Introduction
Environmental risk factors, such as cigarette
smoking and asbestos, lead to increased risk of lung cancer. Before
the 20th century, incidences of lung cancer were very rare, up to
1898, only 140 cases of lung cancer were reported in the world
medical literature (1). Since the
start of tobacco usage, the morbidity and mortality rates of lung
cancer have been gradually rising (2,3), and
according to WHO statistics, lung cancer has the highest incidence
and mortality rate among the 36 most common cancers in the world in
2018 (3). Studies have shown that
smoking is directly related to lung cancer (4). According to the statistical data from
the World Health Organization, lung cancer was the leading cause of
cancer-associated death in 2018; currently there are 300 million
tobacco users worldwide, and there are 8 million deaths every year
(3).
Multiple studies have shown that
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) is the most
potent carcinogen in tobacco that causes lung cancer (5,6). NNK can
induce DNA strand breaks and DNA adduct formation, while its
metabolism results in the generation of hydroxyl and other reactive
oxygen radica9 ls, which in turn causes lung cancer (7). A study by Yeh et al (8) demonstrated that the incubation of A549
lung cancer cells with NNK results in increased levels of reactive
oxygen species (ROS) formation (8).
Furthermore, it has been shown that NNK induces oxidative stress by
increasing ROS level in cells and promotes lung cancer progression,
which may be associated with the changes in the expression of the
genes related to ROS metabolism (9).
The antioxidant defense system in mammalian cells
prevents excessive ROS accumulation (10) and maintains the intracellular redox
balance. The peroxiredoxin (Prdx) family of proteins function in
the cellular oxidative defense system, which eliminates ROS
(11) and affects various cellular
activities, such as cell proliferation, differentiation (12), apoptosis (13) and gene expression (14). Prdx1 inhibits NNK-induced DNA damage
and prevents the development of lung tumors (15–17).
NNK-induced changes in the expression of peroxide redox proteins in
lung cancer cells indicates that Prdx1 may be involved in the
detoxification of ROS during NNK-induced oxidative stress (17). Hence, Prdx1 protects the cells, DNA
and proteins from NNK-induced damage, and thus development of lung
cancer. The interplay between NNK and Prdx1 has recently gained
attention (12,16–18), and
an improved understanding of the role of Prdx1 in the development
of NNK-induced lung cancer may shed new light towards the
development of therapeutic strategies against lung cancer.
NNK
NNK, an aromatic compound, is the most potent
carcinogen in tobacco smoke (19).
In multiple organs, the nicotine in tobacco is rapidly metabolized
by cytochrome (CY) P450. In the liver, it is hydroxylated by
CYP2A26 at the 2′position to form an amino ketone intermediate,
which is subsequently nitrosated to produce NNK (20).
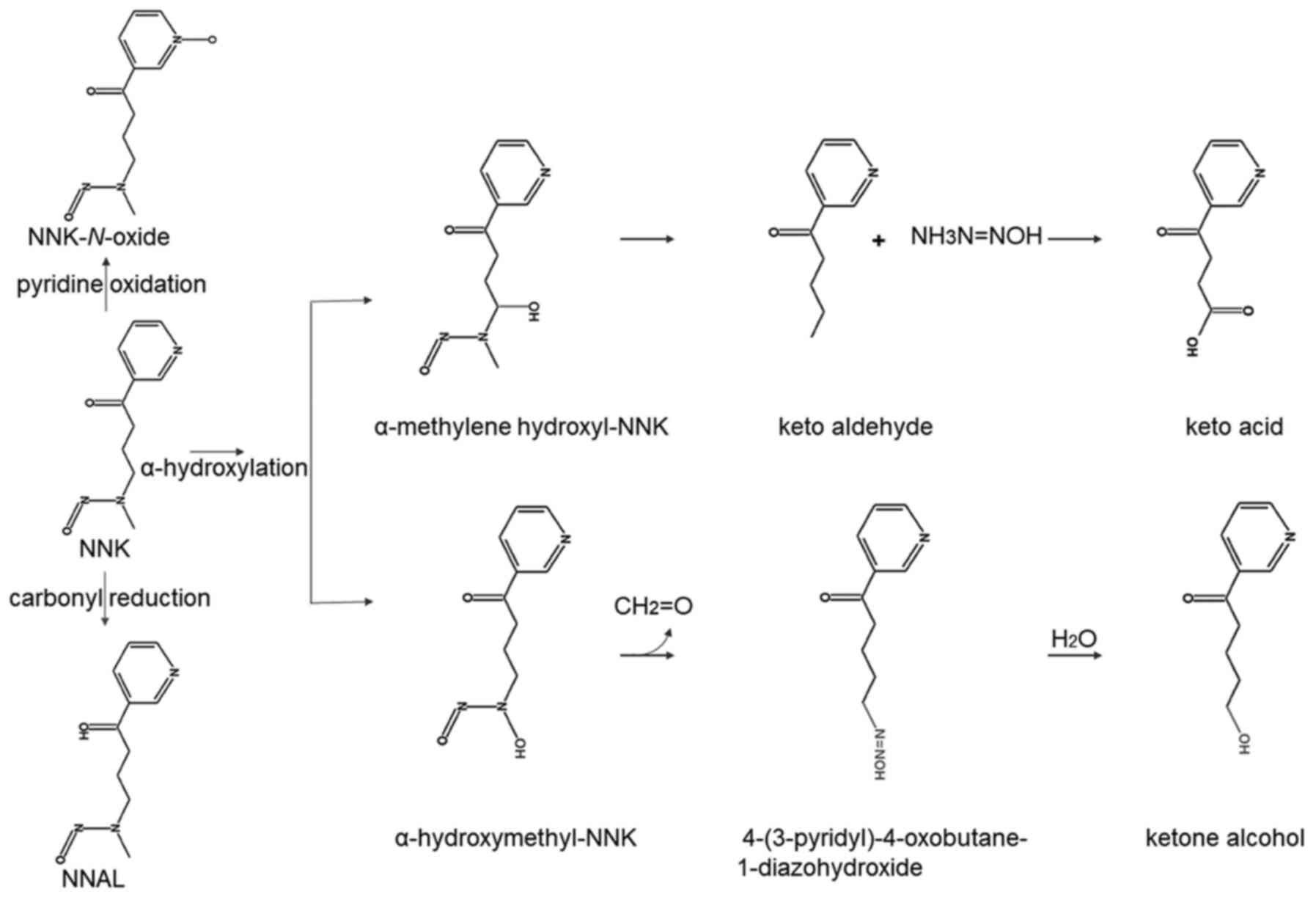
In vivo experiments show that NNK is
metabolized by three methods: Carbonyl reduction, pyridine
oxidation and α-hydroxylation (21).
In the carbonyl reduction process, NNK is carbonylated by
11β-hydroxysteroid dehydrogenase to form
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL), which is then
metabolized by glucuronidation to produce NNAL-glucuronic acid
(22). During oxidation of pyridine
nitrogen, CYP450 2B1 and CYP3A4 metabolize NNK to NNK-N-oxide
(23). The α-hydroxylation process
includes two modes: A-hydroxylation of the methyl carbon adjacent
to the N-nitroso nitrogen and α-hydroxylation of the methylene
carbon adjacent to the N-nitroso nitrogen. NNK is hydroxylated at
the methyl group adjacent to N-nitroso to form α-hydroxymethyl-NNK,
which then decomposes to form formaldehyde and
4-(3-pyridyl)-4-oxobutane-1-diazohydroxide. Finally, the latter
reacts with water to form ketone alcohol. The methylene carbon of
NNK can also be hydroxylated to generate an unstable α-methylene
hydroxyl-NNK, which quickly decomposes to methane diazohydroxide
and keto aldehyde, and finally keto aldehyde oxidizes to form keto
acid (19) (Fig. 1).
Using Syrian golden hamster tissue sections, it has
been shown that lung tissue has a lower NNK total metabolic rate
compared with that of kidney and liver tissues (24). The oxidative metabolism of NNK to
DNA-reactive intermediates by α-hydroxylation accounts for 13–31%,
pyridine nitrogen oxidation accounts for 5–22%, while carbonyl
group reduction of NNK to NNAL accounts for 47–81% of the total
metabolism of NNK in the lung. The total metabolism of NNAL in all
the tissues is ~10 times lower compared with that of NNK (24). The difference in the metabolic rate
of various NNK metabolites is one of the reasons that the lung is
more susceptible to the NNK carcinogen (24).
Prdx1
Prdxs, a class of antioxidant protective proteins,
play an important role in the elimination of ROS and cancer
development (25). Prdx1 is a member
of the Prdxs family of proteins, which is primarily localized in
the cytosol, as well as found in the nucleus, plasma, membrane and
centrosome (11). Prdx1 is
considered to be an important antioxidant protein (15), and exerts an antioxidant effect by
forming a homodimer. The Cys52 sulfhydryl group on one
peptide chain and the Cys172 sulfhydryl group on the
other peptide chain are dehydrogenated to form an intermolecular
disulfide bond, Cys52S-SCys172, which reduces
peroxides by providing hydrogen ions, thereby detoxifying them
(26). Prdx1 is highly sensitive to
hydrogen peroxide (H2O2), among various
peroxides (11).
