Introduction
Colorectal cancer (CRC) is a common malignant tumor
of the gastrointestinal tract that is characterized by high
incidence and mortality rates; as developing countries continue to
progress, the incidence of colorectal cancer is estimated to
increase to 2.5 million new cases worldwide by 2035 (1,2). The
early symptoms of CRC may not be obvious; however, as the cancer
progresses, symptoms such as the presence of blood in the stool,
diarrhea, changes in bowel habits, local abdominal pain and anemia
appear, which seriously affect the health and quality of life of
the patients (2). To improve the
treatment efficacy of CRC, chemotherapy, radiation and surgical
resection are often used in combination. However, conventional
treatments are usually accompanied by severe side effects, toxicity
or chemotherapy resistance (3).
Therefore, identifying novel drugs that are effective in CRC and
have a favorable toxicity profile remains a priority for
researchers.
As well as being a commonly used intravenous
anesthetic for clinical anesthesia, 2,6-diisopropyl phenol
(propofol) has been found to possess considerable non-anesthetic
properties, including being able to prevent nausea and vomiting,
activate γ-aminobutyric acid receptors to enhance analgesia,
regulate nitric oxide synthesis, and exert antioxidant,
neuroprotective, anti-anxiety, immunoregulatory and anti-platelet
aggregation effects (4). Notably,
propofol has also been found to inhibit the proliferation and
migration of tumor cells, promote tumor cell apoptosis and enhance
the sensitivity to chemotherapeutic drugs by modulating microRNA
expression, gene transcription, apoptosis and immune system
function (5). A retrospective cohort
study reported that propofol anesthesia during CRC surgery was
associated with an improved survival rate, irrespective of the
Tumor-Node-Metastasis stage (6).
Another study demonstrated that propofol suppressed CRC
tumorigenesis by promoting cell apoptosis (7). The aforementioned data indicate the
beneficial role of propofol in inhibiting CRC tumorigenesis;
however, to the best of our knowledge, the underlying mechanism
remains poorly understood.
Inducing tumor cell death is the main strategy for
clinical cancer treatment and most anticancer drugs eliminate tumor
cells by triggering apoptosis and cell necrosis (8). However, owing to the acquired or
inherent resistance of cancer cells to apoptosis and necrosis, the
effectiveness of inducing tumor cell death via these mechanisms is
limited (9). Ferroptosis, a type of
cell death that is different to apoptosis and necrosis, has
recently attracted widespread attention due to its observed
effectiveness in killing cancer cells (10). Ferroptosis is an iron-dependent and
reactive oxygen species (ROS)-associated type of cell death,
characterized by the accumulation of ROS and the inactivation of
the cellular antioxidant glutathione (GSH), which leads to redox
dysregulation (11). A large number
of studies reported that the induction of ferroptosis successfully
killed tumor cells and inhibited tumor growth. In CRC, the
activation of ferroptosis has also been suggested to represent a
promising therapeutic option and the underlying mechanisms involved
have been extensively reported (12–14).
Previous studies reported that propofol induced cancer cell death
via ROS-mediated apoptosis (15,16).
Therefore, it was hypothesized that propofol may inhibit CRC
tumorigenesis via the induction of ferroptosis-mediated cell
death.
According to the prediction results from the Search
Tools for Interactions of Chemicals (STITCH) database, propofol
regulates STAT3, which is known to be activated in numerous types
of cancer, including CRC, where it promotes cancer progression
(17). In addition, STAT3 has also
been found to regulate ferroptosis (18). The present study investigated whether
propofol can trigger ferroptosis by downregulating STAT3
expression, thereby inhibiting CRC tumorigenesis.
Materials and methods
Patient studies
A total of 12 paired CRC cancerous and paracancerous
tissues were collected from patients who underwent a radical
resection of colorectal carcinoma at Zhongshan Hospital (Shanghai,
China) between May 2020 and August 2020. Patients were aged 18–90
years (mean age, 73.8 years), with a male to female ratio of 1:1,
and were divided into 12 cases of cancerous tissue and 12 cases of
paracancerous tissue. All the patients provided written, informed
consent and the study was approved by the Ethical Committee of
Zhongshan Hospital (approval no. B2016-014). Exclusion criteria
were as follows: Patients with combined autoimmune diseases; a
history of other malignant tumors; a history of preoperative
radiotherapy; patients under 18 years of age; or older patients who
could not cooperate with the investigation.
Cell culture and treatment
The human normal colonic epithelial cell line,
NCM460, and the human CRC cell line, SW480, were purchased from The
Cell Bank of the Type Culture Collection of The Chinese Academy of
Sciences. Cells were cultured in DMEM (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.), and maintained in a humidified atmosphere at
37°C containing 5% CO2. Propofol (MilliporeSigma) was
diluted in DMSO and then in culture medium to specific
concentrations. NCM460 and SW480 cells were treated with different
concentrations of propofol (12.5, 25 and 50 µm) for 48 h at room
temperature, and cells receiving no treatment were considered as
the controls.
Cell transfection
The human STAT3 cDNA sequence was synthesized by
GenScript and inserted into a pcDNA3.1 vector (Invitrogen; Thermo
Fisher Scientific, Inc.). SW480 cells (70–90% confluence) were
seeded into 6-well plates and transfected with STAT3 gene
recombinant vector pcDNA3.1-STAT3 (pc-STAT3; 10 nM) or pcDNA3.1
blank vector (pcDNA3.1; 10 nM) using Lipofectamine® 2000
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. Following transfection, cells were
subjected to propofol treatment. Following incubation at 37°C for 5
h, untransfected SW480 cells and SW480 cells transfected with
pc-STAT3 or pcDNA3.1 were exposed to DMEM or medium supplemented
with 50 µm propofol. The control group was SW480 cells exposed to
control medium without transfection. Subsequent experimentation was
conducted within 48 h.
MTT assay
An MTT assay was used to determine cell viability.
Briefly, NCM460 and SW480 cells were seeded at a density of
4×103 cells/well into 96-well plates. Following
incubation for 48 h, 10 µl MTT solution (Beyotime Institute of
Biotechnology) was added to each well and further incubated for 4
h. The cell culture medium was subsequently discarded and 150 µl
DMSO was added to the cells. The absorbance was measured at a
wavelength of 570 nM using a high-throughput universal microplate
assay (BMG Labtech GmbH).
