Introduction
Acute myeloid leukemia (AML) is a heterogeneous cancer characterized by the uncontrolled proliferation of abnormal cells that accumulate in the bone marrow and blood (1,2). Acute promyelocytic leukemia (APL) represents 10–12% of AML cases, and is characterized by the presence of abnormal, heavily granulated promyelocytes (3). As a form of acute leukemia, APL progresses rapidly with a median survival time of <1 week without treatment (4). At present, chemotherapeutic agents, including all-trans-retinoic acid (ATRA) and arsenic trioxide (ATO), are widely used for the treatment of APL, with complete remission rates of <90% (5); however, side effects and drug resistance can arise from treatment with ATRA and ATO (6). Therefore, it is necessary to develop novel chemotherapeutic agents for the treatment of AML with high biological activity and low toxicity.
Microtubules, comprising α- and β-tubulin heterodimers, are involved in a variety of biological processes, such as mitosis (7). As microtubules serve key roles in the regulation of the mitotic apparatus, the disruption of microtubules can induce cell cycle arrest in G2/M phase and cell apoptosis (8,9). A set of 4,5-diaryl-3H−1,2-dithiole-3-thiones as combretastatin A-4/oltipraz hybrids (COHs) were designed and synthesized in a previous study. A tubulin polymerization assay, and immunofluorescence and cell cycle analyses suggested that the most active compound COH-203 (Fig. 1), 5-(3-amino-4-methoxyphenyl)-4-(3,4,5-trimethoxyphenyl)-3H−1,2-dithiole-3-one (compound 4d in reference 10), exhibited strong anti-tubulin polymerization activity, in addition to its antiproliferative properties (10,11).
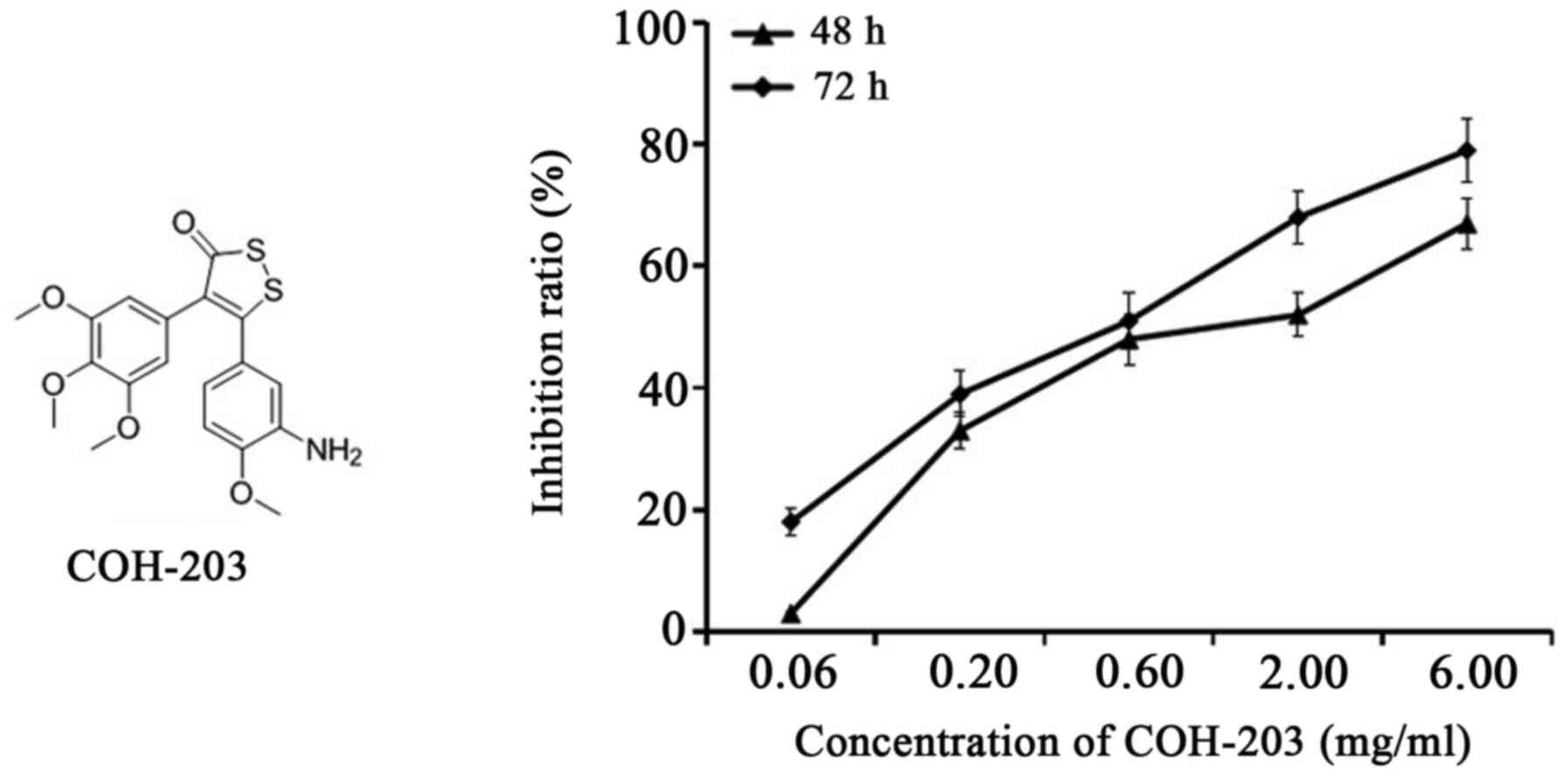 |
Figure 1.
