Introduction
In epigenetics, lysine acetylation has been
considered as a key step of post-translational modifications
(1,2). Histone acetyltransferases (HATs) and
histone deacetylases (HDACs) functions as ‘writers’ and ‘erasers’
respectively by controlling acetyl mark of histone lysine residue
(3). For this acetylation to be
involved in gene expression, we need a ‘reader’ to recognize
acetylated histone. Bromodomain is one of the best known modules to
recognize and bind to acetylated histones (4). In 1992, bromodomain, a protein module
containing approximately 110 amino acids, was identified as a
lysine acetylation reader in Drosophila melanogaster study
(5). In the human genome, there are
46 bromodomain-containing proteins, many of which are HATs,
HAT-associated proteins, helicases, ATP-dependent chromatin
remodeling complexes, transcriptional coactivators, and nuclear
scaffolding proteins. Among them, brd4, one of the BET (bromodmain
and extra-terminal proteins) proteins, was revealed to play a
crucial role in NUT midline carcinoma (NMC) (6). In the majority of NMC patients,
NUT gene, which is located on chromosome 15q14, is
fused with BRD4 or BRD3, creating BRD4-NUT fusion proteins. As
knockdown of BRD4-NUT in NMC caused significant decrease in
BRD4-NUT positive cell proliferation (7), brd4 has been highlighted as a powerful
therapeutic target for NMC. In addition, brd4 knockdown in AML cell
lines causes downregulation of c-myc expression as to induce cell
death (8). Subsequent studies
demonstrated that most of the leukemic and lymphoma cells die by
brd4 inhibition (9). For this
reason, many studies have been conducted to develop potent
bromodomain inhibitors (10). At
present, approximately 40 papers relevant to BET inhibitors have
been published, and 16 inhibitors are on-going in clinical
trial.
Here, we performed mid-throughput screening to
discover a new brd4 bromodomain inhibitor. We setup two biochemical
assays, alpha-screen and Homogeneous Time Resolved Fluorescence
(HTRF), we got hit compound which exhibits excellent efficacy in
vitro and in vivo assay.
Materials and methods
Cell culture
Ty82, and MKN7 cell lines were obtained from JCRB
cell bank (Japan). SNU638, SNU719, SNU668, SNU216, MKN45, MKN74 and
MKN1 cell lines were obtained from Korean cell line Bank (Korea).
All cell lines were cultured with RPMI-1640 supplemented with 10%
fetal bovine serum.
Molecular cloning and protein
expression, and purification
Brd4 cDNA was provided by Dr. Stefan Knapp from the
University of Oxford. N-terminal GST-tagged and C-terminal
His-tagged BD1 (GST-BD1-His6) was expressed in E.
coli and purified. BD1 spans 47–170 amino acids. The pGEX 6P-1
vector was digested with EcoRI and XhoI restriction
enzymes. BD1 PCR was performed with the BD1_Forward primer
(5′-ATCTAGGAATTCCCCCCAGAGACCTCCAACCC-3′) and BD1_Rev primer
(5′-ATCTAGCTCGAGTTAGTGGTGGTGGTGGTGGTGTTCGAGTGCGGCCGCAAGCTCGGTTTCTTCTGTGGGTA-3′).
BL21 Star (DE3) was transformed and induced by 0.1 mM IPTG
overnight at 18°C. The cells were lysed with lysozyme (1 mg/ml) and
sonicated in lysis buffer (50 mM NaH2PO4, 300
mM NaCl, 10 mM imidazole, and adjusted pH to 8.0 by NaOH) and
centrifuged at 8,000 rpm for 30 min. The supernatant was incubated
with Ni-NTA beads (Qiagen) for 2 h at 4°C and proteins were eluted
with elution buffer (50 mM NaH2PO4, 300 mM
NaCl, 250 mM imidazole, and adjusted pH to 8.0 by NaOH). Purified
His-tag proteins were further purified by size exclusion
chromatography on a superdex 16/600 Hiload column (GE Healthcare)
using buffer (50 mM Tris-HCl pH 7.4, 200 mM NaCl)
Alpha-screen biochemical assay
The alpha-screen assay was performed in accordance
with the manufacturer's protocol (PerkinElmer), by using a buffer
(50 mM HEPES, 100 mM NaCl, 0.1% BSA, pH 7.4 supplemented with 0.05%
CHAPS) and OptiPlate™-384 plate (PerkinElmer). Briefly, 2.5 µl of
compound solution and 5 µl of peptide solution
[SGRGK(Ac)GGK(Ac)GLGK(Ac)GGAK(Ac)RHRK-biotin] were added to 5 µl of
glutathione-S-transferase (GST) and His-tagged BD1 in
OptiPlate™-384 plate. Streptavidin-coated donor beads and anti-GST
alpha-screen acceptor beads were added under low-light condition.
Plate was incubated at 25°C for 60 min using a Thermomixer C
(Eppendorf), and read using a Fusion-Alpha™ Multilabel Reader
(PerkinElmer). The alpha-screen results were confirmed by using
alpha-screen TruHit kits (PerkinElmer).