Colony formation assay
For the colony formation assay, 1×103
SW480 cells from the control, propofol, propofol + pcDNA3.1 and
propofol + pc-STAT3 groups were plated into 3.5-cm dished and
maintained in media supplemented with 10% FBS to allow colony
formation, with the medium being replaced every 4 days. Following
~2 weeks of incubation, the colonies were fixed with methanol for
15 min at room temperature and stained with 0.1% crystal violet for
15 min at room temperature. The stained colonies with >50 cells
were subsequently manually counted.
Lipid peroxidation [thiobarbituric
acid reactive substances (TBARS)]
Lipid peroxidation was detected by a TBARS kit assay
(cat. no. STA-330; OxiSelect™; Cell Biolabs Inc.) using RIPA lysis
buffer (Beyotime Institute of Biotechnology) containing SW480 cells
(1×107/ml). SW480 cells treated with propofol and/or
transfected with pc-STAT3 were harvested and transferred to a
centrifuge tube, then centrifuged for 10 min at 1,000 × g at room
temperature. The process was conducted according to a previous
study (19). The absorbance of the
aforementioned solution was detected by a microplate reader at 535
nM.
Measurement of total iron and
Fe2+ levels
An Iron assay kit (cat. no. MAK025; MilliporeSigma)
was used to measure total iron and Fe2+ levels in cells.
A total of 2×106 SW480 cells were quickly homogenized in
4–10X Iron Assay buffer. The sample was subsequently centrifuged at
13,000 × g for 10 min at 4°C to remove the insoluble materials. To
measure total iron, 5 µl iron reducer was added to each sample well
to reduce Fe3+ to Fe2+ and a horizontal
shaker or pipette was used to mix the samples thoroughly, prior to
incubation at room temperature in the dark for 30 min. To measure
ferrous iron levels, 5 µl assay buffer was added to the samples in
one set of wells and 5 µl iron reducer was added to the other set
of wells. Next, 100 µl iron probe was added to each well containing
the standard or test samples. Samples were mixed using a horizontal
shaker or by pipetting, and were then incubated for 60 min at room
temperature in the dark. Finally, the absorbance was measured at a
wavelength of 593 nM.
ROS detection
Changes in intracellular ROS levels were measured
using a 2′,7′-dichlorofluorescein diacetate (DCFDA)-ROS kit (cat.
no. 113851; Abcam). Briefly, SW480 cells were plated into a 6-cm
culture dish at a density of 1×105 cells/ml and allowed
to attach overnight. Cells overexpressing STAT3 or untransfected
cells were subsequently incubated with control media or propofol
for 48 h. The cells were then washed with Hanks' balanced salt
solution and incubated with DCFDA at 37°C for 45 min. The level of
ROS was detected by an inverted fluorescence microscope.
Quantitative analysis was performed using ImageJ software (version
1.52r; National Institutes of Health).
Reverse transcription-quantitative
PCR
Total RNA was extracted from SW480 cells and NCM460
cells using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. Total
RNA was reverse-transcribed into cDNA at 42°C for 30 min using a
PrimeScript™ RT reagent kit (Takara Biotechnology Co., Ltd.). qPCR
was subsequently performed using a SYBR-Green Master PCR mix
(Applied Biosystems; Thermo Fisher Scientific, Inc.) on a TP800
Thermal Cycler Dice™ Real Time system (Takara Biotechnology Co.,
Ltd.). The following thermocycling conditions were used: Initial
denaturation at 95°C for 10 min, followed by 95°C for 15 sec and
60°C for 1 min for 40 cycles, with a final extension at 72°C for 10
min. The primer sequences used for the qPCR were as follows: STAT3
forward, 5′-CATCCTGAAGCTGACCCAGG-3′ and reverse,
5′-TCCTCACATGGGGGAGGTAG-3′; and GAPDH forward,
5′-GAAAGCCTGCCGGTGACTAA-3′ and reverse, 5′-TTCCCGTTCTCAGCCTTGAC-3′.
Relative STAT3 mRNA expression levels were quantified using the
2−ΔΔCq method (20) and
normalized to GAPDH, the internal control.
Western blotting
Total protein was extracted from SW480 cells and
NCM460 cells using RIPA lysis buffer (Beyotime Institute of
Biotechnology) and quantified using a BCA assay kit (Beyotime
Institute of Biotechnology). A total of 30 µg protein/lane was
separated using 12% gels via SDS-PAGE and subsequently transferred
onto PVDF membranes. After blocking with 5% skimmed milk for 2 h at
room temperature, the membranes were then incubated with the
following primary antibodies overnight at 4°C: Anti-STAT3 (cat. no.
ab68153; dilution, 1:1,000; Abcam), anti-Ki67 (cat. no. ab92742;
dilution, 1:5,000; Abcam), anti-proliferating cell nuclear antigen
(PCNA) (cat. no. ab92552; dilution, 1:1,000; Abcam), anti-cation
transport regulator homolog 1 (CHAC1) (cat. no. #PA5-103719;
dilution, 1:1,000; Invitrogen; Thermo Fisher Scientific, Inc.),
anti-prostaglandin-endoperoxide synthase 2 (PTGS2) (cat. no.
ab52237; dilution, 1:1,000; Abcam), anti-glutathione peroxidase 4
(GPX4) (cat. no. ab125066; dilution, 1:1,000; Abcam) and anti-GAPDH
(cat. no. ab9485; dilution, 1:2,500; Abcam). Following the primary
antibody incubation, the membranes were incubated with a goat
anti-rabbit HRP-conjugated antibody (cat. no. ab6721; dilution,
1:2,000; Abcam) for 1 h at room temperature. The protein bands were
imaged on a Tanon Chemiluminescence Imaging system with Pierce™ ECL
Western regent (cat. no. 32209; Thermo Fisher Scientific, Inc.).
The intensity of the bands was semi-quantified using ImageJ
software (version 1.52r; National Institutes of Health).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 6 software (GraphPad Software, Inc.). Data are presented as
the mean ± standard deviation. All experiments were repeated in
triplicate. Statistical differences were determined using a paired
or unpaired Student's t-test between two groups, or one-way ANOVA
followed by Tukey's post hoc test among multiple groups. P<0.05
was considered to indicate a statistically significant
difference.
