Introduction
Lung cancer is the most common cancer and the
leading cause of cancer-associated mortality worldwide (1,2). The
majority of patients with lung cancer are diagnosed during advanced
stages (3). However, for patients
diagnosed at early stages and who receive surgical resection or
definitive chemotherapy, the recurrence rate is as high as 90%
(3). Lung adenocarcinoma (LUAD) is
the most common subtype of lung cancer (4,5).
Currently, increasing evidence suggest that the prognosis of
patients with LUAD is far from ideal, with a 5-year survival rate
of only 15% (6). Thus, it is
important to identify the effective markers for diagnosis and novel
therapeutic targets of LUAD.
Multiple copies in T-cell lymphoma-1 (MCTS1), also
known as MCT-1, is an oncogene that was initially identified to be
upregulated in T-cell lymphoma (7).
Increasing evidence suggest that targeting MCTS1 can promote
genomic instability and tumorigenesis in different types of cancer
(8–10). MCTS1 is associated with
lymphomageneses, including the role of stimulation of cell
proliferation and angiogenesis, and inhibition of apoptosis
(11). Overexpression of MCTS1
attenuates cell doubling time, shortens the period of G1
transition and promotes the expression of cyclin D1 in diffuse
large B-cell lymphomas (12). Weng
et al (13) reported that
MCTS1 expression is a novel prognostic marker in breast cancer, and
upregulation of MCTS1 promotes epithelial-to-mesenchymal transition
(EMT) and activates matrix metalloproteinase in triple-negative
breast cancer cell. Notably, upregulated MCTS1 expression
accelerates the progression of lung cancer by regulating the
YY1-EGFR-MnSOD signaling pathway (13). In addition, MCTS1 mediating
IL-6/Stat3 signaling promotes the stemness of non-small cell lung
cancer (NSCLC) cells (8). However,
the role of MCTS1 in LUAD remains largely unknown.
The E2F family is composed of E2F1-3a and E2F3b,
4–8, which is predominantly regulated by the retinoblastoma family
and plays a key role in various cellular behaviors (14,15).
E2F1 is the most extensively studied transcription factor in
different types of human cancer (16–20). It
contains conserved DNA binding domains and through these putative
domains, E2F1 can closely bind to the promoter of target genes and
modulate the expression of genes participating in the cell cycle
(21). E2F1 has also been identified
as a key transcription factor in NSCLC (22). However, the association between E2F1
and MCTS1 in LUAD remains unknown.
The present study aimed to investigate the effects
of MCTS1 on the progression of LUAD and the potential mechanisms
underlying its effects. The results of the present study
demonstrated that MCTS1 functions as an oncogene in the development
of LUAD. MCTS1 expression was upregulated in patients with LUAD,
which was associated with unfavorable prognosis. Mechanistically,
MCTS1 knockdown notably suppressed LUAD cell viability and
motility. The potential pathomechanism between MCTS1 and E2F1 in
LUAD progression was also investigated. Taken together, the results
of the present study suggest that MCTS1 may be used as a
prospective biomarker to predict the therapeutic outcomes for
patients with LUAD.
Materials and methods
Tumor tissue samples
The expression of MCTS1 in LUAD tissues was analyzed
using the UALCAN cancer OMICs database (http://ualcan.path.uab.edu/index.html) from The Cancer
Genome Atlas Lung Adenocarcinoma (TCGA-LUAD) cohort, including 59
normal tissues and 515 tumor tissues. Patients with sufficient
follow-up information were included in the overall survival
analysis, using the Gene Expression Profiling Interactive Analysis
(GEPIA2, http://gepia2.cancer-pku.cn)
database.
Tumor tissues and paired peripheral normal lung
tissues (2 cm away from the tumor margin) were collected from 30
patients with LUAD (18 men and 12 women; age range, 28–79 years;
median age, 53.5 years) who underwent resection surgery at Qilu
Hospital of Shandong University between January 2018 and December
2019. The inclusion criteria were as follows: i) None of the
patients received neoadjuvant therapy; ii) study patients were
diagnosed with LUAD by histopathology and iii) diagnosis was
confirmed by two independent pathologists. Complete
clinicopathological data for the 30 paired LUAD samples were
available upon request. The present study was approved and
supervised by the Medical Ethics Committee of Qilu Hospital of
Shandong University (Jinan, China; approval no. KYLL-2016-097) and
written informed consent was provided by all patients prior to the
study start.
Cell lines and small interfering
(si)RNA transfection
The human LUAD cell lines, H1573, H23, H1299, A549,
H1703 and H1915, and the normal lung cell line, IMR90, were
purchased from the American Type Culture Collection and maintained
in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum and 1% penicillin and
streptomycin sulphate (both Hyclone; Cytiva), at 37°C with 5%
CO2. The H1573 cell line was authenticated via short
tandem repeat profiling to eliminate cross-contamination.
siRNAs (Guangzhou RiboBio Co., Ltd.) were used to
knockdown MCTS1 expression. pcDNA3.1 vector (Shanghai GenePharma
Co., Ltd.) was used to overexpress E2F1 expression in LUAD cells
(H1573 and H1299). si-MCTS1-1, si-MCTS1-2, negative control siRNA
(si-NC), pcDNA3.1-E2F1 and pcDNA3.1-NC were designed and
synthesized by Shanghai GenePharma Co., Ltd. The following
sequences were used: siMCTS1-1 forward,
5′-CCCUAAGAUUACUUCACAATT-3′and reverse,
5′-UUGUGAAGUAAUCUUAGGCTT-3′; and siMCTS1-2 forward,
5′-GCAAUUUCCAGGUAUUGAATT-3′ and reverse,
5′-UUCAAUACCUGCAAAUUGCTT-3′. For siRNA and pcDNA3.1 transfection,
cells were seeded into 6-well plates at a density of
1×106 cells/well and incubated for 24 h at 37°C. LUAD
cells were transfected with siRNAs (5 nM) or pcDNA3.1 vector (50
nM) using Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol.
Transfection efficiency was assessed via reverse-transcription
quantitative (RT-q)PCR analysis 48 h post-transfection.
MTT assay
A total of 1×104 H1573 and H1299 cells
were seeded into a 96-well plate and incubated for 24 h at room
temperature. When the cells reached 80% confluence, 10 µl MTT
reagent (5 mg/ml; Beijing Solarbio Science & Technology Co.,
Ltd.) was added to each well and cells were incubated for an
additional 2 h with dimethyl sulfoxide (Beijing Solarbio Science
& Technology Co., Ltd.). Cell proliferation was analyzed at a
wavelength of 490 nm under a spectrophotometer (Thermo Fisher
Scientific, Inc.).
EdU assay
H1573 and H1299 cells were initially incubated with
50 µM EdU solution (cat. no. ab222421; Abcam) at 37°C for 2 h.
Cells were subsequently fixed in 4% paraformaldehyde at 37°C for 30
min and treated with 0.5% Triton X-100 (Sigma-Aldrich; Merck KGaA)
at 37°C for 15 min. Cells were washed twice with PBS and 100 µl
Hoechst 33342 stain (cat. no. ab228551; Abcam) was subsequently
added to each well for 30 min at 37°C. EdU positive cells were
observed under a fluorescence microscope (Olympus Corporation,
magnification, ×100).
