Introduction
According to the 2018 Global Cancer Statistics, lung
cancer has the highest morbidity and mortality among all malignant
diseases (1). Although the incidence
of lung cancer is similar in China and the USA, the mortality of
lung cancer in China is 1.4 times greater than that in the USA, and
the number of deaths has been gradually increasing every year
(2). The clinical management of lung
cancer has improved with the in-depth understanding of its
molecular pathogenesis. Targeted therapies have achieved
significant improvement in patient outcomes and quality of life
compared with radiotherapy and chemotherapy (3), especially in patients with epidermal
growth factor receptor (EGFR) mutations (4,5).
Different from the general applicability of radiotherapy and
chemotherapy, targeted therapy is primarily based on the presence
or absence of certain genes and mutations, which are mainly
identified using sequencing technologies (6,7).
Next-generation sequencing (NGS) is an innovative
sequencing technology involving ‘massively parallel’ sequencing. It
has higher sensitivity and is more cost-effective and less
time-consuming compared with the single-gene mutation and/or
partial exon variation analysis, such as PCR-based analysis, Sanger
sequencing or pyrosequencing (8). As
targeted anticancer medications have been included in health
insurance in China, NGS and targeted therapy drugs are becoming
more widely applied in clinical medicine. Activating mutations in
EGFR are the prevalent targetable mutations in lung adenocarcinoma.
Currently, first-, second- and third-generation Food and Drug
Administration (FDA)-approved tyrosine kinase inhibitors (TKIs) are
in use (9,10). For example, in the treatment of
advanced anaplastic lymphoma kinase (ALK)-positive non-small cell
lung cancer (NSCLC), lorlatinib is a potent, brain-penetrant,
third-generation ALK/repressor of silencing 1 TKI with robust
clinical activity (11).
Therapeutic strategies only focus on mutation sites
that can be targeted by drugs recommended by The American Society
of Clinical Oncology (12) or The
Chinese Society of Clinical Oncology (13) guidelines. When no such mutations have
been identified, targeted therapy would be considered not
applicable. Moreover, immunotherapy is also not generally accepted,
owing to high costs, and therefore chemotherapy becomes the only
viable option (14). However, in
clinical practice, the vast majority of patients may eventually
give up chemotherapy, due to severe side effects or
chemoresistance, failure to prolong survival and even death
(15). Thus, it is still of great
importance to further identify genetic mutations that are closely
related to the poor prognosis and clinicopathological
characteristics of patients with lung cancer, thus promoting
research and development of new drugs with target mutation sites
that are complementary to currently applied targets.
The aim of the present study was to identify
mutations that were not concurrent with applicable drug target
sites. A lung cancer-specific panel of 55 genes was established by
multigenic screening in order to analyze gene mutations in 97
patients with NSCLC. Moreover, these 55 genes were further analyzed
using a mutation dataset of NSCLC obtained from The Cancer Genome
Atlas (TCGA) database. By comparing to the Catalog of Somatic
Mutations in Cancer (COSMIC) database, Network of Cancer Genes
(NCG) database and Vogelstein's list (16), 25 driver genes, in which acquired
mutations or expressed aberrantly are causally linked to cancer
progression, were identified out of the 55 genes and subjected to
functional annotation and protein-protein interaction (PPI)
analysis. Subsequently, the associations between mutations in the
25 driver genes and clinicopathological characteristics of 97
patients were examined. Using TCGA, the association between the
mutations in the 25 driver genes and overall survival of 701
patients was analyzed. Furthermore, the relationships between genes
of clinical significance, including EGFR, kirsten rat sarcoma viral
oncogene homolog (KRAS) and phosphatase and tensin homolog (PTEN)
and Notch homolog 2 (NOTCH2), were analyzed in TCGA data and in the
clinical data obtained from the patients. The findings of the
present study may promote the development of new drugs targeting
these mutations and provide new therapeutic options for patients
lacking suitable drug target sites.
Materials and methods
Patients and samples
A total of 97 patients with NSCLC admitted in The
Affiliated Hospital of Chengde Medical University (Hebei, China)
from November 2018 to July 2020 were enrolled in the current study.
The present study was approved by the Research Ethics Review
Committee of Chengde Medical University (Hebei, China; approval no.
2017003) and written informed consent was provided by all patients
prior to the study start. The inclusion criteria were: i) Patients
had a clear clinical diagnosis of NSCLC; ii) detailed clinical data
were recorded accurately and completely, including the patient's
sex, age, smoking history, histological type, clinical stage at
diagnosis, T classification, N classification, M classification,
the levels of tumor markers [carcinoembryonic antigen (CEA),
neuron-specific enolase (NSE), cytokeratin 19 fragment 21-1
(CYFRA21-1), squamous-cell carcinoma antigen (SCC) and cancer
antigen 125 (CA125)]; clinical staging was based on the American
Joint Committee on Cancer (AJCC) Classification (8th edition)
(17); and iii) patients with
available samples, including formalin-fixed and paraffin-embedded
(FFPE) tissues and blood. The patient data (97 patients) used
and/or analyzed during the current study are available from the
corresponding author upon reasonable request.
The tumor tissues were collected and fixed in 4%
formalin at 4°C for 30 min, dehydrated and embedded in paraffin.
Sample sections were cut to 5-µm thickness. Blood samples (5 ml)
were collected in ethylene diamine tetra-acetic acid anticoagulant
tubes and gently inverted 8–10 times. All the patients had no
history of blood transfusion within 4 weeks and fasted for 8–12 h
prior to blood sample collection. None of the blood samples used
for NGS had signs of hemolysis, lipid turbidity or jaundice.
A TCGA dataset (Nat Genet 2016) (18) containing data from 1,144 lung cancer
patient samples were downloaded from cBioPortal database (v3.0.2;
http://www.cbioportal.org/). For
survival analysis, 701 patients among the 1,144 samples in the TCGA
dataset who met the inclusion criteria were included in the present
study. The inclusion criteria were: i) Patients had a clear
clinical diagnosis of NSCLC; ii) detailed clinical data were
recorded accurately and completely, including the patient's sex,
age, smoking history, histologic type, clinical stage at diagnosis,
T classification, N classification, M classification, overall
survival status and time; and iii) the mutations status of the 25
driver genes was described.
In addition, the intersection of the mutated genes
of patients with NSCLC and the mutations in TCGA dataset were
analyzed using Venn software (http://www.bioinformatics.com.cn/static/others/jvenn/index.html).
Design and general performance of the
lung cancer-specific 55-gene NGS panel
A lung cancer-specific 55-gene NGS panel was
established. The inclusion criteria for the gene panel were: i)
Genes from FDA-approved and/or National Comprehensive Cancer
Network-recommended drugs for the treatment of NSCLC; ii) genes
annotated by databases, such as Clinical Knowledgebase (https://ckb.jax.org/; updated in October 2017),
Oncology Knowledge Base (OncoKB; http://oncokb.org/; updated in December 2019;),
cBioPortal, My Cancer Genome database (https://www.mycancergenome.org/; updated in April
2019) and Clinical Trials (https://clinicaltrials.gov/; updated in June 2018);
and iii) genes involved in clinical studies that have been
previously reported in detail.
Genomic DNA was extracted from blood samples using
the QIAamp DNA Blood Mini kit (cat. no. 51106; Qiagen GmbH) and
from FFPE tissues using the GeneRead DNA FFPE kit (cat. no. 180134;
Qiagen GmbH). Concentration of extracted Genomic DNA (gDNA) was
measured using a Qubit 3.0 fluorometer (Thermo Fisher Scientific,
Inc.) and purity was measured by a spectrophotometer. DNA had a
concentration of at least 50 ng/µl with an OD 260/280 between
1.8–2.0. DNA integrity was assessed using 1% agarose gel
electrophoresis.
The gDNA was fragmented randomly by Covaris to
generate gDNA fragments with a maximum of 250 bp and then subjected
to three enzymatic steps: End-repair, A-tailing and adapter
ligation. DNA libraries were purified with Agencourt AMPure XP
beads (Beckman Coulter, Ins.), and PCR was performed. A TrueSeq DNA
library preparation kit (Illumina, Inc.) was used to prepare the
sequencing library according to the manufacturer's protocol. Target
sequences were enriched using commercial hybridization probes
(Nimblegen, Roche, http://www.roche.com) designed for human DNA regions
of our interest. The panel included the following 55 genes:
SLC19A1, ERCC1, TP53, ABCB1, XRCC1, DHFR, PAPD7, MTHFR, CDA, XPC,
RRM1, ESR2, GGH, EGFR, SLC29A1, GSTP1, NT5C2, PTEN, DYNC2H1, DCK,
SOD2, UGT1A1, CMPK1, RB1, CDC5L, KRAS, PIK3CA, CTNNB1, TERT, BRAF,
ERBB2, NOTCH2, ALK, RET, PDGFRA, FBXW7, FGFR3, MET, MUC4, NOS3,
ROS1, KIT, CD3EAP, ERBB4, MTOR, NRAS, APC, NF1, SMAD4, SMO, STK11,
TPMT, VHL, KDM6A and TYMS. The hybridization product was
subsequently purified, amplified and qualified. Finally, sequencing
was performed on the Illumina next500 platform (Illumina, Inc.).
Raw data in FASTQ format were processed to remove reads containing
adapters, reads containing ploy-N, and low-quality reads. All
variants had >99% confidence, as well as an average sequencing
depth of over 500X. Structural variations were annotated using the
Refseq release 96 (http://www.ncbi.nlm.nih.gov/refseq/) and Gencode gene
annotation library (https://www.gencodegenes.org/). Waterfall plots and
Circos plots were generated using R software (v 3.6.1; http://www.R-project.org).
Identification of driver gene
Three databases were used to identify driver genes,
including the COSMIC database, NCG database, and Vogelstein's list.
The COSMIC database is the largest and most comprehensive resource
for exploring the impact of somatic mutations in human cancer in
the world, which contains 576 genes. The NCG database is a manually
curated repository of 2,372 genes whose somatic modifications have
known (711 genes) or predicted (1,661 genes) cancer driver roles
(19), and 125 mutated driver genes
were identified in Vogelstein's study with a total of 294,881
mutations being reported (16). The
intersection of the common mutant genes both in clinical and TCGA
data and the specific genes in the three databases were analyzed
using Venn software (v2.0; http://www.bioinformatics.com.cn/static/others/jvenn/index.html).
Actionable target analysis
The analysis of actionable mutations was carried out
using the Pharmacogenomics Knowledgebase (PharmGKB; http://www.pharmgkb.org/) for single nucleotide
polymorphisms (SNPs) and OncoKB for single nucleotide variants
(SNVs). The variants-drug relationships were compiled by combining
the mutation data and the two annotation databases. The SNPs were
evaluated using the PharmGKB database to identify potentially
actionable variants that could reflect the efficacy and toxic side
effects of chemotherapeutic drugs. The SNV variant sites, which may
guide targeted therapy, were annotated using the OncoKB database,
which is a comprehensive precision oncology database that offers
evidence-based drug information on FDA-approved therapies and other
investigational agents. The final list was filtered based on
previously published preclinical data and clinical trials in
cancer.