Results
Propofol upregulates STAT3 expression
in SW480 CRC cell lines
To determine the role of STAT3 in CRC, the mRNA
expression levels of STAT3 in control normal and CRC tissues were
analyzed. Compared with those in the control tissues, STAT3
expression levels were significantly upregulated in the CRC
tissues, indicating the potential role of STAT3 in CRC (Fig. 1A). The mRNA and protein expression
levels of STAT3 in SW480 cells were all higher than those in NCM460
cells (Fig. 1B and C). The chemical
structural formula of propofol is shown as Fig. 1D. Both cell lines were exposed to 0,
12.5, 25 or 50 µm propofol for 48 h, and then cell viability was
analyzed using an MTT assay. As shown in Fig. 1E, propofol treatment did not
significantly alter NCM460 cell viability, but decreased CRC SW480
cell viability. The expression levels of STAT3 in SW480 cells
following propofol treatment were also analyzed. As shown in
Fig. 1F and G, the mRNA and protein
expression levels of STAT3 were significantly downregulated by
propofol treatment in a concentration-dependent manner. These data
suggested that propofol may decrease CRC cell viability by
downregulating STAT3 expression. As 50 µm propofol exhibited the
highest inhibitory effect on the expression of STAT3, this dose was
selected for use in the following experiments. The dose chosen was
the dose where the cell survival level was >50%. If the dose
were greater than the 50% maximal inhibitory concentration, it
would lead to excessive cell death and would not be conducive to
the cell proliferation observed later in the study.
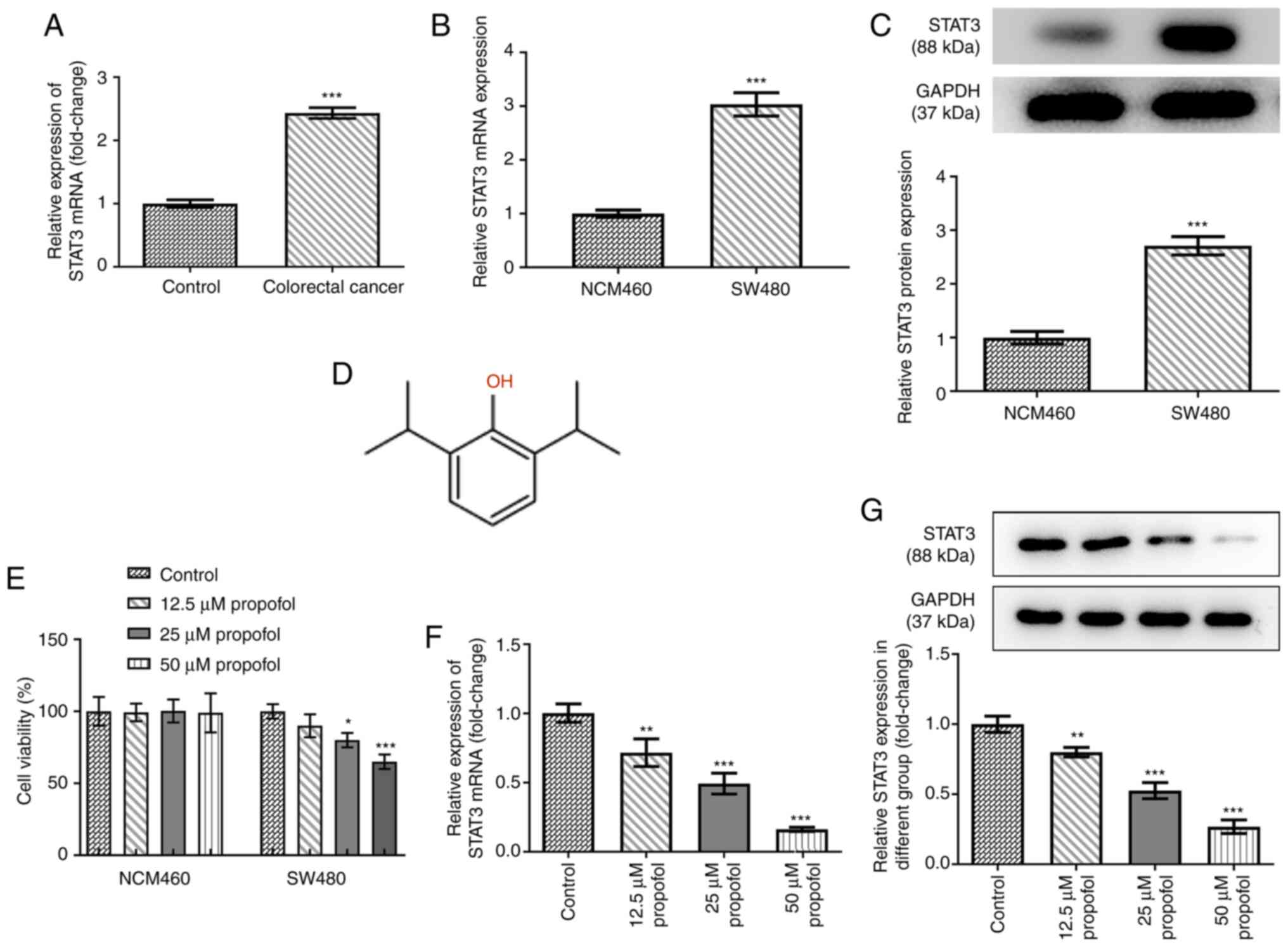 | Figure 1.Effect of propofol on cell viability
and STAT3 expression in SW480 cells. (A) The mRNA level of STAT3 in
colorectal cancer tissues and adjacent control normal tissues after
being normalized to the control group (n=12), as detected by
RT-qPCR. The (B) mRNA and (C) protein expression of STAT3 in SW480
cells and human intestinal epithelial cells (NCM460) was detected
by RT-qPCR and western blotting, respectively. (D) The chemical
structure of propofol. (E) The cell viability of NCM460 and SW480
cells exposed to 0, 12.5, 25 and 50 µm propofol for 48 h was
detected by MTT assay. The (F) mRNA and (G) protein expression of
STAT3 in SW480 cells exposed to 0, 12.5, 25 and 50 µm propofol for
48 h was detected by RT-qPCR and western blotting, respectively.