Colony formation assay
Tumor cells (H1573 and H1299) were seeded into
6-well plates at a density of 500 cells/well and cultured at 37°C
with 5% CO2 for 2 weeks. Cell colonies were fixed in 4%
paraformaldehyde at 37°C for 20 min and subsequently stained with
0.1% crystal violet (Sigma-Aldrich; Merck KGaA) at 37°C for 10 min.
Cell colonies were manually counted using a stereomicroscope
(Thermo Fisher Scientific, Inc., magnification, ×40).
Transwell migration assay
A total of 1×105 H1573 and H1299 cells
were plated in the upper chambers of Transwell plates in 200 µl
RPMI-1640 serum-free medium (Gibco; Thermo Fisher Scientific,
Inc.). RPMI-1640 medium (600 µl, Gibco; Thermo Fisher Scientific,
Inc.) was plated in the lower chambers. Following incubation for 48
h at 37°C, the migratory cells were fixed in 4% paraformaldehyde
for 20 min at 37°C and subsequently stained with 0.1% crystal
violet at 37°C for 10 min. Stained cells were manually counted in
five randomly selected fields using a stereomicroscope
(magnification, ×200; Thermo Fisher Scientific, Inc.).
Wound healing assay
The wound healing assay was performed to assess cell
migration. Cells (H1573 and H1299) were seeded into a 6-well plate
for 24 h at 37°C using serum-free medium. Once the cells reached
~80% confluence (or more) the monolayers were scratched using a
sterile 200 µl pipette tip. Cells were washed with PBS to remove
debris and the attached cells were cultured for an additional 48 h.
The wounded areas were observed under an inverted light microscope
(Olympus Corporation, magnification, ×100).
Flow cytometric analysis
Cell apoptosis was detected via flow cytometric
analysis, using propidium iodide (PI) and Annexin V staining
(Invitrogen; Thermo Fisher Scientific, Inc.). Briefly, transfected
cells (H1573 and H1299) were harvested and resuspended in
pre-cooled PBS buffer. Cells were centrifuged (300 × g for 5 min at
37°C) and resuspended in binding buffer (200 µl, Invitrogen; Thermo
Fisher Scientific, Inc.), and incubated with 5 µl Annexin V-FITC
and PI at room temperature for 15 min in the dark. Apoptotic cells
were subsequently analyzed using the FACS Scan (BD
Biosciences).
RT-qPCR
Total RNA was extracted from H1573, H23, H1299,
A549, H1703, H1915, and IMR90 cells using either TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) or the RNeasy
Mini kit (Qiagen, Inc). Total RNA was reverse transcribed into cDNA
using the PrimeScript™ RT reagent kit (Takara Bio, Inc.). RT was
performed at 16°C for 30 min, 42°C for 30 min and 85°C for 5 min.
qPCR was subsequently performed using the SYBR Green PCR Master Mix
(Takara Bio, Inc.) on the ABI 7900 system. The following
thermocycling conditions were used for qPCR: 95°C for 10 min
followed by 40 cycles at 95°C for 10 sec, 60°C for 20 sec and 72°C
for 30 sec. The following primer sequences were used for qPCR:
MCTS1 forward, 5′-TTCCTCGTGTGAGGGGATCT-3′ and reverse,
5′-ATAGAAAATCGGCCCCTGCT-3′; E2F1 forward,
5′-GTGCTCTCACCGTCCTACAC-3′ and reverse, 5′-CTGCACTTTCGGCCCTTTTG-3′;
and GAPDH forward, 5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse,
5′-ATGGTGGTGAAGACGCCAGT-3′. Relative expression levels were
calculated using the 2−ΔΔCq method (23) and normalized to the internal
reference gene GAPDH.
Western blotting
Total protein was extracted from H1573, H23, H1299,
A549, H1703, H1915, and IMR90 cells using RIPA buffer (Thermo
Fisher Scientific, Inc.). Total protein was quantified using the
BCA Protein Assay kit (Thermo Fisher Scientific, Inc.) and 15 µg
protein/lane was separated by SDS-PAGE on a 10% gel. The separated
proteins were subsequently transferred onto PVDF membranes (Thermo
Fisher Scientific, Inc.) and blocked with 5% non-fat milk at 37°C
for 1 h in the dark. The membranes were incubated with primary
antibodies against MCTS1 (cat. no. ab102678), E-cadherin (cat. no.
ab227639), N-cadherin (cat. no. ab76011), Vimentin (cat. no.
ab92547), B-cell lymphoma 2 (Bcl-2, cat. no. ab182858), Bax (cat.
no. ab32503), E2F1 (cat. no. ab4070) and GAPDH (cat. no. ab181602)
overnight at 4°C (all 1:1,000 and purchased from Abcam). Following
the primary incubation, membranes were incubated with secondary
antibodies (cat. no. ab7090; 1:1,000; Abcam) for 1 h at room
temperature. Protein bands were visualized using the ECL Western
Blotting kit (Thermo Fisher Scientific, Inc.) and analyzed using
Image Lab™ software (version 3.0; Bio-Rad Laboratories, Inc.).
Immunohistochemistry (IHC)
Tissue samples were fixed in 10% neutral buffered
formalin (Thermo Fisher Scientific, Inc.) at 37°C for 24 h,
dehydrated and embedded in paraffin. Tissue sections were incubated
in 3% H2O2 for 15 min at room temperature and
blocked in 10% normal goat serum (Sigma-Aldrich; Merck KGaA) for 30
min at room temperature. Paraffin-embedded tissue samples were cut
into 5-µm-thick sections. Following dewaxing and rinsing, sections
were heated in sodium citrate buffer to retrieve the antigen and
endogenous peroxidase activity was quenched. Tissue sections were
incubated with primary antibody against MCTS1 (1:1,000; Abcam; cat.
no. ab238825) overnight at 4°C. Following the primary incubation,
the sections were incubated with secondary antibody (1:1,000;
Abcam; cat. no. ab150077) at room temperature for 30 min. The
slides were subsequently stained with DAB (Sigma-Aldrich; Merck
KGaA) at room temperature for 30 min and counterstained with 0.02%
hematoxylin (Sigma-Aldrich; Merck KGaA) at room temperature for 30
sec, and observed under a stereomicroscope (magnification, ×200;
Thermo Fisher Scientific, Inc.).
