Introduction
Hepatocellular carcinoma (HCC) is one of the most
common types of primary malignant tumor. HCC usually develops in
the background of chronic liver diseases such as chronic hepatitis
or liver cirrhosis caused by hepatitis B virus (HBV) or hepatitis C
virus (HCV) infection, alcohol intake or metabolic syndrome
(1). The challenging problem of
HCC is its easy recurrence after curative resection; indeed, the
recurrence rate within 5 years is 70% (2). There are two different mechanisms of
HCC recurrence: Intrahepatic metastasis (IM) of primary HCC and
multicentric carcinogenesis (MC) of HCC independent of primary HCC.
Several discriminating factors of IM and MC have been identified
and reviewed (3,4). IM is characterized by early
recurrence of HCC (e.g., within 2 years), pathological similarity
and a more advanced grade of HCC. MC is characterized by late
recurring HCC, different pathological features and relatively
early-stage HCC. However, despite the clinical importance of
discrimination in treatment and prognosis, it is difficult to
precisely discriminate these two types of recurrence. Recently,
molecular omics approaches, such as genomics, transcriptomics,
proteomics and metabolomics, have been used to discriminate between
MC and IM (3,5).
A similar challenging issue regarding carcinogenesis
exists in combined hepatocellular-cholangiocarcinoma (cHCC-CCA).
cHCC-CCA is a primary liver carcinoma with unequivocal features of
both hepatic and cholangiocytic differentiation within the same
tumor based on morphology revealed by hematoxylin and eosin
staining (6). There are three
possible models for the origin and evolution of HCC and
intrahepatic CCA (iCCA) components in cHCC-CCA: i) Hepatocytes and
cholangiocytes synchronously transform to HCC and iCCA,
respectively, even in the same nodule; ii) HCC develops first and
then partial HCC transdifferentiates into iCCA in the same nodule
(6–8); iii) hepatic stem or progenitor cells
(HPCs) expand to progenitor-like tumors and then synchronously
differentiate into HCC and iCCA in the same nodule. HPC directly
develops into cHCC-CCA via a single-cell origin (6,8,9). The
first model involves a different origin of carcinoma, whereas the
second and third models involve the same origin. Molecular
analysis, such as genomics and transcriptomics, is useful for
clarifying the evolution of cHCC-CCA (10,11).
To clarify the cancer origin and evolution of the
two issues described above (i.e., the mechanism of HCC recurrence
and the origin of cHCC-CCA), it is necessary to precisely separate
cancer samples to be used for mutational profiling, including
mutation frequency determination. In the present study, a case of
triple occurrence of liver cancer with a three-year interval was
examined; the first and second liver cancers were diagnosed as HCC;
the third was diagnosed as cHCC-CCA. To precisely separate cancer
samples, laser capture microdissection (LCM) of sample tissues was
performed and they were histopathologically diagnosed. To obtain
precise mutational profiles, next-generation sequencing (NGS) of
409 cancer-associated genes was performed and the mutation
frequency was then quantified by allele-specific quantitative PCR
(qPCR) and digital PCR (dPCR). The two aspects of the phylogenetic
tree of liver cancer components were analyzed as follows: i)
Whether the three metachronous liver cancers were of the same
origin; and ii) whether the two synchronous components of cHCC-CCA,
i.e., HCC and iCCA, were of the same origin.
Materials and methods
Liver tissue samples and DNA
extraction
The patient was a 71-year-old male with a primary
HCC (#1HCC) in 2007 who subsequently developed two recurrent liver
cancers every three years; the second was HCC (#2HCC) and the third
was cHCC-CCA (#3cHCC-CCA). Noncancerous regions of #1N (2 cm away
from the tumor), #2N (1 cm from the tumor) and #3N (2 cm from the
tumor) exhibited chronic hepatitis with a fibrosis score of F2. The
patient was consistently positive for anti-HCV and hepatitis B
surface (HBs) antibodies and negative for serum HBV DNA (Table SI) and received curative resection
as a standard clinical treatment after each diagnosis. The patient
had no history of alcohol abuse and did not receive any antiviral
therapy for HCV infection. Formalin-fixed, paraffin-embedded (FFPE)
liver tissues collected during surgery for pathological diagnosis
were sliced into thin sections of 10 µm in thickness. LCM was
performed with a PALM-MBIII-N (Carl Zeiss AG) to obtain #3HCC and
#3iCCA lesions (Fig. 1).
Macrodissection was performed to obtain #1HCC, #2HCC and #3cHCC-CCA
lesions (Fig. 1). Nontumorous
tissue sections (#1N, #2N and #3N) were also obtained from the
terminal FFPE tissues of resected specimens. DNA was extracted from
the FFPE samples using the RecoverAll Total Nucleic Acid Isolation
Kit for FFPE (Thermo Fisher Scientific, Inc.) with certain
modifications as described previously (12,13).
DNA was also extracted from fresh-frozen liver specimens,
nontumorous liver of a patient with liver metastasis of colon
cancer, to use as controls for TaqMan quantitative PCR (qPCR)
(12,13). The present study was approved by
the Ethics Committee of Nihon University School of Medicine
(approval no. 237-1). Informed consent was obtained from the
patients prior to the start of the study.