*P<0.05, **P<0.01 and ***P<0.001 vs. control or NCM460
group. n=3. STAT3, signal transducer and activator of transcription
3; RT-qPCR, reverse transcription quantitative PCR. |
Propofol inhibits CRC cell
proliferation and colony formation, while STAT3 overexpression
inhibits these effects
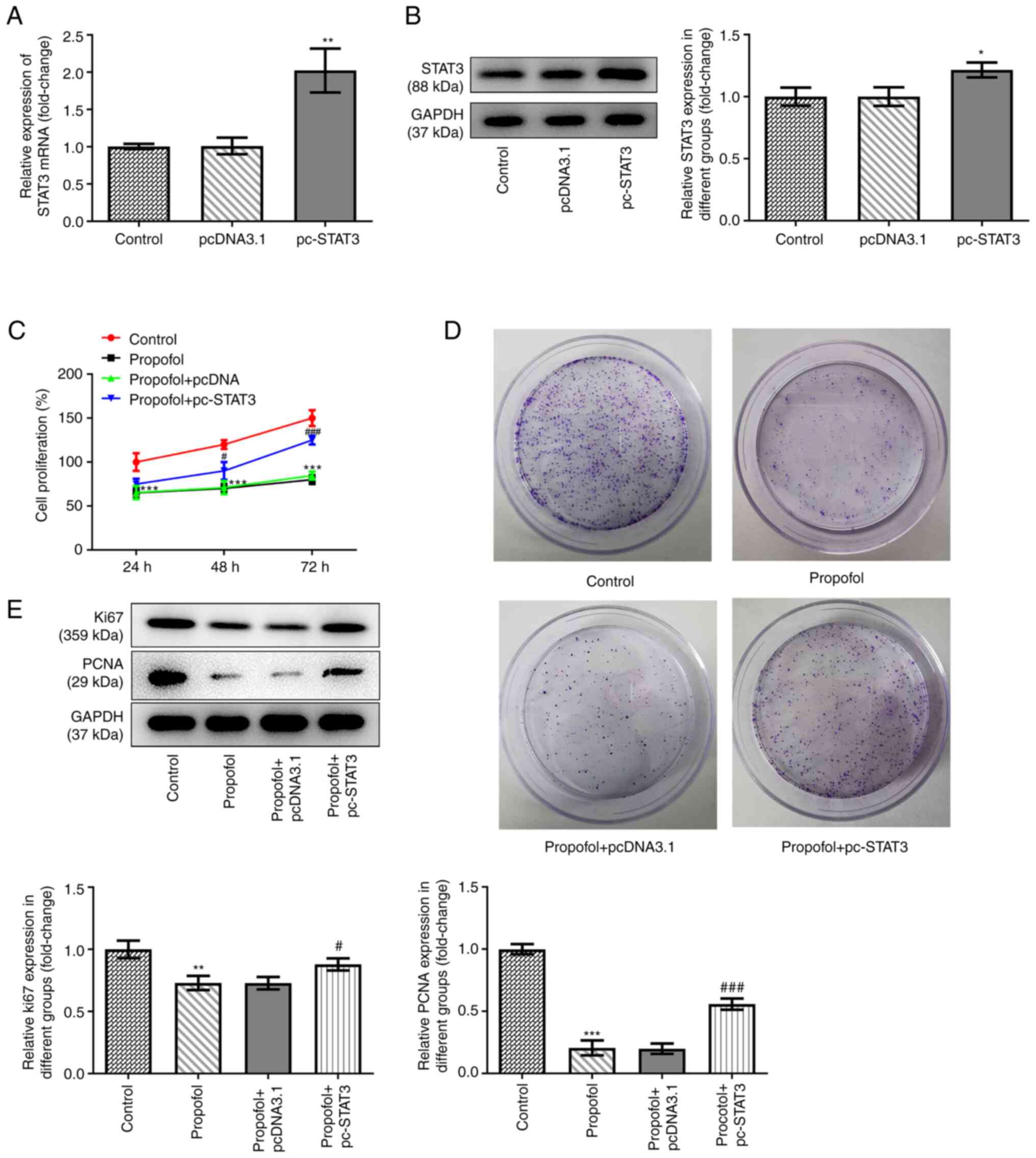
pcDNA3.1-STAT3 overexpression vectors were
transfected into SW480 cells to overexpress STAT3. The data shown
in Fig. 2A and B revealed that STAT3
was successfully overexpressed in the cells, as demonstrated by
significantly upregulated mRNA and protein expression levels of
STAT3 in SW480 cells transfected with the overexpression vector.
Untreated and SW480 cells overexpressing STAT3 were exposed to
control medium or medium supplemented with 50 µm propofol, then
cell proliferation and the colony formation ability were assessed.
The results demonstrated that propofol treatment markedly inhibited
cell proliferation, while STAT3 overexpression promoted cell
proliferation (Fig. 2C). The results
of the colony formation assay demonstrated that the inhibitory
effect of propofol on cell colony formation was also reversed by
STAT3 overexpression (Fig. 2D).
Similarly, propofol treatment downregulated the expression levels
of proteins involved in cell proliferation, including Ki67 and
PCNA, while STAT3 overexpression partially recovered the expression
levels of these proteins (Fig.
2E).