Function annotation analysis
Gene Ontology (GO) annotation and Kyoto Encyclopedia
of Genes and Genomes (KEGG) pathway enrichment analysis were
performed using the DAVID v6.8 online tool (https://david-d.ncifcrf.gov/). GO functional
categories included biological process, cellular component and
molecular function. Statistically significant terms were determined
using a two-sided P<0.05.
Pathogenicity prediction
Variant deleteriousness was predicted using the
Polymorphism Phenotyping version 2 (Polyphen-2) database (v2.2.2;
http://genetics.bwh.harvard.edu/pph2/) (20) and the MutationTaster web-based tool
(v2; http://www.mutationtaster.org/)
(21). The Polyphen-2 score and the
MutationTaster score were calculated. Specifically, the effects of
SNVs and SNPs were evaluated using the Polyphen-2 database and
classified into three categories (probably damaging, possibly
damaging and benign), based on their predicted effect on protein
function. The insertions and deletions (indels) were analyzed by
the MutationTaster tool, which categorizes mutations into one of
four possible types: i) Disease-causing (probably deleterious); ii)
disease-causing automatic (known to be deleterious); iii)
polymorphism (probably harmless); and iv) polymorphism automatic
(known to be harmless).
PPI network construction and mutual
exclusivity analysis
The PPI network was analyzed using the STRING online
tool (v11.0; http://string-db.org/). The list of
official gene names was imported into STRING and the species was
set to Homo sapiens. The medium confidence was set to 0.400.
The MCODE plug-in in Cytoscape (v3.6.2, www.cytoscape.org) was used to screen for PPI network
modules with the following parameters: i) Degree cutoff, 2; ii)
node score cutoff, 0.2; iii) k-core, 2; and iv) max depth, 100. The
hub genes were defined by the maximal clique centrality (MCC)
algorithm in the cytoHubba plug-in in Cytoscape. Mutual exclusivity
analysis was performed on driver genes using the mutual exclusivity
tool in the cBioPortal database.
Statistical analysis
Statistical analysis was performed using SPSS
software (version 19; SPPS, Inc.). All metric and normally
distributed data are presented as mean ± standard deviation,
non-normally distributed data as median (25-75th percentile), and n
(%) for categorical data. Scatter plots were plotted using GraphPad
Prism 8.0 (GraphPad Prism Software lnc.).
The clinicopathological characteristics from the
patients with NSCLC (with or without mutations) were compared using
the χ2 or Fisher's exact test if the expected cell value
was <5. The factors with two-sided P<0.1 were considered
potentially significant. Furthermore, logistic regression was used
to evaluate the correlations between these factors. Logistic,
ordered logistic and multinomial logistic regression analysis were
used for binary, ordered and unordered categorical outcome variable
analysis, respectively. If the two-sided P-value of the parallelism
test was <0.05 in the ordered logistic regression, multinomial
logistic regression was used. Two-sided P<0.05 was considered to
indicate a statistically significant difference.
For survival analysis, 701 patients among the 1,144
samples in the TCGA dataset who met the inclusion criteria were
included in this study. Univariate Cox regression analysis was used
to screen for significant variables (two-sided P<0.05) for
multivariate Cox analysis. The Kaplan-Meier method and log-rank
test were used to generate an analyze the survival curves.
Following the integration of 97 patient data and 701
TCGA data, the correlations among EGFR, PTEN, KRAS and NOTCH2
mutations were assessed using Kendall's τ-b correlation test.
P<0.05 was considered to indicate a statistically significant
difference. Correlation coefficient (CC) values range from +1 to
−1, with positive value representing positive correlation and
negative value representing negative correlation and 0 value
representing no correlation.
Results
Clinicopathological characteristics of
the patients with NSCLC
The clinicopathological characteristics of the 97
patients are presented in Table I. A
total of 46 (47.4%) patients were males and 51 (52.6%) were
females. The patient age ranged from 28–75 years, with a median age
of 59 years. Patients aged >60 years accounted for 42.3% (41/97)
of the cohort, whereas those aged ≤60 years old represented 57.7%
(56/97). Moreover, 21 (21.6%) patients had smoking history, while
76 (78.4%) patients had never smoked. For the histological type, 80
(82.5%) patients had adenocarcinoma, 12 (12.4%) patients had
squamous cell carcinoma and 5 patients (5.1%) had other NSCLC
types. According to the AJCC clinical staging criteria, 10 (10.3%)
patients had stage I or II, 12 (12.4%) had stage III and 75 (77.3%)
had stage IV. In terms of TNM classification, 11 (11.3%) patients
were of T1 classification, 45 (46.4%) were of T2 classification, 21
(21.7%) were of T3 classification and 20 (20.6%) were of T4
classification. In addition, 16 (16.5%) patients had N0
classification, 10 (10.3%) had N1 classification, 43 (44.3%) had N2
classification and 28 (28.9%) had N3 classification. A total of 23
(23.7%) patients were of M0 classification and 74 (76.3%) were of
M1 classification. Moreover, 66 (68.0%), 49 (50.5%), 67 (69.1%), 8
(8.2%), and 52 (53.6%) patients exhibited increased CEA, NSE,
CYFRA21-1, SCC, and CA-125, respectively’.
 | Table I.Clinicopathological characteristics
of patients with non-small cell lung cancer. |
Table I.
Clinicopathological characteristics
of patients with non-small cell lung cancer.
| Clinicopathological
characteristics | n (%) |
|---|
| Total | 97 (100.0) |
| Sex |
|
|
Male | 46 (47.4) |
|
Female | 51 (52.6) |
| Age, years |
|
|
≤60 | 56 (57.7) |
|
>60 | 41 (42.3) |
| Smoking
history |
|
| Never
smoker | 76 (78.4) |
|
Current/Former | 21 (21.6) |
| Histological
type |
|
|
Adenocarcinoma | 80 (82.5) |
|
Squamous cell carcinoma | 12 (12.4) |
|
Other | 5 (5.1) |
| Stage at
diagnosis |
|
|
I/II | 10 (10.3) |
|
III | 12 (12.4) |
| IV | 75 (77.3) |
| T
classification |
|
| T1 | 11 (11.3) |
| T2 | 45 (46.4) |
| T3 | 21 (21.7) |
| T4 | 20 (20.6) |
| N
classification |
|
| N0 | 16 (16.5) |
| N1 | 10 (10.3) |
| N2 | 43 (44.3) |
| N3 | 28 (28.9) |
| M
classification |
|
| M0 | 23 (23.7) |
| M1 | 74 (76.3) |
| CEA, ng/ml |
|
| ≤5 | 31 (32.0) |
|
>5 | 66 (68.0) |
| NSE, ng/ml |
|
|
≤16.3 | 48 (49.5) |
|
>16.3 | 49 (50.5) |
| CYFRA21-1,
ng/ml |
|
|
≤3.3 | 30 (30.9) |
|
>3.3 | 67 (69.1) |
| SCC antigen,
ng/ml |
|
|
≤2.7 | 89 (91.8) |
|
>2.7 | 8 (8.2) |
| CA125, U/ml |
|
|
≤35 | 45 (46.4) |
|
>35 | 52 (53.6) |
Mutational analysis
A lung cancer-specific 55-gene panel was established
and analyzed using NGS. All 97 patients harbored gene mutations and
the mutational profile of 55 genes was summarized in Fig. 1A. The most frequent gene mutations
were found in SLC19A1 (100%), ERCC1 (94%), ABCB1 (92%), TP53 (90%)
and XRCC1 (90%). Overall, five types of mutations were observed,
namely SNVs, SNPs, indels, copy number variants (CNVs) and fusions.
SNVs mostly occurred in the EGFR, PTEN, RB1 and KRAS genes, which
were either confirmed or potential molecular targets, while the
common mutation types of SLC19A1 and ERCC1 genes were identified as
SNPs, which could reflect chemotherapeutic drug efficacy and toxic
side effects. Some genes, such as TP53 and EGFR, displayed both
SNVs and SNPs. The other three mutation types were less observed in
this study. Indels occurred in 13 genes in total. Only EGFR
harbored a CNV and ALK harbored a fusion.
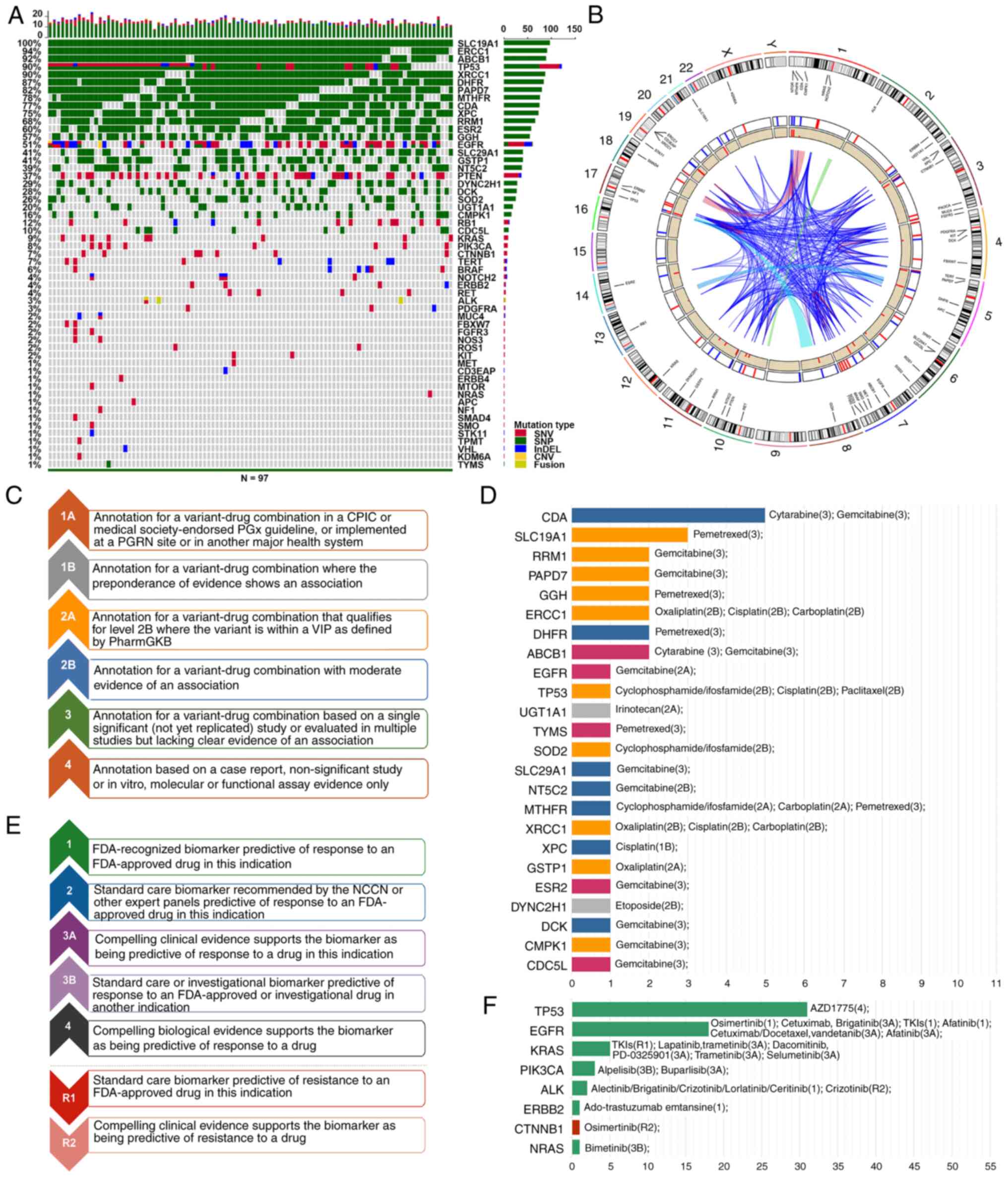 | Figure 1.Mutational analysis of 55 lung
cancer-specific genes in 97 patients with NSCLC. (A) Waterfall plot
of mutational landscape. The 55 mutated genes identified using NGS
were ranked by mutant frequency. Each row represents a gene and
each column represents a patient. (B) Circos view of the 55 mutant
genes on chromosomes. Starting from the outside, the first circle
indicates the distribution of the 55 genes across the human genome
indicated by lines. The second circle represents two therapeutic
modalities: Chemotherapy (blue) and targeted therapy (red). The
third circle represents the gene mutation rates. The lines in the
center area represent interconnections among 55 genes. (C-F)
Actionable analysis of 55 gene mutations. (C) Evidence levels of
the PharmGKB database. (D) Number of SNPs and drug annotation
information. Each horizontal bar represents a gene. The x-axis
indicates the number of mutations. The mutation sites were
annotated using the PharmGKB database, and the corresponding
chemotherapeutic drugs and their evidence levels are shown. The
color of columns represents the drug efficacy and the toxic side
effects. Yellow represents better drug efficacy and purple
represents worse drug efficacy. Gray represents lower toxic side
effects and blue represents higher toxic side effects. (E) Evidence
levels of the OncoKB database. (F) Number of SNVs and drug
annotation information. Each horizontal bar represents a gene. The
x-axis indicates the number of mutations. The mutation sites were
annotated using the OncoKB database, and the corresponding targeted
drugs and their evidence levels are shown. The color of columns
represents drug susceptibility. Green represents sensitivity to
annotated drugs and red represents resistance to annotated drugs.