Bioinformatics analysis
Data on MCTS1 expression in patients with LUAD was
retrieved from TCGA data portal (http://cancergenome.nih.gov). UALCAN was used
(http://ualcan.path.uab.edu/index.html) to plot the
figures of genes expression. Overall survival analysis of patients
with LUAD, based on MCTS1 expression, was performed using GEPIA
(http://gepia2.cancer-pku.cn). Patients
with LUAD were divided into high (MCTS1 expression >median,
n=238) and low (MCTS1 expression ≤median, n=239) MCTS1 expression
groups. Gene Set Enrichment Analysis (GSEA) (24) was performed on TCGA database dataset
(LUAD; http://cancergenome.nih.gov/) of
MCTS1 expression using R package clusterProfiler (25). For GSEA, MCTS1 expression was treated
as a numeric variable. The Pearson correlation coefficient of other
genes and MCTS1 expression was calculated, and then the genes were
sequenced according to the correlation coefficient. Using the
hallmark gene sets deposited in the GSEA Molecular Signatures
Database resource (h.all.v7.1.symbols.gmt, http://www.gsea-msigdb.org/gsea/index.jsp), the
differential pathways between the high-MCTS1 expression and
low-MCTS1 expression specimens were identified. The number of
permutations was 1,000. NES (normalized enrichment score) >1 and
FDR (false discovery rate) q-val<0.05 were set as cut-offs for
significant enrichment.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 7.0 software (GraphPad Software, Inc.) and SPSS 22.0 software
(IBM Corp.). All experiments were performed in triplicate and data
are presented as the mean ± standard deviation. Survival analysis
was performed using the Kaplan-Meier method and log-rank test. The
χ2 test was used to analyze the association between
MCTS1 expression and the clinicopathological characteristics of
patients with LUAD. A two-tailed paired Student's t-test was used
to determine statistical differences in MCTS1 expression between
LUAD tissues and matched adjacent normal tissues. Unpaired
Student's t-test was used to compare differences between two
groups, while one-way ANOVA and Tukey's post hoc test were used to
compare differences between multiple groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
MCTS1 expression is upregulated in
LUAD, which is associated with unfavorable prognosis
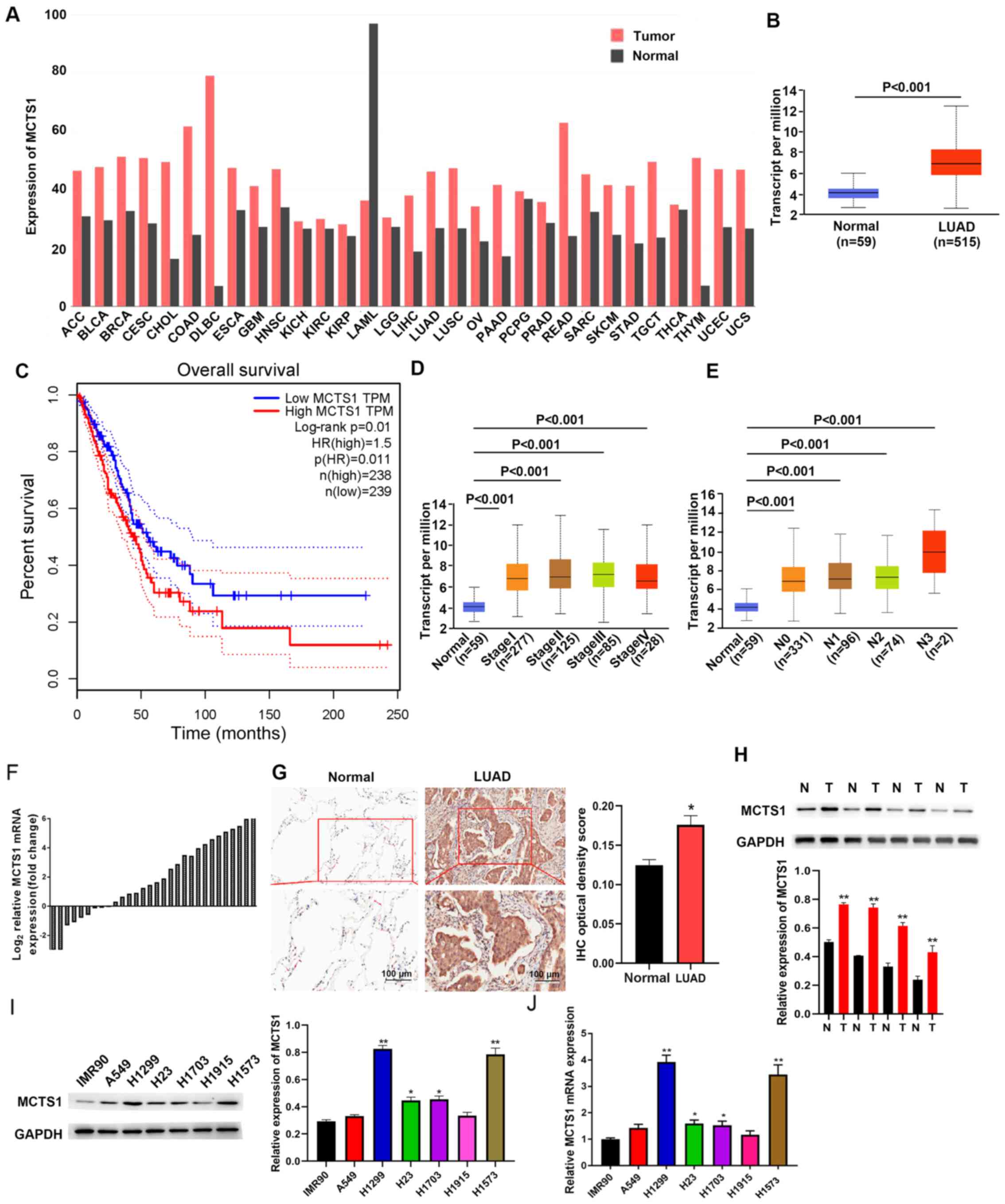
TCGA database was used to determine whether MCTS1
was aberrantly expressed in LUAD tissues compared with normal
tissues. As presented in Fig. 1A,
MCTS1 expression was upregulated in LUAD tissues compared with
normal tissues. To predict the prognostic value of MCTS1, patients
with LUAD were classified into two groups, the high and low MCTS1
expression groups, based on the mean value (cut-off value=0.5,
P=0.01, Fig. 1B). Survival analysis
demonstrated that patients with high MCTS1 expression had a shorter
overall survival time, while low MCTS1 expression was associated
with better prognosis (Fig. 1C).
Taken together, these results suggest that MCTS1 may be involved in
the progression of LUAD, and highlight the need to investigate its
cellular mechanisms.
The association between MCTS1 expression and
clinicopathological characteristics was analyzed using profiles of
patients with LUAD downloaded from TCGA database. As presented in
Fig. 1D, MCTS1 expression was
upregulated in LUAD tissues of patients at different stages.
Furthermore, MCTS1 expression was upregulated in patients with
different node metastasis status compared with the healthy controls
(Fig. 1E). Taken together, these
results suggest that MCTS1 expression is significantly associated
with stage and node metastasis in LUAD.
RT-qPCR and IHC analyses validated upregulated MCTS1
expression in LUAD samples (Fig. 1F and
G). Similarly, tissue samples resected from 30 patients with
LUAD exhibited high MCTS1 expression compared with adjacent normal
tissues (Fig. 1H). High MCTS1
expression levels were detected in H1573 and H1299 cells compared
with the other cell lines (A549, H1703, H23 and H1915) and IMR90
cells (Fig. 1I and J). Thus, H1573
and H1299 cells were selected for subsequent experimentation.