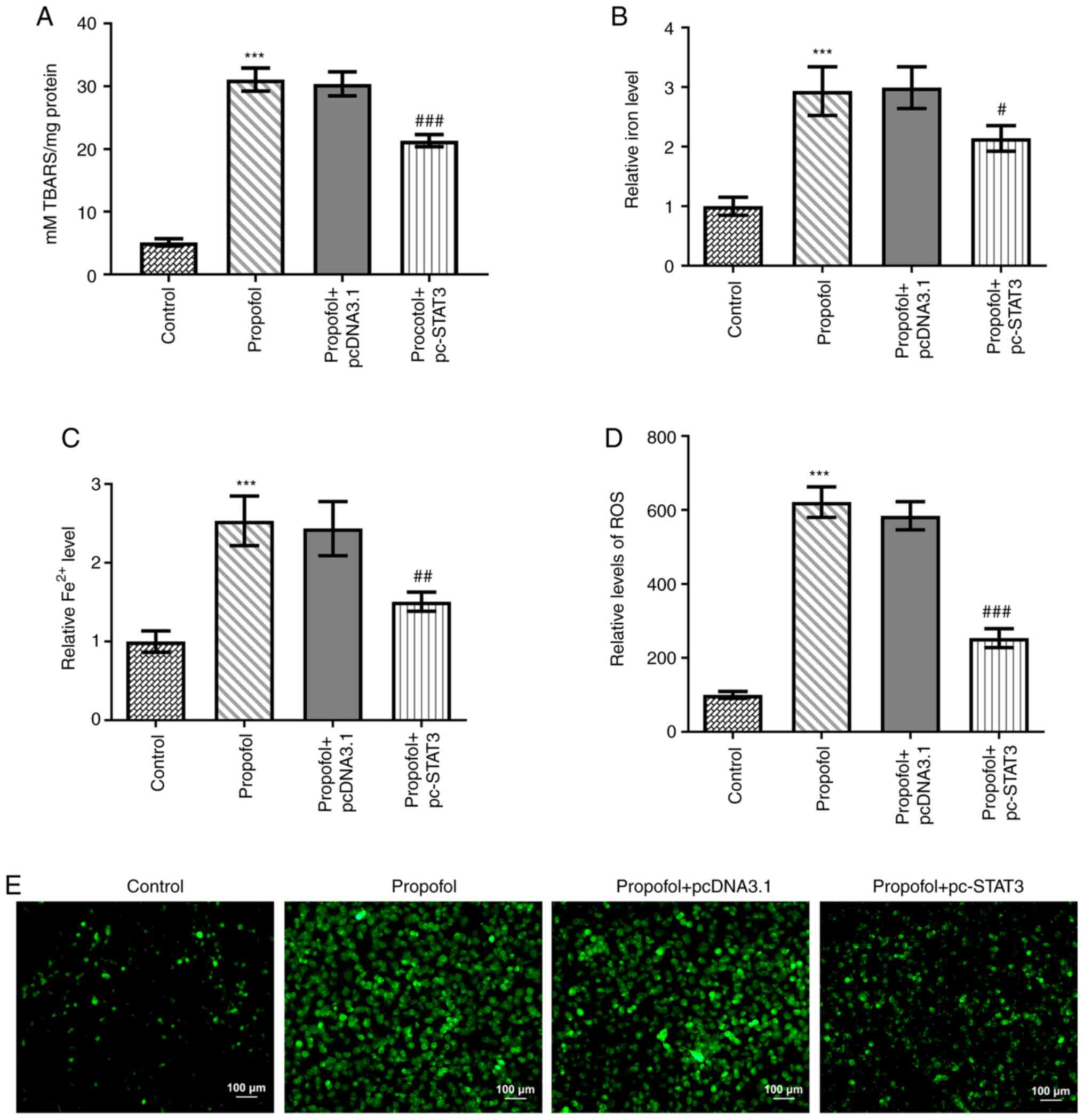
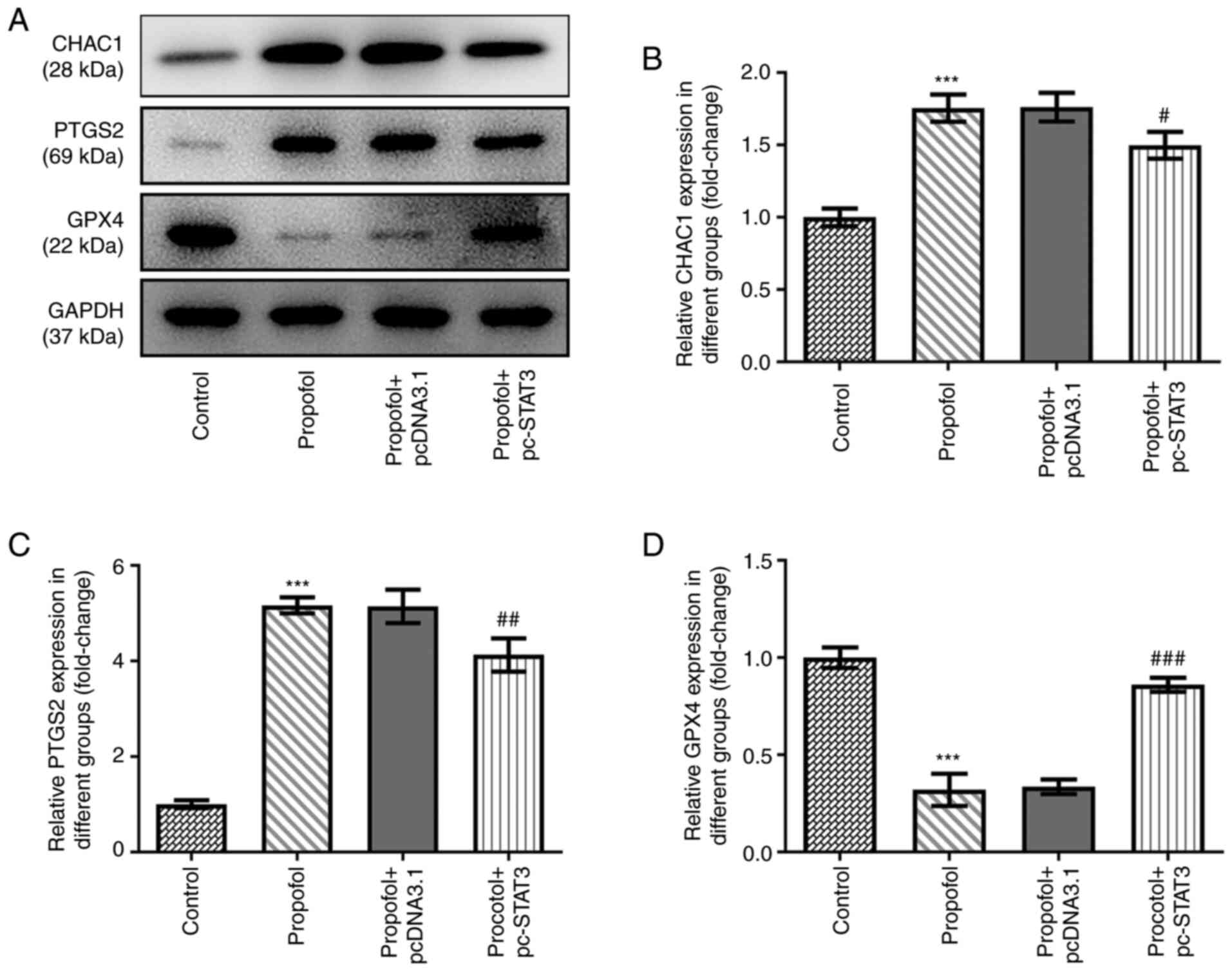
Propofol induces CRC cell ferroptosis,
which is blocked by STAT3 overexpression
Finally, the levels of ferroptosis in CRC cells
following propofol stimulation were evaluated. Propofol treatment
significantly increased TBARS (Fig.
