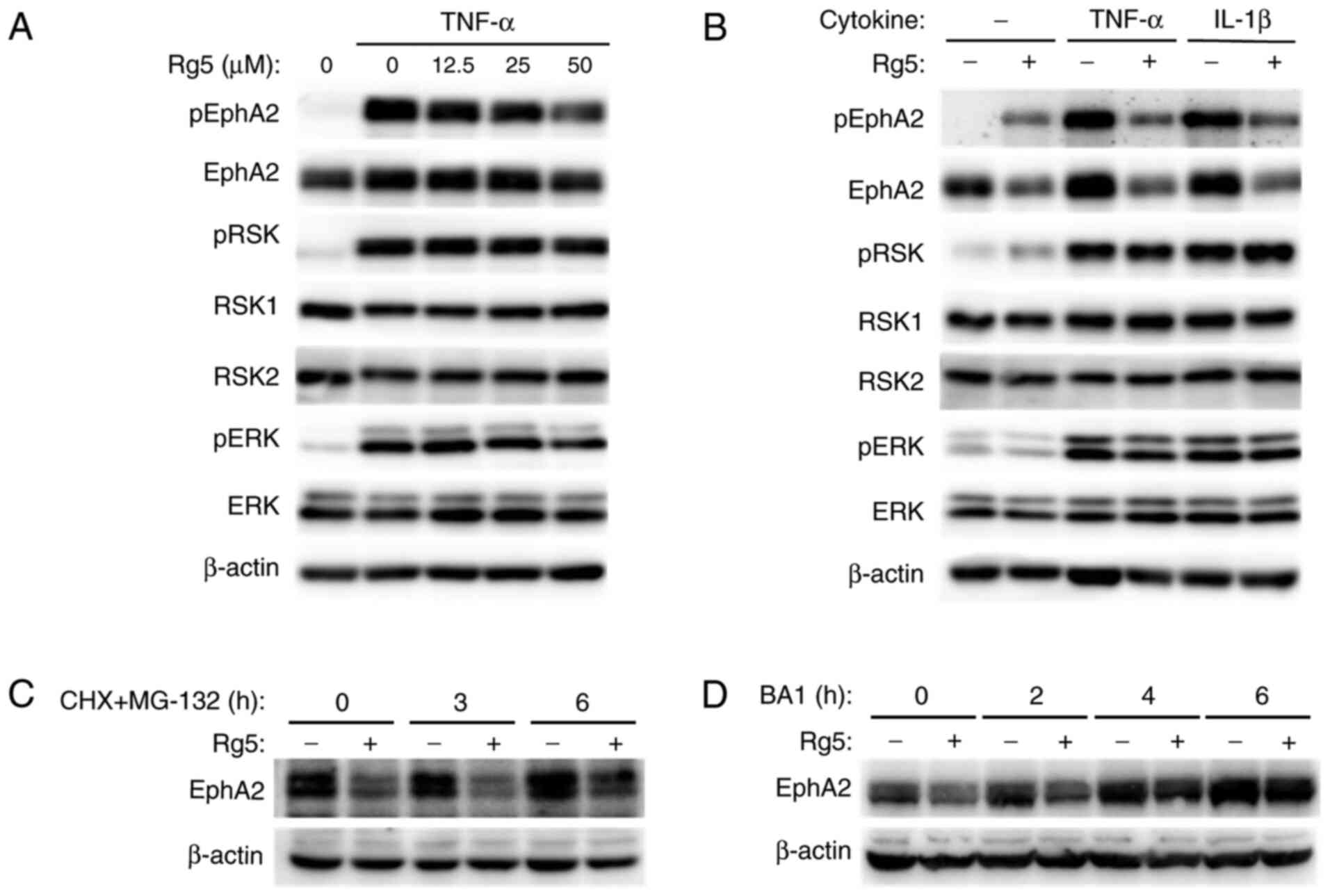
were further investigated. As demonstrated in Figs. 3A and S7A, 50 µM ginsenoside Rg5 inhibited the
levels of TNF-α-induced S897 phosphorylation compared with those in
the DMSO-treated cells. However, the phosphorylation of RSK and its
upstream kinase ERK was not suppressed by ginsenoside Rg5. The
expression levels of EphA2 were slightly suppressed by treatment
with ginsenoside Rg5 compared with those in the DMSO group
(Figs. 3A and S7B). These effects were also observed in
TNF-α- and IL-1β-stimulated A549 cells (Figs. 3B, S7C-E, S8A and
B and S9A-F). To elucidate the
molecular mechanisms underlying the suppression of EphA2 by
ginsenoside Rg5, the mRNA levels of EphA2 were determined in HeLa
(Fig. S8C) and A549 (Fig. S8D) cells, and the results
demonstrated that they were not reduced by ginsenoside Rg5
treatment compared with those in the DMSO-treated cells. A previous
study reported that the EphA2 ligand-mediated tyrosine
phosphorylation of EphA2 promoted EphA2 endocytosis to induce the
proteasomal degradation of EphA2 (6). As demonstrated in Figs. S8E and F, and S9G and H, EphA2 phosphorylation at Tyr-588
was not induced by ginsenoside Rg5. Protein degradation is mediated
through the ubiquitin/proteasome or lysosomal system. To establish
whether ginsenoside Rg5 reduces EphA2 protein expression by these