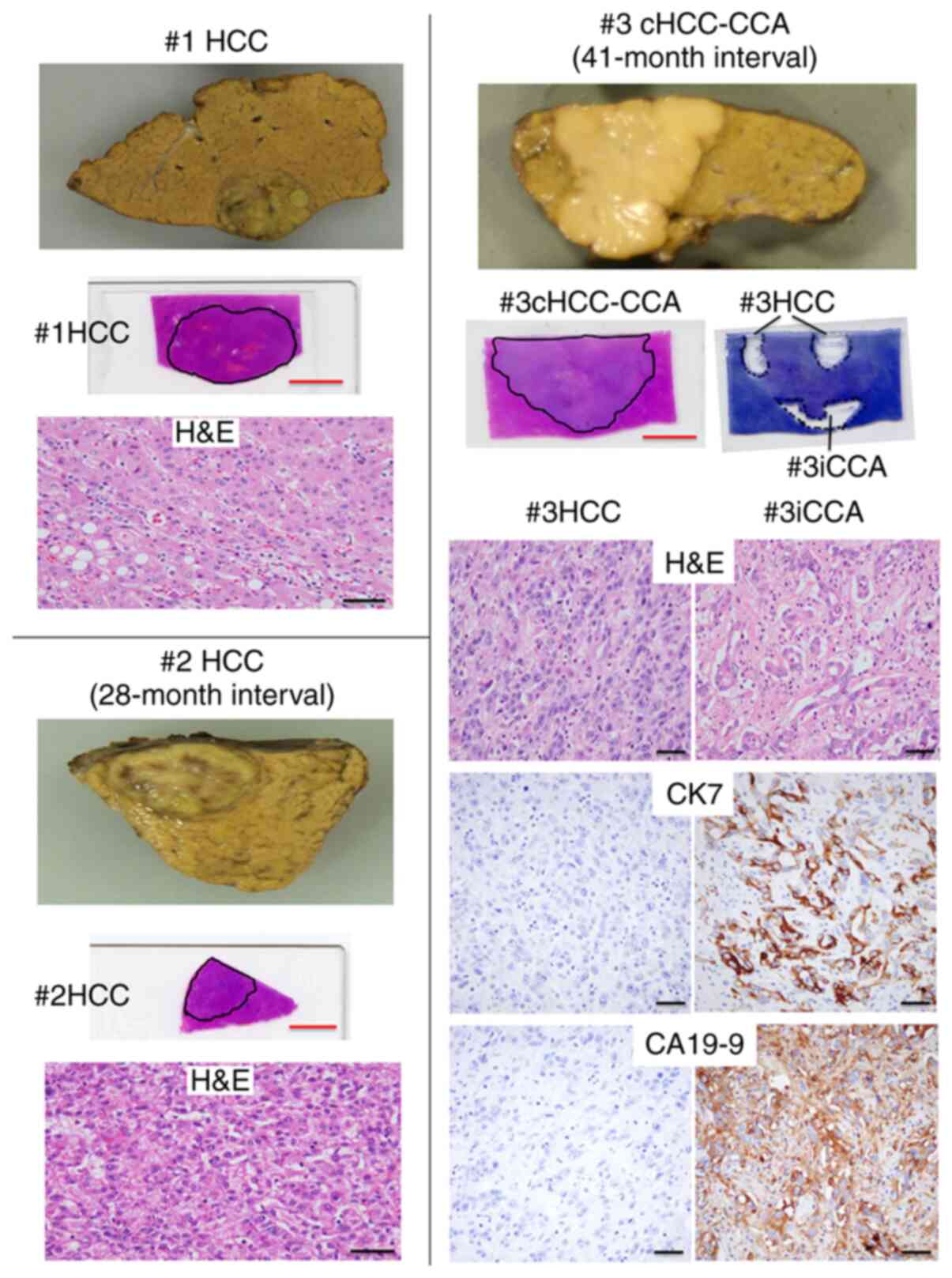 | Figure 1.Gross and histological features of
three metachronous liver cancers curatively resected every three
years (#1 to #3, respectively). #1, well-differentiated HCC,
2.6×1.7 cm in diameter; #2, moderately differentiated HCC, 1.8×1.4
cm in diameter; #3, cHCC-CCA, 4.5×4.5 cm in diameter. The outlined
areas were macroscopically dissected for #1HCC, #2HCC and
#3cHCC-CCA and laser capture microdissected for #3HCC and #3CCA
after staining with toluidine blue (red scale bars, 1 cm). DNA was
then extracted. Histological features of all cancerous regions are
presented by H&E staining (scale bars, 50 µm). #3cHCC-CCA was
subjected to immunohistochemical staining for CK7 and CA19-9 and
each result for the #3HCC and #3iCCA regions is presented (scale
bars, 50 µm). HCC, hepatocellular carcinoma; cHCC-CCA, combined
hepatocellular-cholangiocarcinoma; iCCA, intrahepatic
cholangiocarcinoma; CK7, cytokeratin 7; CA19-9, carbohydrate/cancer
antigen 19-9; H&E, hematoxylin and eosin. |
TaqMan qPCR
Quality assessment of FFPE DNA was performed by
TaqMan qPCR of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and
the ratio of FFPE DNA quality to frozen tissue DNA was calculated
by the ∆ quantification cycle (∆Cq) method, as described previously
(12,13); PCR was performed in a 10-µl
reaction mixture containing Premix Ex Taq (Probe qPCR; Takara Bio,
Inc.) with an initial denaturation step at 95°C for 20 sec,
followed by 45 cycles at 95°C for 1 sec and 60°C for 20 sec using
StepOnePlus (Thermo Fisher Scientific, Inc.). HBV-DNA was also
quantified using a TaqMan Gene Expression Assay (Thermo Fisher
Scientific, Inc.) and QuantStudio 3 System (Thermo Fisher
Scientific, Inc.). Assay IDs for GAPDH and HBV DNA are presented in
Table SII. All assays were
performed in duplicate.
Comprehensive Cancer Panel (CCP)
amplicon sequencing
DNA samples from #3cHCC-CCA and #3N were subjected
to amplicon sequencing using Ion AmpliSeq Comprehensive Cancer
Panel (Thermo Fisher Scientific, Inc.), as described previously
(13). Candidates for cancer
(#3cHCC-CCA)-specific single-nucleotide variants (SNVs) were
identified by Tumor-Normal Pair Analysis version 5.2 of Ion
Reporter (https://ionreporter.thermofisher.com/ir/).
SYBR green allele-specific qPCR
Mutation frequency was determined by allele-specific
qPCR, as described previously (13); allele-specific qPCR was
quantitatively performed using THUNDERBIRD SYBR qPCR Mix (Toyobo
Life Science) with the QuantStudio 3 System (Thermo Fisher
Scientific, Inc.). PCR was performed in a 10-µl reaction in
duplicate by preheating at 95°C for 10 min, followed by 45 cycles
of 95°C for 15 sec and 60 to 64°C for 60 sec. Allele-specific
primers for wild-type (Wt) and mutant (Mu) sequences and a single
opposite-directed primer were designed as previously described
(13) and as presented in Table SII, together with each annealing
temperature. The ratio of the Mu allele to the Wt allele was
calculated as 2−ΔCq, where ΔCq is the result of
subtracting the Cq value of the PCR for the Wt from that of the Mu.
The Mu allele frequency was calculated as
2−ΔCq/(1+2−ΔCq), as described previously
(13). The Mu allele frequency was
used for cluster analysis of cancers with the agglomerative
clustering method and the phylogenetic tree was constructed using
the Ward method (14). To
determine the Mu cell population, the two-hit theory of the
mutation was hypothesized, whereby all Mu cells are heterozygous
with the first-hit SNV when the Mu allele frequency is <0.5 and
all cells consist of Mu heterozygotes and hemizygotes, which
undergo Wt allele loss as the second hit when the Mu allele
frequency is >0.5. In the former case, the Mu heterozygote
population was calculated by the equation of heterozygote
population=2× allele frequency. In the latter case, the populations
of Mu heterozygotes and hemizygotes were calculated by the
equations of Mu heterozygotes population=1/allele frequency-1 and
hemizygote population=2-1/allele frequency, respectively.
