Introduction
In 2020, breast cancer was both the most frequently
diagnosed malignancy worldwide, with 11.7% of all new cancer
diagnoses, and the most common cause of cancer death among women,
with 15.5% of all fatalities; therefore, it remains a public health
problem (1). The identification of
breast cancer subtypes (Luminal A, Luminal B, HER2-enriched, basal
and normal-like) is crucial for the selection of the most effective
therapy (2–4). Depending on the breast cancer
subtype, a specific treatment can be selected from among
antimetabolites, and endocrine, immunological, alkylating or
antimitotic drugs (5,6). Anthracyclines (doxorubicin) and
taxanes (paclitaxel) are widely used for breast cancer
treatment.
Paclitaxel is a taxane that binds to microtubules,
promoting tubulin polymerization and inhibiting its
depolymerization (7,8). The interference in the normal
microtubule dynamics caused by paclitaxel activates the spindle
assembly checkpoint (SAC), therefore inducing mitotic arrest and
apoptosis (8–11). Paclitaxel has been widely used to
treat several types of solid tumors, such as ovarian and breast
cancer (12–15). Resistance to this drug is a major
cause of treatment failure, and in consequence, tumor progression.
Several mechanisms have been associated with paclitaxel resistance;
one mechanism is the overexpression of the efflux protein
P-glycoprotein, which increases the outflow of paclitaxel from the
cells (16). Furthermore, the
overexpression of Aurora A kinase, a regulator of SAC, has been
implicated in paclitaxel resistance in triple-negative breast
cancer cells (17). Another
mechanism that contributes to this resistance is the overexpression
of IκB kinase β (IKKβ), an upstream regulator of nuclear factor-κB
(NF-κB) signaling (18). Although
several mechanisms of paclitaxel chemoresistance have been
described, they are not completely understood.
The transcription factor NF-κB family contains five
members: NF-κB1 (p50), NF-κB2 (p52), Rel A (p65), c-Rel and RelB.
These transcription factors form homo- and hetero-dimers that under
normal conditions reside in the cytoplasm bound to inhibitors,
known as inhibitors of NF-κB (IκBs). The activation of NF-κB
requires the IKK-dependent phosphorylation of IκBs (α, β and ε),
which leads to polyubiquitination and subsequent degradation by the
proteasome. Next, NF-κB is translocated to the nucleus where it
regulates the expression of its target genes (19). NF-κB is activated by a wide variety
of stimuli that include growth factors, cytokines, infectious
agents and chemotherapy drugs, among others. Once activated, NF-κB
is involved in the regulation of biological processes, such as
proliferation, differentiation and apoptosis, and some pathologies,
such as inflammation, cancer and chemotherapy resistance (20). The epithelial-mesenchymal
transition (EMT) is a biological process in which a polarized
epithelial cell normally interacts with the basement membrane via
its basal surface, and undergoes a series of morphological and
biochemical changes that allow it to adopt a mesenchymal phenotype
(21,22). This phenotype increases migratory
capacity, invasiveness, resistance to apoptosis and the production
of extracellular matrix components (21). EMT is one of the multiple cellular
processes that are regulated by NF-κB, since it also regulates the
expression of SNAIL and Twist in breast, renal and colon cancer
(23–25). EMT is an early step of cancer
metastasis. During EMT, cell morphology and shape are modified to
gain motility/invasive features, epithelial markers such as
E-cadherin are downregulated and cells acquire mesenchymal markers
such as N-cadherin and vimentin; these changes are controlled by
the transcription factors SNAIL, Twist and Slug (21,26–28).
Paclitaxel is widely used in breast cancer
treatment. Resistance is the main cause of treatment failure. The
present study aimed to examine the association between paclitaxel
resistance and mesenchymal phenotype, and its putative mechanism,
in a model of primary breast cancer cells. Induction of cell death,
apoptosis and NF-κB activation by paclitaxel were evaluated. To
probe the importance of the NF-κB signaling pathway in paclitaxel
resistance, a proteasome inhibitor (ALLN) was employed to interfere
with the paclitaxel-induced cell death, the activation of NF-κB and
induction of apoptosis. The association of paclitaxel resistance
with the NF-κB signaling pathway in mesenchymal breast cancer cells
will provide a potential target to overcome paclitaxel resistance
in patients with breast cancer.
Materials and methods
Reagents and antibodies
Crystal violet and ALLN were purchased from
MilliporeSigma, and paclitaxel from Intas Pharmaceuticals Ltd.
Antibodies against E-cadherin (cat. no. 3195; clone 24E10;
1:1,000), N-cadherin (cat. no. 13116; clone D4R1H; 1:1,000), SNAIL
(cat. no. 3879; clone C15D3; 1:1,000), vimentin (cat. no. 5741;
clone D21H3; 1:1,000), pp65-Ser536 (cat. no. 3033; clone 93H1;
1:1,000), GAPDH (cat. no. 2118; clone 14C10; 1:2,000), Bcl-2 (cat.
no. 4223; clone D55G8; 1:1,000), Bcl-xL (cat. no. 2764; clone 54H6;
1:1,000) and Bax (cat. no. 5023; clone D2E11; 1:1,000) were
acquired from Cell Signaling Technology, Inc. Anti-Twist (cat. no.
GTX-127310; 1:1,000) was purchased from GeneTex, Inc. Anti-IκBα
(cat. no. SC-371; clone C-21; 1:1,000), anti-IKKα/β (cat. no.
SC-7607; clone H-470; 1:1,000), anti-NF-κB p65 (cat. no. SC-8008;
clone F-6; 1:1,000), anti-NF-κB p50 (cat. no. SC-114; 1:1,000),
anti-β-actin (cat. no. SC-47778; clone C4; 1:2,000), and
anti-caspase-3 (cat. no. 271028; clone B-4; 1:1,000) were obtained
from Santa Cruz Biotechnology, Inc.
Primary breast cancer cell
cultures
The primary breast cancer cell cultures MBCDF,
MBCD17, MBCD3 and MBCDF-D5 were originated from a biopsy obtained
from a radical mastectomy in a patient diagnosed with ductal
infiltrating carcinoma as described previously (29,30)
(protocol approved by the Research Ethics Committee from the
National Institute for Medical Sciences and Nutrition ‘Salvador
Zubirán’ (INCMNSZ); Ref 1549, BQO-008-06/9-1). Biopsies were minced
and seeded as explants in RPMI-1640 (Thermo Fisher Scientific,
Inc.) supplemented with 10% FBS (Thermo Fisher Scientific, Inc.),
and 100 U/ml penicillin and 100 µg/ml streptomycin (Thermo Fisher
Scientific, Inc.). Cells that grew from the explants were left to
fill the plate, and then trypsinized and treated as a regular cell
line and grown over 6 months in RPMI-1640 with 10% FBS, and 100
U/ml penicillin and 100 µg/ml streptomycin at 37°C (5%
CO2). All primary cell cultures were tested for
mycoplasma contamination. Mycoplasma-positive breast cancer cells
were treated with 250 µg/ml tylosin (Merck & Co, Inc.) in
RPMI-1640 with 10% FBS, and 100 U/ml penicillin and 100 µg/ml
streptomycin at 37°C (5% CO2) for 12 days before
performing any experiments. Experiments were performed after
passage 10, when the phenotype observed was homogeneous. After
tylosin treatment, primary breast cancer cells were maintained in
RPMI-1640 with 10% FBS, and 100 U/ml penicillin and 100 µg/ml
streptomycin at 37°C (5% CO2) until experimentation.
MBA-MB-231 (ATCC® HTB-26™) and HCC1937 (ATCC®
CRL-2336™) triple negative breast cancer cell lines were purchased
from American Type Culture Collection. MDA-MB-231 and HCC1937 were
cultured in RPMI-1640 with 10% of FBS, and 100 U/ml penicillin and
100 µg/ml streptomycin at 37°C and 5% CO2.
Western blot analysis
MBCDF, MBCD17, MBCD3 and MBCDF-D5 breast cancer
cells were lysed in a buffer containing 50 mM HEPES (pH 7.4), 250
mM NaCl, 5 mM EDTA, 0.1% Nonidet P-40, 10 mM NaF, 50 mM
β-glycerophosphate, 1 mM Na3VO4 and 1X
protease inhibitor cocktail (Complete EDTA-free; Roche
Diagnostics). Protein was quantified using a Bradford assay
(Bio-Rad Laboratories, Inc.). Whole protein extract (20 µg) was
separated by 9 and 12% SDS-PAGE then transferred to Immobilon-P
PVDF membranes (MilliporeSigma). The membranes were blocked with 5%
skim milk in PBS-Tween (0.1%) for 1 h at room temperature. After
this time, primary antibodies were added and incubated overnight at
4°C. Membranes were washed and then incubated with anti-mouse
horseradish peroxidase--conjugated secondary antibodies (cat. no.