MCTS1 knockdown inhibits cell
proliferation, migration and invasion, and increases the apoptotic
rate in LUAD cancers
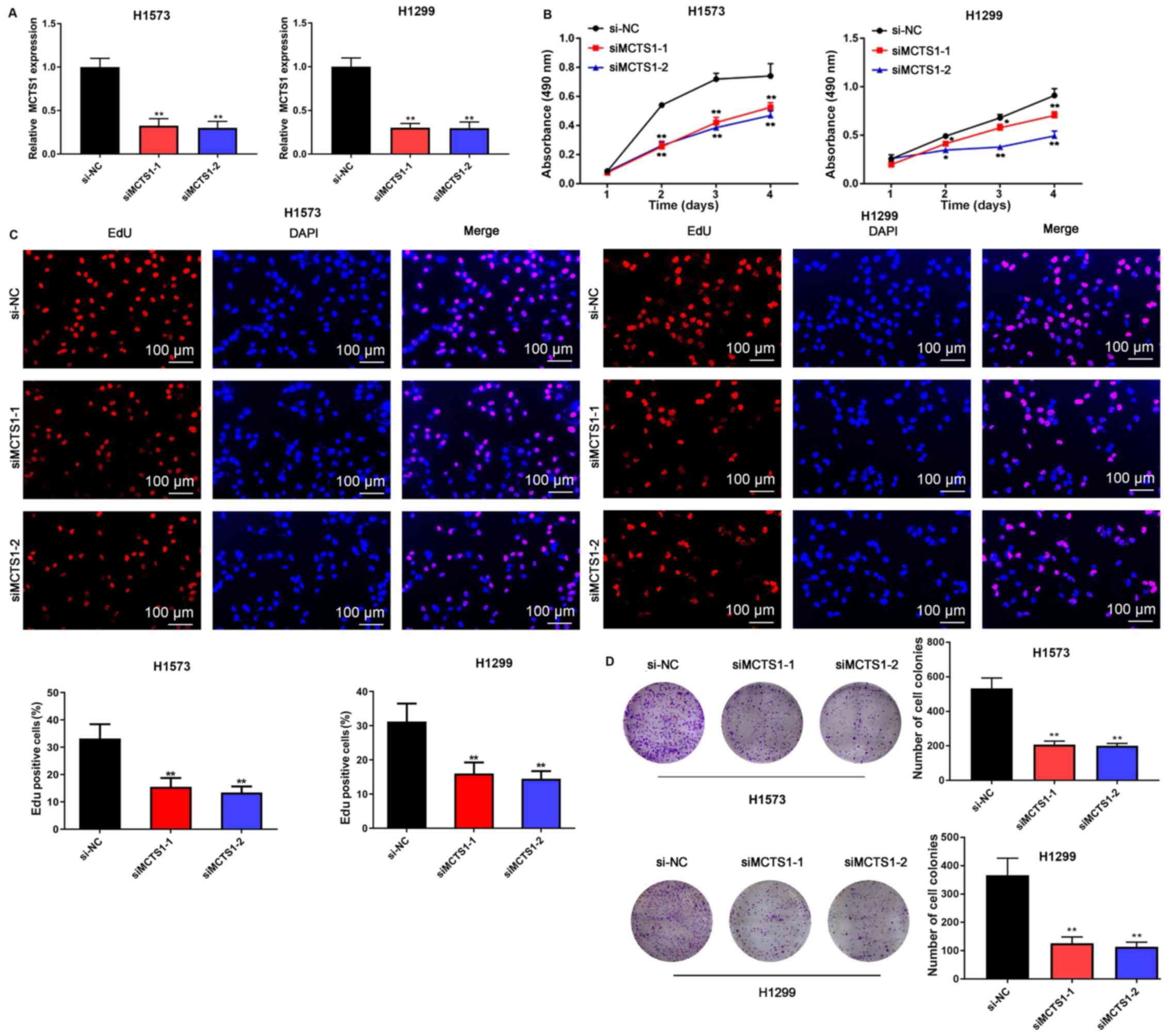
To assess the effect of MCTS1 in LUAD, an in
vitro model of MCTS1 knockdown was established and the changes
in cellular behaviors were investigated. The results demonstrated
that transfection with siMCTS1-1/siMCTS1-2 significantly attenuated
MCTS1 mRNA expression in both H1573 and H1299 cells (Fig. 2A). The MTT, EdU, colony formation,
wound healing, Transwell and flow cytometry assays were
subsequently performed to determine whether aberrant MCTS1
expression affects LUAD cellular malignant behaviors. The results
of the MTT assay demonstrated that transfection with
siMCTS1-1/siMCTS1-2 markedly inhibited the proliferative ability of
H1573 and H1299 cells (Fig. 2B).
Similarly, the results of the EdU assay indicated that the
proportion of proliferative H1573 and H1299 cells decreased
following transfection with siMCTS1-1/siMCTS1-2, suggesting that
the proliferation ability had been impaired (Fig. 2C). The results of the colony
formation assay demonstrated that MCTS1 knockdown impaired
clonogenicity of LUAD cells (Fig.
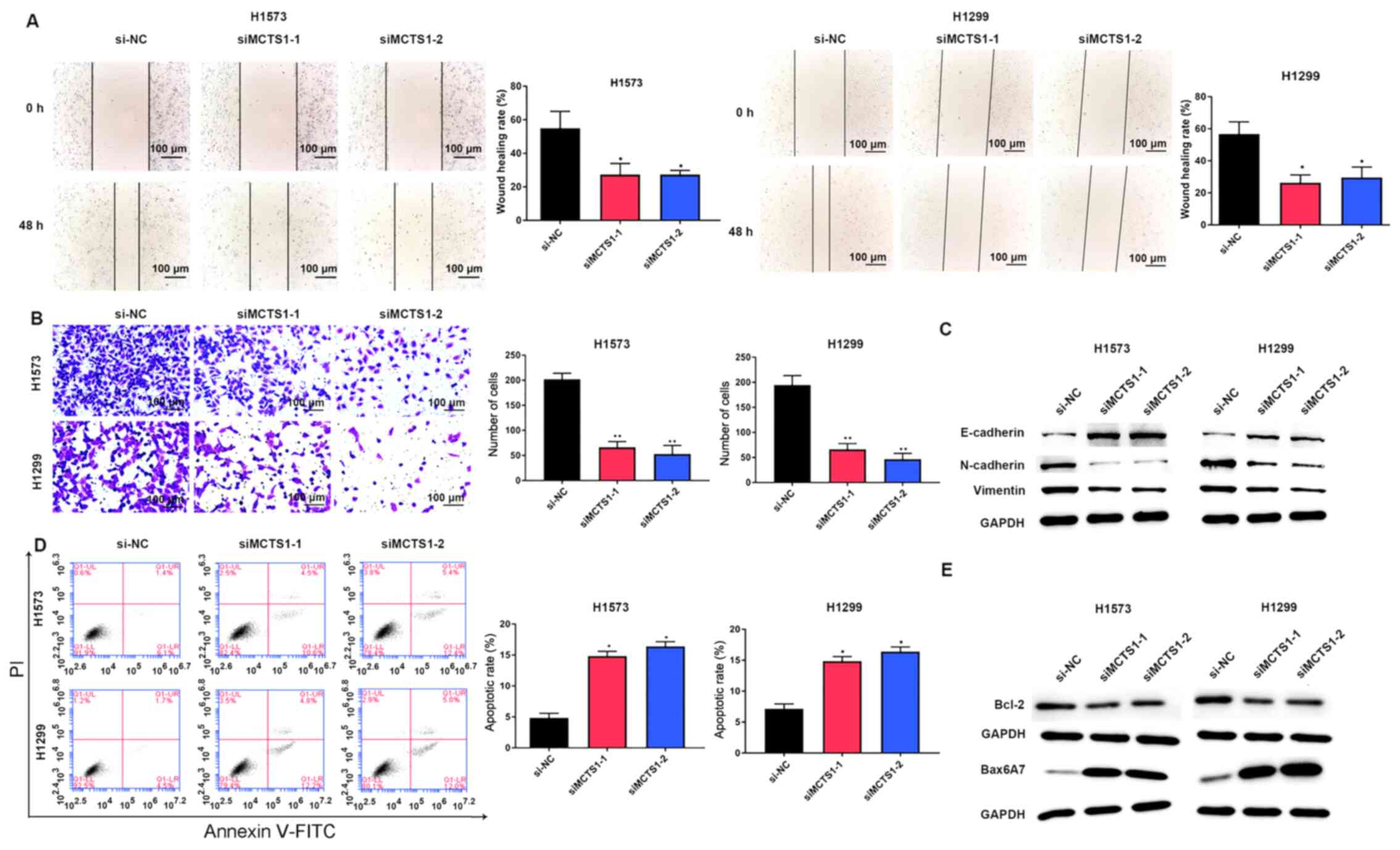
2D). Similarly, the wound healing and Transwell assays
indicated that MCTS1 knockdown significantly suppressed the
migratory and proliferative abilities of LUAD cells (Fig. 3A and B).