In most cancer cells, such as breast, esophageal and
lung cancer, Prdx1 can removes excess intracellular ROS, maintains
ROS balance and protects the cells from oxidative stress-induced
DNA damage (27). Furthermore, by
eliminating ROS, Prdx1 prevents oxidative stress-induced mutations
in the P53 and K-Ras genes, thereby inhibiting tumor
formation, suppressing lung cancer cell proliferation, invasion and
migration, and increasing radiation sensitivity of the cancer cells
(15,28). Cys52 is the active center
of Prdx1 (29), which either reacts
with H2O2 (30) or combines with heme; hence, Prdx1 is
also called as heme-binding protein 23 (31). Prdx1 acts as a scavenger for the
cytoplasmic heme and has an important inhibitory effect on heme
toxicity (32). Furthermore, Prdx1
enhances the immune activity of natural killer (NK) cells against
tumor cells, and is also known as natural killer enhancement factor
(11). It has been shown that Prdx1
can effectively prevent lung cancer progression by enhancing the
tumor killing effects of NK cells (33,34).
Prdx1 is overexpressed in non-small cell lung cancer (NSCLC) cells,
which has been demonstrated to promote transforming growth
factor-β1 (TGF-β1)-induced epithelial mesenchymal transition (EMT)
and A549 cell migration (35). In
addition, the interaction between Prdx1 and nuclear erythroid
2-related factor 2 (Nrf2) can significantly affect the
proliferation of lung cancer cells (28,36). In
a rat acute lung injury model, overexpression of Prdx1 increased
the expression of proinflammatory cytokines interleukin-6 (IL-6),
IL-8 and tumor necrosis factor-α (37). Inflammatory factors play an important
role in the development of lung cancer. In addition, Prdx I affects
the proliferation, migration and invasion of lung cancer cells by
regulating various cytokines, in turn modulating different cell
signaling pathways (38,39).
On one hand, Prdx1 protects macromolecules, such as
proteins and DNA, from oxidative damage and suppresses malignant
transformation of normal cells, thus preventing tumor development.
On the other hand, Prdx1 inhibits ROS-induced apoptosis of cancer
cells and promotes tumor cell survival (27). Hence, understanding its mechanism of
action may provide novel insights into the development of better
therapeutic strategies for lung cancer.
Prdx1 and NNK affect the growth and
development of lung cancer by acting on P53 and K-Ras genes
NNK mainly undergoes metabolic activation through
α-methyl and α-methylene hydroxylation, thereby producing DNA
adducts and promoting cancer development. α-methyl hydroxylated
metabolites of NNK can pyridyloxobutylate DNA and produce DNA
pyridyloxobutyl adducts, whereas α-methylene hydroxylation
generates α-methylenehydroxy-NNK, methane diazohydroxide and
methyldiazonium ions. These react with DNA and yield 7-methyl
guanine, O6−methyl guanine and O4-methyl
thymine adducts (7). NNK induces
oxidative stress by increasing the level of intracellular ROS,
which in turn leads to the mutation of K-Ras and P53
oncogenes. In addition, the NNK metabolites have been shown to
result in the mutation of K-Ras and P53 oncogenes in
the lung. Thus, these deleterious effects of NNK on DNA may promote
the development of lung cancer.
Prdx1 is considered a potential marker for NSCLC,
and the interaction between Prdx1 and ROS plays an important role
in the development of tumors (40).
ROS plays a role in cell growth, differentiation, immune response
and apoptosis (41,42). Increase in intracellular levels of
ROS activates the expression of P53 (15), which in turn induces the expression
of apoptotic factors, such as Bak and Bax, under oxidative stress,
promotes the activation of caspases and finally activates the
mitochondrial apoptotic signaling pathway (27). P53 in its active form suppresses the
proliferation of abnormal cells, thereby exerting a tumor
suppressor effect (43). In
addition, P53 plays an important role in detecting DNA damage. P53
status after reducing the expression of Prdx1 is the major
determinant of tumor growth and response of lung cancer cells to
treatment (15). K-Ras
mutations are known to cause uncontrolled division of human lung
adenocarcinoma cells (44–46). Furthermore, K-Ras mutations
and ROS-induced oxidative stress are the major causes of NSCLC
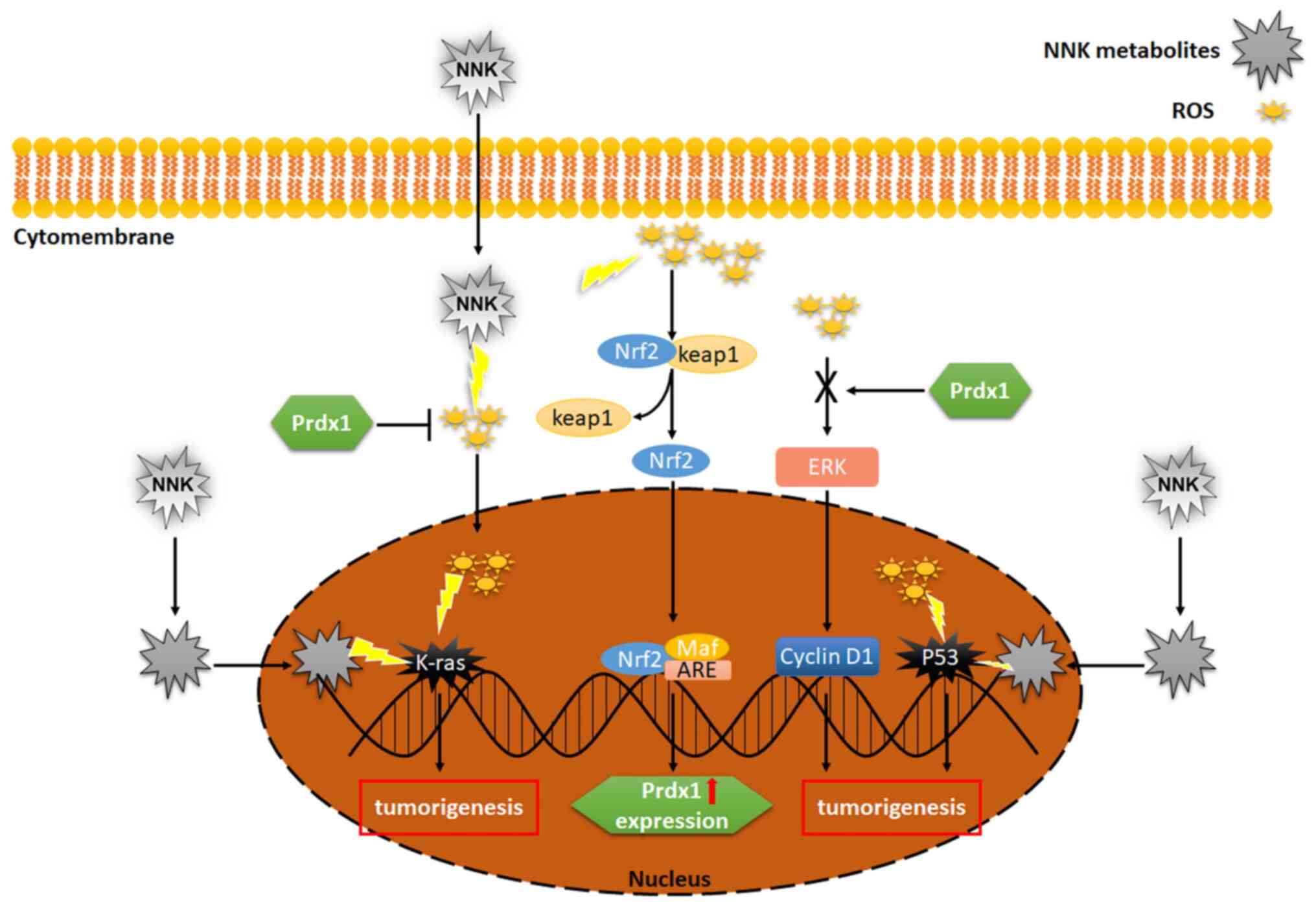
development. Prdx1 inhibits the activation of ROS/ERK/cyclin D1
pathway, thus results in Nrf2-dependent inhibition of
K-Ras-driven lung tumorigenesis (28).
NNK may promote lung cancer development by
increasing the intracellular ROS level and inducing mutations in
important oncogenes. Prdx1 effectively eliminates excess ROS and
prevents gene mutations caused by oxidative damage of DNA. Prdx1
prevents the occurrence of mutations in the P53 gene,
enabling it to detect and repair damaged DNA. Furthermore,
activation of the Nrf2 pathway results in upregulation of Prdx1.