Structure of COH-203 and its antiproliferative activity against HL-60 cell line. HL-60 cells were incubated with 0.06, 0.20, 0.60, 2.00 or 6.00 µg/ml of COH-203 for 48 and 72 h, and the number of viable cells in each well was measured. COH-203, 5-(3-amino-4-methoxyphenyl)-4-(3,4,5-trimethoxyphenyl)-3H−1,2-dithiole-3-one.
|
At present, the antiproliferative activities of 4,5-diaryl-3H−1,2-dithiole-3-thiones (COHs) against several solid tumor cell lines (SGC-7901, KB, HT-1080, HepG2, SMMC-7721 and BEL-7402) have been well reported (10,11). Conversely, to the best of our knowledge, the effects of these COHs on human leukemia cell lines are yet to be investigated. Therefore, the aim of the present study was to determine the sensitivity of the human APL cell line, HL-60, to COH-203 and the mechanism underlying these effects.
Materials and methods
Materials
COH-203 was synthesized by Wang et al (10) (Shenyang Pharmaceutical University); the identity and purity (>98%) of COH-203 were verified by 1H nuclear magnetic resonance and mass spectrometry in the present study. The analyzed compound was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 10 µg/ml, and diluted to the desired concentration prior to use.
Cell culture
Human APL HL-60 cells were obtained from the Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences. HL-60 cells were cultured in RPMI-1640 medium (Sigma-Aldrich; Merck KGaA) supplemented with 10% fetal calf serum (Sigma-Aldrich; Merck KGaA) and penicillin (100 U/ml)/streptomycin (100 µg/ml). Cultures were maintained in a humidified incubator at 37°C in 5% CO2.
Determination of cell proliferation and viability
The antiproliferative activity of COH-203 against HL-60 cells was assessed by an MTT assay. CA-4 served as a positive control. MTT (5.0 mg/ml) was dissolved in PBS and filtered. Cells at a density of 1,000 cells/200 µl medium/well were seeded in a 96-well plate. Cells were then treated with 0.06, 0.20, 0.60, 2.00 and 6.00 µg/ml COH-203 for 48 and 72 h at 37°C in 5% CO2. The five different concentrations were randomly selected. Following incubation, 20.0 µl MTT solution (final concentration, 0.5 mg/ml) was added to each well and incubated at 37°C for a further 4 h. The 96-well plate containing the cells was centrifuged at 1,000 rpm (RCF=166.5 × g) for 5 min at 4°C. The MTT solution was removed from the wells and the formazan crystals were dissolved in 150 µl DMSO. After 10 min, the plates were analyzed with a microplate reader at 570 nm (Sunrise™ RC; Tecan Group Ltd.). The effects of COH-203 on cell viability were assessed and the data were expressed as a percentage of inhibited cell growth. The experiments were repeated in triplicate.
Analysis of DNA content by flow cytometry
Following treatment with various concentrations of COH-203 for 12, 24 or 48 h, cells (1×106) were collected by centrifugation at 1,000 rpm (RCF=166.5 × g) for 5 min at 4°C, washed twice with PBS and then fixed with 70% ethanol at 4°C overnight. After the ethanol was removed the next day, the cells were resuspended in PBS at 4°C. Following treatment with RNase (cat. no. 10109142001; Sigma-Aldrich; Merck KGaA) (20 µg/ml) for 30 min at 37°C, cells (1×106/well) were stained with DNA staining solution propidium iodide (cat. no. P4864; Sigma-Aldrich; Merck KGaA) (50 µg/ml) for 30 min at 4°C in the dark and analyzed with a flow cytometer (FACSCalibur; BD Biosciences). The data regarding the number of cells in different phases of the cell cycle were analyzed by Flowjo® Software V10 (FlowJo LLC). The experiments were repeated in triplicate.
Acridine orange (AO)/ethidium bromide (EB) fluorescence staining to detect apoptosis
In the pilot experiments, 2.0 µg/ml COH-203 exhibited potent G2/M-phase arrest in HL-60 cells and therefore was used in the rest of the study without further optimization. Following treatment with COH-203 (2.0 µg/ml) for 12, 24 or 48 h, cells (1×106) were harvested and stained with acridine orange (AO) (cat. no. A6014; Sigma-Aldrich; Merck KGaA) and ethidium bromide (EB) (cat. no. E1510; Sigma-Aldrich; Merck KGaA) at room temperature for 30 sec, and assessed by fluorescence microscopy (Olympus Corporation). The magnification was ×200 and fields were selected in a random manner. Briefly, 1 µl stock solution containing 100 µg/ml AO and EB, respectively, was added to 25 µl cell suspension. Viable cells were stained green with intact nuclei.
Immunofluorescence microscopy
The results of cell cycle analysis indicated that COH-203-induced G2/M-phase arrest in HL-60 cells firstly at 12 h and subsequently peaked at 24 h. For this reason, the HL-60 cells were treated for 24 h in the analysis of immunofluorescence staining. Following treatment with COH-203 (2.0 µg/ml) for 24 h, cells (3×106 cells were seeded in a 10 mm3 confocal culture dish) were harvested and then fixed in 4% paraformaldehyde for 10 min at room temperature. After washing in PBS for 5 min, cells were permeabilized in 0.5% Triton X-100 in PBS for 30 min. Following blocking with 5% BSA for 2 h at 37°C, cells were incubated with 1:50 mouse anti-β-tubulin monoclonal antibody (cat. no. SAB4200715; Sigma-Aldrich; Merck KGaA) at 4°C overnight. Subsequently, cells were rinsed in PBS three times and incubated with 1:50 fluorescein isothiocyanate (FITC)-conjugated secondary anti-mouse IgG antibody (goat anti-mouse IgG/Alexa-Fluor 488 antibody; cat. no. A28175; Invitrogen; Thermo Fisher Scientific, Inc.) for 2 h at 37°C in the dark. DNA was counterstained with 1:100 Hoechst 33342 (cat. no. B2261; Sigma-Aldrich; Merck KGaA) for 5 min at room temperature in the dark. The stained cells were visualized under a fluorescence microscope (Olympus Corporation). The magnification was ×200 and fields were selected in a random manner.