TERT promoter mutation
TERT promoter mutations (C228T, C225T) were analyzed
by Sanger sequencing of nested PCR products. Nested PCR
amplification of the TERT promoter was performed using PrimeSTAR
GXL DNA polymerase (Takara Bio, Inc.) and the first primer pair
F1-R1 followed by the second primer pair F2-R2 (Table SII). The reaction conditions were
as follows: 94°C for 1 min and 40 cycles of 98°C for 10 sec and
68°C for 1 min. Of the first PCR mix (20 µl), 1 µl was used in the
20-µl reaction for the second PCR. Sanger sequencing of the
products from the second PCR was performed as previously described
(13).
dPCR of the TERT promoter mutation C228T was
performed using the TaqMan dPCR Liquid Biopsy Assay TERT_C228T
(Hs000000092_rm; Thermo Fisher Scientific, Inc.) with the
QuantStudio 3D Digital PCR System (Thermo Fisher Scientific, Inc.).
PCR was performed using 60 to 1,000 ng of sample DNA, with the
following reaction conditions: 96°C for 10 min, 54 cycles of 55°C
for 2 min and 98°C for 30 sec, and two holds of 55°C for 2 min,
followed by 10°C indefinitely. The ramp rate of each temperature
change was 1.6°C/sec, according to the manufacturer's instruction.
The dPCR data were analyzed using QuantStudio 3D AnalysisSuite
version 3.1 (Thermo Fisher Scientific, Inc.). Reproducibility was
confirmed using two different concentrations of DNA from three
representative samples. The negative controls consisted of two DNA
samples, one from nontumorous liver tissue (normal liver) of a
patient with liver metastasis of gallbladder cancer and another
from the blood of a healthy subject using 60 ng DNA/reaction. The
mutation frequency was used for the cluster analysis of cancers as
mentioned above, together with the mutant allele frequency of other
genes. Informed consent was obtained from these subjects.
Immunohistochemistry
Sections of FFPE tissues (4 µm) were treated with 10
mM citrate buffer (pH 6.0) at 120°C for 15 min for antigen
retrieval. After quenching of endogenous peroxidase with 3%
hydrogen peroxide for 5 min at room temperature. The sections were
incubated with a 100-fold diluted mouse monoclonal anti-cytokeratin
(CK)7 antibody (clone OV-TL 12/30; catalog no. M7018; Agilent
Technologies, Inc.) or 50-fold diluted mouse monoclonal
anti-carbohydrate/cancer antigen (CA)19-9 antibody (clone
1116-NS-19-9; catalog no. M3517; Agilent Technologies, Inc.) for 1
h at room temperature. The sections were then incubated with
Histofine Simple Stain MAX-PO (MULTI; Nichirei Biosciences) as
secondary antibody for 30 min at room temperature, visualized with
3,3′-diaminobenzidine chromogen (Wako Pure Chemical Industries) and
counterstained with hematoxylin. The proportion of positive cells
in the HCC and iCCA area was determined by measurement of the
positive area in five fields using cellSens Dimension 1.16 (Olympus
Corporation).
Results
Histological findings for three
metachronous liver cancers
In the present case, three metachronous liver
cancers developed with intervals of 28 and 41 months. The first and
second liver cancers were diagnosed as HCC (#1HCC, #2HCC) and the
third as cHCC-CCA (#3cHCC-CCA) (Fig.
1). All of the metachronous liver cancers were of the nodular
type (Fig. 1). The histological
grade was well-differentiated [Edmondson and Steiner grade I
(15)] for #1HCC and moderately
differentiated (Edmondson and Steiner grade II) for #2HCC.
#3cHCC-CCA mainly consisted of a mixed HCC/iCCA area and partial
HCC and iCCA components were separately located in the same tumor
nodule (Fig. 1).
Immunohistochemistry for CK7 and CA19-9, known as iCCA markers
(16), clearly indicated #3HCC and
#3iCCA to be regionally separated (Figs. 1 and S1).
To perform a precise molecular analysis of cancer
evolution, multiple cancerous and noncancerous regions were
obtained by histological examination of FFPE tissues used in the
pathological diagnostic division of the hospital, followed by
dissection or LCM techniques. DNA was extracted from the dissected
tissue specimens of three metachronous liver cancers, including
noncancerous tissues: #1N, #1HCC, #2N, #2HCC, #3N and #3cHCC-CCA
(whole cancerous region of #3). #3HCC and #3iCCA were extracted
from LCM tissues (Fig. 1). All DNA
samples were negative for HBV DNA (Fig. S2). Therefore, the three
metachronous liver cancers were C-type (HCV-positive) liver
cancers.
Genetic evolution of liver
cancers
DNA quality from the FFPE samples was determined by
GAPDH qPCR to be 0.006 to 0.118 compared with frozen tissue DNA
(set as 1). The DNA quality of #1HCC and #2HCC was 0.006 and 0.008,
respectively, and was <1/10 of that of #3cHCC-CCA and #3N (0.118
and 0.059, respectively), which was due to the higher concentration
of formalin used for tissue fixation (13). Therefore, #3cHCC-CCA and #3N were
subjected to amplicon sequencing to determine tumor-specific
nonsynonymous SNVs of all exons of 409 cancer-associated genes
(Table SIII). A total of 173 SNVs
were detected by Tumor-Normal Pair Analysis version 5.2 of Ion
Reporter. A total of 5 nonsynonymous SNVs [histone-lysine
N-methyltransferase 2D (KMT2D), tumor protein p53 (TP53), DNA
(cytosine-5′)-methyltransferase 3α (DNMT3A), polycystic kidney and
hepatic disease 1 (PKHD1) and toll like receptor 4 (TLR4)] were
detected in #3HCC-CCA after filtering by the following three
factors: i) Allele coverage in the nontumorous sample of 0; ii)
total coverage in the tumor sample of >100; and iii) allele
frequency in the tumor sample of >0.05 (Table SIV). The allele-specific qPCR
results for these SNVs validated the presence of somatic mutations
in not only #3cHCC-CCA but also in both #3HCC and #3iCCA (Table I). Furthermore, all five SNVs were
detected in #2HCC but not in #1HCC. The allele frequency of the
five SNVs varied from 0.063-0.088 for KMT2D to 0.335-0.758 for
PKHD1, but the frequency pattern of the five SNVs was similar among
the three dissected cancer samples, #2HCC, #3HCC and #3iCCA. The
mutant cell populations of variant heterozygotes and hemizygotes
were also calculated when the allele frequency was >0.5; the
PKHD1 variant frequencies of #2HCC and #3HCC were 0.758 and 0.688,
respectively. For HCC cells containing the second hit of wild-type
allele loss of the PKHD1 gene, variant frequencies were predicted
to be 0.64 and 0.54 in the population, respectively. #3iCCA
contained no or a very low number of hemizygous cancer cells.
 | Table I.Allele frequency of five SNVs in five
samples from three metachronous liver cancers. |
Table I.