HTRF assay
The HTRF assay was performed in 384-well black
polystyrene plate, flat bottom, low flange, non-binding surface
(Corning) in assay buffer [50 mM HEPES (pH 7.0), NaN3
0.02%, 0.01% BSA, Orthovanadate 0.1 mM]. 0.5 µM
glutathione-S-transferase (GST) and His-tagged BD1 was co-incubated
with 0.2 µM of Acetylated peptide and compounds. After 30 min
incubation at 25°C, Streptavidine-XL665 and anti-GST-Tb was added
to the reaction and incubated at 25°C for 60 min. The signal was
monitored using a microplate reader (Envision; Perkin-Elmer) using
excitation at 337 nm and dual emission at 665 and 620 nm,
respectively.
Western blotting
For immunoblotting, cells were washed in PBS, lysed
in 1X sample buffer (50 mmol/l Tris-HCl (pH 6.8), 10% glycerol, 2%
SDS, and 3% β-mercaptoethanol), and boiled for 10 min. Lysates were
subjected to SDS-PAGE followed by blotting with the indicated
antibodies and detection by western blotting substrate ECL reagent
(Thermo Fisher Scientific, Inc.). Images were produced using a
SensiQ-2000 and Image software. The following antibodies were
obtained from Cell Signaling Technology: c-Myc (cat. no. 5605).
Tubulin antibody (cat. no. T6199) was purchased from Sigma-Aldrich;
Merck KGaA. HRP-conjugated anti-mouse (cat. no. NCI1430KR), and
HRP-conjugated anti-rabbit (cat. no. NCI1460KR) antibodies were
obtained from Thermo Fisher Scientific, Inc.
Cell cytotoxic assay
For the viability experiments, cells were seeded in
96-well plates at 30% confluency and exposed to chemicals the next
day. After 72 h, WST-1 reagent was added, and absorbance at 450 nm
was measured by using a SpectraMax spectrophotometer (Molecular
Devices) in accordance with the manufacturer's instructions. The
IC50 values were calculated by using GraphPad Prism
version 5 for Windows. The curves were fitted using a nonlinear
regression model with a log (inhibitor) versus response
formula.
In vivo xenograft
Female athymic BALB/c (nu/nu) mice (6 weeks old)
were obtained from Charles River of Japan. Animals were maintained
under clean room conditions in sterile filter top cages and housed
on high efficiency particulate air-filtered ventilated racks.
Animals received sterile rodent chow and water ad libitum. Nude
mice were obtained from Charles River of Japan. Mice were
euthanized by usage of CO2. The CO2 flow rate
for euthanasia was 10–30% of the cage volume per minute. Ethics
approval for Animal experiments were obtained from the Laboratory
Animal Care and Use Committee of Korea Research Institute of
Chemical Technology. (2018-6C-10-02) Ty82 cells (5×106
in 100 µl were implanted subcutaneously (s.c.) into the right flank
region of each mouse and allowed to grow to the designated size.
Once tumors reached an average volume of 200 mm3, mice
were randomized and dosed via oral gavage daily with the indicated
doses of compounds for 14 days. Mice were treated with vehicle,
HIT-A, or A10 compound. The number of mice in each group was 6.
Mice were observed daily throughout the treatment period for signs
of morbidity/mortality. Tumors were measured twice weekly using
calipers, and volume was calculated using the formula: length ×
width2 × 0.5. Body weight was also assessed twice
weekly.
Statistical analysis
Data are presented as the mean ± standard error in 6
mice for each group. Statistical analysis was conducted using
Graphpad Prism 6 software (GraphPad Software). Statistical
comparisons between vehicle-treated and compound-treated groups
were performed using two-way ANOVA with Dunnett's multiple
comparisons test. P≤0.01 was considered to indicate a statistically
significant difference (n=6). The statistical analysis used in an
experiment is described in the figure legend.