3A), total cellular iron (Fig.
3B), Fe2+ (Fig. 3C)
and ROS (Fig. 3D) levels. Notably,
the content of ROS in propofol group was markedly increased
compared with that in the control group (Fig. 3E). In addition, the protein
expression levels of CHAC1 and PTGS2 were significantly
upregulated, while those of GPX4 were significantly downregulated
by propofol treatment (Fig. 4A-D).
However, all these effects were markedly weakened by the
overexpression of STAT3 (Figs. 3 and
4).
Discussion
To the best of our knowledge, the causes of the high
incidence and mortality rates in CRC remain undetermined. Surgical
tumor resection is currently the main clinical treatment method for
CRC (21). However, surgery can
promote cancer metastasis, as the procedure allows cancer cells to
escape and enter the circulatory system, which represents a
significant challenge in the effective treatment of CRC that needs
to be resolved urgently (7). It has
been reported that anesthetics can improve the long-term outcome of
patients after surgery, particularly with regard to recurrence and
metastasis (22). Accumulating
evidence has demonstrated that propofol could inhibit cell
proliferation in different types of cancer, including CRC. However,
to the best of our knowledge, the specific molecular mechanisms
underlying the effects of propofol on the development and
progression of CRC remain to be determined. The results of the
present study demonstrated that propofol treatment significantly
promoted the ferroptosis and inhibited the proliferation of CRC
cells, which suggested that propofol may play a tumor suppressive
role in CRC.
Although the research is currently limited to in
vitro and animal model experiments, a number of small molecular
ferroptosis inducers (e.g., erastin, sorafenib and sulfasalazine)
have shown beneficial antitumor effects (23), and some anticancer drugs approved by
the US Food and Drug Administration have been identified as
inducers of ferroptosis (24–26). For
example, sorafenib was discovered to increase the levels of lipid
oxidation in hepatocellular carcinoma, which led to cell death.
Notably, administration of a specific inhibitor of ferroptosis,
ferrostatin-1, prevented this effect, while inhibitors of apoptosis
and necrosis were unable to prevent this effect (27). In pancreatic cancer, activation of
the heat shock protein A5 (HSPA5)/GPX4 signaling pathway induced
resistance to gemcitabine, while the knockdown of HSPA5 or GPX4
reversed this drug resistance. Ferroptosis was discovered to play
an important role in this process, as GPX4 is known to decrease the
accumulation of lipid-free radicals and prevent the occurrence and
development of ferroptosis (28,29). A
previous study reported that artemisinin and its derivatives
triggered ferroptosis in pancreatic cancer cells with gene
mutations in KRAS, but exerted minimal toxic effects on normal
cells (30). Proanthocyanidin
treatment markedly suppressed the levels of iron, TBAR, acyl-CoA
synthase 4 and arachidonate 15-lipoxygenase type B, while
upregulating the levels of GSH, GPX4, erythroid 2-related factor 2
and heme oxygenase-1 in cases of spinal cord trauma. To the best of
our knowledge, the current study was the first to discover that
propofol could promote ferroptosis in CRC cells. Ferroptosis, which
is characterized by iron accumulation and lipid peroxidation, is
believed to be related to tumor cell death (10), and the present results showed that
TBAR level and both the cellular total iron and Fe2+
levels were significantly increased by propofol treatment. The
accumulation of ROS is a characteristic of ferroptosis, and the
current study also found that ROS levels were significantly
increased in CRC cells upon propofol stimulation. These findings
were consistent with the results of previous studies, indicating
that propofol may induce ROS-mediated apoptosis in cancer cells
(15,16). Ferroptosis is tightly controlled by
GPX4 and certain iron transport regulatory proteins. A previous
study reported that GPX4 inactivation promoted lipid peroxide
accumulation during ferroptosis in CRC (13). The activating transcription factor 4
(ATF4) branch is the predominant signaling pathway activated by
ferroptotic reagents; CHAC1 is downstream of ATF4 and has been
demonstrated to promote the degradation of GSH and subsequently
induce ferroptosis (31,32). PTGS2 is also induced in cells
undergoing ferroptosis (33);
however, the exact role of PTGS2 in the ferroptotic cell death
cascade remains to be elucidated. Previous studies reported that,
inhibiting PTGS2, which is induced by ferroptosis, was an effective
method for alleviating cell death (33,34). The
present study analyzed the expression levels of GPX4, CHAC1 and
PTGS2 in CRC cells following propofol treatment. Consistent with
the aforementioned experimental results, propofol treatment
markedly downregulated GPX4 expression levels, but upregulated
CHAC1 and PTGS2 expression levels. These data suggested that,
consistent with other ferroptosis inducers, propofol may be able to
enhance cellular iron levels and ROS accumulation, as well as
upregulate the expression levels of CHAC1 and PTGS2, while
downregulating those of GPX4.
There are certain limitations to the present study.
For example, cell death assays for detecting PI uptake or LDH
release were not performed to determine if the decreased cell
viability was due to cell death. In addition, experiments using
ferroptosis inhibitor (ferrostatin-1) were not conducted to
distinguish between ferroptosis and apoptosis. Due to limited
funding, not all ferroptosis clarification experiments could be
performed. However, the characteristics of ferroptosis, in which
GPX4 decreases, PTGS2 increases and Fe2+ increases, were
observed and a TBAR lipid peroxidation assay was conducted, which
also indicated the presence of ferroptosis.