Allele frequency of five SNVs in five
samples from three metachronous liver cancers.
|
| Allele
frequency | Mutant cell
population |
|---|
|
|
|
|
|---|
| SNVa/sampleb | CCPc | qPCRd | Heteroe |
Wt-lossf |
|---|
| KMT2D G>C
p.Arg5432Gly | 0.097
(193/1991) |
|
|
|
|
#1HCC |
| 0.000±0.000 | 0.00±0.00 |
|
|
#2HCC |
| 0.063±0.027 | 0.13±0.05 |
|
|
#3cHCC-CCA |
| 0.082±0.006 | 0.16±0.01 |
|
|
#3HCC |
| 0.088±0.011 | 0.18±0.02 |
|
|
#3iCCA |
| 0.085±0.010 | 0.17±0.02 |
|
| TP53 C>A
p.Glu204Ter | 0.096
(192/1994) |
|
|
|
|
#1HCC |
| −0.009±0.009 | 0.00±0.02 |
|
|
#2HCC |
| 0.262±0.043 | 0.52±0.09 |
|
|
#3cHCC-CCA |
| 0.191±0.015 | 0.38±0.03 |
|
|
#3HCC |
| 0.310±0.017 | 0.62±0.03 |
|
|
#3iCCA |
| 0.208±0.010 | 0.42±0.02 |
|
| DNMT3A C>G
p.Glu426Gln | 0.094
(187/1994) |
|
|
|
|
#1HCC |
| 0.000±0.000 | 0.00±0.00 |
|
|
#2HCC |
| 0.248±0.034 | 0.50±0.07 |
|
|
#3cHCC-CCA |
| 0.206±0.025 | 0.41±0.05 |
|
|
#3HCC |
| 0.288±0.012 | 0.58±0.02 |
|
|
#3iCCA |
| 0.236±0.032 | 0.47±0.06 |
|
| PKHD1 G>C
p.leu2479Val | 0.093
(185/1998) |
|
|
|
|
#1HCC |
| 0.000±0.000 | 0.00±0.00 | 0.00 |
|
#2HCC |
| 0.758±0.133 | 0.36±0.24 | 0.64±0.24 |
|
#3cHCC-CCA |
| 0.335±0.039 | 0.67±0.08 | 0.00 |
|
#3HCC |
| 0.688±0.029 | 0.46±0.06 | 0.54±0.06 |
|
#3iCCA |
| 0.460±0.013 | 0.92±0.03 | 0.00 |
| TLR4 C>T
p.His456Tyr | 0.054
(79/1456) |
|
|
|
|
#1HCC |
| 0.000±0.000 | 0.00±0.00 |
|
|
#2HCC |
| 0.172±0.114 | 0.34±0.23 |
|
|
#3cHCC-CCA |
| 0.179±0.037 | 0.36±0.07 |
|
|
#3HCC |
| 0.280±0.019 | 0.56±0.04 |
|
|
#3iCCA |
| 0.116±0.005 | 0.23±0.01 |
|
The TERT promoter mutation C228T but not C250T was
detected in all of these cancers (#1HCC, #2HCC, #3HCC and #3iCCA).
By contrast, no mutations were detected in nontumorous samples
(#1N, #2N and #3N) by Sanger sequencing (Fig. 2A). dPCR of the C228T mutation
indicated a variable mutation frequency from 0.217 to 0.467 in the
individual cancers: #3iCCA<#3HCC<#1HCC<#2HCC (Fig. 2B). Compared with normal liver and
blood, nontumorous regions (#1N, #2N and #3N) all contained TERT
promoter mutations in a small number of cells (Figs. 2B and S3).
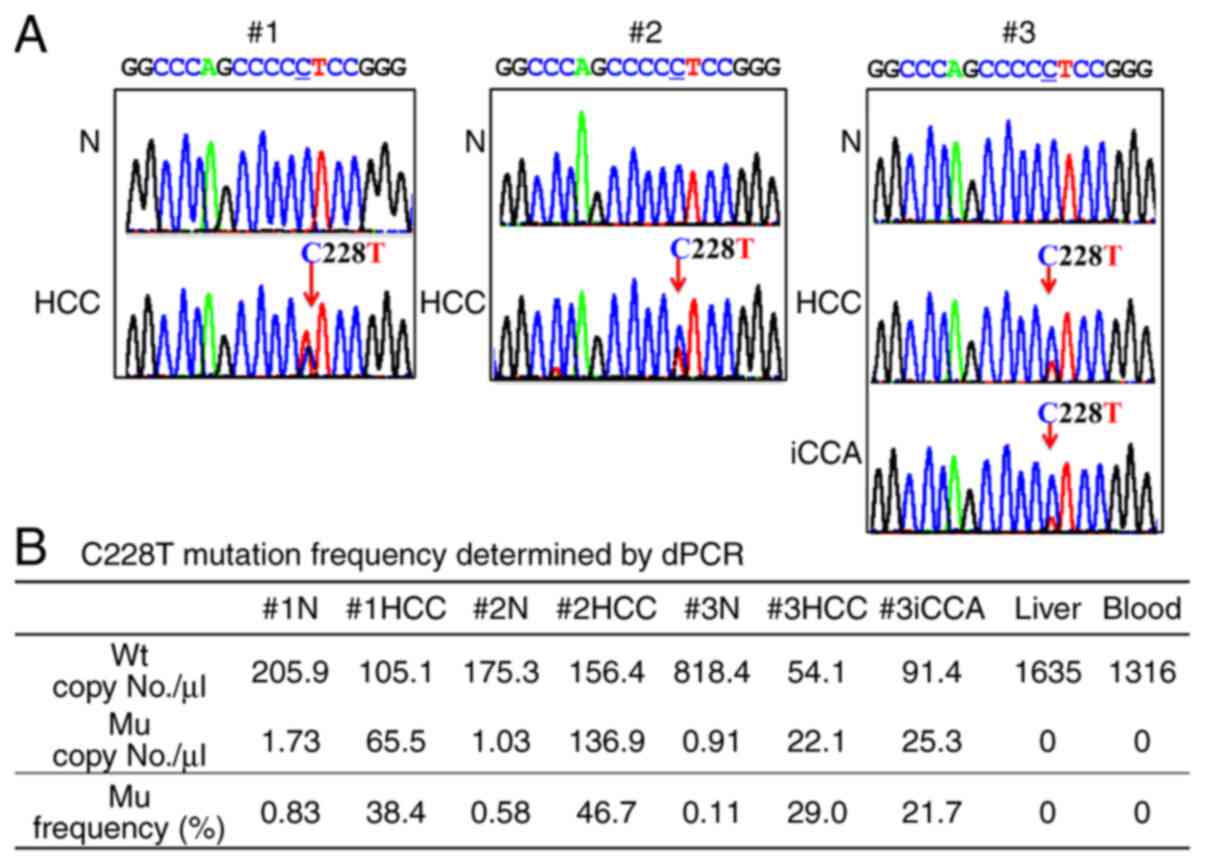 | Figure 2.TERT promoter mutation C228T. (A)
Sanger sequencing of the TERT promoter region. Nested PCR was
performed using seven DNA samples (#1N, #1HCC, #2N, #2HCC, #3N,
#3HCC and #3iCCA), which were then subjected to Sanger sequencing.