Indel, insertion-deletion; FDA, Food and Drug Association; NSCLC,
non-small cell lung cancer; NGS, next-generation sequencing;
PharmGKB, Pharmacogenomics Knowledgebase; SNP, single nucleotide
polymorphism; OncoKB, Oncology Knowledge Base; SNV, single
nucleotide variants; CNV, copy number variants; CPIC, Clinical
Pharmacogenetics Implementation Consortium; PGx, pharmacogenomics;
PGRN, Pharmacogenomics Research Network; VIP, Very Important
Pharmacogene, FDA, Food and Drug Administration; NCCN, National
Comprehensive Cancer Network. |
The chromosomal locations of the 55 genes were then
examined (Fig. 1B). Chromosomes 1
and 7 were enriched in most of the genes (12 in total) followed by
chromosomes 3, 4 and 6. Among the 55 genes, SLC19A1, which had the
highest frequency of the SNPs, was located on chromosome 21 and
EGFR, which had the highest SNV frequencies, was mapped to
chromosome 7 (Fig. 1B; outer
circle).
Moreover, 33 genes were therapeutic targets and 24
genes could reflect efficacy and toxic side effects of
chemotherapeutic drugs. Among them, EGFR and TP53 could be used to
indicate the applicability of both targeted therapy and
chemotherapeutic drugs (Fig. 1B;
middle circle). Mutation rates were consistent with the results
shown in Fig. 1A (Fig. 1B; innermost circle). Additionally, a
total of 386 interactions among 54 genes constituting a gene-gene
network was identified. Only one gene, CDC5L, did not interact with
any other gene.
To evaluate the actionable mutations that could
guide treatment decisions, the annotated information in PharmGKB
and OncoKB databases were added to the lung cancer-specific 55-gene
panel. A total of 24 SNPs that could reflect the efficacy and the
toxic side effects of chemotherapeutic drugs were identified using
the PharmGKB database (Fig. 1C and
D). Among them, 7 genes (CDA, DHFR, SLC29A1, NT5C2, MTHFR, XPC
and DCK) were associated with highly toxic side effects, while
UTG1A1 and DYNC2H1 indicated low toxic side effects. A total of 10
gene mutations (SLC19A1, RRM1, PAPD7, GGH, ERCC1, TP53, SOD2,
XRCC1, GSTP1 and CMPK1) were associated with good drug efficacy.
Among them, the p.Pro72Arg mutation in TP53 suggested that
cisplatin, cyclophosphamide and ifosfamide, might show better
efficacy and lower toxic side effects. ABCB1, EGFR, TYMS, ESR2 and
CDC5L were associated with poor drug efficacy.
In addition, the OncoKB database was used to
annotate confirmed or potential target genes (Fig. 1E). Altogether, 8 genes were
identified as the targets of FDA-approved or other
evidence-supported drugs (Fig. 1F).
Only one actionable mutation in CTNNB1 indicated resistance to
osimertinib, suggesting that osimertinib should not be recommended
for patients with CTNNB1 mutations. The mutations in the remaining
genes reflected sensitivity to the corresponding targeted therapy
drugs. Although TP53 had the most targeted sites, only the drug
AZD1775 was annotated as the lowest supporting level (level 4,
Fig. 1F). Moreover, EGFR had up to
18 actionable mutations targeted by osimertinib, cetuximab,
brigatinib, afatinib, cetuximab and vandetanib (Fig. 1F).
Comparison with TCGA database
In order to evaluate the universality of the lung
cancer-specific 55-gene panel in patients with NSCLC, the
mutational status of the clinical cohort was compared with TCGA
NSCLC cohort (18). The clinical
characteristics of 1,144 patients were obtained, including sex,
diagnosis age, smoking history, histologic types, clinical stage, T
classification, N classification, M classification and overall
survival status (Fig. 2A-I).
Overall, each sample had ≥1 mutation and the 1,144 samples harbored
a total of 17,618 mutations. The top 50-ranked genes are plotted in
Fig. 2J according to the mutation
rate. Mutation frequency was highest for TP53 (67.70%), followed by
TTN (59.60%), CSMD3 (41.70%), MUC16 (40.50 %) and RYR2 (39.20%). In
addition, the intersection of the 55 mutated genes of lung
cancer-specific panel and the 17,618 mutations in TCGA dataset were
analyzed (Fig. 2K). Notably, of the
55 genes, 54 genes presented mutations in both two cohorts, and
only UGT1A1 was not included in the mutation profile of the TCGA
dataset.
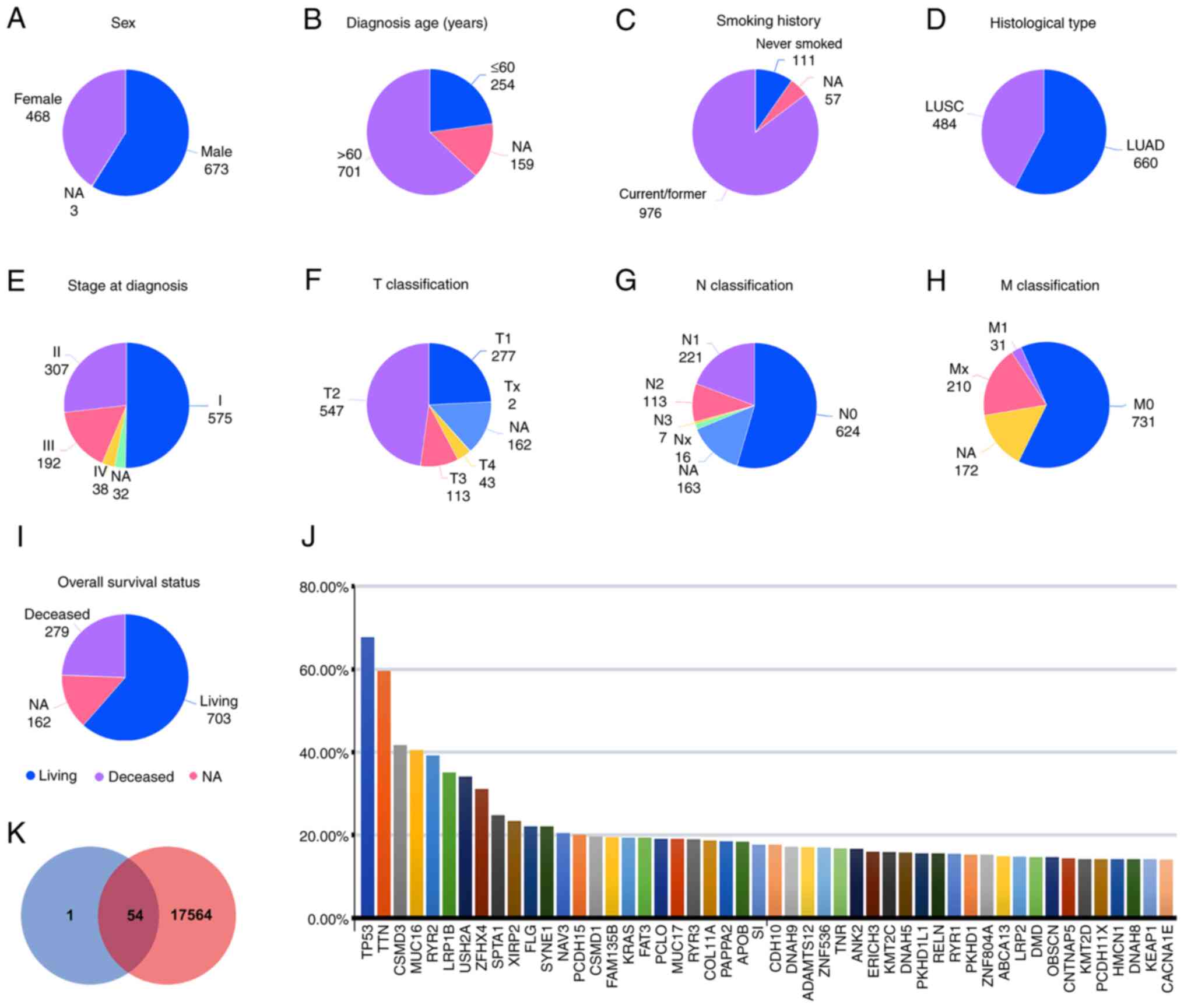 | Figure 2.Comparison with the TCGA database.