The EMT related proteins were investigated, and the
results demonstrated that MCTS1 knockdown significantly increased
E-cadherin expression, while the expression levels of N-cadherin
and Vimentin decreased (Fig. 3C).
Flow cytometric analysis indicated that MCTS1 knockdown increased
the apoptotic rate of LUAD cells (Fig.
3D). Given that Bcl-2 and Bax are the most representative
cellular apoptotic proteins (26),
their relative expression levels were detected. The results
demonstrated that transfection with siMCTS1-1/siMCTS1-2 inhibited
Bcl-2 expression but increased Bax expression (Fig. 3E). Collectively, these results
suggest that MCTS1 is closely associated with the proliferation of
LUAD cells in vitro.
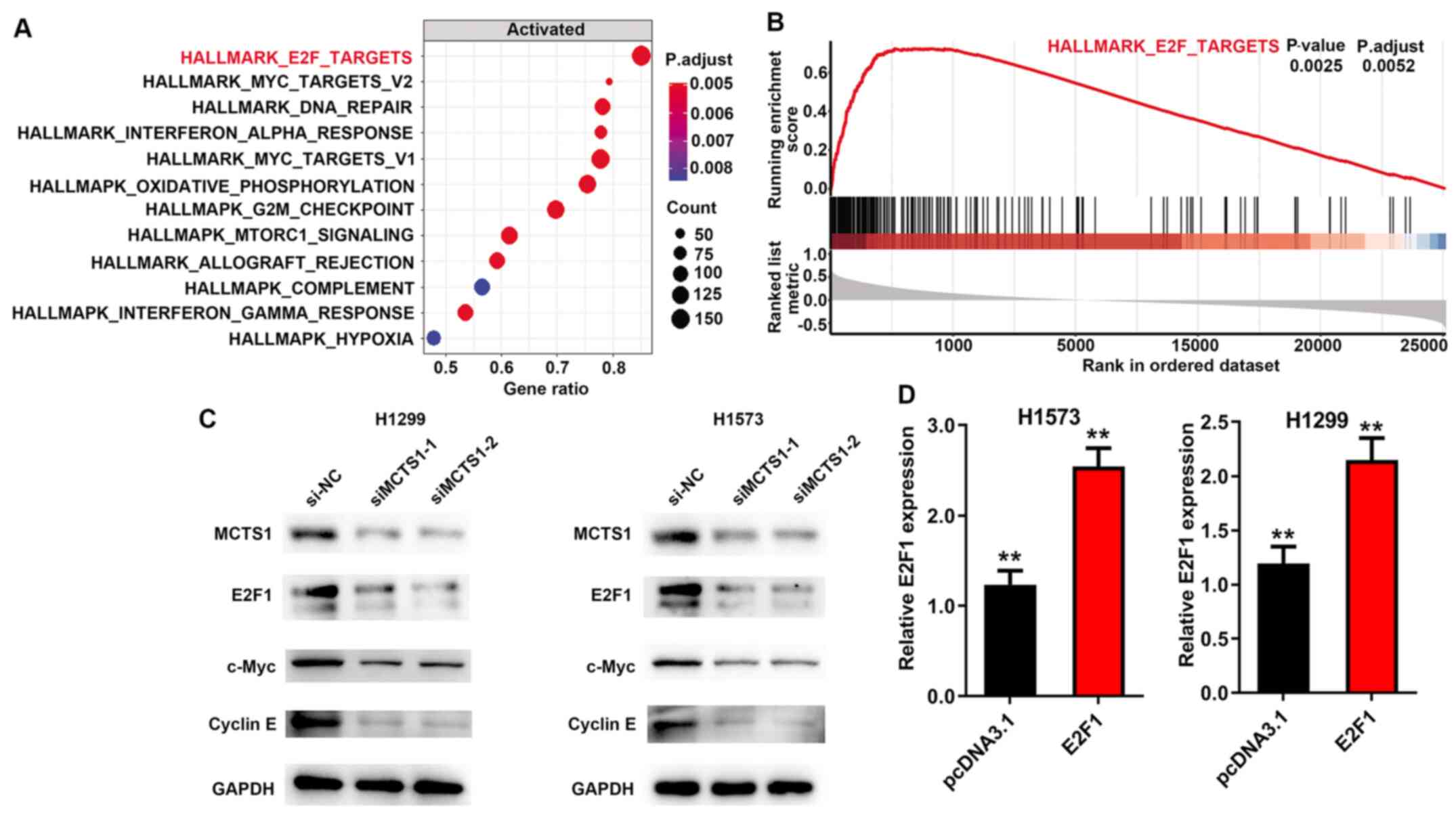
MCTS1 regulates E2F1 expression
GSEA (version 3.0, http://software.broadinstitute.org/gsea/index.jsp) was
performed to determine the potential cellular mechanism of MCTS1 in
LUAD, and to identify the pathways and corresponding biomarkers
between disease and healthy controls with distinct CIMP status in
TCGA cohort. The annotated gene set file (h.all.v7.1.symbols.gmt)
was used as the reference gene. P<0.05 was considered to
indicate statistical significance. The results demonstrated that
MCTS1 was significantly associated with the E2F family (Fig. 4A and B). Notably, a previous study
suggested that upregulation of MCTS1 modulates E2F1 expression at
the translational level (27). Thus,
it was speculated that E2F1 may participate in the regulation of
MCTS1 in LUAD. To verify this hypothesis, RT-qPCR and western blot
analyses were performed to detect E2F1 expression in LUAD cells.