Results
Mid-throughput screening using
alpha-screen assay
We performed MTS to identify molecules that have
inhibitory activity on brd4 bromodomain with the compound library
provided by Korea Chemical Bank (Daejeon, South Korea). We setup 2
different biochemical assays, alpha-screen assay and HTRF. In this
study, we regarded the compound hitting only one biochemical assay
as false positive, and the compound hitting both biochemical assays
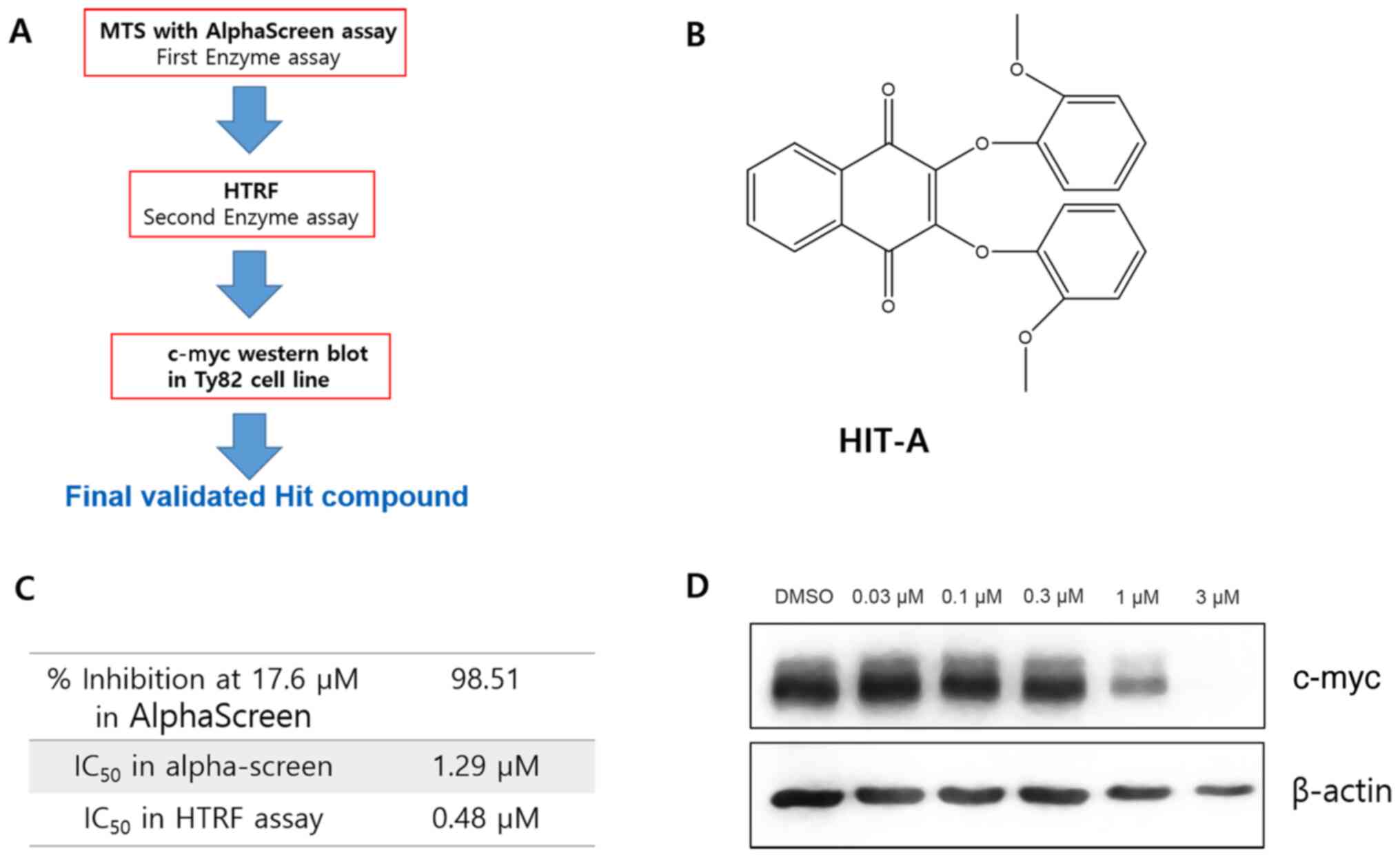
as true hit. The workflow of MTS is shown in Fig. 1A. As a result, we found 1 compound,
called as HIT-A, showing inhibition in both biochemical assays
(Fig. 1B). The IC50 of
HIT-A is 1.29 µM in alpha-screen assay, and 0.48 µM in HTRF assay
(Fig. 1C).
c-myc is known to be highly controlled by brd4
activity, so we checked the c-myc level after compound treatment to
judge whether our hit compound is effective in cells (8). The cellular c-myc level was decreased
by HIT-A in Ty82 cell line (Fig.
1D). These data demonstrate that HIT-A inhibits brd4 activity
in biochemical assay and in cellular assay.