Distribution of clinicopathological characteristics including (A)
sex, (B) diagnosis age, (C) smoking history, (D) histologic type,
(E) stage at diagnosis, (F) T classification, (G) N classification,
(H) M classification and (I) overall survival status. (J) Top 50
genes with the highest mutation frequencies. (K) Venn diagram of
genes shared in the clinical data and TCGA dataset. The lung
cancer-specific 55-gene NGS panel is shown in blue and the
mutational profile of the TCGA dataset is shown in red. TCGA, The
Cancer Genome Atlas; NGS, next-generation sequencing; LUAD, lung
adenocarcinoma; LUSC, lung squamous cell carcinoma; NA, not
available. |
Identification of driver genes
The COSMIC database, NCG database and Vogelstein's
list, which contained 576 genes, 711 known genes and 125 mutated
driver genes, respectively, were used to identify driver genes.
After analyzing the intersection of these three databases and the
above 54 genes, 25 driver genes were screened: TP53, EGFR, PTEN,
RB1, KRAS, PIK3CA, CTNNB1, NOTCH2, BRAF, ERBB2, RET, ALK, PDGFRA,
FBXW7, FGFR3, KIT, MET, NRAS, APC, NF1, SMAD4, SMO, STK11, VHL and
KDM6A (Fig. 3A). The 25 driver genes
were then uploaded to the cBioPortal database. The mutation count,
mutation rates and genetic alterations of 25 driver genes, as well
as sex, smoking history and other clinical characteristics of 1,144
patients with NSCLC are shown in Fig.
3C. The five most frequently mutated genes were TP53 (68%),
KRAS (23%), EGFR (14%), NF1 (12%) and STK11 (10%). To validate the
consistency of the 25 driver genes mutations in 97 patients and
TCGA dataset, scatter plots were generated based on the mutation
rates of the two cohorts (Fig. 3B).
Most points were close to the diagonal line, suggesting that the
mutation rates of the 25 driver genes in the clinical cohort were
highly similar to those of TCGA results. These findings demonstrate
the strong reliability of the 25 driver genes, which may be used
for the subsequent studies.
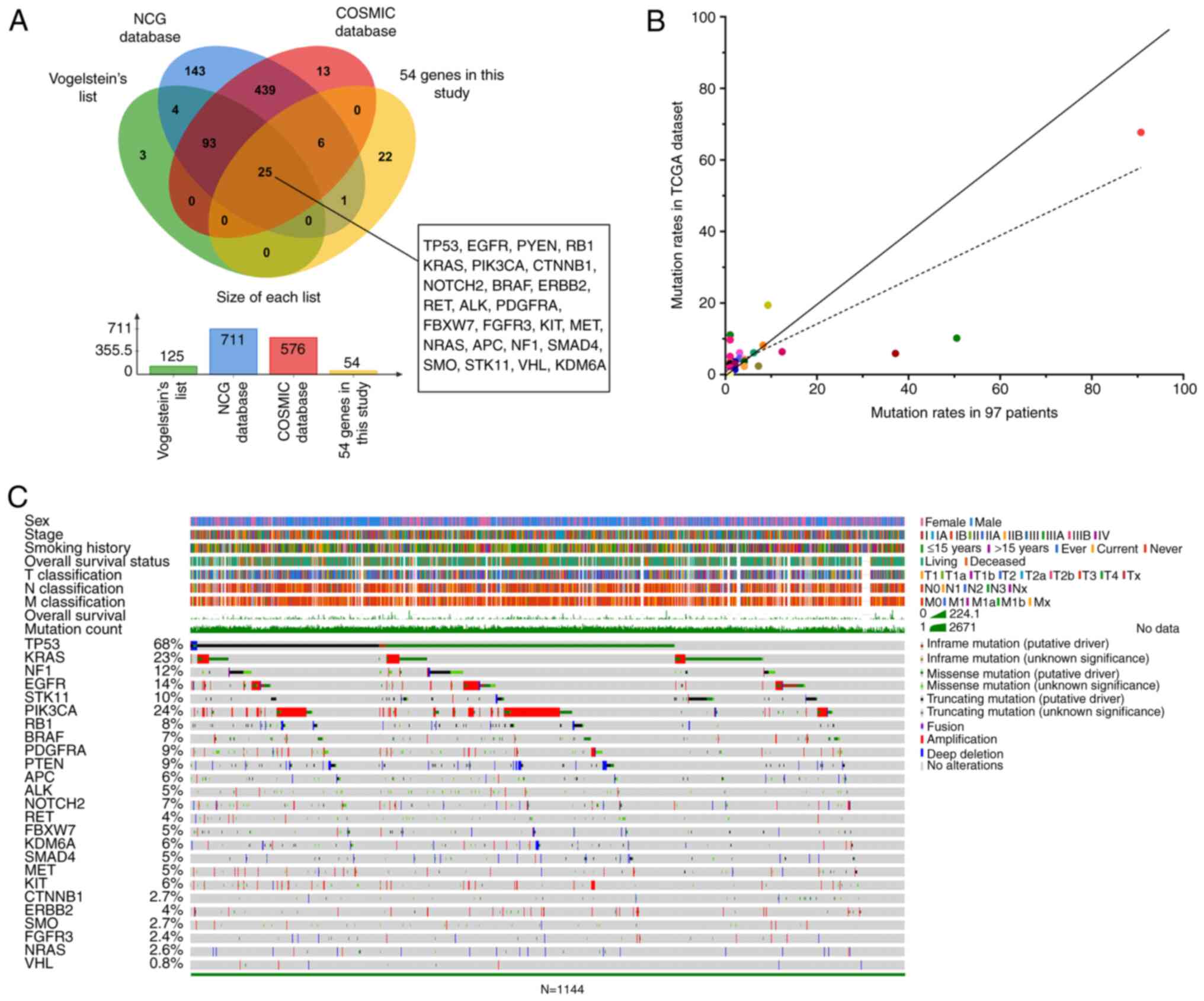 | Figure 3.Identification of 25 driver genes.
(A) Venn diagram of 25 driver genes identified by taking the
intersection of 55 genes and three databases of the COSMIC
database, NCG database and Vogelstein's list. The bar chart
represents the number of genes in each database. (B) OncoPrint map
of the mutational status of 25 driver genes in TCGA dataset
(n=1,144). Each row represents a gene and each column represents a
patient. Summary rows of each case at the top were
clinicopathological characteristics (sex, stage, smoking history,
overall survival status, T classification, N classification, M
classification, overall survival). (C) Mutation rates of 25 driver
genes were compared between 97 patients (abscissa) and the TCGA
dataset (ordinate). The fitting curve is represented by a dashed
line, whereas the ideal curve is shown as a solid, diagonal line.
COSMIC, Catalog of Somatic Mutations in Cancer; NCG, Network of
Cancer Genes; TCGA, The Cancer Genome Atlas. |
Function annotation and PPI network
analysis of 25 driver genes
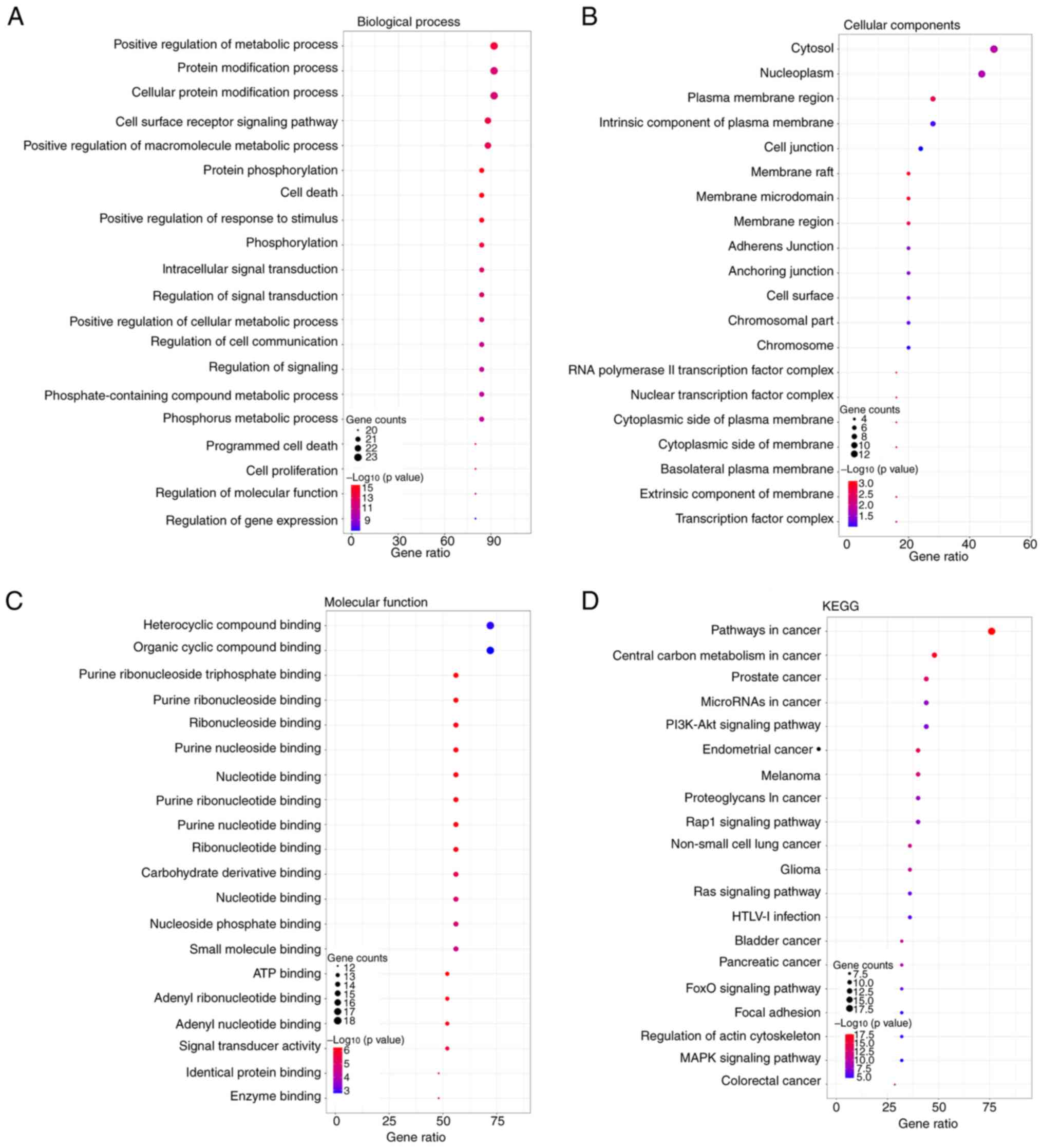
To annotate the 25 driver genes, GO and KEGG pathway
analyses were performed. The biological process category contained
977 terms, of which the major terms were ‘positive regulation of
metabolic process’, ‘protein modification process’ and ‘cellular
protein modification process’ (Fig.
4A). The top enriched terms for cellular component (Fig. 4B) and molecular function (Fig. 4C) were ‘cytosol’ and ‘heterocyclic
compound binding’, respectively. In the KEGG pathway analysis, a
number of pathways related to Oncology were observed, including
‘pathways in cancer’, ‘central carbon metabolism in cancer’,
‘prostate cancer’ and ‘microRNAs in cancer’ (Fig. 4D).