The results demonstrated that transfection with siMCTS1-1/siMCTS1-2
markedly inhibited E2F1 expression in H1573 and H1299 cells
(Fig. 4C). In addition, considering
the importance of E2F1 in the c-Myc pathway (28–30),
c-Myc and cyclin E protein expression were detected in
MCTS1-silencing LUAD cells, and the results demonstrated that MCTS1
knockdown attenuated both c-Myc and cyclin E expression (Fig. 4C). Taken together, these results
suggest that MCTS1 and E2F1 may be involved in LUAD development by
modulating the c-Myc pathway. In addition, transfection with
pcDNA3.1-E2F1 significantly promoted MCTS1 mRNA expression in both
H1573 and H1299 cells (Fig. 4D).
Overexpression of E2F1 reverses the
inhibitory effect of MCTS1 knockdown on LUAD cellular malignant
behaviors
To determine whether the oncogenic role of MCTS1 is
mediated by E2F1, further functional experiments were performed in
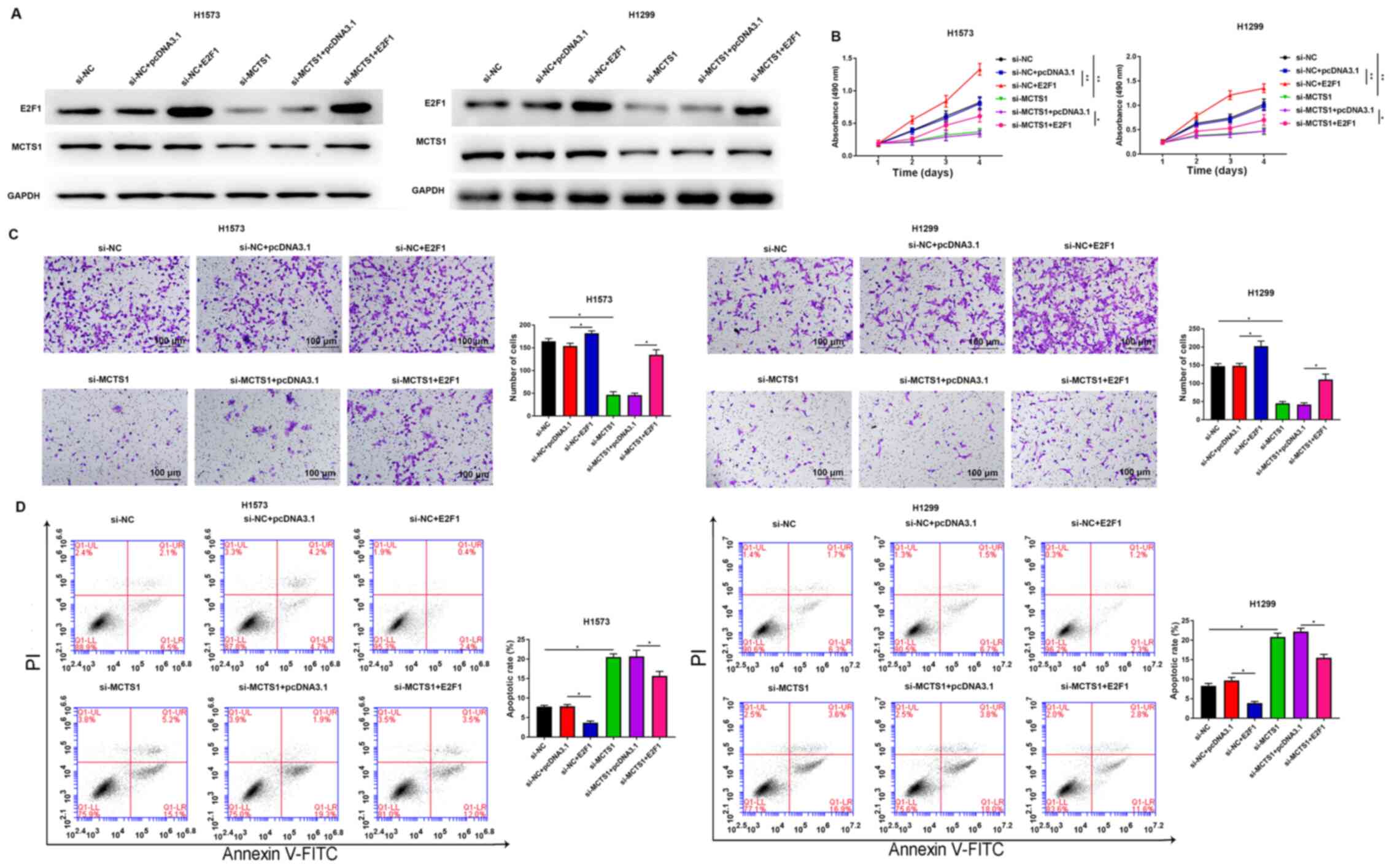
H1573 and H1299 cells. Western blot analyses of E2F1 and MCTS1 are
presented in both E2F1 overexpression and si-MCTS1 groups in
Fig. 5A. The results demonstrated
that MCTS1 knockdown decreased E2F1 expression at the translation
level. The results of the MTT assay demonstrated that
overexpression of E2F1 remarkably reversed the anti-proliferation
ability of MCTS1 knockdown in H1573 and H1299 cells (Fig. 5B). Consistently, the results of the
Transwell assay demonstrated that the suppressive migratory effect
of LUAD cells by MCTS1 knockdown was reversed following
overexpression of E2F1 (Fig. 5C).
Flow cytometric analysis demonstrated that the increasing apoptotic
rate induced by MCTS1 knockdown was markedly attenuated following
overexpression of E2F1 in LUAD cells (Fig. 5D). Collectively, these results
suggest that MCTS1 promotes cellular malignant behaviors of LUAD
cells and is closely associated with E2F1 regulation.
Discussion
Lung cancer is the most common cause of
cancer-associated mortality worldwide, and LUAD is the most common
subtype, accounting for ~40% of all lung cancer cases (31,32).
Increasing evidence suggest that patients with LUAD have
detrimental outcomes owing to the lack of effective therapy
(33). Thus, it is important to
investigate the therapeutic targets for LUAD treatment. Currently,
the variants in EGFR, BRAF, and KRAS have been confirmed as the
primary pro-cancer candidates in lung cancer (34,35).
MCTS1 has received widespread attention due to its involvement in
various malignancies, including lung and colon cancers (36,37). Guo
et al (28) suggested that
MCTS1 and MCTS4 in cancer-endothelial co-culturing
microenvironments elevate the proliferation, migration and invasion
of renal cancer cells. MCTS1 is the main contributor of lactate
uptake in tumor cells and its expression in prostate cancer cells
is associated with upregulated LDH-5 expression (38). High MCTS1 expression stimulates cell
viability, survival and anchorage-independent growth (7,39). The
oncogenicity of MCTS1 is able to overturn the inhibitory effect of
p53, and thus continuously accelerate tumorigenesis (40). In addition, MCTS1 is also associated
with the stem cell property of A549 cells (8), which suggests the important effect of
MCTS1 in lung cancer development. However, to the best of our
knowledge, only a few studies have investigated the role of MCTS1
in LUAD (8,41). The present study analyzed the
expression profiles derived from TCGA database and discovered that
MCTS1 expression is upregulated in LUAD tissues, which was
associated with the clinical stages and metastasis. Furthermore,
functional experiments demonstrated that MCTS1 facilitated
proliferation and migration, while impairing apoptosis of LUAD
cells. Taken together, these results suggest that MCTS1 may promote
the progression of LUAD by modulating tumor cell behaviors.