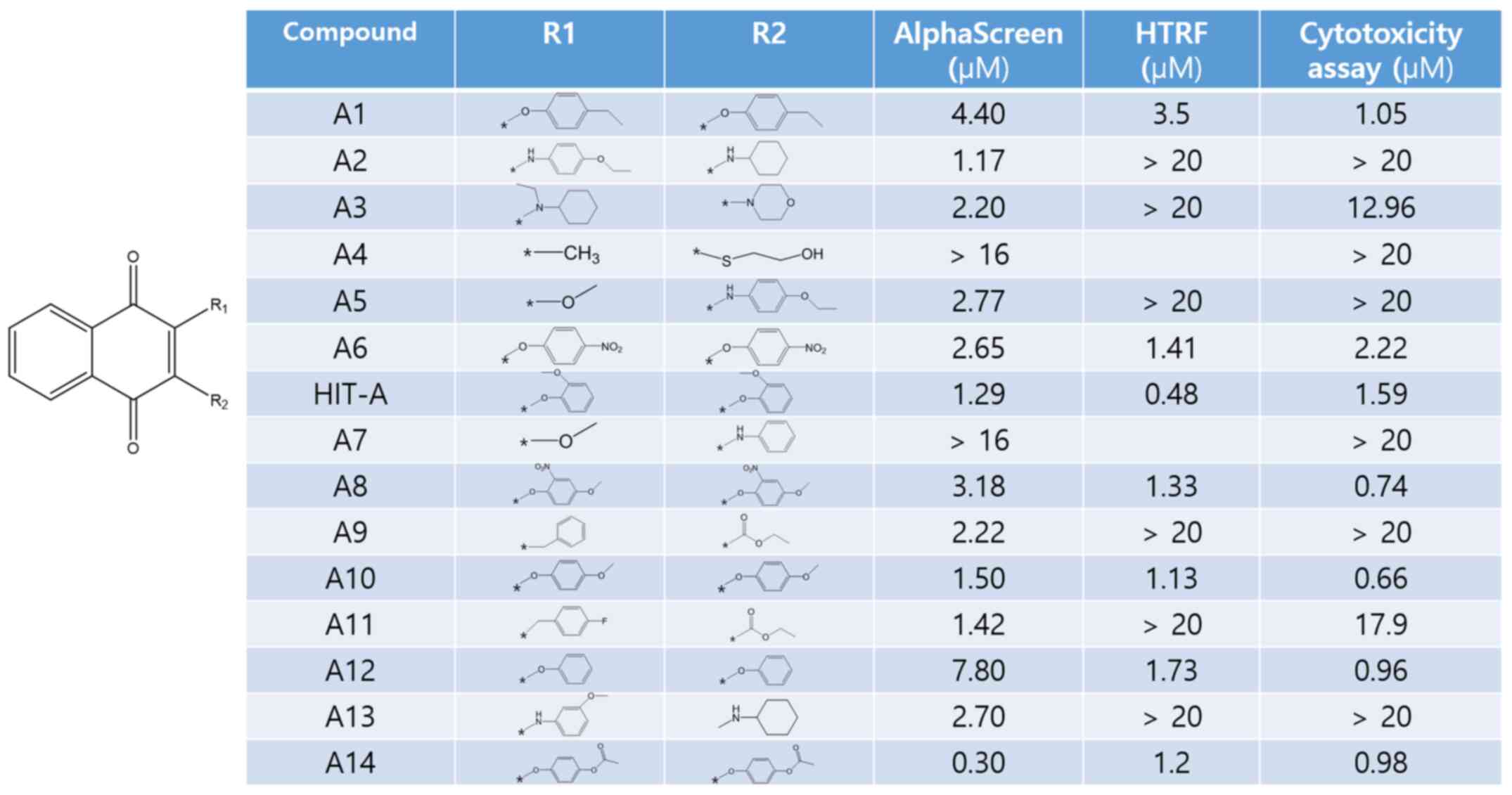
Study on HIT-A derivatives
Out of 400K compounds deposited in Korea Chemical
Bank, there are 16 compounds similar to HIT-A in structure. We have
conducted both biochemical assays and cell cytotoxic assay with all
of these compounds. Interestingly, O-linked compounds (A1, A6,
HIT-A, A8, A10, A12 and A14) showed inhibition in both biochemical
assays, whereas non O-linked compound (A2, A3, A4, A5, A7, A9, A11,
A13 and 1701) showed inhibition only in alpha-screen, not in HTRF
(Fig. 2). Because NUT midline
carcinoma (NMC) cell lines have a NUT-BRD4 fusion protein by
chromosome translocation, the proliferations of NMC cell lines are
dependent on brd4 activity (7). Ty82
is one of the NMC cell lines, and also has BRD4-NUT fusion protein
(11). Cell cytotoxic assay shows
that only O-linked compounds exert cytotoxic effect on Ty82 cells.
Non O-linked compounds don't exert any cytotoxic effect on Ty82
cells. This means that only O-linked compounds, not non O-linked,
are true brd4 bromodomain inhibitor, and non O-linked compounds are