Western blot analysis
Following treatment with COH-203, cells were harvested. A549 cells, seeded in 60-mm dishes at a density of 3×105 cells/well were incubated with or without COH-203 at indicated concentrations for 24 or 48 h. Following incubation, the cells were washed twice with PBS at 4°C and then lysed in RIPA lysis buffer containing 150 mM NaCl, 50 mM Tris (pH 7.4), 1% (w/v) sodium deoxycholate, 1% (v/v) Triton X-100, 0.1% (w/v) SDS, and 1 mm EDTA (Beyotime Institute of Biotechnology). The lysates were incubated at 0°C for 30 min and vortexed every 10 min intermittently, and the total protein was harvested by centrifuging at 12,500 rpm (RCF=26,015.6 × g) for 15 min at 4°C. After the protein concentrations were determined by a BCA Protein Assay Kit (Thermo Fisher Scientific, Inc.), the protein extracts were reconstituted in loading buffer containing 62 mM Tris-HCl, 2% SDS, 10% glycerol, and 5% β-mercaptoethanol (cat. no. 63689; Sigma-Aldrich; Merck KGaA) and boiled at 100°C for 3 min. An equal amount of the proteins (80 µg) was separated by 8–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and was transferred to polyvinylidene fluoride membranes (cat. no. 3010040001, Sigma-Aldrich). After blocking with 5% non-fat dry milk at room temperature for 2 h, the membranes were probed with primary antibodies at 4°C overnight. The following primary antibodies were used: Rabbit anti-caspase-3 polyclonal antibody (1:200; cat. no. H-277), mouse anti-cyclin B monoclonal antibody (1:1,000; cat. no. D-1), mouse anti-p34cdc2 monoclonal antibody (1:200; cat. no. 17), mouse anti-B-cell lymphoma 2 (Bcl-2) monoclonal antibody (1:200; cat. no. 10C4), mouse anti-β-actin monoclonal antibody (1:50; cat. no. C4) and mouse anti-β-tubulin monoclonal antibody (1:200; cat. no. D-10) (all Santa Cruz Biotechnology, Inc.). Then the membranes were incubated with horseradish peroxidase-labeled secondary antibodies (1:800; cat. no. AC111P; Sigma-Aldrich) at room temperature for 2.5 h. The protein band was visualized using BeyoECL Plus (cat. no. P0018M; Beyotime Institute of Biotechnology). Bio-imaging systems (MF-ChemiBIS3.2; version 2.7; DNR Bio-Imaging Systems, Ltd.) was used for quantifying the expression of the target proteins. β-actin (1:1,000; cat. no. 3700S; Cell Signaling Technology, Inc.) was used as the loading control. The experiment was repeated in triplicate.
Statistical analysis
All data were expressed as the mean ± standard deviation and analyzed with SPSS 16.0 software (IBM Corp). Analysis of multiple groups was conducted using one-way analysis of variance. Students-Newman-Keuls was used as a post hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
Analysis COH-203 data
COH-203 was isolated as a yellow solid and assigned the molecular formula C19H19NNaO5S2, as determined by the HRMS-ESI peak at m/z 428.0597 [M+Na]+ (calculated for C19H19NNaO5S2 428.0602). The 1H NMR data displayed 19 hydrogen resonances: δ 3.69 (s, 6H), 3.83 (s, 3H), 3.85 (s, 3H), 6.41 (s, 2H), 6.61 (brs, 1H) and 6.68 (br s, 2H). The 13C NMR data displayed 19 carbon resonances: δ 54.6(s), 55.0 (d), 59.8(s), 106.5 (d), 109.1 (s), 112.8 (s), 117.9 (s), 125.7 (s), 127.4 (s), 128.2 (s), 135.7 (s), 136.8 (s), 148.0 (s), 152.1 (d), 165.5 (s) and 192.8 (s).
Effects of COH-203 on cell proliferation
Following treatment with 0.06, 0.20, 0.60, 2.00 or 6.00 µg/ml COH-203 for 48 and 72 h, the proliferation of HL-60 cells was examined via an MTT assay; CA-4 was used as the positive control. The results demonstrated that COH-203 inhibited the growth of HL-60 cells in a time- and concentration-dependent manner with a half-maximal inhibitory concentration (IC50) of 1.46±0.13 and 0.57±0.05 µg/ml at 48 and 72 h, respectively (Fig. 1). In addition, the IC50 of the positive control CA-4 was determined to be 0.97±0.10 and 0.35±0.12 µg/ml at 48 and 72 h, respectively.
COH-203 arrests HL-60 cells in the G2/M phase
The ability of COH-203 to modulate the cell cycle progression of HL-60 cells was evaluated by using asynchronous cultures, which were continuously exposed to COH-203. Cells were then stained with propidium iodide and analyzed by flow cytometry. Treatment with COH-203 resulted in a time- and concentration-dependent accumulation of HL-60 cells in G2/M phase (Fig. 2). COH-203-induced G2/M phase arrest in HL-60 cells was first observed at 12 h (68.51±6.94%; Fig. 2A). In addition, a characteristic hypodiploid DNA content peak (SubG1), which indicated the presence of apoptotic cells, was observed following treatment with COH-203 for 48 h (14.83±0.02%; Fig. 2A). The results indicated that COH-203 could arrest HL-60 cells in G2/M phase and induce cell apoptosis. Furthermore, the prolongation of G2/M phase of HL-60 cells may induce cell apoptosis.