Prdx1 inhibits the ROS/ERK/cyclin D1 signaling pathway and
suppresses the development of lung tumors (Fig. 2) (42,43,47,48).
Prdx1 and NNK affect the growth and
development of lung cancer by reacting with heme and
hemoglobin
It has been demonstrated that NNK-induced DNA damage
is significantly reduced by antioxidants (BHT), catalase and
superoxide dismutase (SOD) in A549 cells. The order of the
effectiveness has been indicated to be BHT > catalase > SOD
(8). Thus, it is speculated that NNK
mainly induces generation of H2O2 (8,49,50). The
α-methyl hydroxylation of NNK results in the formation of unstable
α-hydroxymethyl NNK. The decomposition of a-hydroxymethyl NNK
results in the formation of electrophilic 4-(3-pyridyl)-4-oxybutyl
diazoxide, which can react with hemoglobin to form hemoglobin
adduct (7,51). NNK α-methyl hydroxylation results in
globulin methylation and pyridyloxybutylation (15), and the hemoglobin adduct formed by
pyridyloxybutylation releases 4-hydroxy-1-(3-pyridyl)-1-butanone by
alkaline hydrolysis (51). Studies
have shown that keto alcohol-releasing adducts were formed by
treatment of hemoglobin with NNK (51–53).
Phenylethyl isothiocyanate treatment can significantly inhibit
NNK-mediated lung tumorigenesis by reducing the release of ketone
alcohol products (54).
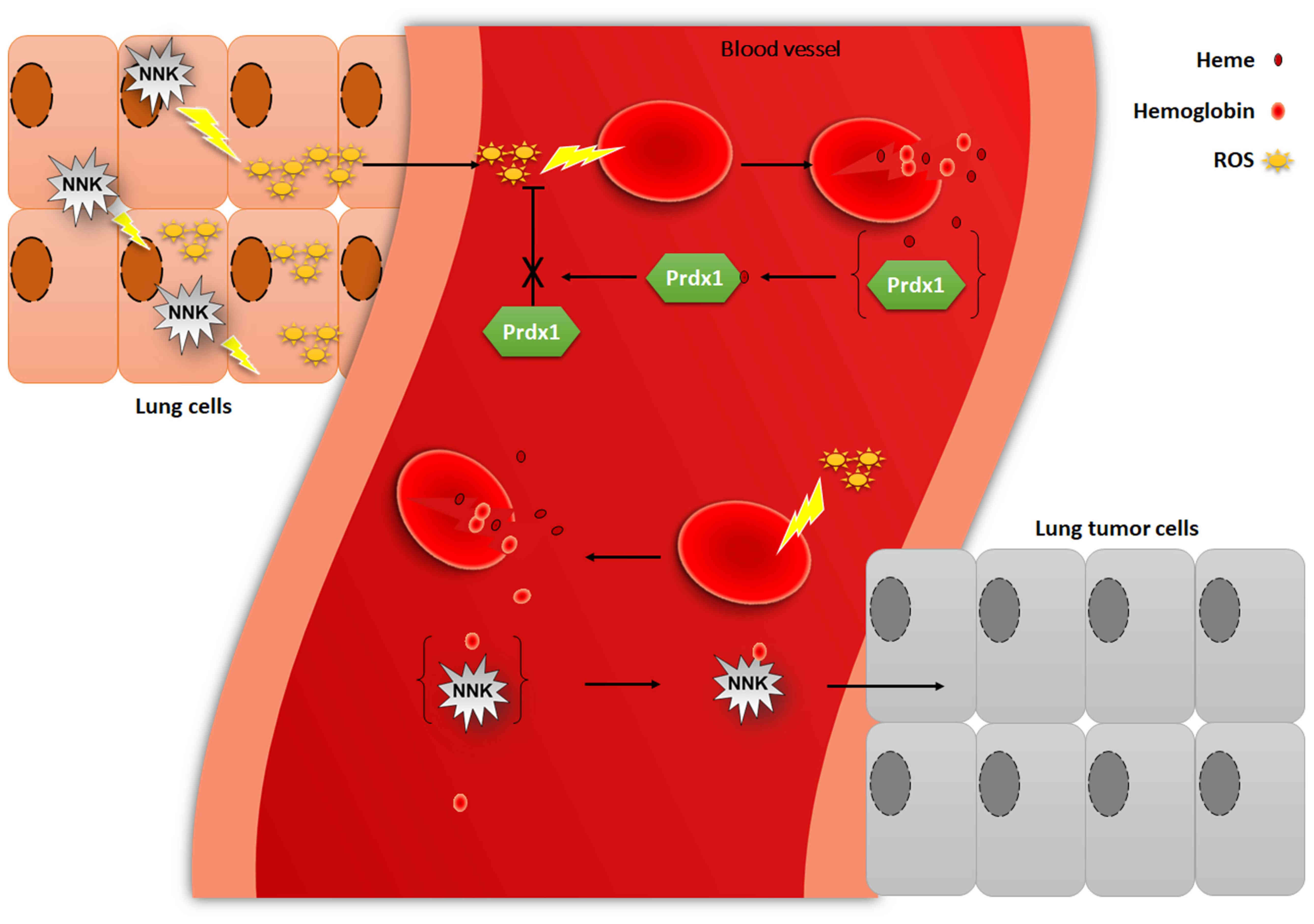
Prdx1 can bind to heme (53), which is abundant in red blood cells.
Furthermore, heme is widely distributed in organelles, such as the
nucleus, endoplasmic reticulum and plasma membrane (55,56), and
it is involved in a processes in mammalian cells, including
respiration, metabolism, transcription, DNA binding and protein
degradation (55,57). Heme is synthesized in mitochondria
and loosely bound to Prdx1 (31),
which is proposed to facilitate the transport of heme to other
organelles (55,56,58).
Heme is insoluble in aqueous solutions and is toxic to the cells
(59), and the toxicity is further
manifested by the generation of ROS (57). However, binding of heme to Prdx1
reduces heme toxicity and promotes
H2O2-mediated heme degradation (60). Thus, Prdx1 protects free heme from
peroxidation but loses its peroxidase activity when bound to heme
(31). NNK induces the generation
H2O2 and Prdx1 is more sensitive to
H2O2. H2O2 causes
erythrocyte lysis, leading to the release of large amounts of heme
and hemoglobin (61). Heme interacts
with oxygen to produce ROS (62),
which further destroys red blood cells. The binding of heme and
Prdx1 reduces heme cytotoxicity; however, Prdx1 loses its ROS
scavenging ability. NNK metabolites produce adducts with
hemoglobin, thereby promoting the development of lung tumors, and
Prdx1 plays a role in ROS scavenging, which in turn protects red
blood cells from oxidative damage and inhibits lung tumorigenesis
(Fig. 3).
Prdx1 and NNK affect the growth and
development of lung cancer by acting on alveolar macrophages (AMs)
and NK cells
NNK metabolites inhibit AM-mediated production of
interleukin-12 (IL-12), nitric oxide and TNF; however, they also
induce the production of IL-10 (63), which may promote the growth and
development of lung tumors (64).
Additionally, NNK has an inhibitory effect on TNF-dependent
cytotoxicity of AMs (49).
Furthermore, metabolites produced by NNK α-methyl hydroxylation may
be involved in the regulation of AM function. For example, keto
acid inhibits IL-12 production by AMs, while keto alcohol may
inhibit the AM production of TNF and IL-12 (65). NNK may induce the expression of
cyclooxygenase 2 (COX-2) and upregulate prostaglandin E2
(PGE2); PGE2 in turn upregulates IL-10 (63,66).
Nicotinic acetylcholine receptors (nAChRs) are present in immune
cells (67), and NNK has high
affinity towards nAChRs. Thus, interaction of NNK and nAChRs may
activate the production of IL-10 (68). Moreover, IL-10 suppresses the
production of IL-12 (69), and
inhibition of IL-12 leads to decreased expression of interferon-γ
(70,71). Furthermore, IL-12 is mainly produced
by phagocytes (monocytes/macrophages and neutrophils) and dendritic
cells (72) and enhances the
cytotoxicity of AM and NK cells (73). NK cells are a group of lymphocytes
that can kill tumor cells (74).
Therefore, it can be speculated that NNK attenuates the toxic
effects of NK cells on tumor cells and further promotes the
development of lung cancer.
All six Prdxs are expressed by human lung cells;
however, AMs mainly express Prdx1 and III (75). Prdx1 may affect the production of
pro-inflammatory cytokines in macrophages (76) and has the ability to enhance NK cell
toxicity in vitro. Furthermore, it has been reported that
free thiol groups are required to maintain NK cell toxicity against
tumor cells (42). The alkylation of
free sulfhydryl groups in Prdx1 upon reduction decreases its
ability to enhance NK cell toxicity, further indicating the
requirement of free thiol groups for enhanced cytotoxicity of NK
cells (16). Therefore, Prdx1 not
only protects the cells from oxidative damage, but also selectively
promotes the killing effect of AM and NK cells in certain
tumors.