115-035-003; 1:10,000; Jackson ImmunoResearch Laboratories, Inc.)
or anti-rabbit horseradish peroxidase-conjugated secondary
antibodies (cat. no. 111-035-003; 1:10,000; Jackson ImmunoResearch
Laboratories, Inc.) for 1 h at room temperature. Proteins were
visualized using the ECL plus western blotting detection system (GE
Healthcare) and blot images were digitized using Chemidoc (Bio-Rad
Laboratories, Inc.). Densitometry of the bands was measured using
ImageLab software v6 (Bio-Rad Laboratories, Inc.).
Viability assays
MBCD3, MBCDF-5, MBCD17 and MBCDF breast cancer cells
were plated at 10,000 cells/cm2 in a 48-well plate. On
the following day, paclitaxel was added at increasing doses (0,
0.05, 0.1, 0.5, 1, 5, 10 and 25 µg/ml) and cells were cultured for
48 h at 37°C (5% CO2). Cell viability was evaluated by
staining with crystal violet for 20 min at room temperature. The
dye was dissolved in 400 µl of 10% acetic acid and the absorbance
was read at 595 nm in a microplate reader (Opsys MR; Dynex
Technologies). The percentage of cell viability was calculated
after normalizing the absorbance of paclitaxel treated cells to the
absorbance of non-treated cells. The plates were seeded in
triplicate in at least three independent experiments. The data are
presented as the mean ± SEM. In the cell viability assays using
ALLN, MBCDF-D5 and MBCD3 cells were seeded at 10,000
cell/cm2 in a 48-well plate. The next day ALLN (10 µM)
was added for 2 h at 37°C (5% CO2), after this time
breast cancer cells were treated with paclitaxel (0, 0.1, 1 and 10
µg/ml). Cells were cultured for a further 48 h at 37°C (5%
CO2). Cell viability was evaluated using the crystal
violet assay as aforementioned.
Statistical analysis
The results are presented as the mean ± SEM of three
independent repeats. Paclitaxel dose-response curves were analyzed
by two-way ANOVA and multiple comparisons were then performed
employing Tukey's Honestly Significant Difference post-hoc test.
Half maximal inhibitory concentration (IC50) was
calculated using a non-linear regression with 95% CI. In
ALLN-paclitaxel combination assays, two-way ANOVA with Bonferroni's
correction for multiple comparisons was used to analyze the
statistical differences in cell viability between ALLN and ALLN
plus paclitaxel. Data were analyzed using GraphPad PRISM v6.01
(GraphPad Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
Resistance to paclitaxel is associated
with a mesenchymal phenotype
We have demonstrated previously that primary breast
cancer cells with a specific pattern of receptor tyrosine kinase
present with resistance to paclitaxel (30); moreover, we have also shown that
cells with a mesenchymal phenotype are resistant to metformin
(29). The present study
investigated whether the mesenchymal phenotype is associated with
paclitaxel resistance, and the putative mechanism. Four different
primary breast cancer cell cultures, two of epithelial phenotype
and two of mesenchymal phenotype, were used. MBCDF and MBCD17 cells
expressed E-cadherin, SNAIL, Slug and Twist, and low levels of
N-cadherin, and no vimentin expression was observed. MBCDF-D5 and
MBCD3 cells expressed low levels of E-cadherin, Slug and Twist, but
expressed N-cadherin and vimentin, and higher levels of SNAIL
(Fig. 1A). These four different
primary breast cancer cells were treated with increasing doses of
paclitaxel (0, 0.05, 0.1, 0.5. 1, 5, 10 and 25 µg/ml) (Fig. 1B). It was found that paclitaxel
induced cell death in a dose-dependent manner, with different
slopes when comparing epithelial and mesenchymal cells. MBCDF-D5
and MBCD3 cells were more resistant to paclitaxel compared with
MBCDF and MBCD17 cells. In the mesenchymal breast cancer cells,
cell viability decreased to ~75% at 0.05 µg/ml of paclitaxel, and
at the highest dose of 25 µg/ml, cell viability fell to 9–18%. By
contrast, MBCDF and MBCD17 epithelial cells were more sensitive to
paclitaxel; viability decreased to 50 and 42%, respectively, at the
lower dose of 0.05 µg/ml, with a downward trend that dropped to 10%
for MBCDF cells and 8% for MBCD17 cells at 25 µg/ml (Fig. 1B). Furthermore, the IC50
for epithelial breast cancer cells varied from 0.01 to 0.046 µg/ml
for the MBCD17 and MBCDF cells, respectively. In the case of
mesenchymal breast cancer cells, the IC50 was higher;
MBCDF-D5 cells exhibited an IC50 of 2.57 µg/ml and MBCD3
cells had an IC50 of 0.93 µg/ml (Fig. 1C). To confirm that the effect of
paclitaxel on primary breast cancer cells was similar to that on
established cell lines, a cytotoxicity assay was performed. Two
breast cancer cell lines (MDA-MB-231 and HCC1937) were selected.
MDA-MB-231 cells resemble a mesenchymal phenotype similar to
MBCDF-D5 cells by expressing vimentin, and HCC1937 cells express
E-cadherin similar to MBCDF cells (31). MDA-MB-231, HCC-1937, MBCDF and
MBCDF-D5 cells were treated with increasing doses of paclitaxel (0,
0.05, 0.1, 0.5, 1, 5, 10 and 25 µg/ml; Fig. S1). MBCDF primary breast cancer
cells were more sensitive to paclitaxel than MBCDF-D5 cells, which
were the most resistant. HCC-1937 and MDA-MB-231 cell lines showed
an intermediate sensitivity compared with the primary breast cancer
cell cultures, with HCC-1937 cells being slightly more resistant to
paclitaxel than MDA-MB-231 cells (Fig. S1).
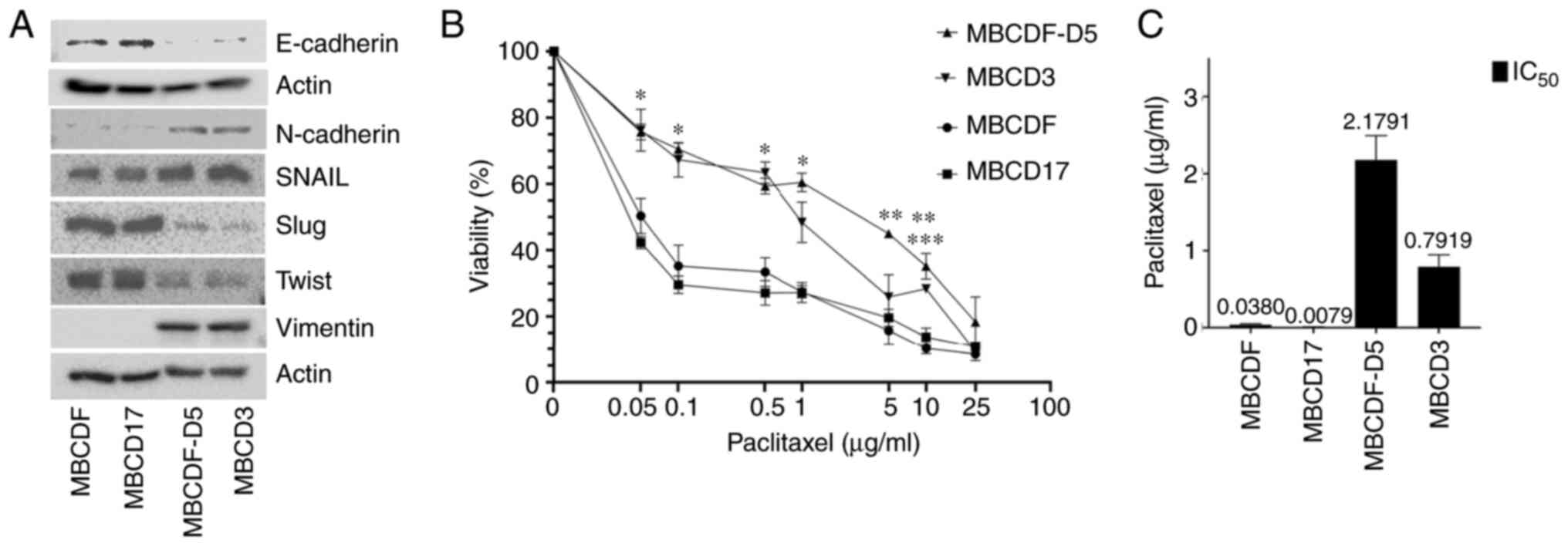 | Figure 1.Mesenchymal phenotype is associated
with paclitaxel resistance in primary breast cancer cells. (A)
Expression of epithelial-mesenchymal transition markers
(E-cadherin, N-cadherin, SNAIL, Slug, Twist and vimentin) was
measured in the primary breast cancer MBCDF, MBCD17, MBCDF-D5 and
MBCD3 cells by western blotting. (B) MBCDF, MBCD17, MBCDF-D5 and
MBCD3 breast cancer cells were treated with increasing doses of
paclitaxel (0, 0.05, 0.1, 0.5. 1, 5, 10 and 25 µg/ml). Cell
viability was evaluated by crystal violet assay 48 h after
treatment. Data represent the mean ± SEM of three independent
experiments performed in triplicate *P<0.05 vs. control (0 µg/ml
paclitaxel). **P<0.05 MBCDF-D5 (5 and 10 µg/ml paclitaxel) vs.