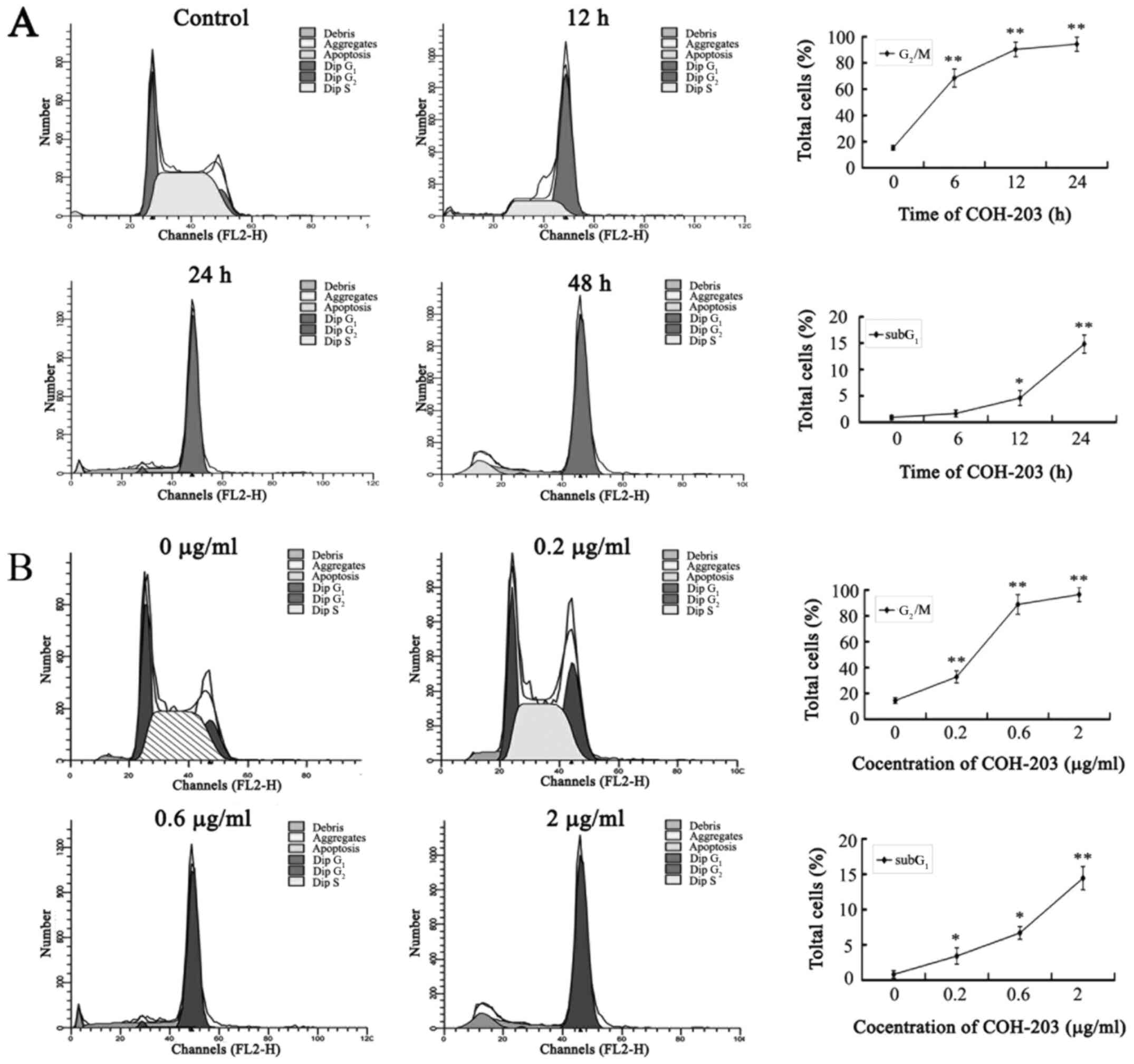 |
Figure 2.
Effect of COH-203 on progression through the cell cycle. HL-60 cells were cultured either (A) in the presence of 2.0 µg/ml COH-203 for the indicated incubation times, or (B) for 48 h with various concentrations of COH-203. Cells were stained with propidium iodide and analyzed by flow cytometry. COH-203, 5-(3-amino-4-methoxyphenyl)-4-(3,4,5-trimethoxyphenyl)-3H−1,2-dithiole-3-one. *P<0.05; **P<0.01 vs. control.
|
COH-203 induces apoptosis in HL-60 cells
To further investigate apoptosis, HL-60 cells were stained with AO/EB and assessed by fluorescence microscopy (Fig. 3). Apoptosis was demonstrated by the appearance of cell shrinkage with condensed and fragmented nuclei, and the formation of apoptotic bodies.
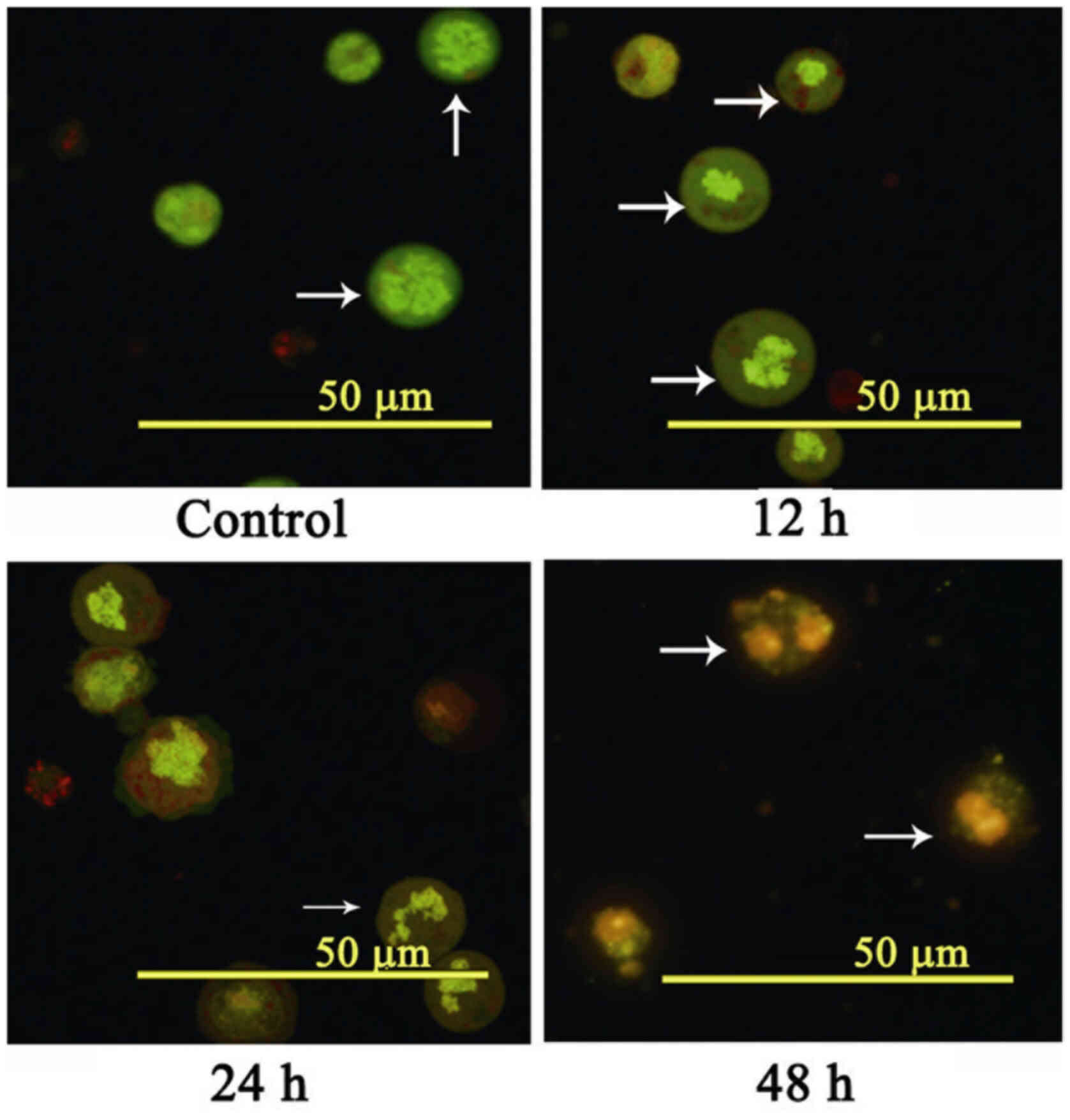 |
Figure 3.