two pathways, HeLa and A549 cells were pre-treated with the
proteasome inhibitor MG-132 and protein synthesis inhibitor
cycloheximide or a specific inhibitor of vacuolar-type H(+)-ATPase
to block lysosomal trafficking bafilomycin A1. Co-treatment with
MG-132 and cycloheximide did not inhibit the reduction of EphA2
expression levels in HeLa compared with that in the ginsenoside
Rg5-treated cells (Figs. 3C and
S7F) or A549 (Figs. S8G and S9I) cells, indicating that the suppression
of EphA2 expression was not dependent on the ubiquitin/proteasome
system. By contrast, following pre-treatment with bafilomycin A1
and treatment with ginsenoside Rg5 in HeLa (Figs. 3D and S7G) or A549 (Figs. S8H and S9J) cells, the reduction of EphA2
expression in ginsenoside Rg5-treated cells was gradually
inhibited, and it was completely inhibited following 6 h
pre-treatment. Therefore, these data suggested that ginsenoside Rg5
induced the degradation of EphA2 through the lysosomal system, and,
as a result, ginsenoside Rg5 inhibited the phosphorylation of EphA2
at S897.
Ginsenoside Rg5 inhibits cell
migration
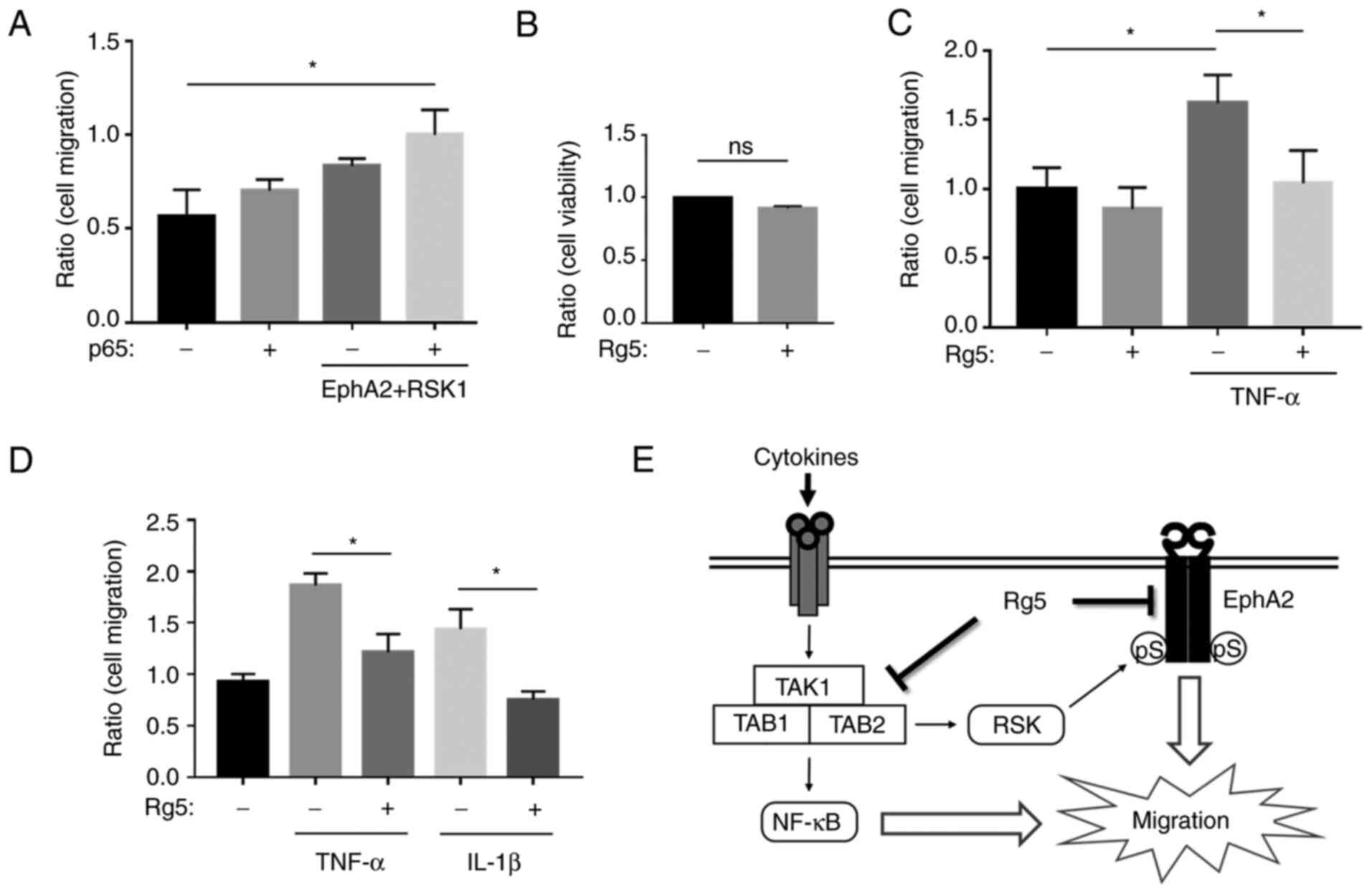
The NF-κB and RSK-EphA2 pathways have been reported
to induce cell migration (3,5,8). We
hypothesized that the activation of the NF-κB and RSK-EphA2
pathways may exert addictive effects on cell migration. To analyze