To further determine the potential underlying
mechanism involved in the effects of propofol, the STITCH database
was used to identify the regulation of STAT3 by propofol. STAT3
expression is known to be upregulated in the majority of cancer
types, including in CRC tissues compared with adjacent control
normal tissues. Notably, it has been widely reported that propofol
can target STAT3-related signaling pathways, thereby playing a key
a role in various diseases. For example, propofol was found to
enhance cisplatin-induced cell apoptosis in cervical cancer cells
via the EGFR/JAK2/STAT3 signaling pathway (35). Propofol also prevented oxidative
stress and apoptosis by regulating iron homeostasis and targeting
the JAK/STAT3 signaling pathway in SH-SY5Y cells (36). In addition, a previous study
demonstrated that propofol inhibited cell invasion and promoted
cell apoptosis by regulating the STAT3/HOX transcript antisense RNA
axis in CRC (37). The findings of
the present study revealed that propofol treatment negatively
regulated STAT3 expression in a concentration-dependent manner.
Notably, STAT3 has also been suggested to regulate ferroptosis. For
example, the administration of small molecules that inhibit STAT3
phosphorylation was reported to increase cellular ROS levels and
decrease the GSH/glutathione disulfide ratio (38). In addition, α6β4 integrin-mediated
STAT3 activation was found to promote resistance to ferroptosis
(39). Cisplatin-resistant
osteosarcoma cells could inhibit ferroptosis after low-dose
cisplatin irradiation; however, the administration of ferroptosis
agonists and STAT3 inhibitors reactivated ferroptosis, thereby
increasing the sensitivity to cisplatin (40). STAT3 overexpression also promoted the
migration and invasion of HT-29 cells (41). Activation and phosphorylation of
STAT3 could markedly promote the proliferation and metastasis of
SW480 cells, and enhance tumorigenesis (42). In one study, the migration and
invasion of colon cancer cells was promoted, possibly via the
activation of the STAT3 pathway (43). Therefore, it was hypothesized that
propofol may promote CRC cell ferroptosis via targeting STAT3. In
the present study, STAT3 was overexpressed in CRC cells to
investigate whether the effects of propofol on CRC cell
proliferation and ferroptosis could be reversed. As expected, the
inhibitory effect of propofol on CRC cell proliferation and colony
formation, together with the stimulatory effect of propofol on
ferroptosis, were all significantly inhibited by STAT3
overexpression. The aforementioned data suggested that propofol may
induce CRC cell ferroptosis by downregulating STAT3 expression. To
the best of our knowledge, the present study is the first to
determine the effect of propofol on ferroptosis in cancer. However,
the study conclusions were based on results obtained from an in
vitro cell model; therefore, further in vivo experiments
are required to verify these findings.
In conclusion, the present study demonstrated that
propofol promoted ferroptosis in CRC cells and that the potential
mechanism may be dependent on targeting STAT3 expression.
Therefore, propofol-induced ferroptosis may represent a promising
therapeutic option for CRC.
Acknowledgements
Not applicable.
Funding
Not applicable.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZ and FC conceived and designed the study. XZ was
responsible for the acquisition analysis and interpretation of
data. FC was responsible for manuscript preparation, writing and
critical revisions. XZ and FC confirm the authenticity of all the
raw data. Both authors have read and approved the manuscript.
Ethics approval and consent to
participate
All the patients provided written, informed consent
and the study was approved by the Ethical Committee of Zhongshan
Hospital (approval no. B2016-014).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dekker E, Tanis PJ, Vleugels JLA, Kasi PM
and Wallace MB: Colorectal cancer. Lancet. 394:1467–1480. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brody H: Colorectal cancer. Nature.
521:S12015. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bateman BT and Kesselheim AS: Propofol as
a transformative drug in anesthesia: Insights from key early
investigators. Drug Discov Today. 20:1012–1017. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jiang S, Liu Y, Huang L, Zhang F and Kang
R: Effects of propofol on cancer development and chemotherapy:
Potential mechanisms. Eur J Pharmacol. 831:46–51. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu ZF, Lee MS, Wong CS, Lu CH, Huang YS,
Lin KT, Lou YS, Lin C, Chang YC and Lai HC: Propofol-based total
intravenous anesthesia is associated with better survival than
desflurane anesthesia in colon cancer surgery. Anesthesiology.
129:932–941. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ren YL and Zhang W: Propofol promotes
apoptosis of colorectal cancer cells via alleviating the
suppression of lncRNA HOXA11-AS on miRNA let-7i. Biochem Cell Biol.
98:90–98. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hickman JA: Apoptosis induced by
anticancer drugs. Cancer Metastasis Rev. 11:121–139. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mohammad RM, Muqbil I, Lowe L, Yedjou C,
Hsu HY, Lin LT, Siegelin MD, Fimognari C, Kumar NB and Dou QP:
Broad targeting of resistance to apoptosis in cancer. Semin Cancer
Biol. 35:S78–S103. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hassannia B, Vandenabeele P and Berghe TV:
Targeting ferroptosis to iron out cancer. Cancer Cell. 35:830–849.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mou Y, Wang J, Wu J, He D, Zhang C, Duan C
and Li B: Ferroptosis, a new form of cell death: Opportunities and
challenges in cancer. J Hematol Oncol. 12:342019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu X, Zhang X, Wei C, Zheng D, Lu X, Yang
Y, Luo A, Zhang K, Duan X and Wang Y: Targeting SLC7A11
specifically suppresses the progression of colorectal cancer stem
cells via inducing ferroptosis. Eur J Pharm Sci. 152:1054502020.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sui X, Zhang R, Liu S, Duan T, Zhai L,
Zhang M, Han X, Xiang Y, Huang X, Lin H and Xie T: RSL3 drives
ferroptosis through GPX4 inactivation and ROS production in
colorectal cancer. Front Pharmacol. 9:13712018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang L, Liu W, Liu F, Wang Q, Song M, Yu
Q, Tang K, Teng T, Wu D, Wang X, et al: IMCA induces ferroptosis
mediated by SLC7A11 through the AMPK/mTOR pathway in colorectal
cancer. Oxid Med Cell Longev. 2020:16756132020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liang WZ, Jan CR and Lu CH: Investigation
of 2,6-diisopropylphenol (propofol)-evoked Ca2+ movement and cell
death in human glioblastoma cells. Toxicol In Vitro. 26:862–871.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang H, Zhao L, Wu J, Hong J and Wang S:
Propofol induces ROS-mediated intrinsic apoptosis and migration in
triple-negative breast cancer cells. Oncol Lett. 20:810–816. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu H, Lee H, Herrmann A, Buettner R and
Jove R: Revisiting STAT3 signalling in cancer: New and unexpected
biological functions. Nat Rev Cancer. 14:736–746. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou B, Liu J, Kang R, Klionsky DJ,
Kroemer G and Tang D: Ferroptosis is a type of autophagy-dependent
cell death. Semin Cancer Biol. 66:89–100. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Y, Zhu M, Mohan SK and Hao Z: Crocin
treatment promotes the oxidative stress and apoptosis in human
thyroid cancer cells FTC-133 through the inhibition of STAT/JAK
signaling pathway. J Biochem Mol Toxicol. 35:e226082021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu Z: Surgical treatment of colorectal
cancer. Chin Oncol. 23:389–398. 2013.(In Chinese).