false positive.
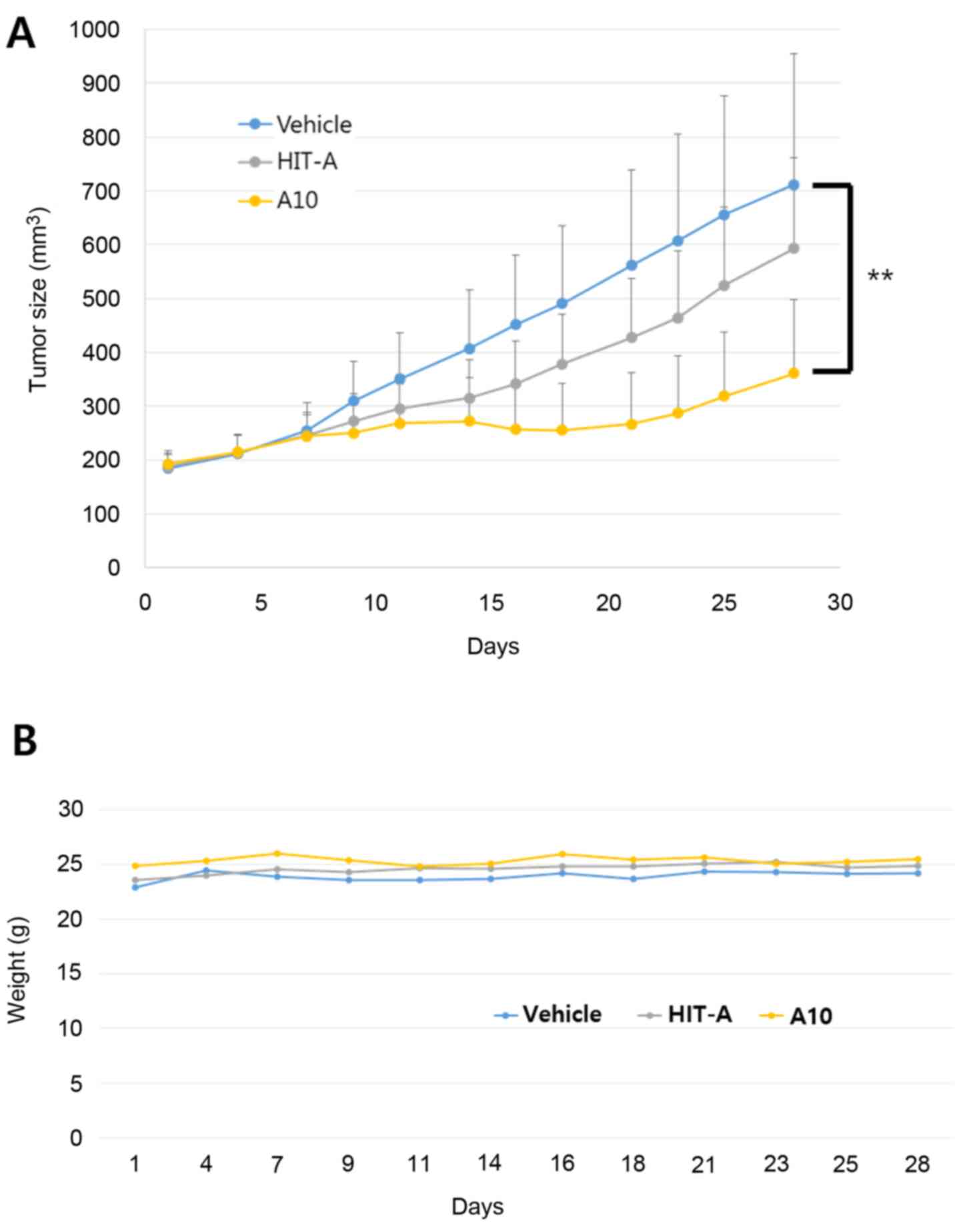
In vivo xenograft assay using
Ty82
To determine whether our hit compounds exhibit tumor
growth inhibition, we conducted an in vivo xenograft assay.
Ty82 cells were implanted in nude mice and allowed to grow to
200mm3 in size. Subsequently, HIT-A and A10 were administered
orally at daily doses of 100 mpk. Tumor volumes were measured for
28 days. As shown in Fig. 3, A10
compound effectively inhibited tumor growth. No weight loss was
shown in the mice administered with the new bromodomain inhibitors
(Fig. 3B). These results suggest
that our compound is a potent bromodomain inhibitor with a unique
scaffold in vivo.
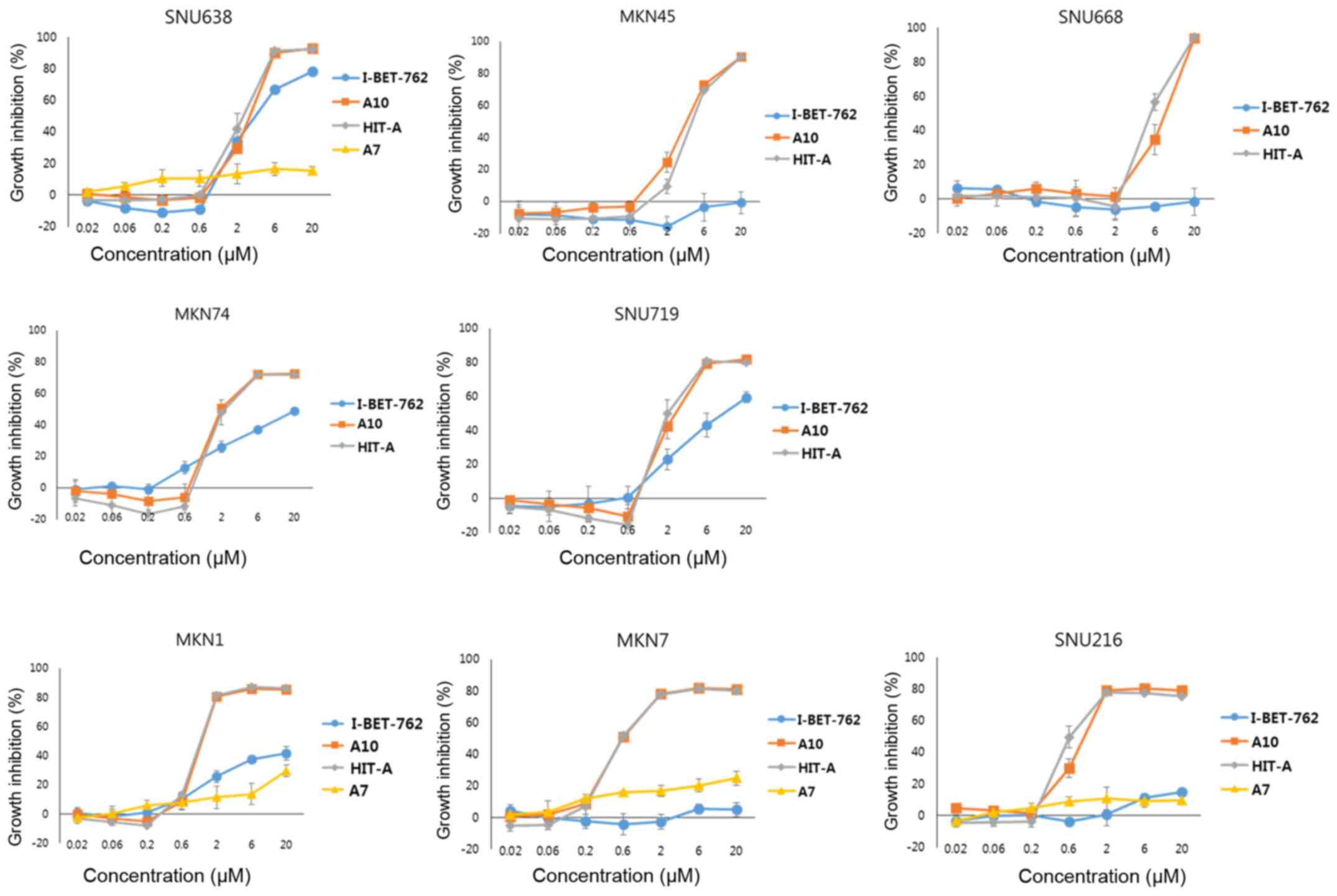
Inhibitory effect of our compound on
gastric cancer cell
Brd4 bromodomain inhibitor is known to suppress the
proliferation of hematological cancer cell. However, it is not well
known if bromodomain inhibitor is effective in solid tumor such as
gastric cancer cell. Here, we performed cytotoxic assay with