this, p65-, RSK- and EphA2-expressing plasmids were transfected
into 293 cells, and cell migration was detected by wound healing
assay. The expression levels of p65, RSK1 and EphA2 are presented
in Fig. S10A. As presented in
Figs. 4A and S10B, cell migration appeared to be
promoted in the p65- and EphA2+RSK1-transfected cells compared with
that in the control group. In addition, migration was strongly
promoted in both p65- and EphA2+RSK1 co-overexpressing cells
compared with that in the control, p65 and EphA2+RSK1 single
transfection groups. To clarify whether NF-κB and RSK-EphA2
signaling induced by inflammatory cytokines promoted cell
migration, and whether ginsenoside Rg5 inhibited these effects,
A549 cells were pre-treated with ginsenoside Rg5 and stimulated
with TNF-α. TNF-α significantly promoted cell migration, whereas
ginsenoside Rg5 pre-treatment suppressed TNF-α-induced cell
migration without affecting cell viability (Figs. 4B and C, and S10C). Similarly, a migration assay using
Transwell chambers revealed that ginsenoside Rg5 inhibited TNF-α-
and IL-1β- induced cell migration (Figs.
4D and S10D). These results
demonstrated that the activation of p65 and the RSK-EphA2 signaling
pathway exerted additive effects on cell migration, whereas
ginsenoside Rg5 attenuated cell migration by inhibiting NF-κB and
EphA2 signaling.