MCTS1 is implicated in multiple molecular mechanisms
with broad cellular activity, which has not yet been fully
elucidated (42,43). MCTS1 protein can interact with the
cap complex and ribosome via the possible RNA-binding motif, a PUA
domain, suggesting that MCTS1 may play a crucial role in mRNA
translation (44). In addition, over
the past decades, several researchers have verified that mRNA
translation functions as an essential control point in cell
proliferation and differentiation (45). The induction of malignant
transformation of cells is associated with the abnormal translation
capacity (46). To determine the
underlying molecular mechanism of MCTS1 in LUAD, GSEA pathway
analyses were performed in the present study. Th e results
demonstrated that MCTS1 expression was associated with E2F1. A
total of 15% of all human genes are regulated by the c-Myc protein,
and it is well-known that aberrant c-Myc expression is closely
associated with tumorigenesis (28,47).
E2F1 also participates in the control of c-Myc signals, depending
on the oncogenic stress (48,49).
Notably, a previous study reported that ectopic MCTS1 expression
can promote translational initiation of tumorigenesis-related
genes, including E2F1 (27). MCTS1
contains the PUA domain, a RNA-binding domain that can interact
with the cap complex through its PUA domain and recruits the
density-regulated protein, containing the SUI1 translation
initiation domain (27). MCTS1 binds
to the cap complex of E2F1 mRNAs and enhance its translation
(1). In the present study, RT-qPCR
and western blot analyses were performed to determine the
transcription and translation relevance between MCTS1 and E2F1 in
LUAD. The results demonstrated that MCTS1 knockdown decreased E2F1
expression at the translation level but no effect was observed at
the transcription level. Furthermore, rescue experiments
demonstrated that the suppressive effect of MCTS1 knockdown on LUAD
cellular behaviors was reversed following overexpression of
E2F1.
In conclusion, the results of the present study
demonstrated that MCTS1 expression was upregulated in LUAD samples,
which was associated with poor outcomes in patients with LUAD.
Functionally, MCTS1 knockdown attenuated cell proliferation and
migration, while inducing apoptosis of LUAD cells. Notably, the
anticancer role of MCTS1 knockdown was reversed following
overexpression of E2F1. Taken together, these results suggest that
MCTS1 may function as a prospective therapeutic target in the
management of LUAD treatment by mediating E2F1 and c-Myc signaling
pathways.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
HT made substantial contributions to conception and
design. CG and RD prepared the experimental materials and performed
the experiments. YL, JL and HT interpreted the data, performed the
statistical analysis and analyzed the results. CG revised and
approved the final version of the manuscript. CG and RD confirm the
authenticity of the data. All authors have read and approved the
manuscript and agree to be accountable for all aspects of the work
in ensuring that the accuracy or integrity of any part of the work
are appropriately investigated and resolved.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Qilu Hospital, Cheeloo College of Medicine, Shandong
University (Ji'nan, China; approval no. KYLL-2016-097) and written
informed consent was provided by all patients prior to the study
start.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tan WL, Jain A, Takano A, Newell EW, Iyer
NG, Lim WT, Tan EH, Zhai W, Hillmer AM, Tam WL and Tan DSW: Novel
therapeutic targets on the horizon for lung cancer. Lancet Oncol.
17:e347–e362. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang H, Guo L and Chen J: Rationale for
lung adenocarcinoma prevention and drug development based on
molecular biology during carcinogenesis. Onco Targets Ther.
13:3085–3091. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ettinger DS: Ten years of progress in
non-small cell lung cancer. J Natl Compr Canc Netw. 10:292–295.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim N, Kim HK, Lee K, Hong Y, Cho JH, Choi
JW, Lee JI, Suh YL, Ku BM, Eum HH, et al: Single-cell RNA
sequencing demonstrates the molecular and cellular reprogramming of
metastatic lung adenocarcinoma. Nat Commun. 11:22852020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mizuno K, Mataki H, Seki N, Kumamoto T,
Kamikawaji K and Inoue H: MicroRNAs in non-small cell lung cancer
and idiopathic pulmonary fibrosis. J Hum Genet. 62:57–65. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Prosniak M, Dierov J, Okami K, Tilton B,
Jameson B, Sawaya BE and Gartenhaus RB: A novel candidate oncogene,
MCT-1, is involved in cell cycle progression. Cancer Res.
58:4233–4237. 1998.PubMed/NCBI
|
|
8
|
Li Y, Wang B, Gui S and Ji J: Multiple
copies in T-cell malignancy 1 (MCT-1) promotes the stemness of
non-small cell lung cancer cells via activating Interleukin-6
(IL-6) signaling through suppressing MiR-34a expression. Med Sci
Monit. 25:10198–10204. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shih HJ, Chen HH, Chen YA, Wu MH, Liou GG,
Chang WW, Chen L, Wang LH and Hsu HL: Targeting MCT-1 oncogene
inhibits Shc pathway and xenograft tumorigenicity. Oncotarget.
3:1401–1415. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu MH, Chen YA, Chen HH, Chang KW, Chang
IS, Wang LH and Hsu HL: MCT-1 expression and PTEN deficiency
synergistically promote neoplastic multinucleation through the
Src/p190B signaling activation. Oncogene. 33:5109–5120. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Levenson AS, Thurn KE, Simons LA,
Veliceasa D, Jarrett J, Osipo C, Jordan VC, Volpert OV, Satcher RL
Jr and Gartenhaus RB: MCT-1 oncogene contributes to increased in
vivo tumorigenicity of MCF7 cells by promotion of angiogenesis and
inhibition of apoptosis. Cancer Res. 65:10651–10656. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dierov J, Prosniak M, Gallia G and
Gartenhaus RB: Increased G1 cyclin/cdk activity in cells
overexpressing the candidate oncogene, MCT-1. J Cell Biochem.
74:544–550. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Weng YS, Tseng HY, Chen YA, Shen PC, Al
Haq AT, Chen LM, Tung YC and Hsu HL: MCT-1/miR-34a/IL-6/IL-6R
signaling axis promotes EMT progression, cancer stemness and M2
macrophage polarization in triple-negative breast cancer. Mol
Cancer. 18:422019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Burkhart DL and Sage J: Cellular
mechanisms of tumour suppression by the retinoblastoma gene. Nat
Rev Cancer. 8:671–682. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen HZ, Tsai SY and Leone G: Emerging
roles of E2Fs in cancer: An exit from cell cycle control. Nat Rev
Cancer. 9:785–797. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang T, Chen X, Qiao W, Kong L, Sun D and
Li Z: Transcription factor E2F1 promotes EMT by regulating ZEB2 in
small cell lung cancer. BMC Cancer. 17:7192017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Singh S, Yennamalli RM, Gupta M and
Changotra H: Identification of nsSNPs of transcription factor E2F1
predisposing individuals to lung cancer and head and neck cancer.