control (0 µg/ml paclitaxel). ***P<0.05 MBCD3 vs. control (0
µg/ml paclitaxel). (C) IC50 was calculated using
non-linear regression. |
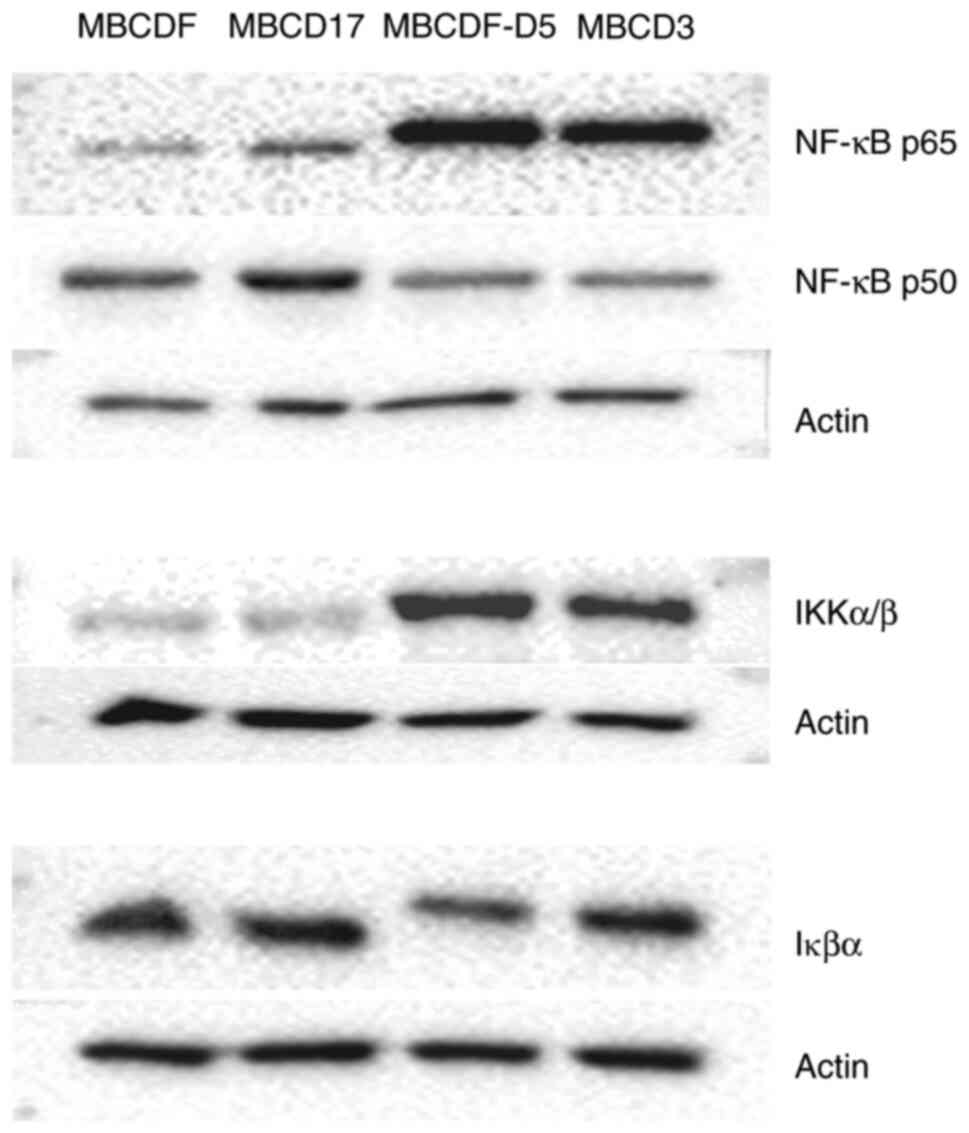
Increased expression of NF-κB
signaling molecules
Since NF-κB has been shown to be involved with EMT
as well as chemoresistance (23),
the putative role of NF-κB in paclitaxel resistance was
investigated in the present study. The expression of certain
members of the NF-κB signaling pathway were evaluated in both
epithelial and mesenchymal primary breast cancer cells. The results
showed that the NF-κB p65 subunit, as well as IKKα/β were
upregulated in the MBCDF-D5 and MBCD3 cells (mesenchymal
phenotype). In contrast to the results observed for NF-κB p65, the
p50 subunit was downregulated in the mesenchymal cells. IκBα did
not show marked changes in its expression levels in both phenotypes
compared with the actin loading control (Fig. 2).
NF-κB activation of mesenchymal
primary breast cells
Given the aforementioned observations, MBCDF-D5 and
MBCD3 were first treated with increasing doses of paclitaxel (0, 1,
5, 10, and 50 µg/ml), and the activation of NF-κB by
phosphorylation at S536 was evaluated. Phosphorylation of p65 at
S536 increased in a dose-dependent manner in response to
paclitaxel. The NF-κB p65 total levels were not changed with
paclitaxel treatment (Fig. 3A).
Secondly, investigations were performed to determine whether there
was IκBα degradation upon different doses of paclitaxel. MBCDF-D5
and MBCD3 cells were treated with 0, 1, 5, 10 and 50 µg/ml
paclitaxel (Fig. 3B). IκBα was
degraded in a paclitaxel dose-dependent manner (Fig. 3B). Next, the effect of paclitaxel
on NF-κB activation over time was evaluated. It was observed that
phosphorylation of NF-κB at Ser536 reached its maximum levels at 1
h after treatment with paclitaxel (5 µg/ml) in MBCDF-D5 cells.
NF-κB activation had a slight decrease at 2 h. In the case of MBCD3
cells, NF-κB phosphorylation at Ser536 increased in an oscillatory
manner (Fig. 3C). Finally,
MBCDF-D5 and MBCD3 were treated at different time points to
evaluate IκBα degradation. Paclitaxel provoked IκBα degradation in
a time-dependent manner. MBCDF-D5 cells exhibited evident IκBα
degradation after 1 h of paclitaxel treatment. Meanwhile, MBCD3
cells showed marked IκBα degradation after 2 h of paclitaxel
addition (Fig. 3D).