Discussion
The results of the present study demonstrated that
ginsenoside Rg5 inhibited p65 and EphA2 phosphorylation by
suppressing TAK1 activity and EphA2 expression levels,
respectively, compared with those in untreated cells. In addition,
the NF-κB and RSK-EphA2 pathways exerted additive effects on cell
migration. To the best of our knowledge, the present study is the
first study to directly demonstrate these effects, suggesting that
the inhibition of these two pathways may be crucial for abolishing
cancer migration. The present results suggest that ginsenoside Rg5
may be a powerful compound for inhibiting cancer migration.
Previous studies have reported that ginsenoside Rg5
inhibits the NF-κB signaling pathway. Lee et al (17) have demonstrated that ginsenoside Rg5
exerts anti-inflammatory effects by inhibiting the phosphorylation
of NF-κB in BV2 cells, and Kim et al (18) reported that ginsenoside Rg5
suppresses the phosphorylation of NF-κB and translocation of p65
into the nucleus in breast cancer cells with inflammation. However,
the molecular mechanisms by which ginsenoside Rg5 inhibits the
NF-κB pathway currently remain unclear. The results of the present
study demonstrated that ginsenoside Rg5 inhibited NF-κB signaling
by suppressing the activation of TAK1. Furthermore, these results
demonstrated that ginsenoside Rg5 directly bound to the TAK1
complex. TAK1 serves multiple functions in inflammation and is
associated with a number diseases, such as tumors and diabetes
(4). The present study not only
revealed the effects of ginsenoside Rg5 on the TAK1 complex, but
also provided an important strategy for the development of TAK1
inhibitors; however, the structural properties of ginsenoside Rg5
binding the TAK1 complex was not elucidated. Further studies are
needed to determine the 3-dimensional structure of the
TAK1/TAB1/TAB2 protein bound to ginsenoside Rg5 using X-ray
crystallography to validate these results in order to develop
specific TAK1 inhibitors for clinical applications.
Accumulating evidence has demonstrated the
importance of EphA2 expression and phosphorylation at S897 in tumor
malignancy, including tumor metastasis, the properties of cancer
stem cells and antitumor drug resistance (6,7).
Regarding the relationship between the NF-κB and EphA2 pathways,
Hong et al (22) have
reported that the expression of EphA2 and its ligand Ephrin-A1 is
induced in mice with lipopolysaccharide-induced lung injury, and an
EphA2 monoclonal antibody inhibits the activation of NF-κB as well
as AKT, SRC proto-oncogene and ribosomal protein S6 kinase B1 in a
mouse model. By contrast, Funk et al (23) have suggested that Ephrin-A1 does not
affect the activation of the NF-κB signaling pathway in human
aortic endothelial cells. The present study also attempted to
clarify the potential crosstalk between the NF-κB and EphA2
signaling pathways. In the cells used in the present study,
TNF-α-induced NF-κB phosphorylation was not affected by the
knockdown of EphA2 (data not shown). Therefore, there appeared not
to be any overlap between NF-κB and EphA2 signaling in these cells.
However, these experiments were limited and may have not provide
enough data to fully elucidate this. The signaling cascade is
complex and exhibits different reactions in various cell types or
stimuli; therefore, to determine the relationship between the NF-κB
and EphA2 signaling pathways, further evidence is needed, for
instance, by using EphA2 and p65 knockout cell lines to analyze the
two signaling cascades.
The results of the present study suggested that the
induction of RSK phosphorylation was promoted by ginsenoside Rg5
(Figs. 3A and B). A previous study
has reported that ginsenoside Rg5 induces the activation of
insulin-like growth factor-1 receptor (IGF-1R) and promotes the
phosphorylation of ERK in human umbilical vein endothelial cells
(24). HeLa and A549 cells also
express IGF-1R (25). Therefore
ginsenoside Rg5 may also induce the phosphorylation of ERK by
activating IGF-1R in these cells. In A549 cells harboring a KRAS
mutation, the basal level of ERK phosphorylation is high (26). Furthermore, in the present study, the
induction of ERK phosphorylation by ginsenoside Rg5 was weak (data
not shown); difficulties were associated with detecting the
induction of ERK phosphorylation in A549 cells. Therefore, its
induction in A549 cells treated with ginsenoside Rg5 was not clear
(Fig. 3B). In the present study, the
induction of ERK phosphorylation following ginsenoside Rg5
treatment was detected in HeLa cells (data not shown). Therefore,
ginsenoside Rg5 may induce the activation of IGF-1R and ERK,
followed by the phosphorylation of its downstream kinase RSK.