Furthermore, the deleteriousness of the 25 driver
genes was predicted using the Polyphen-2 and MutationTaster tools,
and the Polyphen-2 score and the MutationTaster score were
calculated (Fig. 5). The greatest
number of mutation positions was noted in TP53, which was 46. Among
them, 26 variant sites were strongly predicted to be ‘probably
damaging’ or ‘disease causing’, 2 variant sites were considered to
be ‘possibly damaging’, 7 variant sites were predicted to be
‘benign’ or ‘polymorphism’ and the remaining 11 variant sites
failed the deleteriousness prediction because the mutation
positions were not clearly indicated.
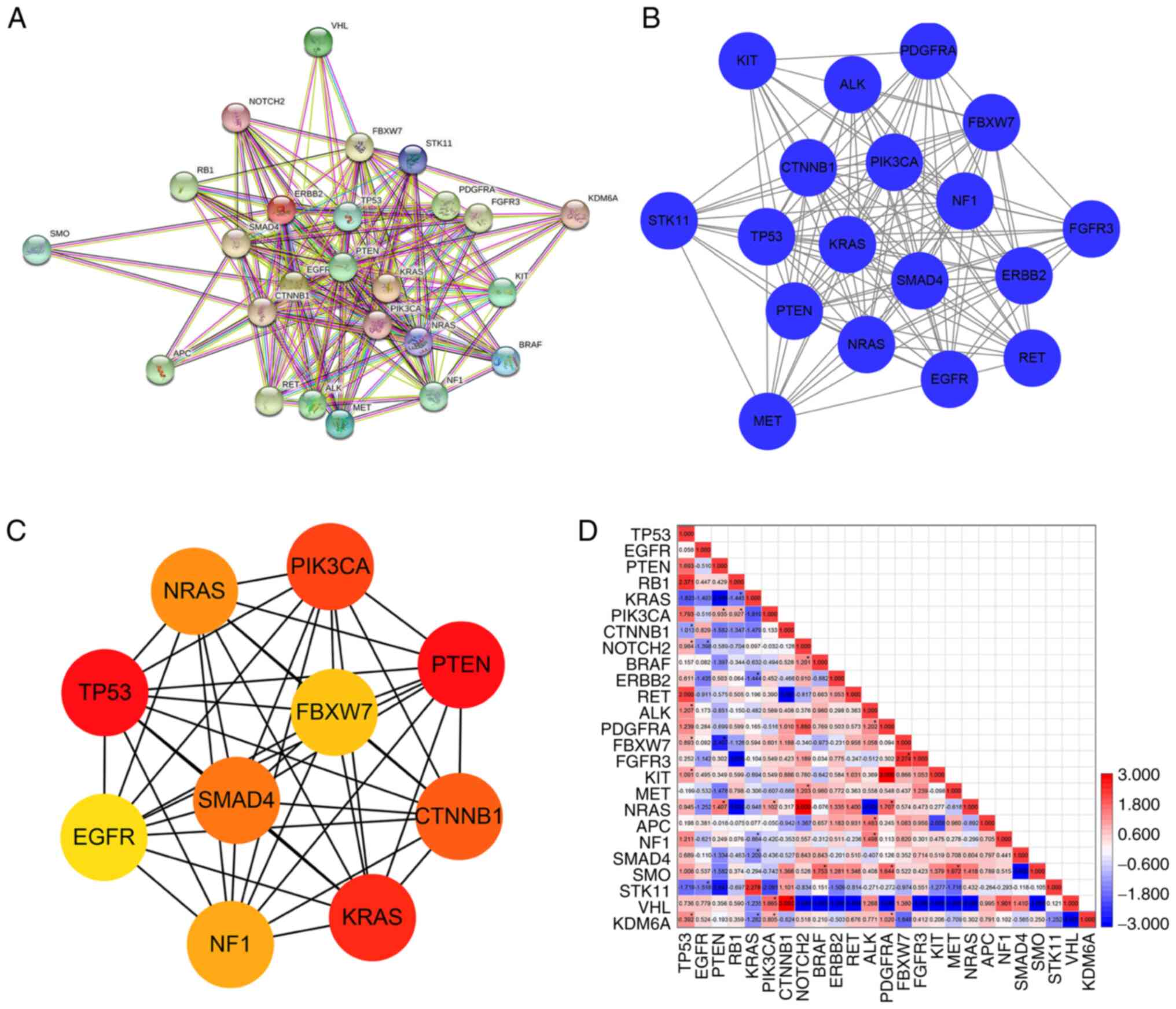
Additionally, in order to explore the potential
relationships between the 25 driver genes, a PPI network was
constructed. As shown in Fig. 6A,
there were 25 nodes and 181 edges in the PPI network. One
significant module had 18 nodes linked via 131 edges with score of
15.412 (Fig. 6B). In this module,
PTEN was identified as the seed gene. The top 10 hub genes were
identified according to MCC ranking, including TP53, PTEN, KRAS,
PIK3CA, CTNNB1, SMAD4, NRAS, NF1, FBXW7 and EGFR (Fig. 6C). Moreover, the mutual exclusivity
tool in cBioPortal database was then used to identify potential
correlations in the frequency of mutations in these 25 driver
genes. Altogether, 300 gene pairs with two relationships,
co-occurrence (177 gene pairs) and mutual exclusivity (123 gene
pairs), were identified (Fig. 6D).
Among them, 32 gene pairs were statistically significant
(P<0.05) in which 23 gene pairs showed co-occurrence and 9 gene
pairs showed mutual exclusivity. According to the log2
odds ratios, the top co-occurrence and mutual exclusivity
gene-pairs were PTEN-FBXW7 and CTNNB1-VHL, respectively.
Associations between gene mutations
and clinicopathological characteristics
The relationships between the 25 driver genes and 13
clinicopathological characteristics were examined in 97 patients
with NSCLC. The results indicated that the mutations of 13 genes
(EGFR, PTEN, KRAS, PIK3CA, BRAF, ERBB2, RET, ALK, FBXW7, KIT, MET,
NRAS and APC) were associated with all of these clinicopathological
characteristics (P<0.1; Table
SI), except smoking history (P>0.1). Subsequently, 13
significant driver genes and 12 clinicopathological characteristics
were further evaluated by logistic regression analysis and the
variables with statistical difference are shown in Table II. The effects of age and sex on
these 13 gene mutations were first examined. As shown in Table II, sex was associated with EGFR and
KRAS mutations. Indeed, the risk of EGFR (P=0.004) and KRAS
(P=0.020) mutation in female patients was 3.428 times and 0.078
times of that in males, respectively. Moreover, the effects of
mutations in the 13 driver genes on 10 clinicopathological
characteristics (including histological type, clinical stage at
diagnosis, T classification, N classification and M classification,
as well as CEA, NSE, CYFRA21-1, SCC and CA125 levels) were also
investigated. Compared with the wild type, patients with EGFR
mutations were more likely to have the features of stage-IV
(P=0.036), metastasis (P=0.007), high CEA (P=0.036) and CYFRA21
(P=0.018) level, and the patients with PTEN mutation had 0.066
times risk of stage III (P=0.032).
| Table II.Logistic regression analysis of
driver genes and clinicopathological characteristics. |
Table II.
Logistic regression analysis of
driver genes and clinicopathological characteristics.
| A, EGFR
mutation |
|---|
|
|---|
| Clinical
characteristics | P-value | OR | 95% CI |
|---|
| Sex | 0.004 | 3.428 | 1.484–7.916 |
| Stage at
diagnosisa | 0.036 | 0.148 | 0.025–0.885 |
| Metastasis | 0.007 | 5.149 |
1.557–17.023 |
| CEA | 0.036 | 3.161 | 1.078–9.264 |
| CYFRA21-1 | 0.018 | 3.289 | 1.227–8.812 |
|
| B, PTEN
mutation |
|
| Clinical
characteristics | P-value | OR | 95% CI |
|
| Sex | – | – | – |
| Stage at
diagnosisb | 0.032 | 0.066 | 0.006–0.791 |
| Metastasis | – | – | – |
| CEA | – | – | – |
| CYFRA21-1 | – | – | – |
|
| C, KRAS
mutation |
|
| Clinical
characteristics | P-value | OR | 95% CI |
|
| Sex | 0.020 | 0.078 | 0.009–0.672 |
| Stage at
diagnosis | – | – | – |
| Metastasis | – | – | – |
| CEA | – |
| – |
| CYFRA21-1 | – | – | – |
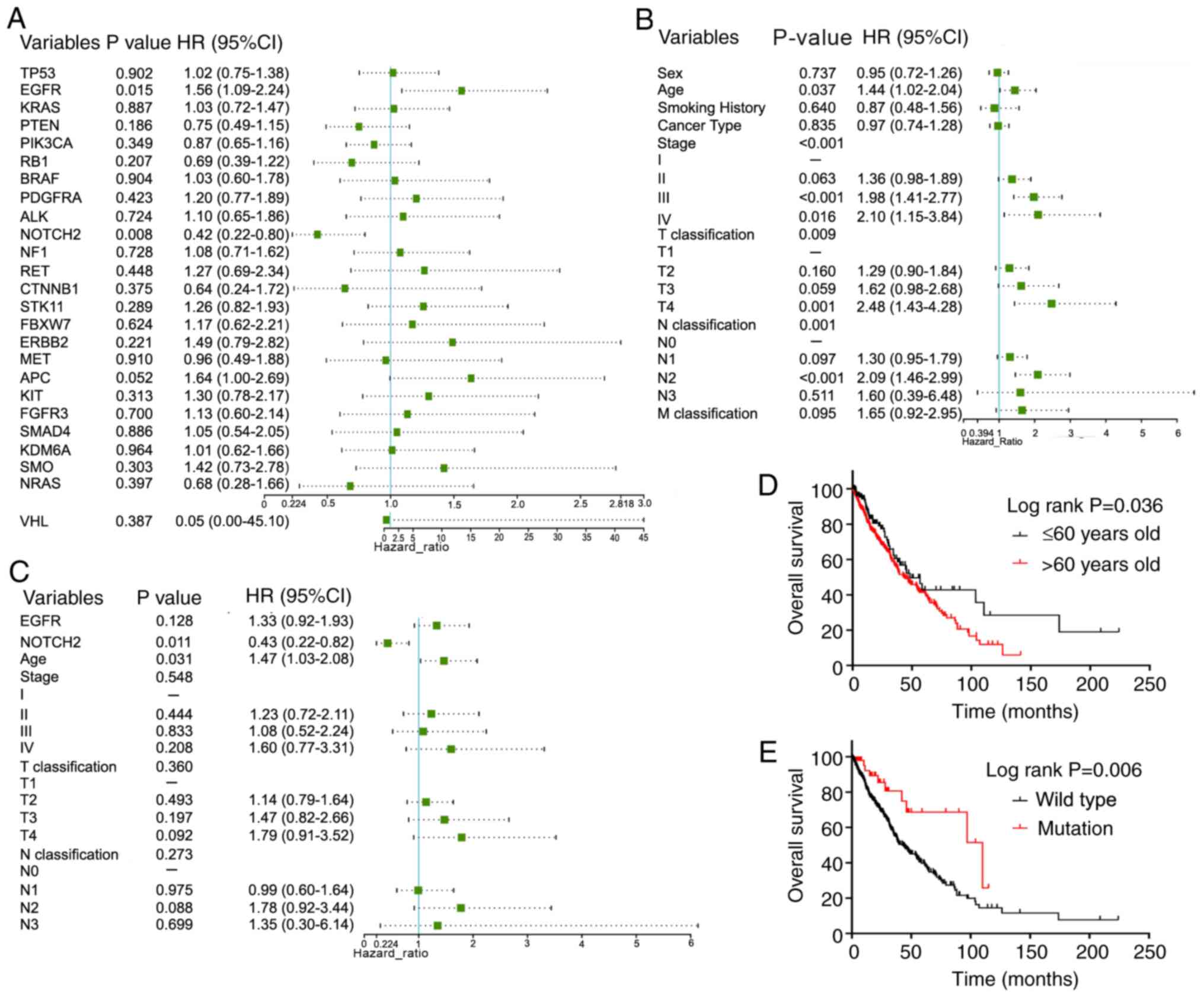
Survival analysis
Univariate and multivariate Cox regression analysis
were carried out on 25 driver genes (Fig. 7A) and 8 clinicopathological
characteristics (Fig. 7B) for the
survival of 701 patients with NSCLC from TCGA. Univariate Cox
regression analysis demonstrated that EGFR mutations (P=0.015),
NOTCH2 mutations (P=0.008), age (P=0.037), clinical stage at
diagnosis (P<0.001), T classification (P=0.009) and N
classification (P<0.001) were significantly associated with
overall survival in patients with NSCLC. These significant factors
were further analyzed using multivariate Cox regression analysis
(Fig. 7C). The results showed that
NOTCH2 mutations (P=0.011) and age (P=0.031) were independent
factors for overall survival. Compared with the wild-type group,
patients with NOTCH2 mutations had a lower risk of death [hazard
ratio (HR), 0.429; 95% CI, 0.224–0.821; P=0.011]. Moreover,
patients with NSCLC aged >60 years had a significantly shorter
survival time than patients aged ≤60 years (HR, 1.47; 95% CI,
1.03–2.08; P=0.031). Kaplan-Meier survival curves were generated
according to NOTCH2 mutation status (P=0.006; Fig. 7D) or age (P=0.036; Fig. 7E), suggesting that NOTCH2 mutation
and age may represent predictive indicators of overall survival in
patients with NSCLC.