Morphological observation after AO/EB staining. HL-60 cells were incubated with 2.0 µg/ml of COH-203 at 12, 24 and 48 h. Cells were stained with AO/EB and analyzed by fluorescence microscopy. Scale bars, 50 µm. The white arrows indicate the area to focus on. COH-203, 5-(3-amino-4-methoxyphenyl)-4-(3,4,5-trimethoxyphenyl)-3H−1,2-dithiole-3-one; AO, Acridine orange; EB, ethidium bromide.
|
In the present study, no obvious alterations in apoptosis were detected in the negative control group. The nucleus of early apoptotic cells (12 h) exhibited yellow-green fluorescence and possessed a granular appearance, which was observed on one side of cells. During the treatment, concentrated and asymmetrically-localized orange nuclear EB staining was also detected (24 and 48 h) in a time-dependent manner. These morphological alterations, including the presence of chromatin condensation and nuclear fragmentation following AO/EB staining further indicated that COH-203 could induce the apoptosis of HL-60 cells.
COH-203 disrupts microtubule polymerization in HL60 cells
To determine whether COH-203 can induce the formation of abnormal mitotic spindles in HL-60 cells, immunofluorescence staining using anti-β-tubulin antibody was performed. Following treatment with COH-203 for 24 h, the normal arrangement and organization of microtubule networks were not observed (Fig. 4).
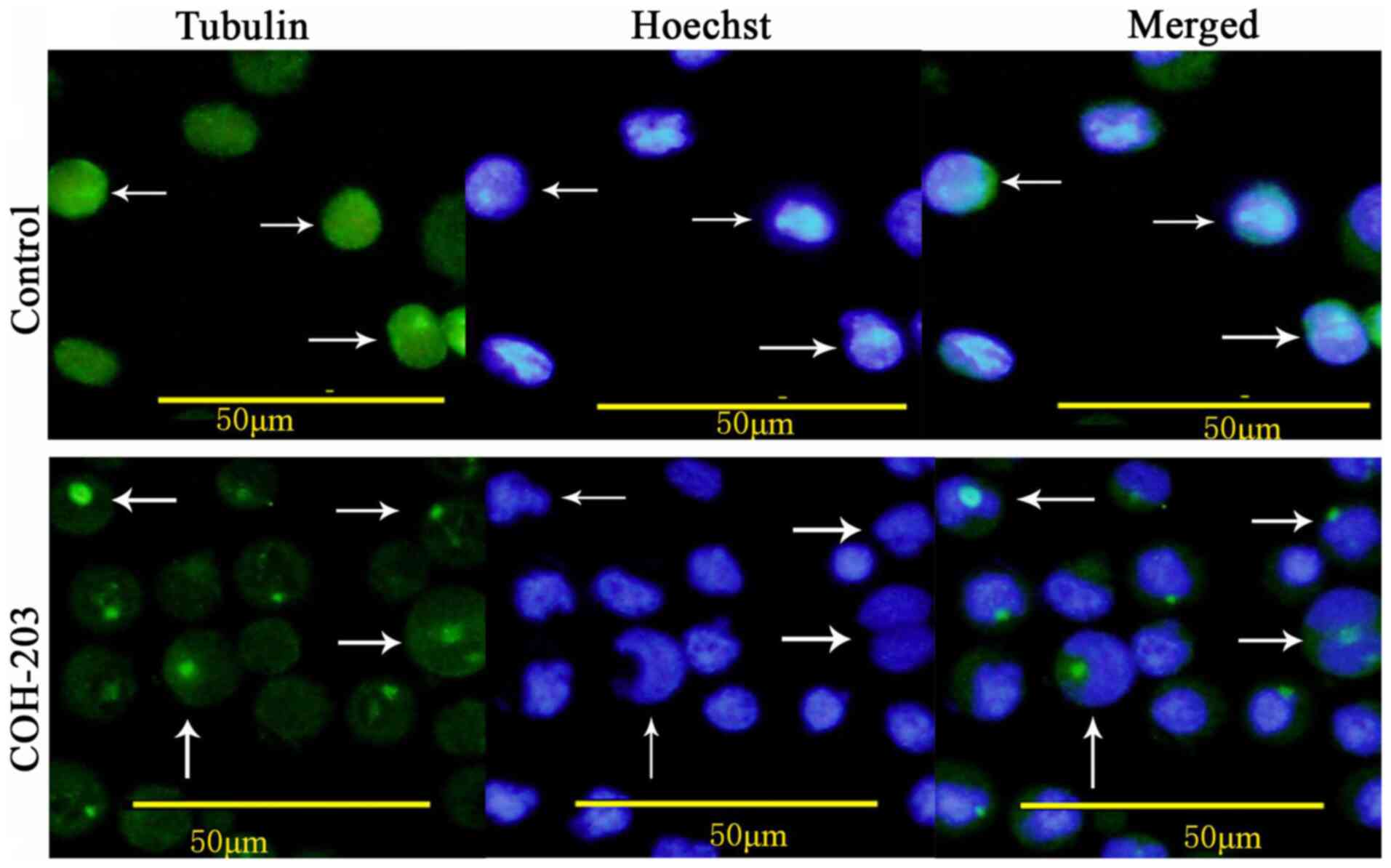 |
Figure 4.