NNK may activate AMs to produce
H2O2. NNK inhibits the production of IL-12
and TNF by AMs, thereby reducing the cytotoxicity of AMs and NKs
against the tumor cells. Prdx1 is expressed mainly in AMs and may
play a role in immune regulation by affecting inflammatory factors.
Prdx1 may inhibit lung tumors by enhancing the killing effect of NK
cells. However, the exact role of NNK and Prdx1 in AM-mediated
killing of tumor cells is still unclear (Fig. 4) (64,70,74–76).
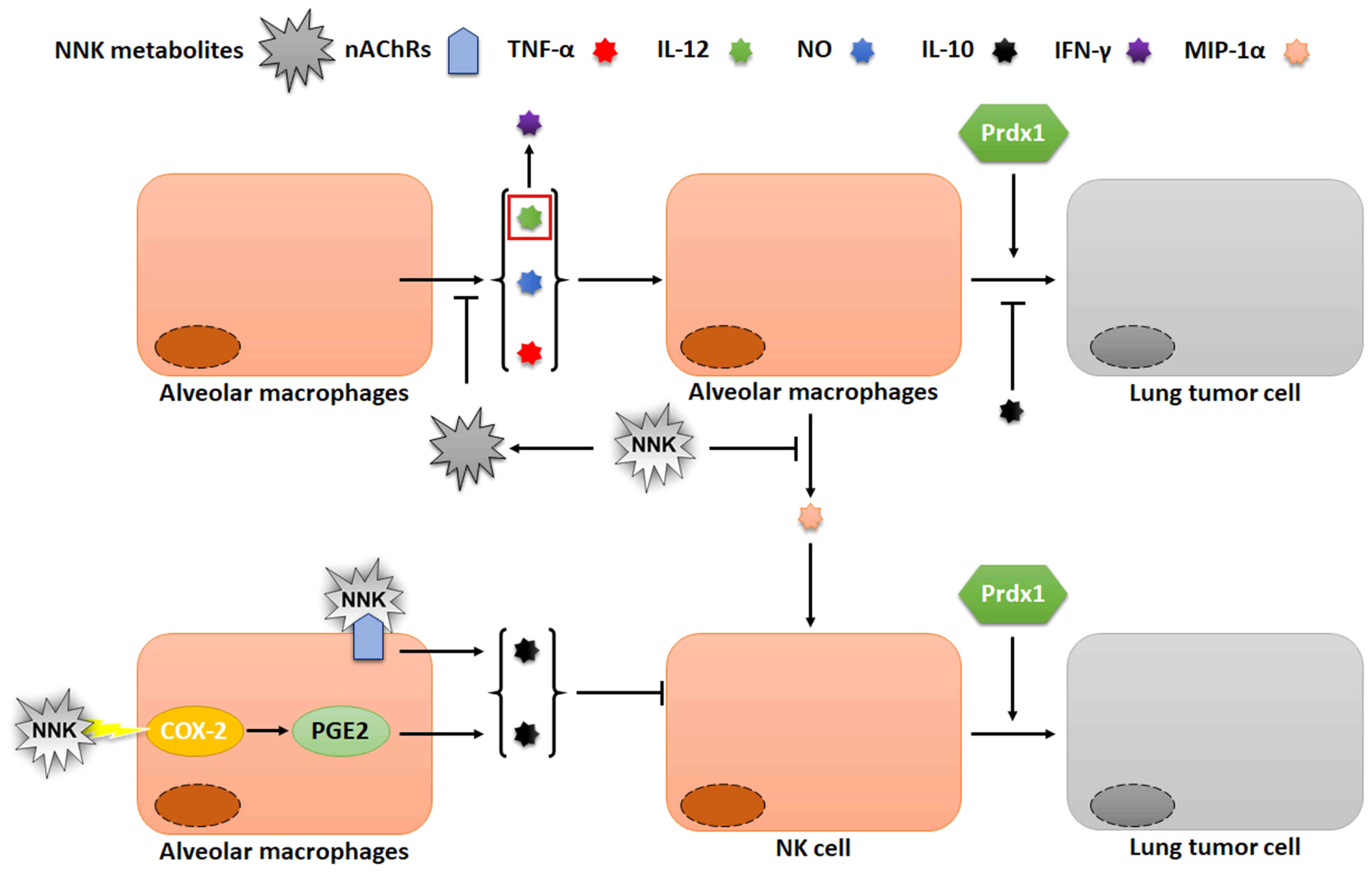 | Figure 4.Effects of Prdx1 and NNK on AM and NK
cells. NNK can reduce the toxicity of AM and NK cells to lung
cancer cells, while Prdx1 can stimulate NK cells to kill lung
cancer cells. Prdx1, peroxiredoxin I; NNK,
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone; AM, alveolar
macrophages; NK, natural killer cells; IL, interleukin; nAChRs,
nicotinic acetylcholine receptors; MIP, macrophage inflammatory
protein; PGE2, prostaglandin E2; COX, cyclooxygenase. |
Prdx1 and NNK affect EMT
Long-term exposure of lung alveolar cells to NNK
results in their proliferation and eventually malignant
transformation (77). After the
cells are exposed to NNK, the expression of intracellular β-catenin
and F-actin decrease, whereas that of fibronectin, vimentin and
matrix metalloproteinase-2 increase. This in turn promotes EMT
(77). EMT is a process by which
epithelial cells are transformed into the mesenchymal phenotype,
which increases their invasion and migration capabilities.
Morphologically, the epithelial cells are loosely connected and the
cytoskeleton structure is reorganized. The EMT process is
accompanied by various changes in protein expression, including a
decrease in E-cadherin and an increase in fibronectin expression
levels (77). Low E-cadherin
expression decreases cell adhesion, and downregulation of
E-cadherin is also considered as a sign of EMT (78). NNK and ROS induce EMT through
different signaling pathways (79).
In cancer cells, NNK may promote the production of ROS, such as
H2O2, which in turn activates c-Src (80,81),
leading to cytoskeletal modifications (79) and initiation of EMT (82). Most tissues usually do not express
COX-2 or express it at a low level (83). The induced expression of COX-2
inhibits apoptosis and increases the migration potential of cancer
cells (84). The combination of NNK
and a7-nAChR can induce the expression of COX-2, which
upregulates fibronectin and promotes EMT (79).
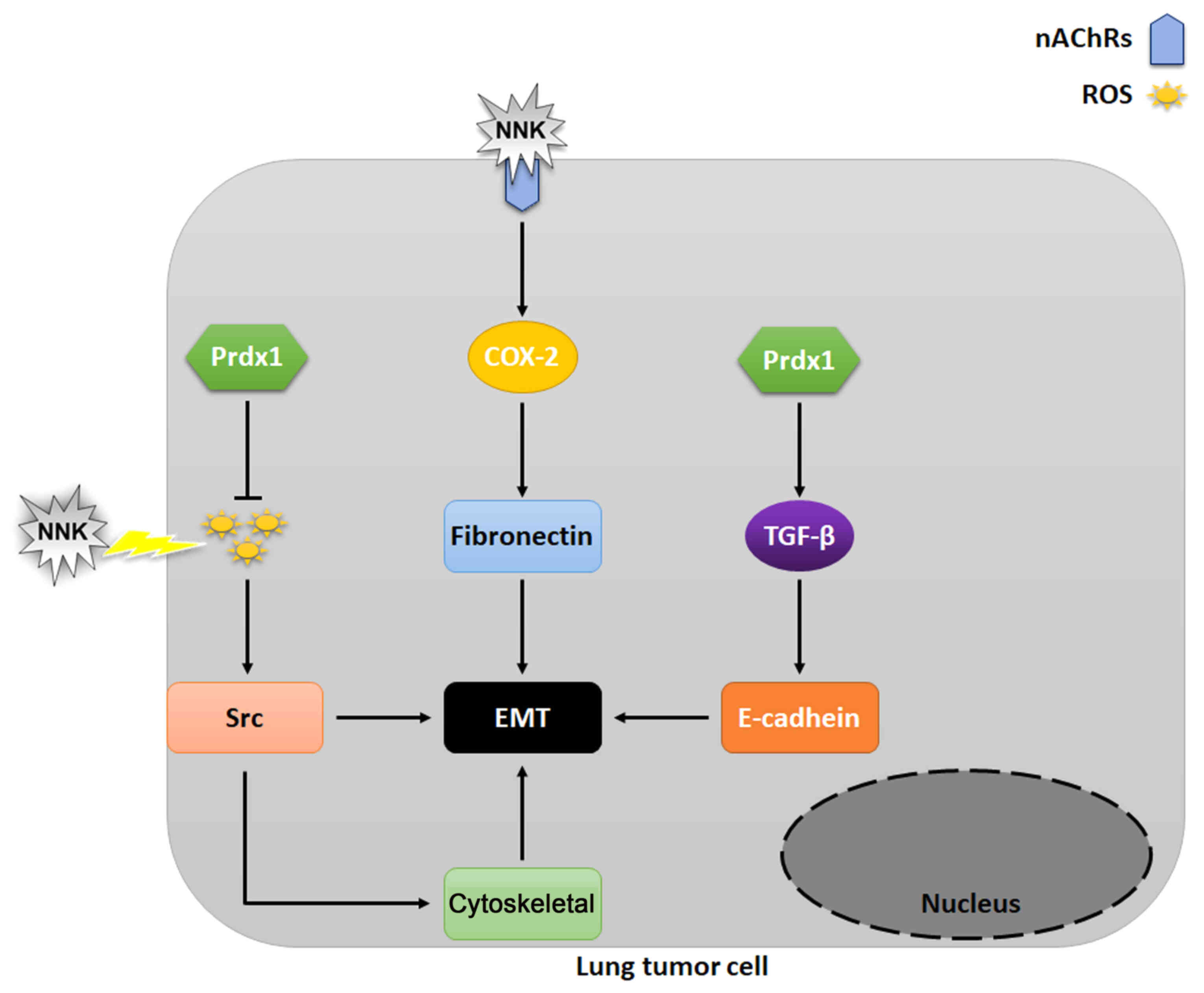
Furthermore, Prdx1 is a type of peroxidase
reductase, which can play the role of scavenging ROS, thus
inhibiting the EMT process. It has been observed that Prdx 1 can
promote EMT in breast (85),
pancreatic (86) and colon cancer
(87). Furthermore, high expression
levels of Prdx1 downregulates E-cadherin, whereas, at lower levels
it upregulates E-cadherin in A549 lung adenocarcinoma cells
(88). TGF-β1 is a pleiotropic
cytokine that is involved in apoptosis, differentiation and
proliferation of cells and is the primary inducer of EMT (83,89–91). It
has been shown that the overexpression of Prdx1 in lung cancer
cells significantly enhances TGF-β1-mediated EMT and cell migration
(92).