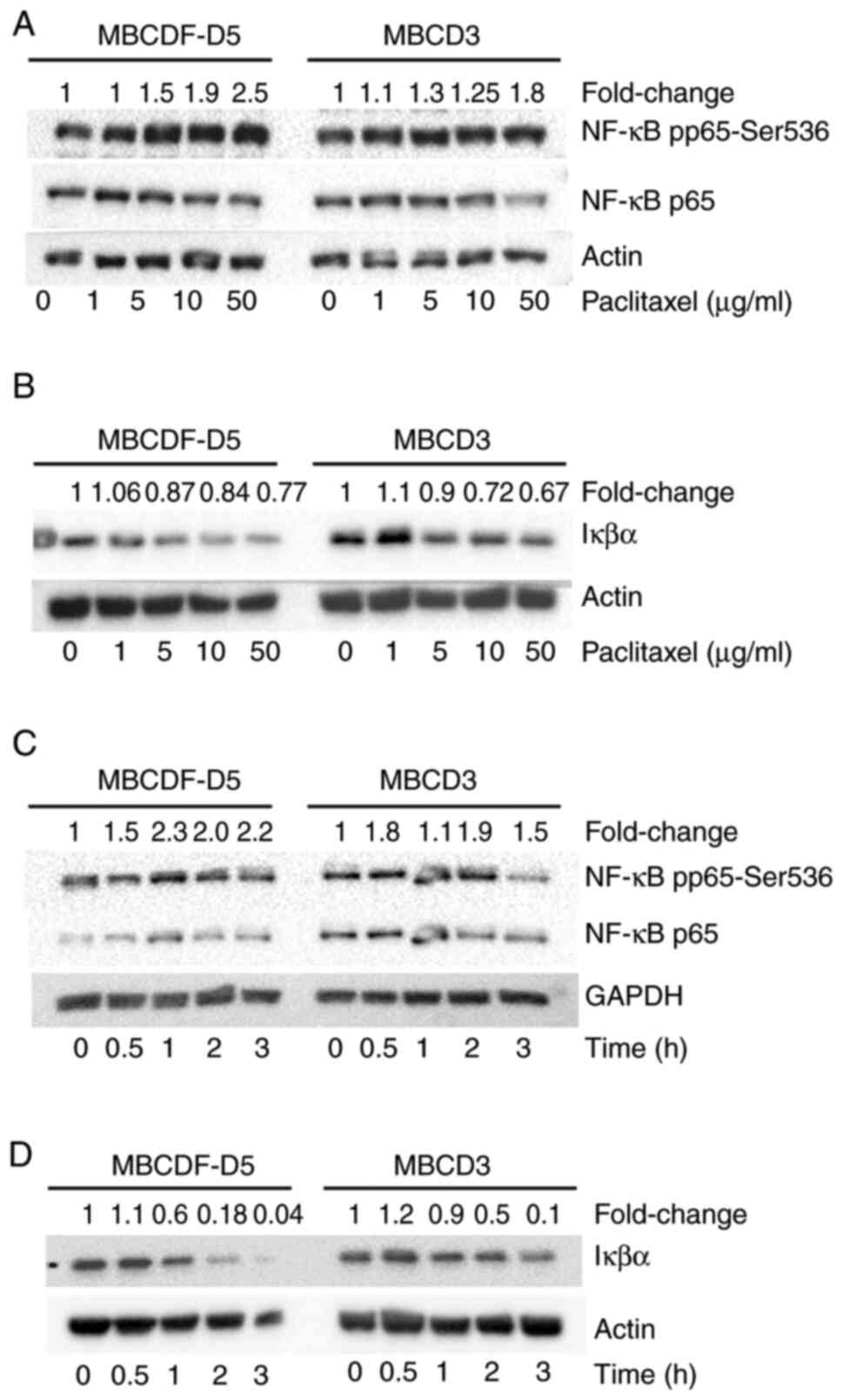 | Figure 3.Paclitaxel induces phosphorylation of
NF-κB at Ser536 and degradation of IκBα. (A) Expression of NF-κB
pp65-Ser536, and NF-κB p65 in MBCDF-D5 and MBCD3 cells treated with
0, 1, 5, 10 and 50 µg/ml paclitaxel. (B) Degradation of IκBα in
MBCDF-D5 and MBCD3 cells treated with 0, 1, 5, 10 and 50 µg/ml
paclitaxel. (C) Expression of NF-κB pp65-Ser536, and NF-κB p65 in
MBCDF-D5 and MBCD3 cells treated with 5 µg/ml paclitaxel for 0,
0.5, 1, 2 and 3 h. (D) Degradation of IκBα in MBCDF-D5 and MBCD3
cells treated with 5 µg/ml paclitaxel for 0, 0.5, 1, 2 and 3 h.
Actin and GAPDH were used as loading controls. Densitometry of the
bands was quantified by ImageLab software. The ratio between NF-κB
pp65-Ser536 and NF-κB p65 was normalized against the loading
control. The fold change of IκBα was calculated by normalizing IκBα
against actin. NF-κB, nuclear factor-κB; IκB, inhibitor of
NF-κB. |
Bcl-2, Bax, Bcl-xL and procaspase-3
expression in response to paclitaxel in paclitaxel-resistant
primary breast cancer cells
To evaluate the role of apoptosis in the
paclitaxel-resistant primary breast cancer cells, the expression of
Bcl-2, Bax, Bcl-xL and procaspase-3 was examined by western
blotting. First, changes in these markers in response to increasing
doses of paclitaxel were measured. In the MBCDF-D5 cells, Bcl-2
expression was elevated by paclitaxel at the concentrations of 1
and 5 µg/ml, and was downregulated at 10 and 50 µg/ml. Bax
expression had a slight fluctuation with concentrations varying
from 1 to 2 fold-change difference. Bcl-xL expression increased in
response to paclitaxel in a dose-dependent manner and procaspase-3
levels decreased in a dose-dependent manner (Fig. 4A; left panel). In the MBCD3 cells,
Bcl-2 expression was elevated by paclitaxel in an increasing trend
from 1 to 10 µg/ml, and at the highest dose (50 µg/ml), dropped to
1.7 fold-change difference. Bax expression was augmented in a
dose-dependent manner, increasing to an 11 fold-change difference
at 5 µg/ml and with the highest level at 50 µg/ml. Bcl-xL was
upregulated, procaspase-3 was downregulated, and both respond in a
dose-dependent manner (Fig. 4A;
right panel). Secondly, Bcl-2, Bax, Bcl-xL, and procaspase-3 were
assessed over a time-course of 3 h. The results showed that Bcl-2
reached its peak at 3 h of paclitaxel treatment in MBCDF-D5 cells.
By contrast, Bax levels were decreased to 0.5-fold at 0.5 h, then
increased up to basal levels at 3 h. Paclitaxel augmented Bcl-xL
expression 3.1-fold at 3 h and procaspase-3 had small fluctuations
during the 3 h period (Fig. 4B).
In the MBCD3 cells, paclitaxel increased Bcl-2 expression levels to
a maximum of 1.9-fold at 0.5 h, which began to diminish by 1 h,
reaching the lowest levels at 3 h. Bax and procaspase-3 expression
exhibited small variations during the 3-h period. Bcl-xL expression
increased up to 1.5-fold at 1 h and then the expression decreased
(Fig. 4B). Since changes were not
observe in some of the markers over this short interval of time,
MBCF-D5 and MBCD3 cell were next treated with paclitaxel for 24 h.
Paclitaxel induced downregulation of Bax and procaspase-3 after 24
h of stimulation (Fig. 4C). In
addition, it was observed that although Bcl-2 and Bcl-xL were
upregulated over a short period of time (Fig. 4B), they returned to their basal
levels after 24 h (Fig. 4C).
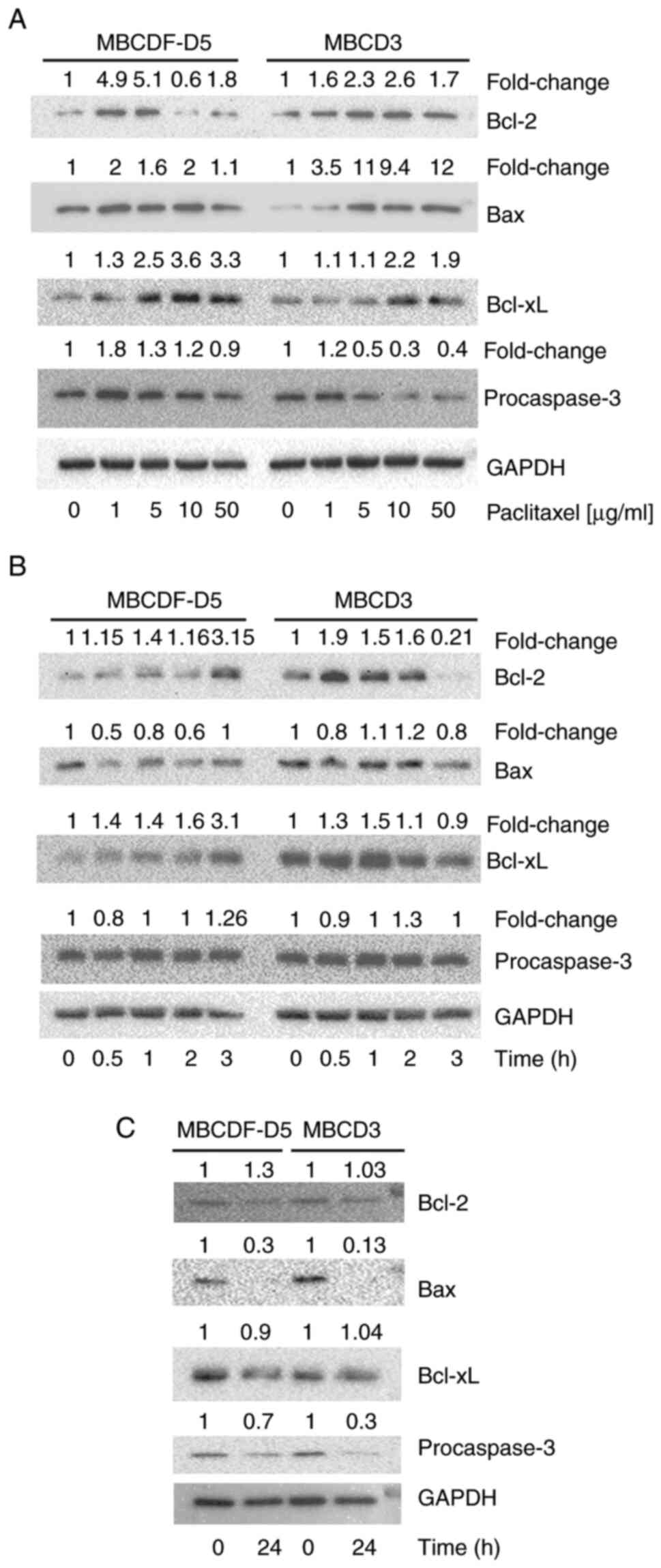 | Figure 4.Expression of Bcl-2, Bax, Bcl-xL and
procaspase-3 in breast cancer cells treated with paclitaxel. (A)
Bcl-2, Bax, Bcl-xL and procaspase-3 were evaluated by western
blotting in MBCDF-D5 and MBCD3 cells treated with 0, 1, 5, 10 and