Effect of COH-203 on the organizations and arrangements of the cellular microtubule network as indicated by the white arrows. HL-60 cells were incubated in the absence and presence of 2.0 µg/ml COH-203 for 24 h and stained for cellular microtubules microtubule network (green) and nuclear stain Hoechst (blue). Scale bars, 50 µm. The white arrows indicated the view to focus on.
|
Alterations in protein expression are associated with G2/M phase
The association between COH-203-induced G2/M phase arrest and alterations in G2/M phase regulatory protein expression was investigated. In the present study, COH-203 induced a significant increase in cyclin B expression (Fig. 5A). Additionally, p34cdc2 was activated via binding with cyclin B and its protein expression levels were unaltered throughout the cell cycle (Fig. 5A). These results indicated that COH-203 may arrest HL-60 cells in G2/M phase by preventing the degradation of cyclin B, which could result in the sustained activation of p34cdc2.
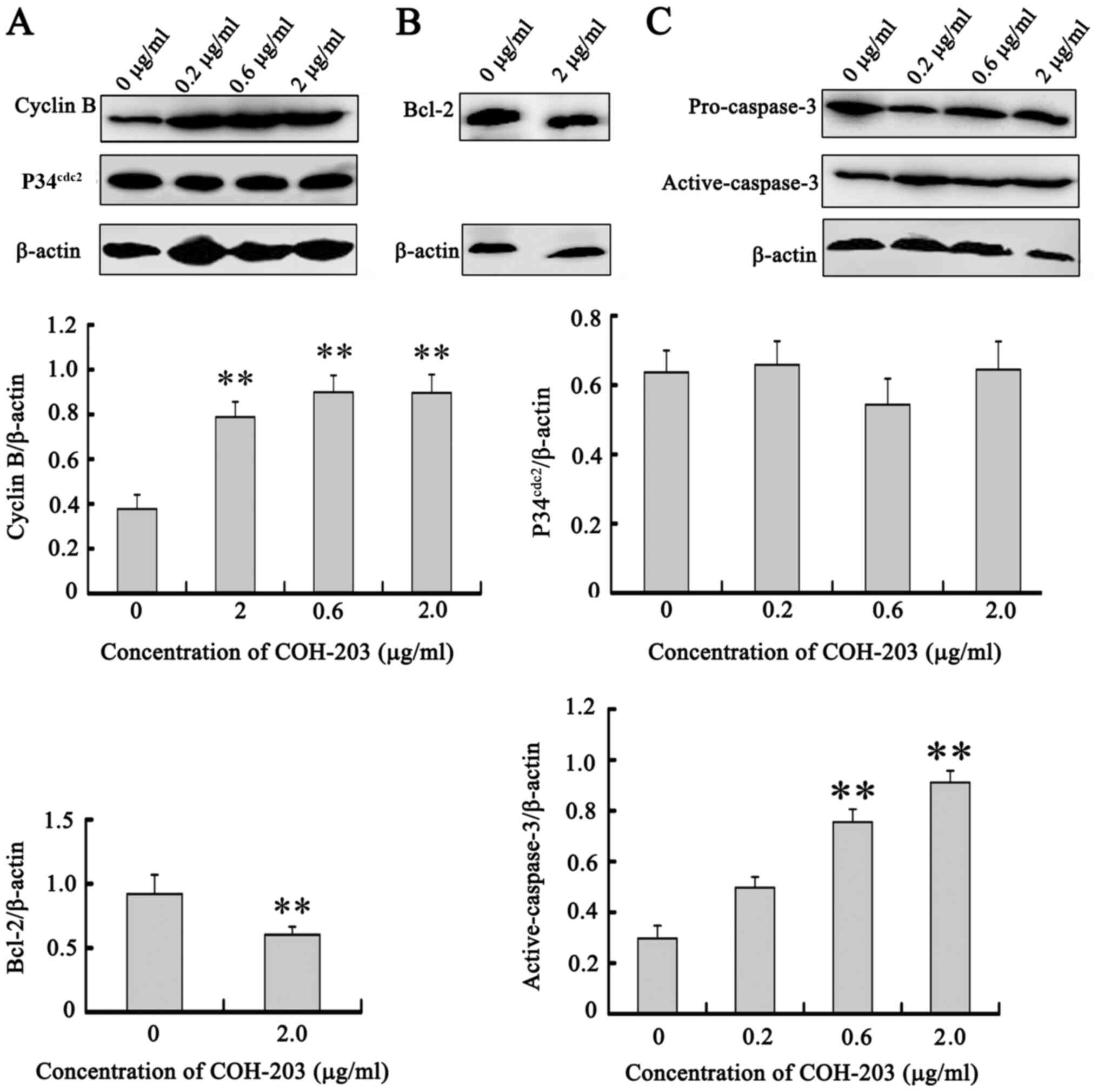 |
Figure 5.