In A549 lung cancer cells, NNK treatment results in
a significant increase in Prdx1 expression. NNK not only results in
ROS production, but also upregulates the expression of fibronectin
via COX-2 and promotes EMT. Prdx1 can play a role in scavenging
ROS, and Prdx1 can also inhibit EMT process by scavenging ROS
caused by NNK. Additionally, high levels of Prdx1 results in
upregulation of E-cadherin, which promotes EMT (Fig. 5) (77–82,84,85).
Signaling pathways associated with
Prdx1 and NNK in lung cancer cells
NADPH oxidase of Nox family is expressed in both
normal and cancer cells, and is related to ROS production and
tumorigenicity in various cancer cells. For example, Nox1 is highly
expressed in human colon cancer and prostate cancer, and lung
cancer A549 cells also express Nox1, 2 and 4 (16,93). NNK
induces the expression of NOX protein and results in the production
of large amount of ROS, which causes damage to protein and DNA
through oxidative stress. In A549 lung cancer cells, NNK-mediated
generation of ROS through NOX activates
phosphatidylinositol-3-kinase (PI3K)/protein kinase B (AKT) and Wnt
signaling pathways, which leads to the development of drug
resistance in lung cancer cells (16) and is associated with lower survival
rate of patients with stage 1 lung tumors in Tumor-Node-Metastasis
staging system (94). NNK increases
the expression of thromboxane A2 (TxA2) and Tx receptor in lung
cancer cells by elevating COX and Tx synthase expression (95). NNK has been shown to promote adhesion
and invasion of CL1.0 cells through α7-nAChR/ERK/Contactin 1
signaling (96). Furthermore, NNK
prevents PH domain leucine-rich repeat-containing protein
phosphatase 2-mediated AKT dephosphorylation, activates AKT to
inhibit E-cadherin expression and promotes lung cancer cell
migration (97). The combination of
NNK and α7-nAChRs activates c-Src and protein kinase C, and
promotes the dissociation of phosphorylated Bad from Bcl-xl, which
in turn inhibits apoptosis (98).
In tumor cells, Prdx1 has been reported to eliminate
large quantities of ROS produced by tumor cell metabolism and thus,
suppresses tumor cell death. Additionally, Prdx1 inhibits the
oxidative stress-induced PI3K/AKT signaling pathway by eliminating
ROS. It is known that the ROS-induced activation of PI3K/AKT is due
to the oxidative inactivation of phosphatase and tensin homolog
(PTEN) protein (99). Furthermore,
Prdx1 protects PTEN lipid phosphatase activity from oxidative
inactivation, thereby preventing AKT from driving tumor cell
proliferation and inducing apoptosis (26). C-Abl plays a vital role in oxidative
stress-induced cell death (100).
Prdx1 can be used as a physiological inhibitor of C-Abl (18) to inhibit apoptosis induced by the
C-Abl/P38/MAPK signaling pathway.
NNK induces Nox protein to produce ROS, and
activates the PI3K/Akt signaling pathway; however, NNK inhibits the
same pathway by removing ROS or preventing oxidative inactivation
of phosphatase and PTEN. In addition, NNK activates α7-nAChRs and
downstream signaling pathways, and hence promotes apoptosis and
migration of lung cancer cells. Furthermore, both NNK and Prdx1 can
regulate apoptosis-related proteins and thus, control the apoptosis
of lung cancer cells (99,101–103)
(Fig. 6).
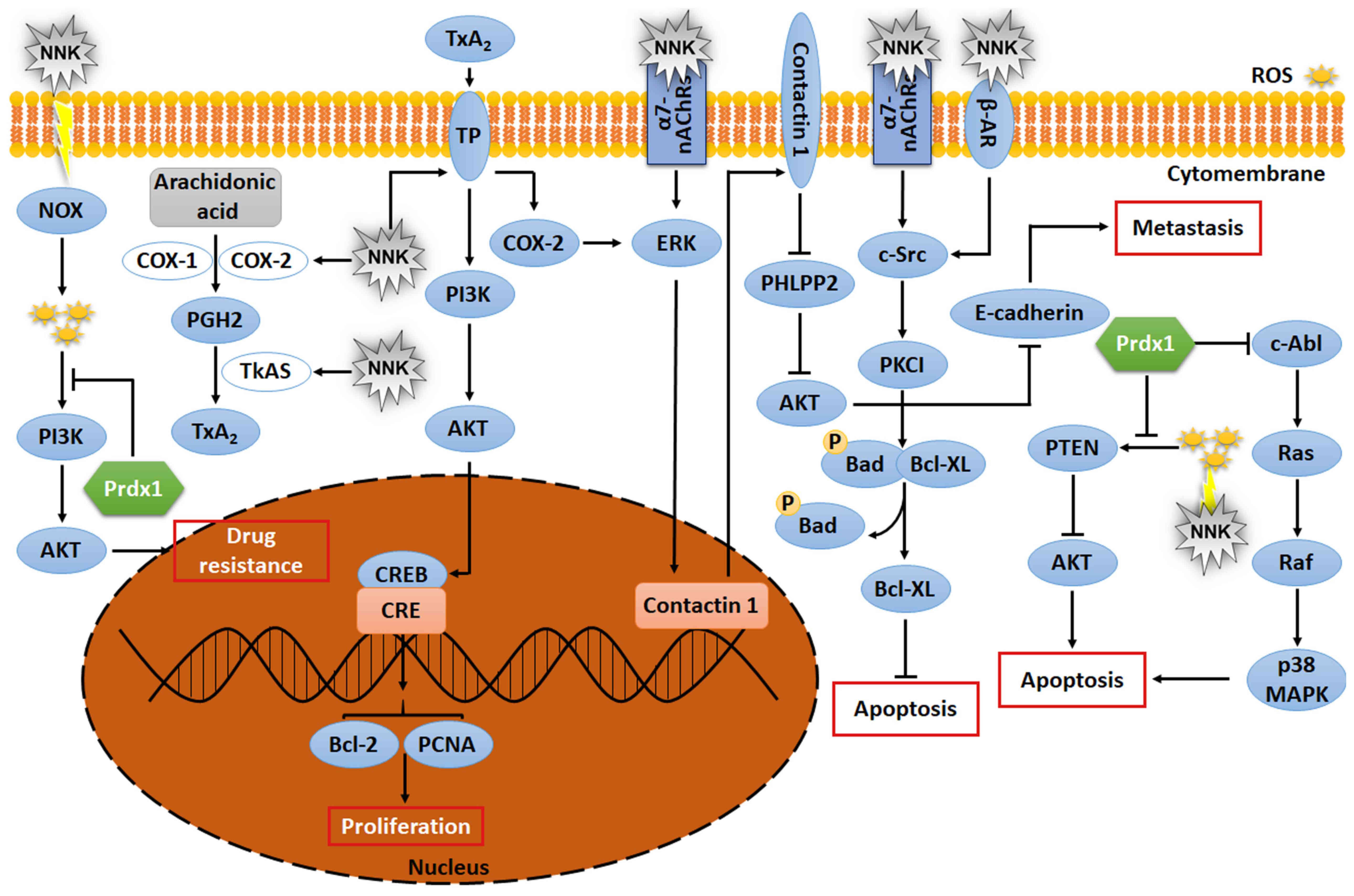 | Figure 6.Signaling pathway of Prdx1 and NNK.