Correlation analysis of the genes with
clinical significance
Based on the results of logistic regression and
multivariate Cox regression analysis, four genes of clinical
significance (EGFR, KRAS, PTEN and NOTCH2) were subjected to
correlation analysis in 798 patients, consisting of 97 patients
from the clinical cohort and 701 patients from TCGA. As indicated
by Kendall's τ-b correlation test, there was a negative correlation
between EGFR and NOTCH2 mutations (correlation coefficient, −0.078;
P=0.027) and a positive correlation between EGFR mutation and KRAS
mutation (correlation coefficient, 0.136; P<0.001) (Fig. 8). However, the correlations between
other gene pairs were not statistically significant.
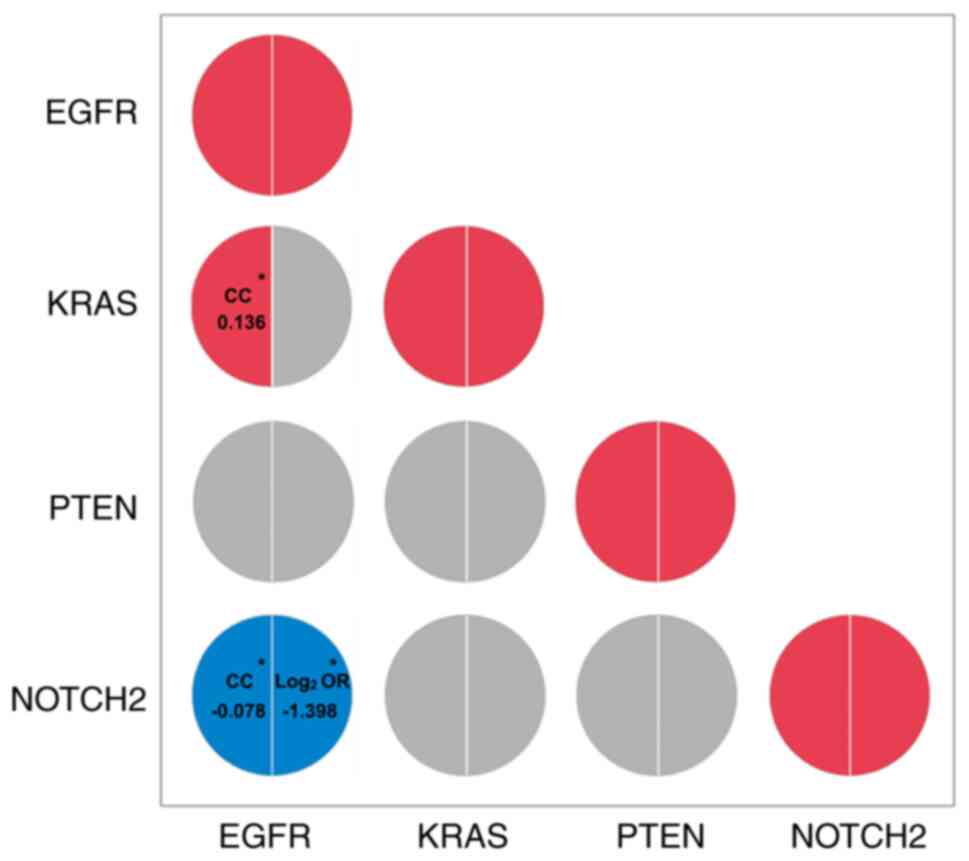 | Figure 8.Correlation analysis of the genes
with clinical significance. The correlations among EGFR, KRAS, PTEN
and NOTCH2 mutations in 798 patients (97 patients from the clinical
cohort and 701 from TCGA) are indicated on the left semi-circles,
while the results from cBioPortal database were shown on the right
semi-circles. Red represents a positive correlation, blue
represents a negative correlation, grey represents a no
correlation. Statistically significant CC and log2 ORs
are displayed. *P<0.05. EGFR, epidermal growth factor receptor;
KRAS, kirsten rat sarcoma viral oncogene homolog; PTEN, phosphatase
and tensin homolog; CC, correlation coefficient; OR, odds
ratio. |
The negative correlation between EGFR and NOTCH2
mutations was consistent with the predicted results of the mutual
exclusivity analysis in the cBioPortal database (log2
odds ratio, −1.398; P=0.017). However, a correlation between EGFR
and KRAS mutations was not observed in cBioPortal (P=0.215).
Discussion
NGS technology is now widely available, making it
possible to test multiple genes simultaneously. Several genomic
studies based on NGS technology have been performed to broadly
assess the molecular profile of the tumor (22,23).
Guibert et al (22) performed
plasma NGS using enhanced tagged amplicon sequencing of hotspots
and coding regions from 36 genes and demonstrated the ability of
amplicon-based plasma NGS to detect a full range of targetable
genotypes in NSCLC, including fusion genes, with high accuracy.
Craig et al (23)
demonstrated that a panel of 11 lung cancer-specific driver genes
used in competitive multiplex PCR amplicon NGS library preparation
for Standardized Nucleic Acid Quantification for Sequencing could
measure mutations in the 0.05–1.00% variant allele frequency range
and enable the identification of an airway epithelial cell somatic
mutation ‘field of injury’ associated with lung cancer risk. In the
present study, a lung cancer-specific panel of 55 genes was
established in order to examine driver gene mutations. To the best
of the authors' knowledge, such a panel is considered relatively
large in NGS studies of NSCLC. Moreover, in the 55-gene panel, the
mutational status of 54 genes was largely consistent with TCGA
data, except that UGT1A1 mutation was not observed in TCGA cohorts.
Although previous studies have reported that UGT1A1 polymorphisms
are associated with irinotecan-induced toxicity and treatment
outcome in lung cancer (24,25), UGT1A1 was excluded from the present
study, because the SNP frequency of UGT1A1 ranked third from the
bottom in the SNP-containing 97 patient gene list and UGT1A1
mutations were absent from TCGA.
Of the 54 aforementioned 54 genes, 25 driver genes
were identified by taking the intersection of three databases,
including the COSMIC database, the NCG database and Vogelstein's
list, which are commonly used to identify driver genes. Choi et
al (26) performed whole-exome
sequencing of a Pseudomyxoma peritonei case secondary to an
ovarian mucinous tumor whose genome harbored 28 somatic non-silent
mutations. Of these, eight putative driver gene mutations were
further identified using COSMIC database. In the present study, the
aforementioned databases were combined to improve screening
accuracy. Moreover, the co-occurrence of mutations in the 25 driver
genes was compared in 97 patients from a clinical cohort and TCGA
samples in order to validate their reliability in different
cohorts. Ample evidence has shown that mutations of the 25 driver
genes, which were identified in the present study, could promote
oncogenic transformation and all of them have been detected as
diagnostic NGS to facilitate precision therapeutic approaches in
lung cancer (27–30). Among them, EGFR mutations are one of
the best-characterized, which have been implicated in pathogenesis
of NSCLC (31). Centeno et al
(32) demonstrated that increased
autophosphorylation in the p.Arg776Gly mutation in EGFR might be
associated with a proliferative advantage, suggesting that germline
mutations in EGFR may contribute to oncogenesis.
The other gene that has been studied extensively is
TP53. It has been reported that nondisruptive mutations in TP53
negatively affect responsiveness to crizotinib and are associated
with shorter progression-free survival in ALK-rearranged patients
with NSCLC (33). Notably, the
present study indicated that TP53 was the most frequently mutated
driver gene in the TCGA dataset. Halvorsen et al (34) suggested that TP53 point mutations
were found in ~47.2% of the NSCLC samples, with the highest
mutation frequency (65%) in squamous cell carcinoma. Despite the
high frequency of mutant TP53 in tumor cells, the development and
clinical application of TP53 targeting drugs have been unsuccessful
to date. In the present study, TP53 had the greatest number of
mutation sites at 46. Only the p.Pro72Arg mutation suggested that
the chemotherapeutic agents, cisplatin, cyclophosphamide and
ifosfamide, might show better efficacy and lower toxic side
effects. All the remaining mutation sites were subjected to
targeted drug AZD1775, a WEE1 kinase inhibitor that has been
evaluated in phase-I clinical trials since 2016 (35). Unexpectedly, TP53 mutations were not
associated with clinicopathological characteristics or poor
prognosis in the current study. These might be one of the reasons
why TP53-targeted drug has not been applied in clinic.