Upregulation of cyclin B, downregulation of Bcl-2, and activation of caspase-3 by COH-203. HL-60 cells were treated with indicated concentrations of COH-203 for 24 h. Cells were harvested and total extracts derived from cells were analyzed by western blot analysis for (A) cyclin B/P34cdc2, (B) Bcl-2 and (C) caspase-3 expression. **P<0.01 vs. 0 µg/ml. COH-203, 5-(3-amino-4-methoxyphenyl)-4-(3,4,5-trimethoxyphenyl)-3H-1,2-dithiole-3-one; Bcl-2, B-cell lymphoma 2.
|
Mechanism of COH-203-induced apoptosis
Cell apoptosis is an active process mediated by various signaling pathways, such as caspase-mediated pathways, mitochondrial pathway, death receptor pathway, KRAS and the PI3K/AKT/mTOR pathway (12). To determine the mechanisms of COH-203-induced apoptosis, the activation of caspases and the protein expression of Bcl-2 in HL-60 cells were determined by western blot analysis. Bcl-2 located on the outer mitochondrial membrane is important for the inhibition of mitochondrial manifestations of apoptosis (12). Following treatment with COH-203, the expression of Bcl-2 was downregulated (Fig. 5B). By monitoring the cleavage of caspase-3, the present study aimed to determine whether caspases were involved in COH-203-mediated HL60 cell death. Treatment with various concentrations of COH-203 for 24 h revealed the cleavage of caspase-3 to be notably increased compared with the control (Fig. 5C). These findings suggested that COH-203 induced apoptosis via downregulation of Bcl-2 and the activation of caspase-3.
Discussion
As a COH compound, COH-203 may be a promising tubulin-binding agent (10). In the present study, numerous biological assays and techniques were performed to identify the antiproliferative effects of COH-203 on human APL HL-60 cells and the associated mechanism.
According to the MTT assay, it was reported that COH-203 could inhibit cell proliferation in a time- and dose-dependent manner. The present study demonstrated that COH-203 arrested HL-60 cells in G2/M phase and induced apoptosis via flow cytometry and AO/EB fluorescence double staining. In addition, it was observed in the present study that treatment with COH-203 could disrupt microtubule polymerization, which may be an anti-mitotic mechanism. Tubulin-binding agents activate the mitotic spindle checkpoint, which monitors chromosome attachment to the mitotic spindle, and delays chromosome segregation during anaphase, until defects in the mitotic spindle apparatus are corrected (13). Cell cycle arrest can occur at checkpoints for DNA repair in response to cellular damage; however, providing that repair mechanisms fail or the damage is irreparable, then apoptosis is induced to eliminate cells carrying DNA lesions and to preserve genomic integrity (14). The results of the present study indicated that the proportion of apoptotic cells significantly increased by prolonging the duration of HL-60 cell arrest in G2/M phase. Therefore, COH-203-induced cell death may be associated with G2/M arrest and this may be the reason as to why these damaged cells could not be restored to progress through the cell cycle, therefore undergoing apoptosis.
The progression of the cell cycle has been widely investigated (15). p34cdc2 along with cyclin B serve a crucial role in the regulation of the cell cycle in G2/M phase (16). Eukaryotic cell entry into mitosis and subsequent exit into G1 requires the sequential entry of cyclin B into the nucleus during prophase, where it activates p34cdc2 kinase, followed by the degradation of cyclin B; thus, p34cdc2 activity is reduced during anaphase, which allows the cell to complete mitosis (17). Increasing evidence has indicated that sustained activation of p34cdc2 has an important role in anti-tubulin agent-induced mitotic arrest and apoptosis (18). The inactivation of p34cdc2 depends upon degradation of cyclin B; however, providing this mechanism fails, sustained activation of p34cdc2 may induce the cell to arrest in mitosis (16). To determine whether the cyclin B/p34cdc2 complex was involved in COH-203-induced G2/M phase arrest, the protein expression levels of cyclin B were analyzed. Compared with the control group, the protein expression levels of cyclin B were significantly increased following treatment with COH-203. The accumulation of cyclin B prolonged the activity of p34cdc2. These results suggested that increased expression of cyclin B, which prolonged the activity of p34cdc2, was involved in COH-203-induced G2/M phase arrest.
Apoptosis has been reported to be associated with a variety of cellular signaling pathways (12). The mitochondrial and death receptor pathways are two major apoptotic signaling pathways (19). In addition, two important groups of proteins involved in apoptotic cell death are the members of the Bcl-2 family and a class of cysteine proteases, known as caspases (12). The Bcl-2 family of proteins can be classified into two functionally distinct groups: Anti-apoptotic and pro-apoptotic proteins (12). Bcl-2, an anti-apoptotic protein, has been reported to regulate the apoptotic pathways and protect against cell death (20). The Bcl-2 family of proteins regulate cell apoptosis via numerous pathways and mainly affects the mitochondrial pathways (20–23). Bcl-2 can prevent mitochondrial membrane disruption and the release of cytochrome c and other pro-apoptotic factors (24,25). The results of the present study revealed a significant decrease in Bcl-2 expression following treatment with COH-203 in HL-60 cells. The data indicated that COH-203-induced apoptosis could be associated with the downregulation of Bcl-2 protein expression, which according to a previous study may induce the release of cytochrome c (26). Released cytochrome c complexes with Apaf-1 and caspase-9 to form the apoptosome, which subsequently cleaves caspase-3 (27). The caspases are aspartate-specific cysteine proteases that have been reported as the central executioners of apoptosis (28). The present study investigated whether caspases are involved in HL-60 cell death, mediated by COH-203, by analyzing the cleavage of caspase-3. The results demonstrated that the cleavage of caspase-3 was significantly increased following treatment with COH-203 compared with untreated cells. These results indicated that COH-203-induced apoptosis in HL-60 cells may occur via the mitochondrial pathways in a caspase-dependent manner.