NNK and Prdx1 are involved in the regulation of signaling pathways
in the development of lung cancer cells. NOX, NADPH oxidase;
TxA2, thromboxane A2; PGH2, prostaglandin H2; COX,
cyclooxygenase; NNK,
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone; TP, thromboxane
receptor; PCNA, proliferating cell nuclear antigen; CREB, cyclic
AMP response element-binding protein; CRE, cyclic AMP response
element; ROS, reactive oxygen species; PHLPP2, pleckstrin homology
domain leucine-rich repeat protein phosphatase 2; PKCI, protein
kinase C interacting protein. |
Conclusions
In conclusion, on the one hand, Prdx1 has the
ability to protect erythrocytes and DNA from NNK-induced oxidative
damage, prevent malignant transformation of cells and promote
cytotoxicity of NK cells, suppressing tumor formation. In addition,
Prdx1 prevents NNK-induced generation of large amount of ROS and
hence, ROS-induced apoptosis, and promotes tumor cell survival. On
the other hand, together with NNK, Prdx1 promotes EMT and migration
of lung tumor cells. The signaling pathways of NNK and Prdx1 in
lung cancer cells are intricate, and the associated mechanisms are
yet to be explored.
Acknowledgments
Not applicable.
Funding
This research was supported by the Basic Science
Research Program through the National Research Foundation of Korea
funded by the Ministry of Education (grant no. 2020R1I1A2052417),
The Korean Research Institute of Bioscience and Biotechnology
Research Information System (grant no. RBM0112112) and The Project
of Sanzong (grant no. ZRCPY202030) by Heilongjiang Bayi
Agricultural University.
Availability of data and materials
Data sharing is not applicable to this article, as
no datasets were generated or analyzed during the current
study.
Authors' contributions
HNS, CXR, YXG and TK conceived and designed the
review. HNS, CXR, YXG, DPX and TK wrote the manuscript and prepared
the figures. HNS and TK reviewed and edited the manuscript. TK
acquired the funding. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
NNK
|
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
|
|
Prdx1
|
peroxiredoxin I
|
|
ROS
|
reactive oxygen species
|
|
Prdx
|
peroxiredoxins
|
|
NK cells
|
natural killer cells
|
|
NSCLC
|
non-small cell lung cancer
|
|
TGF-β1
|
transforming growth factor-β1
|
|
EMT
|
epithelial mesenchymal transition
|
|
Nrf2
|
nucleosome 2-related factor 2
|
|
SOD
|
superoxide dismutase
|
|
H2O2
|
hydrogen peroxide
|
|
AM
|
alveolar macrophages
|
|
IL-12
|
interleukin-12
|
|
TNF
|
tumor necrosis factor
|
|
IL-10
|
interleukin 10
|
|
COX-2
|
cyclooxygenase 2
|
|
PGE2
|
prostaglandin E2
|
|
nAChRs
|
nicotinic acetylcholine receptors
|
|
PI3K
|
phosphatidylinositol-3-kinase
|
|
AKT
|
protein kinase B
|
|
TxA2
|
thromboxane A2
|
|
PTEN
|
phosphatase and tensin homolog
|
References
|
1
|
Proctor RN: Tobacco and the global lung
cancer epidemic. Nat Rev Cancer. 1:82–86. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thun M, Peto R, Boreham J and Lopez AD:
Stages of the cigarette epidemic on entering its second century.
Tob Control. 21:96–101. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Witschi H: Carcinogenic activity of
cigarette smoke gas phase and its modulation by beta-carotene and
N-acetylcysteine. Toxicol Sci. 84:81–87. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jin Z, Gao F, Flagg T and Deng X:
Tobacco-specific nitrosamine
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone promotes functional
cooperation of Bcl2 and c-Myc through phosphorylation in regulating
cell survival and proliferation. J Biol Chem. 279:40209–40219.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Maser E: Significance of reductases in the
detoxification of the tobacco-specific carcinogen NNK. Trends
Pharmacol Sci. 25:235–237. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yalcin E and de la Monte S: Tobacco
nitrosamines as culprits in disease: Mechanisms reviewed. J Physiol
Biochem. 72:107–120. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yeh SL, Wang WY, Huang CS and Hu ML:
Flavonoids suppresses the enhancing effect of beta-carotene on DNA
damage induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
(NNK) in A549 cells. Chem Biol Interact. 160:175–182. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lehtonen ST, Svensk AM, Soini Y, Pääkkö P,
Hirvikoski P, Kang SW, Säily M and Kinnula VL: Peroxiredoxins, a
novel protein family in lung cancer. Int J Cancer. 111:514–521.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gorrini C, Harris IS and Mak TW:
Modulation of oxidative stress as an anticancer strategy. Nat Rev
Drug Discov. 12:931–947. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rhee SG and Kil IS: Multiple functions and
regulation of mammalian peroxiredoxins. Annu Rev Biochem.
86:749–775. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bajor M, Zych AO, Graczyk-Jarzynka A,
Muchowicz A, Firczuk M, Trzeciak L, Gaj P, Domagala A, Siernicka M,
Zagozdzon A, et al: Targeting peroxiredoxin 1 impairs growth of
breast cancer cells and potently sensitises these cells to
prooxidant agents. Br J Cancer. 119:873–884. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Han YH, Zhang YQ, Jin MH, Jin YH, Qiu MY,
Li WL, He C, Yu LY, Hyun JW, Lee J, et al: Peroxiredoxin I
deficiency increases keratinocyte apoptosis in a skin tumor model
via the ROS-p38 MAPK pathway. Biochem Biophys Res Commun.
529:635–641. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hampton MB, Vick KA, Skoko JJ and Neumann
CA: Peroxiredoxin involvement in the initiation and progression of
human cancer. Antioxid Redox Signal. 28:591–608. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen MF, Chen WC, Wu CT, Lin PY, Shau H,
Liao SK, Yang CT and Lee KD: p53 status is a major determinant of
effects of decreasing peroxiredoxin I expression on tumor growth
and response of lung cancer cells to treatment. Int J Radiat Oncol
Biol Phys. 66:1461–1472. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hirata N, Yamada S, Sekino Y and Kanda Y:
Tobacco nitrosamine NNK increases ALDH-positive cells via ROS-Wnt
signaling pathway in A549 human lung cancer cells. J Toxicol Sci.
42:193–204. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shi GQ, Zhou WS, Li M, Ren F and Han YW:
Characterization and expression analysis of peroxiredoxin genes in
NNK-induced V79 cells. Biomed Environ Sci. 30:224–228.
2017.PubMed/NCBI
|
|
18
|
Immenschuh S and Baumgart-Vogt E:
Peroxiredoxins, oxidative stress, and cell proliferation. Antioxid
Redox Signal. 7:768–777. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Akopyan G and Bonavida B: Understanding
tobacco smoke carcinogen NNK and lung tumorigenesis. Int J Oncol.
29:745–752. 2006.PubMed/NCBI
|
|
20
|
Hecht SS, Hochalter JB, Villalta PW and
Murphy SE: 2′-Hydroxylation of nicotine by cytochrome P450 2A6 and
human liver microsomes: Formation of a lung carcinogen precursor.
Proc Natl Acad Sci USA. 97:12493–12497. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Peterson LA: Context matters: Contribution
of specific DNA adducts to the genotoxic properties of the
tobacco-specific nitrosamine NNK. Chem Res Toxicol. 30:420–433.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ashmore JH, Luo S, Watson CJW and Lazarus
P: Carbonyl reduction of NNK by recombinant human lung enzymes:
Identification of HSD17β12 as the reductase important in (R)-NNAL
formation in human lung. Carcinogenesis. 39:1079–1088. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Leslie EM, Ghibellini G, Nezasa K and
Brouwer KL: Biotransformation and transport of the tobacco-specific
carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in
bile duct-cannulated wild-type and Mrp2/Abcc2-deficient (TR) Wistar
rats. Carcinogenesis. 28:2650–2656. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Richter E, Friesenegger S, Engl J and
Tricker AR: Use of precision-cut tissue slices in organ culture to
study metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
(NNK) and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) by
hamster lung, liver and kidney. Toxicology. 144:83–91. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rhee SG, Kang SW, Chang TS, Jeong W and
Kim K: Peroxiredoxin, a novel family of peroxidases. IUBMB Life.
52:35–41. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Neumann CA, Cao J and Manevich Y:
Peroxiredoxin 1 and its role in cell signaling. Cell Cycle.