An arrow indicates the C228T single nucleotide variant. (B) dPCR of
the C228T mutation. The numbers of Mu/WT/both allele amplifications
in each nontumorous DNA sample (#1N, #2N and #3N) were as follows:
19/2429/3, 14/2233/0 and 10/8007/2, respectively. The levels in two
negative control DNA samples, liver (nontumorous liver of a patient
with liver metastasis of gallbladder cancer) and blood (healthy
donor blood) were 0/9942/0 and 0/9318/0, respectively. TERT,
telomerase reverse transcriptase; Wt, wild-type; Mu,
mutant/mutation; N, nontumorous liver section; HCC, hepatocellular
carcinoma; iCCA, intrahepatic cholangiocarcinoma; dPCR, digital
PCR. |
To summarize, the molecular evolution of three
metachronous liver cancers was illustrated, as presented in
Figs. 3 and S4, based on the phylogenetic principle
that the truncal mutations present in the common ancestor are
present in all descendants at higher frequencies. The different
allele frequencies of six SNVs, including the TERT promoter
mutation, are presented in doughnut charts in Fig. 3 and were subjected to cluster
analysis (Fig. S4). Mutant cancer
cell populations were predicted and presented in Table I and cancer cell evolution by
accumulation of somatic mutations is illustrated from right to left
in ellipses in Fig. 3. The
phylogenetic relationships suggest the following evolutionary
cancer history: i) #1HCC was not related to #2HCC and #3cHCC-CCA;
ii) #2HCC was generated independently of #1HCC in MC mode; iii)
#3cHCC-CCA originated from #2HCC; #2HCC already intrahepatically
metastasized prior to curative resection and developed into
#3cHCC-CCA in IM mode; iv) #3iCCA was generated directly from #2HCC
or indirectly from #3HCC.
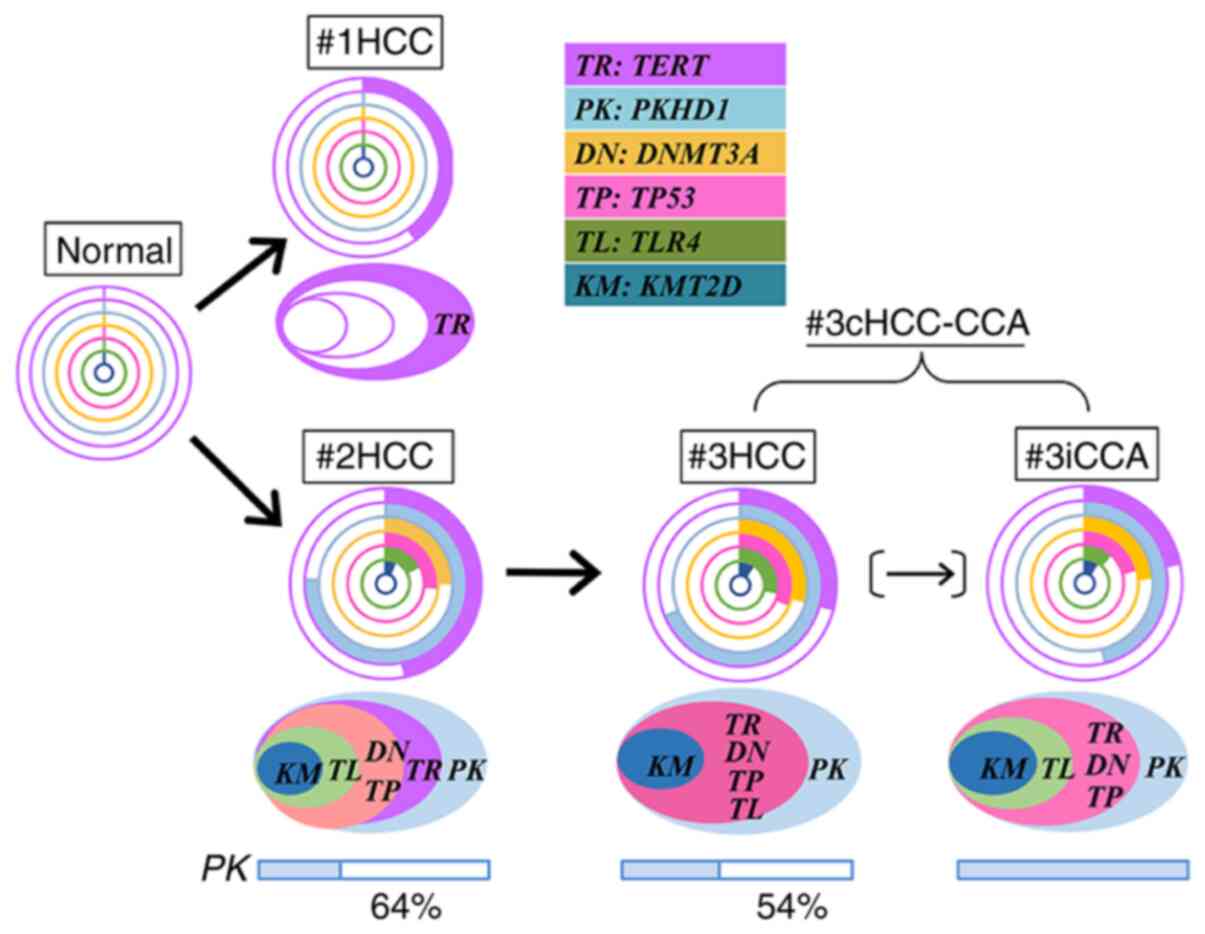 | Figure 3.Schematic of the molecular evolution
of three metachronous liver cancers. Different allele frequencies
of six SNVs, including the TERT promoter mutation, are presented in
doughnut charts and used for the cluster analysis (Fig. S4). Mutant cancer cell populations
were predicted as indicated in Table
I and cancer cell evolution by accumulation of somatic
mutations is presented from right to left in ellipses. Bar charts
indicate the population of two types of PKDH1-mutant cells:
Heterozygous with SNV (colored) and hemizygous with wild-type loss
as the second hit (blank); the % population of the mutant
hemizygote is also presented. There are two possibilities for
#3iCCA generation: #3iCCA was directly generated from #2HCC or
indirectly generated from #2HCC through #3HCC, as indicated by a
narrow arrow in parentheses. SNV, single-nucleotide variant; HCC,
hepatocellular carcinoma; cHCC-CCA, combined
hepatocellular-cholangiocarcinoma; iCCA, intrahepatic
cholangiocarcinoma; TERT, telomerase reverse transcriptase; KMT2D,
histone-lysine N-methyltransferase 2D; TP53, tumor protein p53;
DNMT3A, DNA (cytosine-5′)-methyltransferase 3α; PKHD1, polycystic
kidney and hepatic disease 1; TLR4, toll like receptor 4. |
Discussion
In the present study, three major findings were
obtained from the phylogenetic analysis of three metachronous liver
cancers with three-year intervals: i) There were two modes of
cancer occurrence, MC and IM, in a single case; ii) it is possible
that iCCA is generated from HCC in cHCC-CCA; iii) the TERT promoter
mutation C228T was not only a common event in the three cancers but
also an early event in the noncancerous chronic hepatitis liver
without liver cirrhosis. The mutant cell population was 0.22-1.66%
in chronic hepatitis liver cells, which probably existed prior to
carcinogenesis. Of note, these results were obtained through the
combination of two methods: i) Precise dissection of target tissue
samples based on histopathology; and ii) the mutation frequency of
multiple gene mutations determined by previously established
allele-specific qPCR (13) and
dPCR protocols.