To the best of our knowledge, the present study was
the first to demonstrate that ginsenoside Rg5 suppressed the
expression of EphA2 by promoting its degradation using the
lysosomal system. Although other ginsenosides that have been
demonstrated to exert anti-inflammatory effects were also tested in
the present study (data not shown), only ginsenoside Rg5 promoted
the degradation of EphA2. In addition, the expression of other
receptor tyrosine kinases, such as epidermal growth factor
receptor, was detected in ginsenoside Rg5-treated cells (data not
shown), and the results revealed that their expression was not
reduced, suggesting that ginsenoside Rg5 specifically induced the
degradation of EphA2. Previous studies have reported that the
ubiquitin/proteasome system degrades specific proteins; by
contrast, the lysosomal system performs non-specific protein
degradation (27,28). Therefore, further studies are needed
to fully elucidate the molecular mechanisms of action of
ginsenoside Rg5 on the lysosomal system.
Ginseng and its main components ginsenosides have
been reported to exert antimetastatic effects; however, further
evidence is needed to elucidate the underlying mechanisms. Although
in vivo studies are required, the results of the present
study provide a scientific basis for the potential development of
ginsenoside Rg5 as an antimetastatic drug.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Professor Hiroaki
Sakurai (University of Toyama, Toyama, Japan) for providing the
transfection plasmids.
Funding
This work was supported by the National Natural
Science Foundation of China (grant nos. 81603156 and 81920108033),
the Young Eastern Scholar Program (grant no. QD2016038), the
Chenguang Program (grant no. 16CG49), the Shanghai Sailing Program
(grant no. 18YF1421700) and the MSD Life Science Foundation, Public
Interest Incorporated Foundation.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding authors on reasonable
request.
Authors' contributions
LS conducted the experiments and wrote the
manuscript. ZW and LY designed the study. FY and YZ designed the
study, conducted the experiments and wrote the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mehlen P and Puisieux A: Metastasis: A
question of life or death. Nat Rev Cancer. 6:449–458. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Crusz SM and Balkwill FR: Inflammation and
cancer: Advances and new agents. Nat Rev Clin Oncol. 12:584–596.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Karin M: Nuclear factor-kappaB in cancer
development and progression. Nature. 441:431–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sakurai H: Targeting of TAK1 in
inflammatory disorders and cancer. Trends Pharmacol Sci.
33:522–530. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bharti AC and Aggarwal BB: Nuclear
factor-kappa B and cancer: Its role in prevention and therapy.
Biochem Pharmacol. 64:883–888. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou Y and Sakurai H: Emerging and diverse
functions of the EphA2 noncanonical pathway in cancer progression.
Biol Pharm Bull. 40:1616–1624. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wykosky J and Debinski W: The EphA2
receptor and ephrinA1 ligand in solid tumors: Function and
therapeutic targeting. Mol Cancer Res. 6:1795–1806. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou Y, Yamada N, Tanaka T, Hori T,
Yokoyama S, Hayakawa Y, Yano S, Fukuoka J, Koizumi K and Sakurai H:
Crucial roles of RSK in cell motility by catalyzing serine
phosphorylation of EphA2. Nat Commun. 6:76792015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Song H and Lee YJ: Inhibition of
hypoxia-induced cyclooxygenase-2 by Korean Red Ginseng is dependent
on peroxisome proliferator-activated receptor gamma. J Ginseng Res.