various gastric cancer cells (Fig.
4). A7, which doesn't inhibit bromodomain and is similar to hit
compound in structure, didn't suppress the proliferation of any
gastric cancer cell lines tested. I-BET-762 exerts inhibition only
on limited cell lines. However, our hit compounds, A10 and HIT-A,
show cytotoxic effect on all the gastric cancer cell lines
tested.
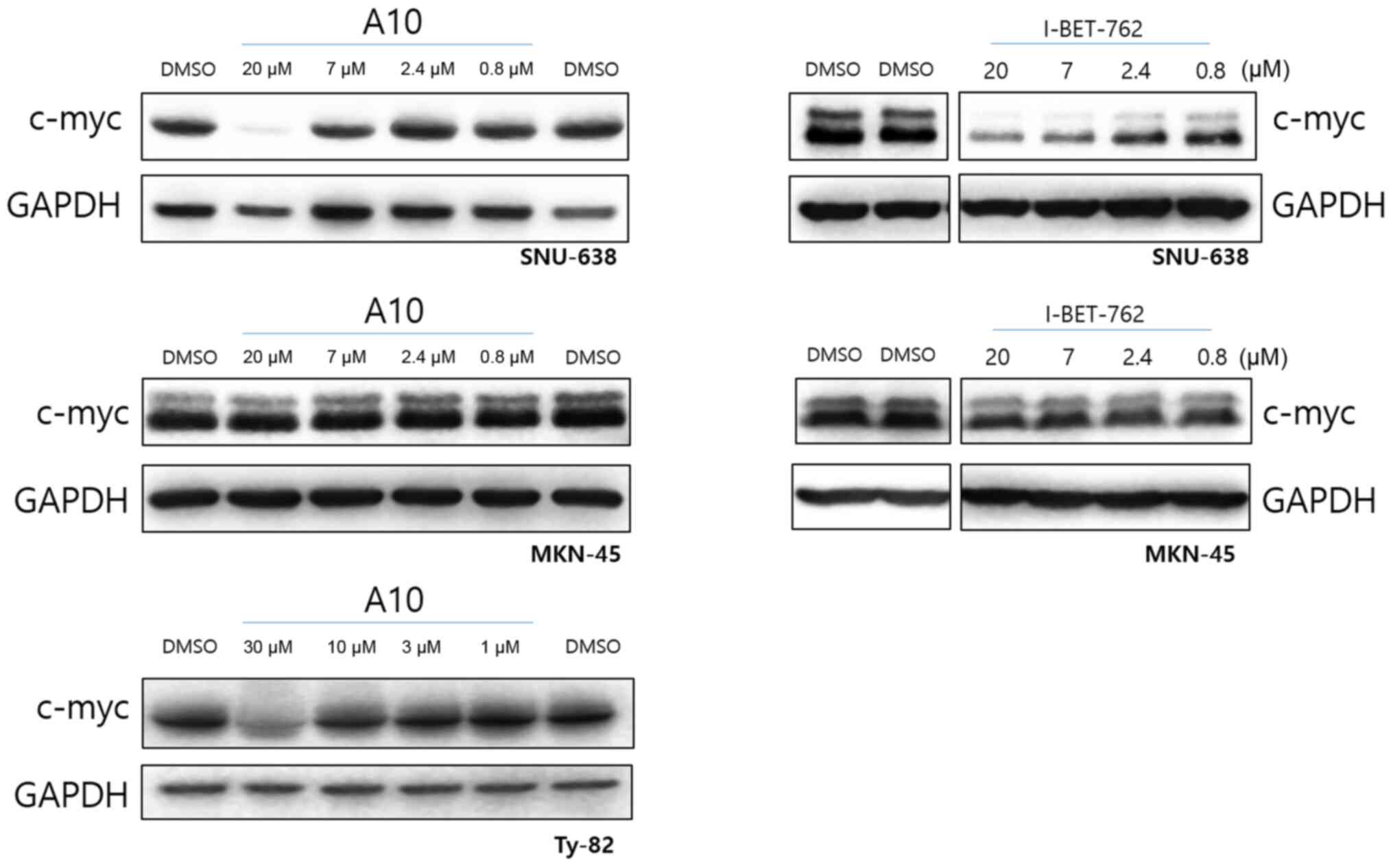
To see the c-myc protein level, we performed western
blotting with the cell lysates treated with compounds (Fig. 5). SNU-638, MNK-45 and Ty82 cells were
treated with A10 or I-BET-762. As we expected, c-myc in Ty82 cell
line is downregulated by A10. I-BET-762 downregulated the c-myc
level in SNU-638 to which I-BET-762 had cytotoxic effect. I-BET-762
did not downregulate the c-myc level of MKN-45 to which I-BET-762
had no cytotoxic effect. Interestingly, although A10 compound had
cytotoxic effect to both SNU-638 and MKN-45, it downregulated the
c-myc level only in SNU-638. It means that A10 compound has another
cytotoxic mechanism other than c-myc signaling in MKN-45.