50 µg/ml paclitaxel. (B) Bcl-2, Bax, Bcl-xL and procaspase-3
expression was assessed by western blotting in MBCDF-D5 and MBCD3
cells as in (A) following treatment with paclitaxel for 0, 0.5, 1,
2 and 3 h. (C) MBCDF-D5 and MBCD3 cells were treated with 5 µg/ml
for 24 h. Bcl-2, Bax, Bcl-xL and procaspase-3 expression was
evaluated by western blotting following treatment with paclitaxel
for 0 or 24 h. GAPDH was used as a loading control. Densitometry of
the bands was quantified by ImageLab software. Fold change of
Bcl-2, Bax, Bcl-xL and procaspase-3 expression was calculated as
the ratio of paclitaxel-treated cells over untreated cells and
normalized against the loading control. |
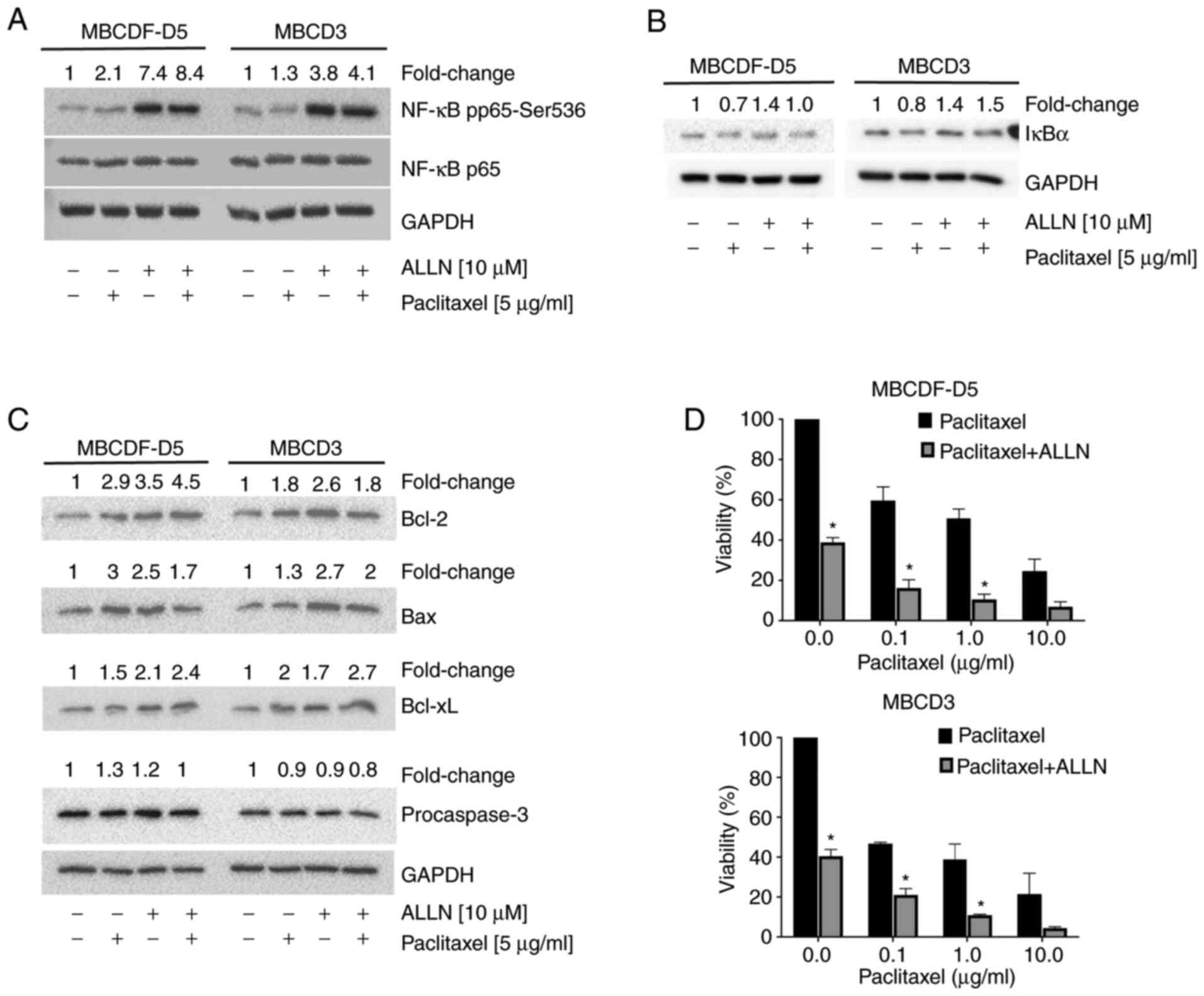
ALLN enhances phosphorylation of NF-κB
at Ser536 and cell death in paclitaxel-resistant primary breast
cancer cells
ALLN is a proteasome inhibitor that functions by
stabilizing the degradation of IκBα and then producing inhibition
of NF-κB (32,33). To investigate whether ALLN could
interfere with paclitaxel-induced NF-κB and, for instance,
overcoming the resistance to paclitaxel, MBCDF-D5 and MBCD3 cells
were pre-treated with 10 µM ALLN 2 h before paclitaxel addition.
Proteasome inhibition with ALLN and ALLN plus paclitaxel provoked a
7.4- and 8.4-fold increase, respectively, in NF-κB pp65 expression,
with no changes in total NF-κB p65 in the MBCDF-D5 cells (Fig. 5A). In the MBCD3 cells, ALLN
produced similar results, with a 3.8- and 4.1-fold increase in
NF-κB pp65 expression for ALLN and ALLN plus paclitaxel,
respectively, while the amount of NF-κB p65 was not affected by
treatment with ALLN, paclitaxel or ALLN plus paclitaxel (Fig. 5A). Second, the study examined
whether ALLN prevented IκBα degradation. MBCDF-D5 and MBCD3 cells
were stimulated with 10 µM ALLN for 2 h before the addition of 5
µg/ml of paclitaxel (Fig. 5B). The
results showed that ALLN blocked IκBα degradation and produced its
accumulation due to proteasome inhibition in MBCD3 breast cancer
cells. In the case of MBCDF-D5 cells, ALLN plus paclitaxel
prevented degradation of IκBα by only returning to basal levels but
was not associated with its accumulation (Fig. 5B). Next, Bcl-2, Bax, Bcl-xL, and
procaspase-3 expression were evaluated under the aforementioned
conditions in both MBCDF-D5 and MBCD3 cells. ALLN alone increased
Bcl-2 expression, 3.5-fold in MBCDF-D5 cells and 2.6-fold in MBCD3
cells (Fig. 5C). The combination
of ALLN plus paclitaxel resulted in a 4.5-fold increase in
expression in MBCDF-D5 cells; meanwhile, expression in MBCD3 cells
decreased to 1.8-fold. Bcl-xL expression was also increased
following treatment with ALLN alone in both primary breast cancer
cell cultures, and ALLN plus paclitaxel further raised Bcl-xL
expression. ALLN and the combination of ALLN plus paclitaxel had
few effects on procaspase-3 expression (Fig. 5C). Finally, the study evaluated
whether ALLN could increase paclitaxel-induced cell death.
Cytotoxicity assays were performed by adding 10 µM ALLN at 2 h
before the addition of increasing doses of paclitaxel. Cell
viability was evaluated at 48 h after paclitaxel stimulation using
a crystal violet assay. It was found that ALLN potentiates
paclitaxel-induced cell death in both MBCDF-D5 and MBCD3 breast
cancer cells in a dose-dependent manner (Fig. 5D).
Discussion
Paclitaxel is employed for breast cancer treatment;
however, chemoresistance is often a challenge to its use (34). This chemoresistance has been
attributed to several mechanisms, including the upregulation of
P-glycoprotein (35) and mutations
in β-tubulin that affect the binding of paclitaxel (36,37).