Subsequently, the associations of 25 driver genes
with clinicopathological characteristics were assessed using
logistic regression analysis, which indicated three driver genes
were significantly associated with certain clinical factors.
Indeed, EGFR and KRAS mutations were associated with sex. Carriers
of EGFR mutations showed sex skewing, in accordance with the
findings by Minamimoto et al (36) that females were more likely to harbor
EGFR mutations. The report that estrogen receptor α expression
correlated with EGFR mutation in lung adenocarcinomas might
indicate the reasons for sex difference (37). The mutation rate of KRAS in male
patients is reported to be remarkably high (38), and similar results were also observed
in the present study. Carey et al (39) reported that expression of Ras or its
effector-loop mutants reduced the androgen levels required for the
growth of LNCaP prostate cancer cells, whereas high androgen level
in males increased tumorigenicity.
Furthermore, the present study identified other
distinct clinical characteristics between the EGFR mutant and
wild-type groups. Patients with EGFR mutations were more likely to
have the features of metastatic and stage-IV than patients with
nonmutated tumors. The results were in line with other studies. For
example, patients with NSCLC with EGFR mutation have a higher
incidence of brain metastasis, compared to EGFR wild type (40). Zhang et al (41) demonstrated that the migration and
invasiveness of A549 lung cancer cells were promoted by enhancing
the EGFR and ERK signaling pathway following filamin A expression
silencing, which could also explain the relationship between EGFR
mutation and metastasis. Additionally, the present findings
indicated that EGFR mutation was linked to higher CEA and CYFRA21-1
levels. CEA is a secreted glycoprotein biomarker, the levels of
which can reflect tumor growth, recurrence and metastasis (41). A previous study has reported a
positive correlation between serum CEA levels and EGFR mutation
rates in patients with NSCLC; specifically, the EGFR
mutation-positive rate increased with increasing CEA levels within
a certain range (42). CYFRA21-1 is
cytokeratin 19 (CK 19) fragment, a member of type 0I epithelial
cytokeratins, which can contribute to the mechanical integrity of
the cell and participate in cell division, motility and
cell-to-cell contact (43). It has
been confirmed that activating EGFR mutations were correlated with
increased CK 19 expression in human lung cancer (44).
Cox regression analysis demonstrated that age and
NOTCH2 mutation were independent prognostic factors for overall
survival of patients with NSCLC in the present study. Molinier
et al (45) confirmed that
patients with lung adenocarcinoma aged >70 years experienced
shorter survival. NOTCH2 is a member of the NOTCH family of
receptors (46). The current study
identified NOTCH2 mutation as an independent prognostic factor in
NSCLC. Several studies have reported high-frequency mutations of
NOTCH2 in lung cancer patients, further highlighting the importance
of this molecule (47,48). Chen et al (49) indicated that NOTCH2 expression was
higher in patients with lung adenocarcinoma than other histology
types of NSCLC, which was accompanied by high recurrence rates. It
has also been reported that NOTCH2 can suppress apoptosis of H69AR
small cell lung carcinoma cells (50) and that concomitant upregulation of
NOTCH2 and SIX1 contributed to preinvasive-to-invasive
adenocarcinoma progression by inducing epithelial-mesenchymal
transition and nuclear atypia (51).
The aforementioned findings imply that NOTCH2 could
promote tumor progression and affect the prognosis of patients.
However, the role of NOTCH2 in cancer is both complex and
paradoxical. Other reports have shown that NOTCH2 deletion could
result in markedly increased carcinogenesis and increased MAPK
activity, which may ultimately lead to death in
KrasG12D-driven endogenous NSCLC model mice, due to high
tumor burden (52). This
contradiction is likely due to the crosstalk between the NOTCH
signaling pathway and other pathways, such as the PTEN pathway and
MAPK pathway. For instance, delta-like ligand 4, a vascular ligand
of NOTCH1-4, upregulates PTEN expression by activating NOTCH1 in
NSCLC cells (53). NOTCH2
significantly increases phosphorylated ERK1 and ERK2 levels in
DMS53 small cell lung cancer cells, leading to activation of the
MAPK pathway (54).
A key finding of the present study was that NOTCH2
and EGFR mutations were mutually exclusive. It has been
demonstrated that EGFR mutations enabled the constitutive
activation of the downstream MAPK signaling pathway (55), which was also considered as the
downstream of NOTCH. Therefore, the NOTCH and EGFR signaling
pathways might influence each other, resulting in active and
complex crosstalk, rather than independent function, during NSCLC
progression. Understanding these interactions will greatly broaden
our knowledge about NSCLC diseases, thus promoting the development
of treatment options and personalized approaches. For patients with
EGFR mutations, EGFR TKIs are the treatment of choice (56); however, a large proportion of
patients (40-60%) does not present EGFR mutations (57). According to the results of mutual
exclusivity analysis, the patients with wild-type EGFR tended to
present NOTCH2 mutations, highlighting the importance of NOTCH2
mutational status and suggesting that the patients without EGFR
mutation might benefit from NOTCH pathway inhibitors, such as
γ-secretase inhibitors (58).
In summary, the present study demonstrated the
ability of NGS to detect a wide range of mutation types in NSCLC
and affirmed the value of some underappreciated mutations in tumor
progression. These results may help provide additional therapeutic
possibilities for patients with NSCLC. However, further studies are
warranted to elucidate the underlying mechanisms or develop new
drugs.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding was provided by The National Natural Science
Foundation of China (grant no. 81703001), Chengde Medical
University Scientific Research Major Projects (grant no.
KY2020005), Hebei Province Key Research and Development Projects
(grant no. 19277783D), Hebei Province Talent Engineering Training
Funded Research Projects (grant no. A2016002085), Hebei Province
Pathogenic Biology Emphasis Subject Projects, Project for Science
and Technology Innovation Guidance Fund of Hebei Provincial
Department of Science and Technology.
Availability of data and materials
The patient data used and/or analyzed during the
current study are available from the corresponding author on
reasonable request. TCGA dataset analyzed for this study can be
found in the cBioPortal database (http://www.cbioportal.org/study/summary?id=nsclc_tcga_broad_2016).
Authors' contributions
LeL and LN designed the study, analyzed the data and
wrote and revised the manuscript. CD and LiL contributed to sample
collection, quality control and NGS analysis. NG, YX and XL
performed experiments on tumor tissue and blood and collected and
analyzed patient data. LC, QX and LZ acquired and analyzed the
public database data, recruited the patients and supervised the
study. All authors have read and approved the final manuscript. LeL
and LN confirmed the authenticity of the data shown in the present
manuscript.
Ethics approval and consent to
participate
This study was approved by the Research Ethics
Review Committee of Chengde Medical University (Hebei, China;
approval no. 2017003). All participants provided written informed
consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Feng RM, Zong YN, Cao SM and Xu RH:
Current cancer situation in China: Good or bad news from the 2018
Global Cancer Statistics? Cancer Commun. 39:222019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang TJ, Saad S, Qureshi YH, Jani A, Nanda
T, Yaeh AM, Rozenblat T, Sisti MB, Bruce JN, McKhann GM, et al:
Does lung cancer mutation status and targeted therapy predict for
outcomes and local control in the setting of brain metastases
treated with radiation? Neuro Oncol. 17:1022–1028. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, et al: Erlotinib versus
chemotherapy as first-line treatment for patients with advanced
EGFR mutation-positive non-small-cell lung cancer (OPTIMAL,
CTONG-0802): A multicentre, open-label, randomised, phase 3 study.
Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tsang YH, Dogruluk T, Tedeschi PM,
Wardwell-Ozgo J, Lu H, Espitia M, Nair N, Minelli R, Chong Z, Chen
F, et al: Functional annotation of rare gene aberration drivers of
pancreatic cancer. Nat Commun. 7:105002016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu J, Lee W, Jiang Z, Chen Z,
Jhunjhunwala S, Haverty PM, Gnad F, Guan Y, Gilbert HN, Stinson J,
et al: Genome and transcriptome sequencing of lung cancers reveal
diverse mutational and splicing events. Genome Res. 22:2315–2327.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Metzker ML: Sequencing technologies-the
next generation. Nat Rev Genet. 11:31–46. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhai H, Zhong W, Yang X and Wu YL:
Neoadjuvant and adjuvant epidermal growth factor receptor tyrosine
kinase inhibitor (EGFR-TKI) therapy for lung cancer. Transl Lung
Cancer Res. 4:82–93. 2015.PubMed/NCBI
|
|
10
|
Yuan J, Zhang N, Yin L, Zhu H, Zhang L,
Zhou L and Yang M: Clinical implications of the autophagy core gene
variations in advanced lung adenocarcinoma treated with gefitinib.
Sci Rep. 7:178142017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shaw AT, Solomon BJ, Besse B, Bauer TM,
Lin CC, Soo RA, Riely GJ, Ou SI, Clancy JS, Li S, et al: ALK
resistance mutations and efficacy of lorlatinib in advanced
anaplastic lymphoma kinase-positive non-small-cell lung cancer. J
Clin Oncol. 37:1370–1379. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kalemkerian GP, Narula N, Kennedy EB,
Biermann WA, Donington J, Leighl NB, Lew M, Pantelas J, Ramalingam
SS, Reck M, et al: Molecular testing guideline for the selection of
patients with lung cancer for treatment with targeted tyrosine
kinase inhibitors: American society of clinical oncology
endorsement of the college of American pathologists/international
association for the study of lung cancer/association for molecular
pathology clinical practice guideline update. J Clin Oncol.
36:911–919. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu YL, Planchard D, Lu S, Sun H, Yamamoto
N, Kim DW, Tan DSW, Yang JC, Azrif M, Mitsudomi T, et al: Pan-Asian
adapted Clinical Practice Guidelines for the management of patients
with metastatic non-small-cell lung cancer: A CSCO-ESMO initiative
endorsed by JSMO, KSMO, MOS, SSO and TOS. Ann Oncol. 30:171–210.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Savas P, Hughes B and Solomon B: Targeted
therapy in lung cancer: IPASS and beyond, keeping abreast of the
explosion of targeted therapies for lung cancer. J Thorac Dis. 5
(Suppl 5):S579–S92. 2013.PubMed/NCBI
|
|
15
|
Tian L, Chen Q, Yi X, Wang G, Chen J, Ning
P, Yang K and Liu Z: Radionuclide I-131 labeled albumin-paclitaxel
nanoparticles for synergistic combined chemo-radioisotope therapy
of cancer. Theranostics. 7:614–623. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LJ and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Amin MB, Greene FL, Edge SB, Compton CC,
Gershenwald JE, Brookland RK, Meyer L, Gress DM, Byrd DR and
Winchester DP: The eighth edition AJCC Cancer Staging Manual:
Continuing to build a bridge from a population-based to a more
‘personalized’ approach to cancer staging. CA Cancer J Clin.
67:93–99. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Campbell JD, Alexandrov A, Kim J, Wala J,
Berger AH, Pedamallu CS, Shukla SA, Guo G, Brooks AN, Murray BA, et
al: Distinct patterns of somatic genome alterations in lung
adenocarcinomas and squamous cell carcinomas. Nat Genet.