There have been several reports regarding how to
discriminate between the two different mechanisms of recurrence, IM
and MC (3,4); early recurrence is indicative of IM
and late recurrence is indicative of MC occurrence. The
recurrence-free time is the most differentiating factor between IM
and MC, and 18 months is the best cutoff time point (17). Recently, somatic SNV information
has provided accurate diagnosis of MC and IM using whole-genome
sequencing of multiple liver cancers, including synchronous and
metachronous cancers (5). In
particular, this comprehensive molecular diagnosis is powerful
compared to previous techniques due to its high sensitivity and it
is useful for cases with inconsistent clinicopathological
diagnoses. It was also demonstrated here that mutational profiling
is the most definitive approach for discriminating between IM and
MC recurrence. Despite an approximate three-year interval from
#1HCC (well-differentiated) to #2HCC (moderately differentiated)
(28-month interval) and from #2HCC to #3cHCC-CCA (41-month
interval), the former recurrence was MC and the latter was IM. SNVs
specifically detected in only #1HCC are convincing. Unfortunately,
#1HCC and #2HCC were not subjected to NGS using a comprehensive
cancer panel, but the following information supports a rational
explanation for the conclusion in the absence of #1HCC-specific
SNVs. Intratumor heterogeneity and branched evolution have been
reported and the evolutionary history of cancer metastasis has been
established by several articles, including those by Gerlinger et
al (18) and Gundem et
al (19). Cancer evolutionary
genomics using clinical samples from metastatic cancers, including
the cancer cell fraction (CCF) plot, indicates that ubiquitous
mutations are present in all primary and metastatic (metachronous
and synchronous) cancers, constructing a trunk phylogenetic tree.
The CCF (the fraction of cancer cells within a sample containing a
mutation) plot also indicates that the mutation frequency of
truncal mutations is higher than that of branch and leaf mutations.
Therefore, if #1HCC had been the origin of #2HCC and #3HCC-CCA, the
top 5 SNVs detected in #2HCC and #3cHCC-CCA would have also been
detected in #1HCC. The top 5 SNVs were at least ubiquitous
mutations in #2HCC and #3HCC-CCA. Thus, truncal mutations were not
detected in #1HCC; i.e., #1HCC has no relationship with #2HCC.
Furuta et al (5) used
whole-genome sequencing analyses to demonstrate similar results for
three metachronous HCCs with intervals of 21 and 30 months; the
second HCC was MC and the third was IM from the second HCC.
Yamamoto et al (20) also
performed whole-exome sequencing of 41 multiple HCCs: 18 genomic
IMs and 23 genomic MCs. Among 10 clinical MCs with recurrence >2
years after initial resection, 3 were genomic IMs. Thus, the
recurrence-free time is ambiguous when attempting to discriminate
MC and IM. The present study indicated that patients with MC
occurrence have a lower risk of death following surgery than those
with IM (4,20). In the present case, the patient
died at 6 months after the third operation due to multiple bone
metastases and recurrent liver cancer in S4. Thus, the precise
discrimination of IM and MC by cancer genomics is valuable for
prognosis.
The carcinogenesis of cHCC-CCA is also debated with
regard to cancer clonality. Wang et al (11), Joseph et al (21) and Xue et al (22) investigated the sequence-based
molecular pathogenesis of cHCC-CCA by separately sequencing two
components, HCC and iCCA, of 6, 9 and 41 cases, respectively. All
cases carried ubiquitous mutations shared by HCC and iCCA, as well
as substantial individual mutations, suggesting the monoclonal
origin of cHCC-CCA and intratumor heterogeneity. In particular, Xue
et al (22) performed not
only genomic and but also transcriptomic profiling of multiple
cHCC-CCA cases classified into three subtypes: Separate, combined
(corresponding to the present case) and mixed. All cases were of
monoclonal origin, except for 2 of 6 separate subtype cases. Both
HCC and iCCA components of the combined subtype also exhibited a
similar global gene expression pattern, and of note, high Nestin
expression in both components was indicated to be a biomarker for
cHCC-CCA. Thus, the combined type cHCC-CCA is certainly monoclonal,
as in the present case. Next, the genesis of monoclonal but
heterologous cHCC-CCA may be debated with respect to two
mechanisms: Transdifferentiation of HCC or HPC origin (6,8).
Regarding the two mechanisms of monoclonal origin, Wang et
al (11) suggested the HPC
origin theory, whereby two components of cHCC-CCA originate from a
common cell with stem cell-like features. On the other hand, Joseph
et al (21) demonstrated
that the genetics of cHCC-CCA are similar to those of HCC and
distinct from iCCA and described a possible mechanism of
transdifferentiation of partial HCC to iCCA via interplay between
genetic factors and the surrounding tumor microenvironment
(23,24). The novel finding reported in the
present study based on mutational profiles is a potential mechanism
of transdifferentiation from HCC to iCCA in cHCC-CCA. The present
study is the first, to the best of our knowledge, to indicate
transdifferentiation from HCC to iCCA in clinical samples based on
the natural history of cancer development, as it is difficult to
obtain direct evidence from clinical samples. The present genetic
analysis of metachronous liver cancers revealed that the iCCA
component in #3cHCC-CCA evolved from the preceding #2HCC or #3HCC.