However, the exact mechanism of paclitaxel resistance is still
elusive. The goal of the present study was to examine the
association of paclitaxel resistance with mesenchymal phenotype,
and its putative mechanism in primary breast cancer cells. Primary
breast cancer cells with mesenchymal phenotype showed resistance to
paclitaxel, and this was associated with increased levels of NF-κB
p65 and IKKα/β. Moreover, paclitaxel induced NF-κB phosphorylation
of NF-κB p65 at Ser536 and degradation of IκBα. It was observed
that upon paclitaxel-induced NF-κB activation, there was
upregulated expression of Bcl-2 and Bcl-xL, which are NF-κB target
genes. Bax and procaspase-3 expression was negatively regulated
after paclitaxel treatment. Furthermore, NF-κB inhibition using the
proteasome inhibitor ALLN provoked accumulation of phosphorylated
NF-κB and IκBα, and increased susceptibility to paclitaxel.
EMT is considered the first step for metastasis and
is characterized by dissolution of tight junctions, loss of cell
polarity and epithelial markers such as E-cadherin, and gain in
expression of mesenchymal markers such as N-cadherin and vimentin
(21,22). Furthermore, EMT has been associated
with resistance to conventional therapies (38,39).
Our previous study have shown that primary breast cancer cells with
expression of HER1, HER3, c-Met and VEGFR2 are resistant to
paclitaxel (30). The data from
the present study indicated that primary breast cancer cells with
expression of the aforementioned receptor tyrosine kinases
presented mesenchymal features and were resistant to paclitaxel
compared with breast cancer cells with an epithelial phenotype.
These data correlate with previous investigations that have
suggested that EMT produces chemotherapy resistance (38–40).
The EMT process is controlled by several
transcription factors, including SNAIL, Slug and Twist. NF-κB is
another transcription factor that is important for EMT via the
regulation of SNAIL and Twist expression in breast, renal and colon
cancer (23–25,41).
Several reports have associated NF-κB with certain pathologies such
as inflammation, cancer and chemotherapy resistance (20,23).
The elevated expression of NF-κB p65 has been observed in ovarian
and breast cancer, and has been associated with the clinical stage
and the histological subtype (42,43).
The present study found increased levels of IKKs and NF-κB p65 in
mesenchymal primary breast cancer cells. These results were
consistent with a previous study where NF-κB p65 has been shown to
be required in the late stages of tumorigenesis, to mediate and
maintain the EMT phenotype (28);
these authors also showed that inhibition of IKKβ/IκBα/NF-κB axes
using the super-repressor of IκBα, which contains mutations in
Ser32/36Ala, inhibited NF-κB DNA-binding activity and TGF-β-induced
EMT (28). By contrast, expression
of constitutively active IKKβ increased NF-κB DNA-binding activity
and EMT markers independently of TGFβ stimulation (28). Other investigations demonstrated
that IKKβ is an upstream regulator of NF-κB activation that has
been involved in the regulation of proliferation and migration of
cisplatin-resistant head and neck squamous carcinoma cells
(23,44). Expression of IKKβ has also been
associated with higher levels of N-cadherin and cisplatin
resistance in head and neck squamous carcinoma cells (23). In accordance with the
aforementioned results, the present study observed elevated levels
of IKKβ and N-cadherin in mesenchymal breast cancer cells and
paclitaxel resistance. In melanoma, NF-κB provokes tumor
aggressiveness by activating MMP9 (45). Moreover, integrin β-like 1 induces
metastasis, invasion and EMT in prostate cancer through activation
of NF-κB (46). In the breast
cancer MDA-MD-231 and HCC-1937 cell lines, inhibition of NF-κB
decreased levels of EMT markers such as Slug, Sip1, Twist and
MMP11, and decreased aggressiveness measured by migration and
invasion assays (47). These
results suggest a correlation between the high expression of
NF-κB/IKK, mesenchymal phenotype and paclitaxel resistance in
primary breast cancer cells. In addition, paclitaxel induced
activation of NF-κB in mesenchymal primary breast cancer cells that
agreed with previous reports showing paclitaxel-induced NF-κB
activation in breast and ovarian cancer cells (48,49).
Furthermore, NF-κB has been implicated in the regulation of other
mechanisms of paclitaxel resistance, such as the induction of
P-glycoprotein expression. In accordance with these findings, we
have observed high expression of MDR in MBCF-D5 and MBCD3
mesenchymal breast cancer cells that have elevated levels of NF-κB
p65 concomitant with paclitaxel resistance (data not shown). In
this context, Abdin et al (50) demonstrated that breast cancer MCF-7
and MDA-MB-231 cell lines resistant to doxorubicin exhibited
increased levels of NF-κB p65, which is linked to high expression
of MDR. The study showed that the combination of doxorubicin and
NF-κB inhibitors sensitized breast cancer cell lines to
chemotherapy and decreased expression of MDR (50).
By contrast, the present study found that levels of
NF-κB p50 were higher in epithelial cells compared with those in
mesenchymal cells. NF-κB p50 or NF-κB1 is a product of proteasomal
processing of p105. Structurally, NF-κB p50 lacks the
transcriptional domain, and thus its function as a transcription
factor depends on its heterodimerization with other members of the
family. Functionally, NF-κB p50 participates in the reprogramming
of genes involved in inflammation, the inhibition of apoptosis,
angiogenesis, cell survival and cell proliferation (51). Overexpression of NF-κB p50 induces
tumor growth with an increase in the formation of p50-p50
homodimers, while p50-p65 heterodimers are diminished (52). Normal expression levels of NF-κB
p50 have been found in breast cancer, melanoma and lung cancer
(51). By contrast, high
expression of NF-κB p50 in gastric cancer has been shown to
correlate with tumor size and metastasis (53). According to the aforementioned
findings, the role of NF-κB p50 seems contradictory in different
types of cancer. The reason why less expression of NF-κB p50 was
observed in the present study is not clear, and further studies
must be developed to understand the role of NF-κB p50 during EMT in
breast cancer cells.
NF-κB resides inactive in the cytoplasm; its
activation requires phosphorylation of IκBα by IKKs targeting IκBα
for degradation by the proteasome. Proteasome inhibitor ALLN is a
potent inhibitor of NF-κB activation that functions by preventing
IκBα degradation (32,33). In the present study, ALLN
pre-treatment before paclitaxel treatment induced degradation of
IκBα and accumulation of phosphorylated NF-κB p65 in mesenchymal
breast cancer cells, and increased paclitaxel-induced cell death.
These results suggest that inhibition of NF-κB activation reverses
paclitaxel resistance in mesenchymal breast cancer cells.
NF-κB has been associated with the transcription of
antiapoptotic genes such as Bcl-2 and Bcl-xL (54). The present results showed that
paclitaxel increased the expression of these antiapoptotic markers,
most likely through NF-κB activation. These results agreed with a
previous study where paclitaxel upregulated NF-κB (55). In the present study, the
proapoptotic marker Bax was downregulated by paclitaxel and
procaspase-3 was not cleaved, suggesting that apoptosis was
inhibited in these cells. Notably, ALLN did not abrogate the high
levels of Bcl-2 and Bcl-xL induced by paclitaxel, and when used
alone actually induced the expression of both antiapoptotic
markers. Bcl-2 is known to be regulated by the proteasome and this
could explain the accumulation of Bcl-2 in the presence of ALLN
(56). The fact that ALLN
sensitized resistant breast cancer cells to paclitaxel suggested
that the cell death caused by ALLN plus paclitaxel was through a
mechanism independent of Bcl-2 family members.
Although other breast cancer cell lines had similar
behavior in their sensitivity to paclitaxel (Fig. S1), the present primary breast
cancer cell cultures model retains some features of the original
tumor, such as intratumor heterogeneity, as previously demonstrated
(57). The primary breast cancer
cells have less accumulated mutations due to continuing passaging
in vitro compared with established cell lines such as
MDA-MB-231 and HCC1937 (58). Even
though this system turns out to be extraordinarily reproducible and
permitted the exploration of different aspects of cell signaling
and the mechanisms for drug resistance, the translation from an
in vitro model to humans has required further research. Two
models have been used to closer approach what occurs in humans. One
is the use of 3-dimensional (3D) cultures and tumor xenografts in
an immunosuppressed mouse model. Organoids are produced by
culturing embryonic stem cells, tissues, or induced pluripotent
stem cells in a 3D extracellular matrix, typically using Matrigel;
this method has received special attention since it recreates the
spatial conformation of a tumor mass trying to overcome the
limitations of monolayer cell culture (59,60).