48:607–616. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Repana D, Nulsen J, Dressler L,
Bortolomeazzi M, Venkata SK, Tourna A, Yakovleva A, Palmieri T and
Ciccarelli FD: The Network of Cancer Genes (NCG): A comprehensive
catalogue of known and candidate cancer genes from cancer
sequencing screens. Genome Biol. 20:12019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guibert N, Hu Y, Feeney N, Kuang Y,
Plagnol V, Jones G, Howarth K, Beeler JF, Paweletz CP and Oxnard
GR: Amplicon-based next-generation sequencing of plasma cell-free
DNA for detection of driver and resistance mutations in advanced
non-small cell lung cancer. Ann Oncol. 29:1049–1055. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Craig DJ, Morrison T, Khuder SA, Crawford
EL, Wu L, Xu J, Blomquist TM and Willey JC: Technical advance in
targeted NGS analysis enables identification of lung cancer
risk-associated low frequency TP53, PIK3CA, and BRAF mutations in
airway epithelial cells. BMC Cancer. 19:10812019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen X, Liu L, Guo Z, Liang W, He J, Huang
L, Deng Q, Tang H, Pan H, Guo M, et al: UGT1A1 polymorphisms with
irinotecan-induced toxicities and treatment outcome in Asians with
Lung Cancer: A meta-analysis. Cancer Chemother Pharmacol.
79:1109–1117. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bai Y, Wu HW, Ma X, Liu Y and Zhang YH:
Relationship between UGT1A1*6/*28 gene polymorphisms and the
efficacy and toxicity of irinotecan-based chemotherapy. Onco
Targets Ther. 10:3071–3081. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Choi YJ and Lee SH, Kim MS, Jung SH, Hur
SY, Chung YJ and Lee SH: Whole-exome sequencing identified the
genetic origin of a mucinous neoplasm in a mature cystic teratoma.
Pathology. 48:372–376. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tan AC, Lai GGY, Tan GS, Poon SY, Doble B,
Lim TH, Aung ZW, Takano A, Tan WL, Ang MK, et al: Utility of
incorporating next-generation sequencing (NGS) in an Asian
non-small cell lung cancer (NSCLC) population: Incremental yield of
actionable alterations and cost-effectiveness analysis. Lung
Cancer. 139:207–215. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
DiBardino DM, Rawson DW, Saqi A, Heymann
JJ, Pagan CA and Bulman WA: Next-generation sequencing of non-small
cell lung cancer using a customized, targeted sequencing panel:
Emphasis on small biopsy and cytology. Cytojournal. 14:72017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Preusser M, Berghoff AS, Koller R,
Zielinski CC, Hainfellner JA, Liebmann-Reindl S, Popitsch N, Geier
CB, Streubel B and Birner P: Spectrum of gene mutations detected by
next generation exome sequencing in brain metastases of lung
adenocarcinoma. Eur J Cancer. 51:1803–1811. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sonmezler O, Boga I and Bisgin A:
Integration of liquid biopsies into clinical laboratory
applications via NGS in cancer diagnostics. Clin Lab. doi:
10.7754/Clin.Lab.2019.190836.
|
|
31
|
Wang Y, Lai H, Fan X, Luo L, Duan F, Jiang
Z, Wang Q, Leung ELH, Liu L and Yao X: Gossypol inhibits non-small
cell lung cancer cells proliferation by targeting
EGFRL858R/T790M. Front Pharmacol. 9:7282018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Centeno I, Blay P, Santamaría I, Astudillo
A, Pitiot AS, Osorio FG, González-Arriaga P, Iglesias F, Menéndez
P, Tardón A, et al: Germ-line mutations in epidermal growth factor
receptor (EGFR) are rare but may contribute to oncogenesis: A novel
germ-line mutation in EGFR detected in a patient with lung
adenocarcinoma. BMC Cancer. 11:1722011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Song P, Zhang F, Li Y, Yang G, Li W, Ying
J and Gao S: Concomitant TP53 mutations with response to crizotinib
treatment in patients with ALK-rearranged non-small-cell lung
cancer. Cancer Med. 8:1551–1557. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Halvorsen AR, Silwal-Pandit L, Meza-Zepeda
LA, Vodak D, Vu P, Sagerup C, Hovig E, Myklebost O, Børresen-Dale
AL, Brustugun OT and Helland Å: TP53 mutation spectrum in smokers
and never smoking lung cancer patients. Front Genet. 7:852016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Leijen S, van Geel RM, Pavlick AC, Tibes
R, Rosen L, Razak AR, Lam R, Demuth T, Rose S, Lee MA, et al: Phase
I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in
combination with gemcitabine, cisplatin, or carboplatin in patients
with advanced solid tumors. J Clin Oncol. 34:4371–4380. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Minamimoto R, Jamali M, Gevaert O,
Echegaray S, Khuong A, Hoang CD, Shrager JB, Plevritis SK, Rubin
DL, Leung AN, et al: Prediction of EGFR and KRAS mutation in
non-small cell lung cancer using quantitative 18F
FDG-PET/CT metrics. Oncotarget. 8:52792–52801. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Raso MG, Behrens C, Herynk MH, Liu S,
Prudkin L, Ozburn NC, Woods DM, Tang X, Mehran RJ, Moran C, et al:
Immunohistochemical expression of estrogen and progesterone
receptors identifies a subset of NSCLCs and correlates with EGFR
mutation. Clin Cancer Res. 15:5359–5368. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhao J, Gao J, Guo L, Hu X, Liu Q, Zhao J,
Liu L, Jiang J, Wang M, Liang Z, et al: EGFR and KRAS gene
mutations in 754 patients with resectable stage I–IIIa non-small
cell lung cancer and its clinical significance. Zhongguo Fei Ai Za
Zhi. 20:617–622. 2017.(In Chinese). PubMed/NCBI
|
|
39
|
Carey AM, Pramanik R, Nicholson LJ, Dew
TK, Martin FL, Muir GH and Morris JD: Ras-MEK-ERK signaling cascade
regulates androgen receptor element-inducible gene transcription
and DNA synthesis in prostate cancer cells. Int J Cancer.
121:520–527. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hsu F, De Caluwe A, Anderson D, Nichol A,
Toriumi T and Ho C: EGFR mutation status on brain metastases from
non-small cell lung cancer. Lung Cancer. 96:101–107. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang Y, Zhu T, Liu J, Liu J, Gao D, Su T
and Zhao R: FLNa negatively regulated proliferation and metastasis
in lung adenocarcinoma A549 cells via suppression of EGFR. Acta
Biochim Biophys Sin. 50:164–170. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang ZM, Ding XP, Pen L, Mei L and Liu T:
Analysis of CEA expression and EGFR mutation status in non-small
cell lung cancers. Asian Pac J Cancer Prev. 15:3451–3455. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Barak V, Goike H, Panaretakis KW and
Einarsson R: Clinical utility of cytokeratins as tumor markers.
Clin Biochem. 37:529–540. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cho A, Hur J, Moon YW, Hong SR, Suh YJ,
Kim YJ, Im DJ, Hong YJ, Lee HJ, Kim YJ, et al: Correlation between
EGFR gene mutation, cytologic tumor markers, 18F-FDG uptake in
non-small cell lung cancer. BMC Cancer. 16:2242016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Molinier O, Goupil F, Debieuvre D, Auliac
JB, Jeandeau S, Lacroix S, Martin F and Grivaux M: Five-year
survival and prognostic factors according to histology in 6101
non-small-cell lung cancer patients. Respir Med Res. 77:46–54.
2020.PubMed/NCBI
|
|
46
|
Artavanis-Tsakonas S, Rand MD and Lake RJ:
Notch signaling: Cell fate control and signal integration in
development. Science. 284:770–776. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Oshita F, Kasajima R and Miyagi Y:
Multiplex genomic test of mutation and fusion genes in small biopsy
specimen of lung cancer. J Exp Ther Oncol. 11:189–194.
2016.PubMed/NCBI
|
|
48
|
Liao L, Ji X, Ge M, Zhan Q, Huang R, Liang
X and Zhou X: Characterization of genetic alterations in brain
metastases from non-small cell lung cancer. FEBS Open Bio.
8:1544–1552. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chen CY, Chen YY, Hsieh MS, Ho CC, Chen
KY, Shih JY and Yu CJ: Expression of notch gene and its impact on
survival of patients with resectable Non-small cell lung cancer. J
Cancer. 8:1292–1300. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Motooka Y, Fujino K, Sato Y, Kudoh S,
Suzuki M and Ito T: Pathobiology of Notch2 in lung cancer.
Pathology. 49:486–493. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Mimae T, Okada M, Hagiyama M, Miyata Y,
Tsutani Y, Inoue T, Murakami Y and Ito A: Upregulation of notch2
and six1 is associated with progression of early-stage lung
adenocarcinoma and a more aggressive phenotype at advanced stages.
Clin Cancer Res. 18:945–955. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Baumgart A, Mazur PK, Anton M, Rudelius M,
Schwamborn K, Feuchtinger A, Behnke K, Walch A, Braren R, Peschel
C, et al: Opposing role of Notch1 and Notch2 in a Kras(G12D)-driven
murine non-small cell lung cancer model. Oncogene. 34:578–588.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ding XY, Ding J, Wu K, Wen W, Liu C, Yan
HX, Chen C, Wang S, Tang H, Gao CK, et al: Cross-talk between
endothelial cells and tumor via delta-like ligand 4/Notch/PTEN
signaling inhibits lung cancer growth. Oncogene. 31:2899–2906.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sriuranpong V, Borges MW, Ravi RK, Arnold
DR, Nelkin BD, Baylin SB and Ball DW: Notch signaling induces cell
cycle arrest in small cell lung cancer cells. Cancer Res.
61:3200–3205. 2001.PubMed/NCBI
|
|
55
|
Baumgartner U, Berger F, Hashemi Gheinani
A, Burgener SS, Monastyrskaya K and Vassella E: MiR-19b enhances
proliferation and apoptosis resistance via the EGFR signaling
pathway by targeting PP2A and BIM in non-small cell lung cancer.
Mol Cancer. 17:442018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Karachaliou N, Cardona AF, Bracht JWP,
Aldeguer E, Drozdowskyj A, Fernandez-Bruno M, Chaib I, Berenguer J,
Santarpia M, Ito M, et al: Integrin-linked kinase (ILK) and src
homology 2 domain-containing phosphatase 2 (SHP2): Novel targets in
EGFR-mutation positive non-small cell lung cancer (NSCLC).
EBioMedicine. 39:207–214. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yu S, Liu D, Shen B, Shi M and Feng J:
Immunotherapy strategy of EGFR mutant lung cancer. Am J Cancer Res.
8:2106–2115. 2018.PubMed/NCBI
|
|
58
|
Katoh M and Katoh M: Precision medicine
for human cancers with Notch signaling dysregulation (Review). Int
J Mol Med. 45:279–297. 2020.PubMed/NCBI
|