In conclusion, the anti-leukemic effects of COH-203 on HL-60 cells were investigated in the present study. COH-203 exhibited antiproliferative activity in a dose-dependent manner, which may be attributed to the induction of cell cycle arrest in G2/M phase and apoptosis. COH-203 induced G2/M phase arrest via inhibiting microtubule depolymerization and promoting the expression of G2/M-phase regulatory proteins. Apoptosis induced by COH-203 may occur via the mitochondrial pathways in a caspase-dependent manner. Therefore, the results of the present study indicate that COH-203 may serve as a potential anticancer drug for the treatment of leukemia.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Shenyang Science and Technology Bureau Item (grant nos., 18-014-4-01 and 20-205-4-026).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
LW designed the study, and reviewed and edited the manuscript. LZ, JL and ZW performed the experiments. JL wrote the paper. KB and QG analyzed and interpreted the data for this work. LJ, LZ, ZW and LW confirm the authenticity of all the raw data. All authors read and approved the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
1
|
Kadia TM, Ravandi F, O'Brien S, Cortes J and Kantarjian HM: Progress in acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 15:139–151. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Al-Hussaini M and DiPersio JF: Small molecule inhibitors in acute myeloid leukemia: from the bench to the clinic. Expert Rev Hematol. 7:439–464. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ng CH and Chng WJ: Recent advances in acute promyelocytic leukaemia. F1000Res. 6:12732017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Coombs CC, Tavakkoli M and Tallman MS: Acute promyelocytic leukemia: Where did we start, where are we now, and the future. Blood Cancer J. 5:e3042015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McCulloch D, Brown C and Iland H: Retinoic acid and arsenic trioxide in the treatment of acute promyelocytic leukemia: Current perspectives. Onco Targets Ther. 10:1585–1601. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou GB, Zhang J, Wang ZY, Chen SJ and Chen Z: Treatment of acute promyelocytic leukaemia with all-trans retinoic acid and arsenic trioxide: A paradigm of synergistic molecular targeting therapy. Philos Trans R Soc Lond B Biol Sci. 362:959–971. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou J and Giannakakou P: Targeting microtubules for cancer chemotherapy. Curr Med Chem Anticancer Agents. 5:65–71. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen SM, Meng LH and Ding J: New microtubule-inhibiting anticancer agents. Expert Opin Investig Drugs. 19:329–343. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jordan MA and Wilson L: Microtubules as a target for anticancer drugs. Nat Rev Cancer. 4:253–265. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang Z, Qi H, Shen Q, Lu G, Li M, Bao K, Wu Y and Zhang W: 4,5-Diaryl-3H-1,2-dithiole-3-thiones and related compounds as combretastatin A-4/oltipraz hybrids: Synthesis, molecular modelling and evaluation as antiproliferative agents and inhibitors of tubulin. Eur J Med Chem. 122:520–529. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qi H, Zuo DY, Bai ZS, Xu JW, Li ZQ, Shen QR, Wang ZW, Zhang WG and Wu YL: COH-203, a novel microtubule inhibitor, exhibits potent anti-tumor activity via p53-dependent senescence in hepatocellular carcinoma. Biochem Biophys Res Commun. 455:262–268. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pistritto G, Trisciuoglio D, Ceci C, Garufi A and D'Orazi G: Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging (Albany NY). 4:603–619. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Greene LM, Campiani G, Lawler M, Williams DC and Zisterer DM: BubR1 is required for a sustained mitotic spindle checkpoint arrest in human cancer cells treated with tubulin-targeting pyrrolo-1,5-benzoxazepines. Mol Pharmacol. 73:419–430. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Haring SJ, Mason AC, Binz SK and Wold MS: Cellular functions of human RPA1. Multiple roles of domains in replication, repair, and checkpoints. J Biol Chem. 283:19095–19111. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schafer KA: The cell cycle: A review. Vet Pathol. 35:461–478. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ling YH, El-Naggar AK, Priebe W and Perez-Soler R: Cell cycle-dependent cytotoxicity, G2/M phase arrest, and disruption of p34cdc2/cyclin B1 activity induced by doxorubicin in synchronized P388 cells. Mol Pharmacol. 49:832–841. 1996.PubMed/NCBI
|
|
17
|
Knudsen KE, Knudsen ES, Wang JY and Subramani S: p34cdc2 kinase activity is maintained upon activation of the replication checkpoint in Schizosaccharomyces pombe. Proc Natl Acad Sci USA. 93:8278–8283. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang SW, Pan SL, Huang YC, Guh JH, Chiang PC, Huang DY, Kuo SC, Lee KH and Teng CM: CHM-1, a novel synthetic quinolone with potent and selective antimitotic antitumor activity against human hepatocellular carcinoma in vitro and in vivo. Mol Cancer Ther. 7:350–360. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Peng X, Yu Z, Liang N, Chi X, Li X, Jiang M, Fang J, Cui H, Lai W, Zhou Y and Zhou S: The mitochondrial and death receptor pathways involved in the thymocytes apoptosis induced by aflatoxin B1. Oncotarget. 7:12222–12234. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kang MH and Reynolds CP: Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 15:1126–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Du X, Fu XF, Yao K, Lan ZW, Xu H, Cui QH and Yang E: Bcl-2 delays cell cycle through mitochondrial ATP and ROS. Cell Cycle. 16:707–713. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gupta S: Molecular signaling in death receptor and mitochondrial pathways of apoptosis. Int J Oncol. 22:15–20. 2003.PubMed/NCBI
|
|
23
|
Hatok J and Racay P: Bcl-2 family proteins: Master regulators of cell survival. Biomol Concepts. 7:259–270. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gupta S, Kass GE, Szegezdi E and Joseph B: The mitochondrial death pathway: A promising therapeutic target in diseases. J Cell Mol Med. 13:1004–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sun BB, Fu LN, Wang YQ, Gao QY, Xu J, Cao ZJ, Chen YX and Fang JY: Silencing of JMJD2B induces cell apoptosis via mitochondria-mediated and death receptor-mediated pathway activation in colorectal cancer. J Dig Dis. 15:491–500. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science. 275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dou C, Han M, Zhang B, Sun L, Jin X and Li T: Chrysotoxene induces apoptosis of human hepatoblastoma HepG2 cells in vitro and in vivo via activation of the mitochondria-mediated apoptotic signaling pathway. Oncol Lett. 15:4611–4618. 2018.PubMed/NCBI
|
|
28
|
Thornberry NA and Lazebnik Y: Caspases: Enemies within. Science. 281:1312–1316. 1998. View Article : Google Scholar : PubMed/NCBI
|