The use of embryonic or adult stem cells is crucial for
self--organization, self-renewal and differentiation. Organoids
provide a better representation of solid tumors when compared with
monolayers, since they can resemble a tumor mass where the exterior
cells will be better exposed to nutrients, oxygen and, importantly,
to chemotherapy agents, and by contrast, cells in the interior of
the organoid will have little exposure. When investigating
paclitaxel resistance in organoids originating from primary breast
cancer cells, the organoid will represent a tumor mass in which the
paclitaxel will have little access to the cells in the center.
Organoids are not as complex as tumors, since they lack the
components of the tumor microenvironment. By contrast, a tumor
xenograft is a piece of tumor tissue, a cell suspension from a
disaggregated tumor or a cell line that is injected into a
subcutaneous position or an orthotopic site (61,62).
The tumor xenograft is subjected to a selective pressure in its new
niche, and its successful growth will depend on different factors,
including the mouse strain, the aggressiveness of the tumor and the
interactions that are established within the new microenvironment
(63,64). The use of a tumor xenografted model
with primary breast cancer cells will closely resemble the original
tumor. An orthotopic xenograft model will provide relevant
information on the in vivo efficacy of the combination of
paclitaxel and a proteasome inhibitor such as bortezomib (Velcade)
to inhibit NF-κB.
The present study demonstrated that primary breast
cancer cells with mesenchymal phenotype have high levels of
IKK/IκBα/NF-κB p65 and that this is associated with the resistance
to paclitaxel. We previously demonstrated that these
paclitaxel-resistant breast cancer cells present a specific pattern
of receptor tyrosine kinase and the present study showed that these
cells had a mesenchymal phenotype and elevated expression of the
NF-κB/IKK axis. Together, this research highlights the importance
of targeting the IKK/IκBα/NF-κB p65 axes in mesenchymal breast
cancer cells resistant to paclitaxel and the putative use of a
proteasome inhibitor as a regimen to overcome paclitaxel
resistance.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Alberto Huberman
(Biochemistry Unit, Salvador Zubirán National Institute of Health
Sciences and Nutrition, Mexico City, Mexico) for providing a
critical review of the manuscript. The authors would also like to
thank Ms. Elizabeth Guadarrama (National Medical Sciences and
Nutrition Institute Salvador Zubirán, Mexico City, Mexico) for
kindly donating the paclitaxel. Triple negative breast cancer cell
lines (MDA-MB-231 and HCC-1937) were kindly donated by Dr Leticia
Rocha-Zavaleta (Biomedical Research Institute, National Autonomous
University of Mexico, Mexico City, Mexico).
Funding
This study was supported by the Biochemistry Unit, Salvador
Zubirán National Institute of Health Sciences and Nutrition
(INCMNSZ) and Research Support Network (RAI), National Autonomous
University of Mexico, Mexico City, Mexico.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
Design and coordination of the study: MDJIS, JEL and
ELR. Generation of primary breast cancer cell cultures: MDJIS and
JEL. Paclitaxel cytotoxicity assays: OL and JCGD. Western blot
assays: JEL and ENDLCE. ALLN assays: ENDLCE. Statistical analysis:
MDJIS and OL. Data analysis and discussion: MDJIS, JEL and ELR.
Writing of the manuscript: MDJIS and JEL. MDJIS, JEL and OL confirm
the authenticity of all raw data. All authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Primary breast cancer cells were generated from a
tissue biopsy obtained during surgery in patients with breast
cancer. All patients provided written informed consent for a
protocol approved by the Ethics and Research Committee of the
National Medical Sciences and Nutrition Institute Salvador Zubirán
(approval no. 1549, BQO-008-06/9-1).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
NF-κB
|
nuclear factor-κB
|
|
IKK
|
IκB kinase
|
|
IκB
|
inhibitor of NF-κB
|
|
EMT
|
epithelial-mesenchymal transition
|
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Allison KH, Hammond ME, Dowsett M,
McKernin SE, Carey LA, Fitzgibbons PL, Hayes DF, Lakhani SR,
Chavez-MacGregor M, Perlmutter J, et al: Estrogen and progesterone
receptor testing in breast cancer: ASCO/CAP guideline update. J
Clin Oncol. 38:1346–1366. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wolff AC, Hammond ME, Allison KH, Harvey
BE, Mangu PB, Bartlett JM, Bilous M, Ellis IO, Fitzgibbons P, Hanna
W, et al: Human epidermal growth factor receptor 2 testing in
breast cancer: American society of clinical oncology/college of
american pathologists clinical practice guideline focused update. J
Clin Oncol. 36:2105–2122. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Voduc KD, Cheang MC, Tyldesley S, Gelmon
K, Nielsen TO and Kennecke H: Breast cancer subtypes and the risk
of local and regional relapse. J Clin Oncol. 28:1684–1691. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tong CW, Wu M, Cho WC and To KK: Recent
advances in the treatment of breast cancer. Front Oncol. 8:2272018.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Falzone L, Salomone S and Libra M:
Evolution of cancer pharmacological treatments at the turn of the
third millennium. Front Pharmacol. 9:13002018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Samaan TM, Samec M, Liskova A, Kubatka P
and Büsselberg D: Paclitaxel's mechanistic and clinical effects on
breast cancer. Biomolecules. 9:7892019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weaver BA: How taxol/paclitaxel kills
cancer cells. Mol Biol Cell. 25:2677–2681. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rowinsky EK, Cazenave LA and Donehower RC:
Taxol: A novel investigational antimicrotubule agent. J Natl Cancer
Inst. 82:1247–1259. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Weaver BA and Cleveland DW: Decoding the
links between mitosis, cancer, and chemotherapy: The mitotic
checkpoint, adaptation, and cell death. Cancer Cell. 8:7–12. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nezi L and Musacchio A: Sister chromatid
tension and the spindle assembly checkpoint. Curr Opin Cell Biol.
21:785–795. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Raab M, Kobayashi NF, Becker S,
Kurunci-Csacsko E, Krämer A, Strebhardt K and Sanhaji M: Boosting
the apoptotic response of high-grade serous ovarian cancers with
CCNE1 amplification to paclitaxel in vitro by targeting APC/C and
the pro-survival protein MCL-1. Int J Cancer. 146:1086–1098. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Falzone L, Scandurra G, Lombardo V,
Gattuso G, Lavoro A, Distefano AB, Scibilia G and Scollo P: A
multidisciplinary approach remains the best strategy to improve and
strengthen the management of ovarian cancer (Review). Int J Oncol.
59:2021. View Article : Google Scholar
|
|
14
|
Falchook G, Coleman RL, Roszak A, Behbakht
K, Matulonis U, Ray-Coquard I, Sawrycki P, Duska LR, Tew W,
Ghamande S, et al: Alisertib in combination with weekly paclitaxel
in patients with advanced breast cancer or recurrent ovarian
cancer: A randomized clinical trial. JAMA Oncol. 5:e1837732019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu L and Chen L: Progress in research on
paclitaxel and tumor immunotherapy. Cell Mol Biol Lett. 24:402019.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Němcová-Fürstová V, Kopperová D,
Balušíková K, Ehrlichová M, Brynychová V, Václavíková R, Daniel P,
Souček P and Kovář J: Characterization of acquired paclitaxel
resistance of breast cancer cells and involvement of ABC
transporters. Toxicol Appl Pharmacol. 310:215–228. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang N, Wang C, Wang J, Wang Z, Huang D,
Yan M, Kamran M, Liu Q and Xu B: Aurora kinase A stabilizes FOXM1
to enhance paclitaxel resistance in triple-negative breast cancer.
J Cell Mol Med. 23:6442–6453. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang Y, Chen G, Wang Y, He R, Du J, Jiao
X and Tai Q: Inhibition of microRNA-16 facilitates the paclitaxel
resistance by targeting IKBKB via NF-κB signaling pathway in
hepatocellular carcinoma. Biochem Biophys Res Commun.
503:1035–1041. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Karin M and Ben-Neriah Y: Phosphorylation
meets ubiquitination: The control of NF-[kappa]B activity. Annu Rev
Immunol. 18:621–663. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Courtois G and Smahi A: NF-κB-related
genetic diseases. Cell Death Differ. 13:843–851. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wendt MK, Taylor MA, Schiemann BJ and
Schiemann WP: Down-regulation of epithelial cadherin is required to
initiate metastatic outgrowth of breast cancer. Mol Biol Cell.
22:2423–2435. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liao J, Yang Z, Carter-Cooper B, Chang ET,
Choi EY, Kallakury B, Liu X, Lapidus RG, Cullen KJ and Dan H:
Suppression of migration, invasion, and metastasis of
cisplatin-resistant head and neck squamous cell carcinoma through
IKKβ inhibition. Clin Exp Metastasis. 37:283–292. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM
and Zhou BP: Stabilization of snail by NF-kappaB is required for
inflammation-induced cell migration and invasion. Cancer Cell.