|
|
22
|
Cassinello F, Prieto I, del Olmo M, Rivas
S and Strichartz GR: Cancer surgery: How may anesthesia influence
outcome? J Clin Anesth. 27:262–272. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liang C, Zhang X, Yang M and Dong X:
Recent progress in ferroptosis inducers for cancer therapy. Adv
Mater. 31:e19041972019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Trujillo-Alonso V, Pratt EC, Zong H,
Lara-Martinez A, Kaittanis C, Rabie MO, Longo V, Becker MW, Roboz
GJ, Grimm J and Guzman ML: FDA-approved ferumoxytol displays
anti-leukaemia efficacy against cells with low ferroportin levels.
Nat Nanotechnol. 14:616–622. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lo M, Ling V, Low C, Wang YZ and Gout PW:
Potential use of the anti-inflammatory drug, sulfasalazine, for
targeted therapy of pancreatic cancer. Curr Oncol. 17:9–16. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Y, Shi J, Liu X, Feng L, Gong Z,
Koppula P, Sirohi K, Li X, Wei Y, Lee H, et al: BAP1 links
metabolic regulation of ferroptosis to tumour suppression. Nat Cell
Biol. 20:1181–1192. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R
and Tang D: Activation of the p62-Keap1-NRF2 pathway protects
against ferroptosis in hepatocellular carcinoma cells. Hepatology.
63:173–184. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu S, Zhang Q, Sun X, Zeh HJ III, Lotze
MT, Kang R and Tang D: HSPA5 regulates ferroptotic cell death in
cancer cells. Cancer Res. 77:2064–2077. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tang H, Chen D, Li C, Zheng C, Wu X, Zhang
Y, Song Q and Fei W: Dual GSH-exhausting sorafenib loaded
manganese-silica nanodrugs for inducing the ferroptosis of
hepatocellular carcinoma cells. Int J Pharm. 572:1187822019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang K, Zhang Z, Wang M, Cao X, Qi J, Wang
D, Gong A and Zhu H: Role of GRP78 inhibiting artesunate-induced
ferroptosis in KRAS mutant pancreatic cancer cells. Drug Des Dev
Ther. 13:2135–2144. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W and
Wang J: Molecular mechanisms of ferroptosis and its role in cancer
therapy. J Cell Mol Med. 23:4900–4912. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dixon SJ, Patel DN, Welsch M, Skouta R,
Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS
and Stockwell BR: Pharmacological inhibition of cystine-glutamate
exchange induces endoplasmic reticulum stress and ferroptosis.
Elife. 3:e025232014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xiao X, Jiang Y, Liang W, Wang Y, Cao S,
Yan H, Gao L and Zhang L: miR-212-5p attenuates ferroptotic
neuronal death after traumatic brain injury by targeting Ptgs2. Mol
Brain. 12:782019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Q, Han X, Lan X, Gao Y, Wan J, Durham
F, Cheng T, Yang J, Wang Z, Jiang C, et al: Inhibition of neuronal
ferroptosis protects hemorrhagic brain. JCI Insight. 2:e907772017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li H, Lu Y, Pang Y, Li M, Cheng X and Chen
J: Propofol enhances the cisplatin-induced apoptosis on cervical
cancer cells via EGFR/JAK2/STAT3 pathway. Biomed Pharmacother.
86:324–333. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang Y, Zuo Y, Li B, Xie J, Ma Z,
Thirupathi A, Yu P, Gao G, Shi M, Zhou C, et al: Propofol prevents
oxidative stress and apoptosis by regulating iron homeostasis and
targeting JAK/STAT3 signaling in SH-SY5Y cells. Brain Res Bull.
153:191–201. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang YF, Li CS, Zhou Y and Lu XH: Effects
of propofol on colon cancer metastasis through STAT3/HOTAIR axis by
activating WIF-1 and suppressing wnt pathway. Cancer Med.
9:1842–1854. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hu S, Sechi M, Singh PK, Dai L, McCann S,
Sun D, Ljungman M and Neamati N: A novel redox modulator induces a
GPX4-mediated cell death that is dependent on iron and reactive
oxygen species. J Med Chem. 63:9838–9855. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Brown CW, Amante JJ, Goel HL and Mercurio
AM: The α6β4 integrin promotes resistance to ferroptosis. J Cell
Biol. 216:4287–4297. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu Q and Wang K: The induction of
ferroptosis by impairing STAT3/Nrf2/GPx4 signaling enhances the
sensitivity of osteosarcoma cells to cisplatin. Cell Biol Int.
43:1245–1256. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang GY, Yang WH and Chen Z: Upregulated
STAT3 and RhoA signaling in colorectal cancer (CRC) regulate the
invasion and migration of CRC cells. Eur Rev Med Pharmacol Sci.
20:2028–2037. 2016.PubMed/NCBI
|
|
42
|
Zhao FL and Qin CF: EGF promotes HIF-1α
expression in colorectal cancer cells and tumor metastasis by
regulating phosphorylation of STAT3. Eur Rev Med Pharmacol Sci.
23:1055–1062. 2019.PubMed/NCBI
|
|
43
|
Zhang WJ, Hu CG, Luo HL and Zhu ZM:
Activation of P2×7 receptor promotes the invasion and migration of
colon cancer cells via the STAT3 signaling. Front Cell Dev Biol.
8:5865552020. View Article : Google Scholar : PubMed/NCBI
|