The former evolution, directly from #2HCC to #3iCCA, is more likely
due to the Euclidean distance and clustering of cancers; #3HCC-iCCA
developed from IM-#2HCC in three years, with #2HCC evolving to
#3HCC and partial #2HCC transdifferentiating to #3iCCA.
C228T is the most frequent TERT promoter mutation
(25). The frequency of TERT
promoter mutations in HCV-positive HCC is high, at 64–72%, compared
to that of HBV, at 37–39% (26,27).
By contrast, TERT promoter mutations are rare in iCCA (5%)
(28). In addition, considering
this point of view, the iCCA component of cHCC-CCA in the present
study was not similar to iCCA but was similar to HCC. The mutant
cell population was predicted from the mutation frequency. The
C228T mutant cells comprised 77% in #1HCC and 93% in #2HCC; on the
other hand, they were 48% in the #3HCC component and 43% in the
#3iCCA component. Most cancer cells in #1HCC and #2HCC were
positive for the C228T mutation, but the mutant cell population
similarly decreased in both components of #3cHCC-CCA, resulting in
similar mutant cell populations with TP53 and DNMT3A mutations.
These results suggest that after cancer evolution, #2HCC contained
an early HCC clone harboring the C228T mutation but not the other
mutations. The early clone was probably not metastatic and was
filtered out, resulting in the absence of the early clone in
#3cHCC-CCA. Digital PCR of C228T clearly indicated no mutation in
the negative control consisting of normal liver and normal blood
samples but the presence of mutation signals in nontumorous
tissues. This is a novel finding and suggests that small foci with
TERT promoter mutations may be generated in nontumor lesions.
Carcinogenesis may even be initiated in the chronic hepatitis state
without cirrhosis. Recently, Kim et al (29) performed deep sequencing of various
tumor-related genes, including the TERT promoter region, in
regenerative nodules of liver cirrhosis, with an average sequence
depth of 958; low-abundance mutations of TP53 and ARID1A were
detected in independent nodules, but no TERT promoter mutation was
observed in any of the 205 regenerative nodules from 10 cirrhotic
livers examined. Although it is a novel finding that clonal
expansion within a cirrhotic regenerative nodule occurs in the
absence of TERT promoter mutation, it does not exclude the present
results for the presence of TERT promoter mutation with lower
frequency, 0.11 to 0.83%, which was determined by dPCR based on
2,247-8,021 amplifications. Thus, it is still possible that the
TERT promoter mutation occurs followed by small subclonal expansion
during chronic liver inflammation, even without cirrhosis.
Finally, the present study was a retrospective
analysis of comprehensive cancer genomics by separate sequencing of
each component of liver cancers, including cHCC-CCA, suggesting the
usefulness not only for clonal evolution analysis but also for
prognosis through the discrimination of IM or MC. In the future,
targeted therapy based on cancer driver mutations may be
useful.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported in part by a grant from the Program for
Promoting Advanced Medical Research in Nihon University School of
Medicine (Tokyo, Japan; approval no. 2016).
Availability of data and materials
All the data analyzed during the present study are
included in this published article. The datasets collected using
the Ion AmpliSeq Comprehensive Cancer Panel are not publicly
available as the patient died and it is therefore impossible to
obtain consent. The patient consent for publication obtained
contains the genetic analysis results but not NGS data containing
the personal information, as NGS technology had not yet been
included in the genetic analysis at that time consent was given.
However, the dataset used for analysis in the present study is
available from the corresponding author upon reasonable
request.
Authors' contributions
SO, HY, YH and YN performed the experimental work.
SO and MS performed pathological diagnoses of the tissue samples.
YM was involved in the surgical treatment and diagnosis. SO, HY, HN
and TN performed the molecular genetic studies and analyzed the
data. SO drafted the manuscript. ME designed the study and edited
the manuscript. MM reviewed the study design, interpreted data and
critically revised the manuscript for scientific content. ME and MM
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Nihon University School of Medicine (Tokyo, Japan;
approval no. 237-1). Written informed consent for use and analysis
of samples was obtained from the patients and a healthy subject
including those for control samples prior to the start of the
study.
Patient consent for publication
Written informed consent for publication of data was
obtained from the patient.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
HBV
|
hepatitis B virus
|
|
HCV
|
hepatitis C virus
|
|
IM
|
intrahepatic metastasis
|
|
MC
|
multicentric carcinogenesis
|
|
cHCC-CCA
|
combined
hepatocellular-cholangiocarcinoma
|
|
iCCA
|
intrahepatic cholangiocarcinoma
|
|
HPCs
|
hepatic stem or progenitor cells
|
|
LCM
|
laser capture microdissection
|
|
qPCR
|
quantitative PCR
|
|
dPCR
|
digital PCR
|
|
FFPE
|
formalin-fixed, paraffin-embedded
|
|
SNV
|
single-nucleotide variant
|
|
GAPDH
|
glyceraldehyde-3-phosphate
dehydrogenase
|
|
KMT2D
|
histone-lysine N-methyltransferase
2D
|
|
TP53
|
tumor protein p53
|
|
DNMT3A
|
DNA (cytosine-5′)-methyltransferase
3α
|
|
PKHD1
|
polycystic kidney and hepatic disease
1
|
|
TLR4
|
toll like receptor 4
|
References
|
1
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vilarinho S and Calvisi DF: New advances
in precision medicine for hepatocellular carcinoma recurrence
prediction and treatment. Hepatology. 60:1812–1814. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Feo F and Pascale RM: Multifocal
hepatocellular carcinoma: Intrahepatic metastasis or multicentric
carcinogenesis? Ann Transl Med. 3:42015.PubMed/NCBI
|
|
4
|
Yang SL, Luo YY, Chen M, Zhou YP, Lu FR,
Deng DF and Wu YR: A systematic review and meta-analysis comparing
the prognosis of multicentric occurrence and vs. intrahepatic
metastasis in patients with recurrent hepatocellular carcinoma
after hepatectomy. HPB (Oxford). 19:835–842. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Furuta M, Ueno M, Fujimoto A, Hayami S,
Yasukawa S, Kojima F, Arihiro K, Kawakami Y, Wardell CP, Shiraishi
Y, et al: Whole genome sequencing discriminates hepatocellular
carcinoma with intrahepatic metastasis from multi-centric tumors. J
Hepatol. 66:363–373. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sempoux C, Kakar S, Kondo F and
Schirmacher P: Combined hepatocellular-cholangiocarcinoma and
undifferentiated primary liver carcinoma. WHO classification of
tumours of the digestive system. Bosman FT, Garneiro F, Hruban RH
and Theise ND: IARC; Lyon: pp. 260–262. 2019
|
|
7
|
Nagahama Y, Sone M, Chen X, Okada Y,
Yamamoto M, Xin B, Matsuo Y, Komatsu M, Suzuki A, Enomoto K and
Nishikawa Y: Contributions of hepatocytes and bile ductular cells
in ductular reactions and remodeling of the biliary system after
chronic liver injury. Am J Pathol. 184:3001–3012. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sia D, Villanueva A, Friedman SL and
Llovet JM: Liver cancer cell of origin, molecular class, and
effects on patient prognosis. Gastroenterology. 152:745–761. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chiba T, Zheng YW, Kita K, Yokosuka O,
Saisho H, Onodera M, Miyoshi H, Nakano M, Zen Y, Nakanuma Y, et al:
Enhanced self-renewal capability in hepatic stem/progenitor cells
drives cancer initiation. Gastroenterology. 133:937–950. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fujimoto A, Furuta M, Totoki Y, Tsunoda T,
Kato M, Shiraishi Y, Tanaka H, Taniguchi H, Kawakami Y, Ueno M, et
al: Whole-genome mutational landscape and characterization of
noncoding and structural mutations in liver cancer. Nat Genet.