Discussion
Compound screening such as high-throughput or
mid-throughput screening is a key step of the early stage in drug
development, to identify molecules which have activity on specific
targets. However, this step is easily weakened by a high incidence
of false-positives, which are not active toward the biological
target of interest, but active in an assay (12). False positives result from the
compound interference in assay system (13). These compound interference can be
produced solely by compounds themselves, such as fluorescent
compounds, or by their interaction with biological components in
assay system (14). One of the
powerful method to solve this problem is to use orthogonal assay
systems (15–17). Here, we setup 2 orthogonal assays for
bromodomain inhibitor screening, alpha-screen and homogeneous time
resolved fluorescence assay. In this study, alpha-screen was used
as primary screening assay for MTS. We identified more than 70 hits
in alpha-screen assay. Subsequently, HTRF assay revealed that HIT-A
compound is a true hit among 70 hits. To confirm HIT- A is a true
hit, we checked c-myc expression level after HIT-A treatment in
Ty82 cell line. Jang et al (18) reported that c-myc promoter is
regulated by brd4 protein. Yang et al (19) also reported that c-myc expression is
clearly impaired by brd4 knockdown. JQ-1, the first brd4 inhibitor,
has demonstrated significant downregulation of c-myc protein
(9). Therefore, various brd4
inhibitors were confirmed to be true hits by showing suppression of
c-myc expression in cancer cell lines (20–23).
Western blot data shows that HIT-A compound downregulates c-myc
expression in Ty82 cell line, which means that HIT-A is a true hit.
With 16 derivatives of HIT-A, we performed both biochemical assays.
Interestingly, O-linked compounds exert inhibition in both assay
systems, however, non O-linked compounds exert inhibition only in
alpha-screen. We anticipated that only O-linked compounds are real
hit, because they exerted inhibition in both assay systems. As we
expected, cell cytotoxic assay with Ty82, which is addicted to
bromodomain activity, showed that only O-linked compounds exerted
cytotoxicity in Ty82. This data reflects that our orthogonal assay
system is very effective to remove false positives. In vivo
assay, one of the hit derivatives, A10, showed excellent tumor
growth inhibition without body weight change. We tested whether our
hit compound is working on gastric cancer cells. O-linked
compounds, A10 and HIT-A, shows cytotoxic effect against gastric
cancer cells. Because A7, which didn't inhibit brd4 at all, exerted
no cytotoxicity, it is sure that the cytotoxic effect of O-linked
compound is due to the bromodomain inhibition. In addition,
O-linked compounds showed cell growth inhibition even in
I-BET-762-resistant cell lines, MKN45, SH-10-TC, SNU668, MKN7 and
SNU216. Therefore, we anticipate that our compound is more powerful
for cancer therapy than I-BET-762.
Acknowledgements
The chemical library used in the present study was
kindly provided by Korea Chemical Bank (http://www.chembank.org/) of the Korea Research
Institute of Chemical Technology (Daejeon, South Korea).
Funding
The present study was supported by the Korea
Research Institute of Chemical Technology Research Fund (grant nos.