Mutat Res. 821:1117042020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Farra R, Grassi G, Tonon F, Abrami M,
Grassi M, Pozzato G, Fiotti N, Forte G and Dapas B: The role of the
transcription factor E2F1 in hepatocellular carcinoma. Curr Drug
Deliv. 14:272–281. 2017.PubMed/NCBI
|
|
19
|
Farra R, Dapas B, Grassi M, Benedetti F
and Grassi G: E2F1 as a molecular drug target in ovarian cancer.
Expert Opin Ther Targets. 23:161–164. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bi XC, Pu XY, Liu JM and Huang S: Effect
of transcription factor E2F1 expression on the invasion of prostate
cancer. Zhonghua Yi Xue Za Zhi. 97:2856–2859. 2017.(In Chinese).
PubMed/NCBI
|
|
21
|
Tsantoulis PK and Gorgoulis VG:
Involvement of E2F transcription factor family in cancer. Eur J
Cancer. 41:2403–2414. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yin J, Fu W, Dai L, Jiang Z, Liao H, Chen
W, Pan L and Zhao J: ANKRD22 promotes progression of non-small cell
lung cancer through transcriptional up-regulation of E2F1. Sci Rep.
7:44302017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Porebska I, Wyrodek E, Kosacka M, Adamiak
J, Jankowska R and Harłozińska-Szmyrka A: Apoptotic markers p53,
Bcl-2 and Bax in primary lung cancer. In Vivo. 20:599–604.
2006.PubMed/NCBI
|
|
27
|
Reinert LS, Shi B, Nandi S, Mazan-Mamczarz
K, Vitolo M, Bachman KE, He H and Gartenhaus RB: MCT-1 protein
interacts with the cap complex and modulates messenger RNA
translational profiles. Cancer Res. 66:8994–9001. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guo C, Huang T, Wang QH, Li H, Khanal A,
Kang EH, Zhang W, Niu HT, Dong Z and Cao YW: Monocarboxylate
transporter 1 and monocarboxylate transporter 4 in
cancer-endothelial co-culturing microenvironments promote
proliferation, migration, and invasion of renal cancer cells.
Cancer Cell Int. 19:1702019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Stewart MJ, Litz-Jackson S, Burgess GS,
Williamson EA, Leibowitz DS and Boswell HS: Role for E2F1 in p210
BCR-ABL downstream regulation of c-myc transcription initiation.
Studies in murine myeloid cells. Leukemia. 9:1499–1507.
1995.PubMed/NCBI
|
|
30
|
Matsumura I, Tanaka H and Kanakura Y: E2F1
and c-Myc in cell growth and death. Cell Cycle. 2:333–338. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wei S, Zhang ZY, Fu SL, Xie JG, Liu XS, Xu
YJ, Zhao JP and Xiong WN: Correction to: Hsa-miR-623 suppresses
tumor progression in human lung adenocarcinoma. Cell Death Dis.
9:8292018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xiong DD, Li ZY, Liang L, He RQ, Ma FC,
Luo DZ, Hu XH and Chen G: The LncRNA NEAT1 accelerates lung
adenocarcinoma deterioration and Binds to Mir-193a-3p as a
competitive endogenous RNA. Cell Physiol Biochem. 48:905–918. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Martin LW, D'Cunha J, Wang X, Herzan D, Gu
L, Abraham N, Demmy TL, Detterbeck FC, Groth SS, Harpole DH, et al:
Detection of occult micrometastases in patients with clinical stage
I non-small-cell lung cancer: A prospective analysis of mature
results of CALGB 9761 (Alliance). J Clin Oncol. 34:1484–1491. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ali SA, Justilien V, Jamieson L, Murray NR
and Fields AP: Protein Kinase Cι Drives a NOTCH3-dependent
Stem-like phenotype in mutant KRAS lung adenocarcinoma. Cancer
Cell. 29:367–378. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Calvayrac O, Pradines A, Pons E, Mazières
J and Guibert N: Molecular biomarkers for lung adenocarcinoma. Eur
Respir J. 49:16017342017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kasiappan R, Shih HJ, Chu KL, Chen WT, Liu
HP, Huang SF, Choy CO, Shu CL, Din R, Chu JS and Hsu HL: Loss of
p53 and MCT-1 overexpression synergistically promote chromosome
instability and tumorigenicity. Mol Cancer Res. 7:536–548. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sprowl-Tanio S, Habowski AN, Pate KT,
McQuade MM, Wang K, Edwards RA, Grun F, Lyou Y and Waterman ML:
Lactate/pyruvate transporter MCT-1 is a direct Wnt target that
confers sensitivity to 3-bromopyruvate in colon cancer. Cancer
Metab. 4:202016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Giatromanolaki A, Koukourakis MI,
Koutsopoulos A, Mendrinos S and Sivridis E: The metabolic
interactions between tumor cells and tumor-associated stroma (TAS)
in prostatic cancer. Cancer Biol Ther. 13:1284–1289. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shi B, Hsu HL, Evens AM, Gordon LI and
Gartenhaus RB: Expression of the candidate MCT-1 oncogene in B- and
T-cell lymphoid malignancies. Blood. 102:297–302. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kasiappan R, Shih HJ, Wu MH, Choy C, Lin
TD, Chen L and Hsu HL: The antagonism between MCT-1 and p53 affects
the tumorigenic outcomes. Mol Cancer. 9:3112010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Huang HK, Lee SY, Huang SF, Lin YS, Chao
SC, Huang SF, Lee SC, Cheng TH, Loh SH and Tsai YT: Isoorientin
decreases cell migration via decreasing functional activity and
molecular expression of proton-linked monocarboxylate transporters
in human lung cancer cells. Am J Chin Med. 48:201–222. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Aravind L and Koonin EV: Novel predicted
RNA-binding domains associated with the translation machinery. J
Mol Evol. 48:291–302. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Johannsson S, Neumann P and Ficner R:
Crystal structure of the human tRNA guanine transglycosylase
catalytic subunit QTRT1. Biomolecules. 8:812018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fleischer TC, Weaver CM, McAfee KJ,
Jennings JL and Link AJ: Systematic identification and functional
screens of uncharacterized proteins associated with eukaryotic
ribosomal complexes. Genes Dev. 20:1294–1307. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Holcik M and Sonenberg N: Translational
control in stress and apoptosis. Nat Rev Mol Cell Biol. 6:318–327.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Andersen G, Busso D, Poterszman A, Hwang
JR, Wurtz JM, Ripp R, Thierry JC, Egly JM and Moras D: The
structure of cyclin H: Common mode of kinase activation and
specific features. EMBO J. 16:958–967. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dang CV, O'Donnell KA, Zeller KI, Nguyen
T, Osthus RC and Li F: The c-Myc target gene network. Semin Cancer
Biol. 16:253–264. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bell LA and Ryan KM: Life and death
decisions by E2F-1. Cell Death Differ. 11:137–142. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wu Z, Zheng S and Yu Q: The E2F family and
the role of E2F1 in apoptosis. Int J Biochem Cell Biol.
41:2389–2397. 2009. View Article : Google Scholar : PubMed/NCBI
|