48:500–509. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang A, Wu L, Lin J, Han L, Bian J, Wu Y,
Robson SC, Xue L, Ge Y, Sang X, et al: Whole-exome sequencing
reveals the origin and evolution of hepato-cholangiocarcinoma. Nat
Commun. 9:8942018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nakayama Y, Yamaguchi H, Einaga N and
Esumi M: Pitfalls of DNA quantification using DNA-binding
fluorescent dyes and suggested solutions. PLoS One.
11:e01505282016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Einaga N, Yoshida A, Noda H, Suemitsu M,
Nakayama Y, Sakurada A, Kawaji Y, Yamaguchi H, Sasaki Y, Tokino T
and Esumi M: Assessment of the quality of DNA from various
formalin-fixed paraffin-embedded (FFPE) tissues and the use of this
DNA for next-generation sequencing (NGS) with no artifactual
mutation. PLoS One. 12:e01762802017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ward JH Jr: Hierarchical grouping to
optimize an objective function. J Am Stat Assoc. 58:236–244. 1963.
View Article : Google Scholar
|
|
15
|
Edmondson HA and Steiner PE: Primary
carcinoma of the liver: A study of 100 cases among 48,900
necropsies. Cancer. 7:462–503. 1954. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nakanuma Y, Klimstra DS, Komuta M and Zen
Y: Intrahepatic cholangiocarcinoma. WHO Classification of Tumours
of the Digestive System. Bosman FT, Carneiro F, Hruban RH and
Theise ND: IARC; Lyon: pp. 254–259. 2019
|
|
17
|
Huang Zy, Liang By, Xiong M, Zhan DQ, Wei
S, Wang GP, Chen YF and Chen XP: Long-term outcomes of repeat
hepatic resection in patients with recurrent hepatocellular
carcinoma and analysis of recurrent types and their prognosis: A
single-center experience in China. Ann Surg Oncol. 19:2515–2525.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gerlinger M, Rowan AJ, Horswell S, Math M,
Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N and
Stewart A: Intratumor heterogeneity and branched evolution revealed
by multiregion sequencing. N Engl J Med. 366:883–892. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gundem G, Van Loo P, Kremeyer B,
Alexandrov LB, Tubio JM, Papaemmanuil E, Brewer DS, Kallio HM,
Högnäs G, Annala M, et al: The evolutionary history of lethal
metastatic prostate cancer. Nature. 520:353–357. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yamamoto S, Midorikawa Y, Nagae G, Tatsuno
K, Ueda H, Moriyama M, Takayama T and Aburatani H: Spatial and
temporal expansion of intrahepatic metastasis by
molecularly-defined clonality in multiple liver cancers. Cancer
Sci. 111:601–609. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Joseph NM, Tsokos CG, Umetsu SE, Shain AH,
Kelley RK, Onodera C, Bowman S, Talevich E, Ferrell LD, Kakar S and
Krings G: Genomic profiling of combined
hepatocellular-cholangiocarcinoma reveals similar genetics to
hepatocellular carcinoma. J Pathol. 248:164–178. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xue R, Chen L, Zhang C, Fujita M, Li R,
Yan SM, Ong CK, Liao X, Gao Q, Sasagawa S, et al: Genomic and
transcriptomic profiling of combined hepatocellular and
intrahepatic cholangiocarcinoma reveals distinct molecular
subtypes. Cancer Cell. 35:932–947 e938. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hill MA, Alexander WB, Guo B, Kato Y,
Patra K, O'Dell MR, McCall MN, Whitney-Miller CL, Bardeesy N and
Hezel AF: Kras and Tp53 mutations cause cholangiocyte- and
hepatocyte-derived cholangiocarcinoma. Cancer Res. 78:4445–4451.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Seehawer M, Heinzmann F, D'Artista L,
Harbig J, Roux PF, Hoenicke L, Dang H, Klotz S, Robinson L, Doré G,
et al: Necroptosis microenvironment directs lineage commitment in
liver cancer. Nature. 562:69–75. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vinagre J, Almeida A, Populo H, Batista R,
Lyra J, Pinto V, Coelho R, Celestino R, Prazeres H, Lima L, et al:
Frequency of TERT promoter mutations in human cancers. Nat Commun.
4:21852013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nault JC, Mallet M, Pilati C, Calderaro J,
Bioulac-Sage P, Laurent C, Laurent A, Cherqui D, Balabaud C and
Zucman-Rossi J: High frequency of telomerase reverse-transcriptase
promoter somatic mutations in hepatocellular carcinoma and
preneoplastic lesions. Nat Commun. 4:22182013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Totoki Y, Tatsuno K, Covington KR, Ueda H,
Creighton CJ, Kato M, Tsuji S, Donehower LA, Slagle BL, Nakamura H,
et al: Trans-ancestry mutational landscape of hepatocellular
carcinoma genomes. Nat Genet. 46:1267–1273. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fujimoto A, Furuta M, Shiraishi Y, Gotoh
K, Kawakami Y, Arihiro K, Nakamura T, Ueno M, Ariizumi SI, Nguyen
HH, et al: Whole-genome mutational landscape of liver cancers
displaying biliary phenotype reveals hepatitis impact and molecular
diversity. Nat Commun. 6:61202015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim SK, Takeda H, Takai A, Matsumoto T,
Kakiuchi N, Yokoyama A, Yoshida K, Kaido T, Uemoto S, Minamiguchi
S, et al: Comprehensive analysis of genetic aberrations linked to
tumorigenesis in regenerative nodules of liver cirrhosis. J
Gastroenterol. 54:628–640. 2019. View Article : Google Scholar : PubMed/NCBI
|