8:4072–4078. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ding C, Fan X and Wu G: Peroxiredoxin 1-an
antioxidant enzyme in cancer. J Cell Mol Med. 21:193–202. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Park YH, Kim SU, Lee BK, Kim HS, Song IS,
Shin HJ, Han YH, Chang KT, Kim JM, Lee DS, et al: Prx I suppresses
K-ras-driven lung tumorigenesis by opposing redox-sensitive
ERK/cyclin D1 pathway. Antioxid Redox Signal. 19:482–496. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chang TS, Jeong W, Choi SY, Yu S, Kang SW
and Rhee SG: Regulation of peroxiredoxin I activity by
Cdc2-mediated phosphorylation. J Biol Chem. 277:25370–25376. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Poole LB: The basics of thiols and
cysteines in redox biology and chemistry. Free Radic Biol Med.
80:148–157. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Watanabe Y, Ishimori K and Uchida T: Dual
role of the active-center cysteine in human peroxiredoxin 1:
Peroxidase activity and heme binding. Biochem Biophys Res Commun.
483:930–935. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gozzelino R, Jeney V and Soares MP:
Mechanisms of cell protection by heme oxygenase-1. Annu Rev
Pharmacol Toxicol. 50:323–354. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guo Y, Patil NK, Luan L, Bohannon JK and
Sherwood ER: The biology of natural killer cells during sepsis.
Immunology. 153:190–202. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Aktaş ON, Öztürk AB, Erman B, Erus S,
Tanju S and Dilege Ş: Role of natural killer cells in lung cancer.
J Cancer Res Clin Oncol. 144:997–1003. 2018. View Article : Google Scholar
|
|
35
|
Li S, Wang R, Zhang M, Wang L and Cheng S:
Proteomic analysis of non-small cell lung cancer tissue
interstitial fluids. World J Surg Oncol. 11:1732013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen MF, Keng PC, Shau H, Wu CT, Hu YC,
Liao SK and Chen WC: Inhibition of lung tumor growth and
augmentation of radiosensitivity by decreasing peroxiredoxin I
expression. Int J Radiat Oncol Biol Phys. 64:581–591. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu D, Mao P, Huang Y, Liu Y, Liu X, Pang
X and Li Y: Proteomic analysis of lung tissue in a rat acute lung
injury model: Identification of PRDX1 as a promoter of
inflammation. Mediators Inflamm. 2014:4693582014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Brenner DR, Fanidi A, Grankvist K, Muller
DC, Brennan P, Manjer J, Byrnes G, Hodge A, Severi G, Giles GG, et
al: Inflammatory cytokines and lung cancer risk in 3 prospective
studies. Am J Epidemiol. 185:86–95. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
DeCotiis C, Hu Y, Greenberg AK, Huie M,
Tsay JC, Pass H, Goldberg JD and Rom WN: Inflammatory cytokines and
non-small cell lung cancer in a CT-scan screening cohort:
Background review of the literature. Cancer Biomark. 16:219–233.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chang JW, Lee SH, Jeong JY, Chae HZ, Kim
YC, Park ZY and Yoo YJ: Peroxiredoxin-I is an autoimmunogenic tumor
antigen in non-small cell lung cancer. FEBS Lett. 579:2873–2877.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Finkel T and Holbrook NJ: Oxidants,
oxidative stress and the biology of ageing. Nature. 408:239–247.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Moloney JN and Cotter TG: ROS signalling
in the biology of cancer. Semin Cell Dev Biol. 80:50–64. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Macip S, Igarashi M, Berggren P, Yu J, Lee
SW and Aaronson SA: Influence of induced reactive oxygen species in
p53-mediated cell fate decisions. Mol Cell Biol. 23:8576–8585.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Moll HP, Pranz K, Musteanu M, Grabner B,
Hruschka N, Mohrherr J, Aigner P, Stiedl P, Brcic L, Laszlo V, et
al: Afatinib restrains K-RAS-driven lung tumorigenesis. Sci Transl
Med. 10:eaao23012018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Aran V and Omerovic J: Current approaches
in NSCLC targeting K-RAS and EGFR. Int J Mol Sci. 20:57012019.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Scheffler M, Ihle MA, Hein R,
Merkelbach-Bruse S, Scheel AH, Siemanowski J, Brägelmann J, Kron A,
Abedpour N, Ueckeroth F, et al: K-ras mutation subtypes in NSCLC
and associated Co-occuring mutations in other oncogenic pathways. J
Thorac Oncol. 14:606–616. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Peeper DS, Upton TM, Ladha MH, Neuman E,
Zalvide J, Bernards R, DeCaprio JA and Ewen ME: Ras signalling
linked to the cell-cycle machinery by the retinoblastoma protein.
Nature. 386:177–181. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Slebos RJ, Kibbelaar RE, Dalesio O,
Kooistra A, Stam J, Meijer CJ, Wagenaar SS, Vanderschueren RG, van
Zandwijk N, Mooi WJ, et al: K-ras oncogene activation as a
prognostic marker in adenocarcinoma of the lung. N Engl J Med.
323:561–565. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Proulx LI, Paré G and Bissonnette EY:
Alveolar macrophage cytotoxic activity is inhibited by
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), a
carcinogenic component of cigarette smoke. Cancer Immunol
Immunother. 56:831–838. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Rioux N and Castonguay A: The induction of
cyclooxygenase-1 by a tobacco carcinogen in U937 human macrophages
is correlated to the activation of NF-kappaB. Carcinogenesis.
21:1745–1751. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang Y, Narayanapillai SC, Hu Q, Fujioka N
and Xing C: Detection and quantification of
4-hydroxy-1-(3-pyridyl)-1-butanone (HPB) from smoker albumin and
its potential as a surrogate biomarker of tobacco-specific
nitrosamines exposure and bioactivation. Toxicol Lett. 311:11–16.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Peterson LA, Carmella SG and Hecht SS:
Investigations of metabolic precursors to hemoglobin and DNA
adducts of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone.
Carcinogenesis. 11:1329–1333. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hecht SS, Stepanov I and Carmella SG:
Exposure and metabolic activation biomarkers of carcinogenic
tobacco-specific nitrosamines. Acc Chem Res. 49:106–114. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hecht SS, Trushin N, Rigotty J, Carmella
SG, Borukhova A, Akerkar S, Desai D, Amin S and Rivenson A:
Inhibitory effects of 6-phenylhexyl isothiocyanate on
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone metabolic activation
and lung tumorigenesis in rats. Carcinogenesis. 17:2061–2067. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tsiftsoglou AS, Tsamadou AI and
Papadopoulou LC: Heme as key regulator of major mammalian cellular
functions: Molecular, cellular, and pharmacological aspects.
Pharmacol Ther. 111:327–345. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Severance S and Hamza I: Trafficking of
heme and porphyrins in metazoa. Chem Rev. 109:4596–4616. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Shimizu T, Lengalova A, Martínek V and
Martínková M: Heme: Emergent roles of heme in signal transduction,
functional regulation and as catalytic centres. Chem Soc Rev.
48:5624–5657. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Khan AA and Quigley JG: Control of
intracellular heme levels: Heme transporters and heme oxygenases.
Biochim Biophys Acta. 1813:668–682. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Roumenina LT, Rayes J, Lacroix-Desmazes S
and Dimitrov JD: Heme: Modulator of plasma systems in hemolytic
diseases. Trends Mol Med. 22:200–213. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Vlasova II: Peroxidase activity of human
hemoproteins: Keeping the fire under control. Molecules.
23:25612018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Peralta IN, Cogoi L, Filip R and Anesini
C: Prevention of hydrogen peroxide-induced red blood cells lysis by
Ilex paraguariensis aqueous extract: Participation of phenolic and
xanthine compounds. Phytother Res. 27:192–198. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hahl P, Hunt R, Bjes ES, Skaff A,
Keightley A and Smith A: Identification of oxidative modifications
of hemopexin and their predicted physiological relevance. J Biol
Chem. 292:13658–13671. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Therriault MJ, Proulx LI, Castonguay A and
Bissonnette EY: Immunomodulatory effects of the tobacco-specific
carcinogen, NNK, on alveolar macrophages. Clin Exp Immunol.
132:232–238. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Liu Y and Cao X: The origin and function
of tumor-associated macrophages. Cell Mol Immunol. 12:1–4. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Proulx LI, Castonguay A and Bissonnette
EY: Cytokine production by alveolar macrophages is down regulated
by the alpha-methylhydroxylation pathway of
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK).
Carcinogenesis. 25:997–1003. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Proulx LI, Gaudreault M, Turmel V, Augusto
LA, Castonguay A and Bissonnette EY:
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone, a component of
tobacco smoke, modulates mediator release from human bronchial and
alveolar epithelial cells. Clin Exp Immunol. 140:46–53. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Mashimo M: Dual roles of α7 nicotinic
acetylcholine receptors expressed in immune cells in T cell
differentiation-α7 nAChRs exert different actions between
antigen-presenting cells and CD4(+) T cells. Yakugaku Zasshi.