41:240–246. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kee JY, Han YH, Mun JG, Park SH, Jeon HD
and Hong SH: Effect of Korean Red Ginseng extract on colorectal
lung metastasis through inhibiting the epithelial-mesenchymal
transition via transforming growth
factor-β1/Smad-signaling-mediated Snail/E-cadherin expression. J
Ginseng Res. 43:68–76. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Poudyal D, Cui X, Le PM, Hofseth AB,
Windust A, Nagarkatti M, Nagarkatti PS, Schetter AJ, Harris CC and
Hofseth LJ: A key role of microRNA-29b for the suppression of colon
cancer cell migration by American ginseng. PLoS One. 8:e750342013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gao Q and Zheng J: Ginsenoside Rh2
inhibits prostate cancer cell growth through suppression of
microRNA-4295 that activates CDKN1A. Cell Prolif. 51:e124382018.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jang KJ, Choi SH, Yu GJ, Hong SH, Chung
YH, Kim CH, Yoon HM, Kim GY, Kim BW and Choi YH: Anti-inflammatory
potential of total saponins derived from the roots of Panax
ginseng in lipopolysaccharide-activated RAW 264.7 macrophages.
Exp Ther Med. 11:1109–1115. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mao H, Jiang C, Xu L, Chen D, Liu H, Xu Y,
Ma K and Wang M: Ginsenoside protects against AKI via activation of
HIF-1α and VEGF-A in the kidney-brain axis. Int J Mol Med.
45:939–946. 2020.PubMed/NCBI
|
|
15
|
Nguyen NH and Nguyen CT: Pharmacological
effects of ginseng on infectious diseases. Inflammopharmacology.
27:871–883. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liang LD, He T, Du TW, Fan YG, Chen DS and
Wang Y: Ginsenoside-Rg5 induces apoptosis and DNA damage in human
cervical cancer cells. Mol Med Rep. 11:940–946. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee YY, Park JS, Jung JS, Kim DH and Kim
HS: Anti-inflammatory effect of ginsenoside Rg5 in
lipopolysaccharide-stimulated BV2 microglial cells. Int J Mol Sci.
14:9820–9833. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim SJ and Kim AK: Anti-breast cancer
activity of Fine Black ginseng (Panax ginseng Meyer) and
ginsenoside Rg5. J Ginseng Res. 39:125–134. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Waiwut P, Shin MS, Inujima A, Zhou Y,
Koizumi K, Saiki I and Sakurai H: Gomisin N enhances TNF-α-induced
apoptosis via inhibition of the NF-κB and EGFR survival pathways.
Mol Cell Biochem. 350:169–175. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jafari R, Almqvist H, Axelsson H,
Ignatushchenko M, Lundbäck T, Nordlund P and Martinez Molina D: The
cellular thermal shift assay for evaluating drug target
interactions in cells. Nat Protoc. 9:2100–2122. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hong JY, Shin MH, Douglas IS, Chung KS,
Kim EY, Jung JY, Kang YA, Kim SK, Chang J, Kim YS and Park MS:
Inhibition of EphA2/EphrinA1 signal attenuates
lipopolysaccharide-induced lung injury. Clin Sci (Lond).
130:1993–2003. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Funk SD, Finney AC, Yurdagul A Jr,
Pattillo CB and Orr AW: EphA2 stimulates VCAM-1 expression through
calcium-dependent NFAT1 activity. Cell Signal. 49:30–38. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cho YL, Hur SM, Kim JY, Kim JH, Lee DK,
Choe J, Won MH, Ha KS, Jeoung D, Han S, et al: Specific activation
of insulin-like growth factor-1 receptor by ginsenoside Rg5
promotes angiogenesis and vasorelaxation. J Biol Chem. 290:467–477.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
The Human Protein Atlas. Available from, .
https://www.proteinatlas.org/.
|
|
26
|
The Catalogue Of Somatic Mutations In
Cancer (COSMIC). COSMIC v92, released 27-AUG-20, . https://cancer.sanger.ac.uk/cosmic
|
|
27
|
Kwon YT and Ciechanover A: The ubiquitin
code in the ubiquitin-proteasome system and autophagy. Trends
Biochem Sci. 42:873–886. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ganley IG: Autophagosome maturation and
lysosomal fusion. Essays Biochem. 55:65–78. 2013. View Article : Google Scholar : PubMed/NCBI
|