SI1706 and SKO1706H01).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YHK and CHP confirm the authenticity of all the raw
data. CHP developed this project. YHK performed the mid-throughput
screening and found the HIT compound. JEK and MYY performed the
enzyme assay to measure the inhibitory activities. MK performed the
cell-based assay. HKL and COL performed and analyzed the in
vivo experiment. KYJ and MJY synthesized the compounds. YK
performed the cell-based assay. SUC analyzed the data. CHP designed
the study, planned the experiments, analyzed the data and wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Animal experiments were approved by the Laboratory
Animal Care and Use Committee of Korea Research Institute of
Chemical Technology (Daejeon, South Korea).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yang XJ and Seto E: Lysine acetylation:
Codified crosstalk with other post-translational modifications. Mol
Cell. 31:449–461. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kouzarides T: Chromatin modifications and
their function. Cell. 128:693–705. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jenuwein T and Allis CD: Translating the
histone code. Science. 293:1074–1080. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sanchez R, Meslamani J and Zhou MM: The
bromodomain: From epigenome reader to druggable target. Biochim
Biophys Acta. 1839:676–685. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tamkun JW, Deuring R, Scott MP, Kissinger
M, Pattatucci AM, Kaufman TC and Kennison JA: brahma: A regulator
of Drosophila homeotic genes structurally related to the
yeast transcriptional activator SNF2/SWI2. Cell. 68:561–572. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
French CA: NUT midline carcinoma. Cancer
Genet Cytogenet. 203:16–20. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
French CA, Ramirez CL, Kolmakova J,
Hickman TT, Cameron MJ, Thyne ME, Kutok JL, Toretsky JA, Tadavarthy
AK, Kees UR, et al: BRD-NUT oncoproteins: A family of closely
related nuclear proteins that block epithelial differentiation and
maintain the growth of carcinoma cells. Oncogene. 27:2237–2242.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zuber J, Shi J, Wang E, Rappaport AR,
Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al:
RNAi screen identifies Brd4 as a therapeutic target in acute
myeloid leukaemia. Nature. 478:524–528. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mertz JA, Conery AR, Bryant BM, Sandy P,
Balasubramanian S, Mele DA, Bergeron L and Sims RJ III: Targeting
MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl
Acad Sci USA. 108:16669–16674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brand M, Measures AR, Wilson BG,
Cortopassi WA, Alexander R, Höss M, Hewings DS, Rooney TP, Paton RS
and Conway SJ: Small molecule inhibitors of
bromodomain-acetyl-lysine interactions. ACS Chem Biol. 10:22–39.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
French CA, Miyoshi I, Kubonishi I, Grier
HE, Perez-Atayde AR and Fletcher JA: BRD4-NUT fusion oncogene: A
novel mechanism in aggressive carcinoma. Cancer Res. 63:304–307.
2003.PubMed/NCBI
|
|
12
|
Thorne N, Auld DS and Inglese J: Apparent
activity in high-throughput screening: Origins of
compound-dependent assay interference. Curr Opin Chem Biol.
14:315–324. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Simeonov A, Yasgar A, Klumpp C, Zheng W,
Shafqat N, Oppermann U, Austin CP and Inglese J: Evaluation of
micro-parallel liquid chromatography as a method for HTS-coupled
actives verification. Assay Drug Dev Technol. 5:815–824. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shapiro AB, Walkup GK and Keating TA:
Correction for interference by test samples in high-throughput
assays. J Biomol Screen. 14:1008–1016. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Weber E, Rothenaigner I, Brandner S,
Hadian K and Schorpp K: A high-throughput screening strategy for
development of RNF8-Ubc13 protein-protein interaction inhibitors.
SLAS Discov. 22:316–323. 2017.PubMed/NCBI
|
|
16
|
Carter DM, Specker E, Przygodda J,
Neuenschwander M, von Kries JP, Heinemann U, Nazaré M and Gohlke U:
Identification of a novel benzimidazole pyrazolone scaffold that
inhibits KDM4 lysine demethylases and reduces proliferation of
prostate cancer cells. SLAS Discov. 22:801–812. 2017.PubMed/NCBI
|
|
17
|
Dahlin JL, Nissink JW, Strasser JM,
Francis S, Higgins L, Zhou H, Zhang Z and Walters MA: PAINS in the
assay: Chemical mechanisms of assay interference and promiscuous
enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J
Med Chem. 58:2091–2113. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jang MK, Mochizuki K, Zhou M, Jeong HS,
Brady JN and Ozato K: The bromodomain protein Brd4 is a positive
regulatory component of P-TEFb and stimulates RNA polymerase
II-dependent transcription. Mol Cell. 19:523–534. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang Z, He N and Zhou Q: Brd4 recruits
P-TEFb to chromosomes at late mitosis to promote G1 gene expression
and cell cycle progression. Mol Cell Biol. 28:967–976. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Raux B, Voitovich Y, Derviaux C, Lugari A,
Rebuffet E, Milhas S, Priet S, Roux T, Trinquet E, Guillemot JC, et
al: Exploring selective inhibition of the first bromodomain of the
human bromodomain and extra-terminal domain (BET) proteins. J Med
Chem. 59:1634–1641. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao L, Wang Y, Cao D, Chen T, Wang Q, Li
Y, Xu Y, Zhang N, Wang X, Chen D, et al: Fragment-based drug
discovery of 2-thiazolidinones as BRD4 inhibitors: 2.
Structure-based optimization. J Med Chem. 58:1281–1297. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tanaka M, Roberts JM, Seo HS, Souza A,
Paulk J, Scott TG, DeAngelo SL, Dhe-Paganon S and Bradner JE:
Design and characterization of bivalent BET inhibitors. Nat Chem
Biol. 12:1089–1096. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang Y, Fang L, Chen P, Zhang H and Zhou
J: Identification of 3,5-dimethylisoxazole derivatives as BRD4
inhibitors for the treatment of colorectal cancer. ACS Med Chem
Lett. 11:2174–2181. 2020. View Article : Google Scholar : PubMed/NCBI
|