140:1421–1425. 2020.(In Japanese). View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Schuller HM, Jull BA, Sheppard BJ and
Plummer HK: Interaction of tobacco-specific toxicants with the
neuronal alpha(7) nicotinic acetylcholine receptor and its
associated mitogenic signal transduction pathway: Potential role in
lung carcinogenesis and pediatric lung disorders. Eur J Pharmacol.
393:265–277. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Rahim SS, Khan N, Boddupalli CS, Hasnain
SE and Mukhopadhyay S: Interleukin-10 (IL-10) mediated suppression
of IL-12 production in RAW 264.7 cells also involves c-rel
transcription factor. Immunology. 114:313–321. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zirnheld AL, Villard M, Harrison AM,
Kosiewicz MM and Alard P: β-Catenin stabilization in NOD dendritic
cells increases IL-12 production and subsequent induction of
IFN-γ-producing T cells. J Leukoc Biol. 106:1349–1358. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Chakraborty K, Zhou Z, Wakamatsu N and
Guerrero-Plata A: Interleukin-12p40 modulates human
metapneumovirus-induced pulmonary disease in an acute mouse model
of infection. PLoS One. 7:e371732012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Guo Y, Cao W and Zhu Y: Immunoregulatory
functions of the IL-12 family of cytokines in antiviral systems.
Viruses. 11:7722019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Trinchieri G: Interleukin-12 and the
regulation of innate resistance and adaptive immunity. Nat Rev
Immunol. 3:133–146. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Caligiuri MA: Human natural killer cells.
Blood. 112:461–469. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Kinnula VL, Lehtonen S, Kaarteenaho-Wiik
R, Lakari E, Pääkkö P, Kang SW, Rhee SG and Soini Y: Cell specific
expression of peroxiredoxins in human lung and pulmonary
sarcoidosis. Thorax. 57:157–164. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Tae Lim Y, Sup Song D, Joon Won T, Lee YJ,
Yoo JS, Eun Hyung K, Won Yoon J, Park SY and Woo Hwang K:
Peroxiredoxin-1, a possible target in modulating inflammatory
cytokine production in macrophage like cell line RAW264.7.
Microbiol Immunol. 56:411–419. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Mennecier G, Torres LN, Cogliati B,
Sanches DS, Mori CM, Latorre AO, Chaible LM, Mackowiak II, Nagamine
MK, Da Silva TC, et al: Chronic exposure of lung alveolar
epithelial type II cells to tobacco-specific carcinogen NNK results
in malignant transformation: A new in vitro lung carcinogenesis
model. Mol Carcinog. 53:392–402. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Yilmaz M and Christofori G: EMT, the
cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev.
28:15–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Vu T, Jin L and Datta PK: Effect of
cigarette smoking on epithelial to mesenchymal transition (EMT) in
lung cancer. J Clin Med. 5:442016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Mehdi MZ, Pandey NR, Pandey SK and
Srivastava AK: H2O2-induced phosphorylation of ERK1/2 and PKB
requires tyrosine kinase activity of insulin receptor and c-Src.
Antioxid Redox Signal. 7:1014–1020. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Shen J, Xu L, Owonikoko TK, Sun SY, Khuri
FR, Curran WJ and Deng X: NNK promotes migration and invasion of
lung cancer cells through activation of c-Src/PKCι/FAK loop. Cancer
Lett. 318:106–113. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zhang H, Liu H, Borok Z, Davies KJ, Ursini
F and Forman HJ: Cigarette smoke extract stimulates
epithelial-mesenchymal transition through Src activation. Free
Radic Biol Med. 52:1437–1442. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Shintani Y, Okimura A, Sato K, Nakagiri T,
Kadota Y, Inoue M, Sawabata N, Minami M, Ikeda N, Kawahara K, et
al: Epithelial to mesenchymal transition is a determinant of
sensitivity to chemoradiotherapy in non-small cell lung cancer. Ann
Thorac Surg. 92:1794–1804. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Tseng YC, Tsai YH, Tseng MJ, Hsu KW, Yang
MC, Huang KH, Li AF, Chi CW, Hsieh RH, Ku HH and Yeh TS:
Notch2-induced COX-2 expression enhancing gastric cancer
progression. Mol Carcinog. 51:939–951. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Ocaña OH, Córcoles R, Fabra A,
Moreno-Bueno G, Acloque H, Vega S, Barrallo-Gimeno A, Cano A and
Nieto MA: Metastatic colonization requires the repression of the
epithelial-mesenchymal transition inducer Prrx1. Cancer Cell.
22:709–724. 2012. View Article : Google Scholar
|
|
86
|
Reichert M, Takano S, von Burstin J, Kim
SB, Lee JS, Ihida-Stansbury K, Hahn C, Heeg S, Schneider G, Rhim
AD, et al: The Prrx1 homeodomain transcription factor plays a
central role in pancreatic regeneration and carcinogenesis. Genes
Dev. 27:288–300. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelial-mesenchymal transition: New insights in
signaling, development, and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Ha B, Kim EK, Kim JH, Lee HN, Lee KO, Lee
SY and Jang HH: Human peroxiredoxin 1 modulates TGF-β1-induced
epithelial-mesenchymal transition through its peroxidase activity.
Biochem Biophys Res Commun. 421:33–37. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Yang Y, Pan X, Lei W, Wang J and Song J:
Transforming growth factor-beta1 induces epithelial-to-mesenchymal
transition and apoptosis via a cell cycle-dependent mechanism.
Oncogene. 25:7235–7244. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Derynck R and Akhurst RJ: Differentiation
plasticity regulated by TGF-beta family proteins in development and
disease. Nat Cell Biol. 9:1000–1004. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Gotzmann J, Huber H, Thallinger C,
Wolschek M, Jansen B, Schulte-Hermann R, Beug H and Mikulits W:
Hepatocytes convert to a fibroblastoid phenotype through the
cooperation of TGF-beta1 and Ha-Ras: Steps towards invasiveness. J
Cell Sci. 115:1189–1202. 2002.PubMed/NCBI
|
|
93
|
Ushio-Fukai M and Nakamura Y: Reactive
oxygen species and angiogenesis: NADPH oxidase as target for cancer
therapy. Cancer Lett. 266:37–52. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Jiang F, Qiu Q, Khanna A, Todd NW, Deepak
J, Xing L, Wang H, Liu Z, Su Y, Stass SA and Katz RL: Aldehyde
dehydrogenase 1 is a tumor stem cell-associated marker in lung
cancer. Mol Cancer Res. 7:330–338. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Huang RY, Li MY, Hsin MK, Underwood MJ, Ma
LT, Mok TS, Warner TD and Chen GG: 4-Methylnitrosamino-
1-3-pyridyl-1-butanone (NNK) promotes lung cancer cell survival by
stimulating thromboxane A2 and its receptor. Oncogene. 30:106–116.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Hung YH and Hung WC:
4-(Methylnitrosamino)-1- (3-pyridyl)-1-butanone (NNK) enhances
invasiveness of lung cancer cells by up-regulating contactin-1 via
the alpha7 nicotinic acetylcholine receptor/ERK signaling pathway.
Chem Biol Interact. 179:154–159. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Yan J, Wong N, Hung C, Chen WX and Tang D:
Contactin-1 reduces E-cadherin expression via activating AKT in
lung cancer. PLoS One. 8:e654632013. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Jin Z, Xin M and Deng X: Survival function
of protein kinase C{iota} as a novel nitrosamine
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-activated bad
kinase. J Biol Chem. 280:16045–16052. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Wang KC, Liu YC, El-Shazly M, Shih SP, Du
YC, Hsu YM, Lin HY, Chen YC, Wu YC, Yang SC and Lu MC: The
antioxidant from ethanolic extract of Rosa cymosa fruits
activates phosphatase and tensin homolog in vitro and in vivo: A
new insight on its antileukemic effect. Int J Mol Sci. 20:19352019.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Neumann CA and Fang Q: Are peroxiredoxins
tumor suppressors? Curr Opin Pharmacol. 7:375–380. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Nauseef WM: Biological roles for the NOX
family NADPH oxidases. J Biol Chem. 283:16961–16965. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Ge GZ, Xu TR and Chen C: Tobacco
carcinogen NNK-induced lung cancer animal models and associated
carcinogenic mechanisms. Acta Biochim Biophys Sin (Shanghai).
47:477–487. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Hong WG, Kim JY, Cho JH, Hwang SG, Song
JY, Lee E, Chang TS, Um HD and Park JK: AMRI-59 functions as a
radiosensitizer via peroxiredoxin I-targeted ROS accumulation and
apoptotic cell death induction. Oncotarget. 8:114050–114064. 2017.
View Article : Google Scholar : PubMed/NCBI
|