15:416–428. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu ST, Sun GH, Hsu CY, Huang CS, Wu YH,
Wang HH and Sun KH: Tumor necrosis factor-α induces
epithelial-mesenchymal transition of renal cell carcinoma cells via
a nuclear factor kappa B-independent mechanism. Exp Biol Med
(Maywood). 236:1022–1029. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Geiger TR and Peeper DS: Metastasis
mechanisms. Biochim Biophys Acta. 1796:293–308. 2009.PubMed/NCBI
|
|
27
|
Craene BD and Berx G: Regulatory networks
defining EMT during cancer initiation and progression. Nat Rev
Cancer. 13:97–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huber MA, Azoitei N, Baumann B, Grünert S,
Sommer A, Pehamberger H, Kraut N, Beug H and Wirth T: NF-κB is
essential for epithelial-mesenchymal transition and metastasis in a
model of breast cancer progression. J Clin Invest. 114:569–581.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Esparza-Lopez J, Alvarado-Munoz JF,
Escobar-Arriaga E, Ulloa-Aguirre A and Ibarra-Sanchez MJ: Metformin
reverses mesenchymal phenotype of primary breast cancer cells
through STAT3/NF-κB pathways. BMC Cancer. 19:7282019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Esparza-Lopez J, Ramos-Elias PA,
Castro-Sanchez A, Rocha-Zavaleta L, Escobar-Arriaga E,
Zentella-Dehesa A, León-Rodríguez E, Medina-Franco H and
Ibarra-Sánchez MJ: Primary breast cancer cell culture yields
intra-tumor heterogeneous subpopulations expressing exclusive
patterns of receptor tyrosine kinases. BMC Cancer. 16:7402016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Navarrete-Bernal MG, Cervantes-Badillo MG,
Martinez-Herrera JF, Lara-Torres CO, Gerson-Cwilich R,
Zentella-Dehesa A, Ibarra-Sánchez MJ, Esparza-López J, Montesinos
JJ, Cortés-Morales VA, et al: Biological landscape of triple
negative breast cancers expressing CTLA-4. Front Oncol.
10:12062020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Murray RZ and Norbury C: Proteasome
inhibitors as anti-cancer agents. Anticancer Drugs. 11:407–417.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ibarra-Sanchez MJ, Wagner J, Ong MT,
Lampron C and Tremblay ML: Murine embryonic fibroblasts lacking
TC-PTP display delayed G1 phase through defective NF-kappaB
activation. Oncogene. 20:4728–4739. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen J, Tian W, He H, Chen F, Huang J,
Wang X and Chen Z: Downregulation of miR200c3p contributes to the
resistance of breast cancer cells to paclitaxel by targeting SOX2.
Oncol Rep. 40:3821–3829. 2018.PubMed/NCBI
|
|
35
|
Greenberger LM, Lothstein L, Williams SS
and Horwitz SB: Distinct P-glycoprotein precursors are overproduced
in independently isolated drug-resistant cell lines. Proc Natl Acad
Sci USA. 85:3762–3766. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mozzetti S, Ferlini C, Concolino P,
Filippetti F, Raspaglio G, Prislei S, Gallo D, Martinelli E,
Ranelletti FO, Ferrandina G and Scambia G: Class III beta-tubulin
overexpression is a prominent mechanism of paclitaxel resistance in
ovarian cancer patients. Clin Cancer Res. 11:298–305.
2005.PubMed/NCBI
|
|
37
|
Dumontet C, Isaac S, Souquet PJ,
Bejui-Thivolet F, Pacheco Y, Peloux N, Frankfurter A, Luduena R and
Perol M: Expression of class III beta tubulin in non-small cell
lung cancer is correlated with resistance to taxane chemotherapy.
Bull Cancer. 92:E25–E30. 2005.PubMed/NCBI
|
|
38
|
Li X, Lewis MT, Huang J, Gutierrez C,
Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC,
et al: Intrinsic resistance of tumorigenic breast cancer cells to
chemotherapy. J Natl Cancer Inst. 100:672–679. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ren H, Du P, Ge Z, Jin Y, Ding D, Liu X
and Zou Q: TWIST1 and BMI1 in cancer metastasis and
chemoresistance. J Cancer. 7:1074–1080. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang L, Zhang F, Cui JY, Chen L, Chen YT
and Liu BW: CAFs enhance paclitaxel resistance by inducing EMT
through the IL6/JAK2/STAT3 pathway. Oncol Rep. 39:2081–2090.
2018.PubMed/NCBI
|
|
41
|
Liao SJ, Luo J, Li D, Zhou YH, Yan B, Wei
JJ, Tu JC, Li YR, Zhang GM and Feng ZH: TGF-β1 and TNF-α
synergistically induce epithelial to mesenchymal transition of
breast cancer cells by enhancing TAK1 activation. J Cell Commun
Signal. 13:369–380. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Espinoza-Sánchez NA, Győrffy B,
Fuentes-Pananá EM and Götte M: Differential impact of classical and
non-canonical NF-κB pathway-related gene expression on the survival
of breast cancer patients. J Cancer. 10:5191–5211. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Prajoko YW and Aryandono T: Expression of
nuclear factor kappa B (NF-κB) as a predictor of poor pathologic
response to chemotherapy in patients with locally advanced breast
cancer. Asian Pac J Cancer Prev. 15:595–598. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nottingham LK, Yan CH, Yang X, Si H,
Coupar J, Bian Y, Cheng TF, Allen C, Arun P, Gius D, et al:
Aberrant IKKα and IKKβ cooperatively activate NF-κB and induce
EGFR/AP1 signaling to promote survival and migration of head and
neck cancer. Oncogene. 33:1135–1147. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Guarneri C, Bevelacqua V, Polesel J,
Falzone L, Cannavò PS, Spandidos DA, Malaponte G and Libra M: NFκB
inhibition is associated with OPN/MMP9 downregulation in cutaneous
melanoma. Oncol Rep. 37:737–746. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li W, Li S, Yang J, Cui C, Yu M and Zhang
Y: ITGBL1 promotes EMT, invasion and migration by activating NF-κB
signaling pathway in prostate cancer. Onco Targets Ther.
12:3753–3763. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pires BR, Mencalha AL, Ferreira GM, de
Souza WF, Morgado-Díaz JA, Maia AM, Corrêa S and Abdelhay ES:
NF-kappaB is involved in the regulation of EMT genes in breast
cancer cells. PLoS One. 12:e01696222017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Momeny M, Eyvani H, Barghi F, Ghaffari SH,
Javadikooshesh S, Jamadi RH, Esmaeili F, Alishahi Z, Zaghal A,
Bashash D, et al: Inhibition of bromodomain and extraterminal
domain reduces growth and invasive characteristics of
chemoresistant ovarian carcinoma cells. Anticancer Drugs.
29:1011–1020. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang Y, Yang B, Zhao J, Li X, Zhang L and
Zhai Z: Proteasome inhibitor
carbobenzoxy-L-Leucyl-L-Leucyl-L-Leucinal (MG132) enhances
therapeutic effect of paclitaxel on breast cancer by inhibiting
nuclear factor (NF)-κB signaling. Med Sci Monit. 24:294–304. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Abdin SM, Tolba MF, Zaher DM and Omar HA:
Nuclear factor-κB signaling inhibitors revert multidrug-resistance
in breast cancer cells. Chem Biol Interact. 340:1094502021.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Concetti J and Wilson CL: NFKB1 and
cancer: Friend or Foe? Cells. 7:1332018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kravtsova-Ivantsiv Y, Shomer I,
Cohen-Kaplan V, Snijder B, Superti-Furga G, Gonen H, Sommer T, Ziv
T, Admon A, Naroditsky I, et al: KPC1-mediated ubiquitination and
proteasomal processing of NF-κB1 p105 to p50 restricts tumor
growth. Cell. 161:333–347. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Long YM, Ye S, Rong J and Xie WR: Nuclear
factor kappa B: A marker of chemotherapy for human stage IV gastric
carcinoma. World J Gastroenterol. 14:4739–4744. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kim JH, Gupta SC, Park B, Yadav VR and
Aggarwal BB: Turmeric (Curcuma longa) inhibits inflammatory nuclear
factor (NF)-κB and NF-κB-regulated gene products and induces death
receptors leading to suppressed proliferation, induced
chemosensitization, and suppressed osteoclastogenesis. Mol Nutr
Food Res. 56:454–465. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kim SH, Park HJ and Moon DO: Sulforaphane
sensitizes human breast cancer cells to paclitaxel-induced
apoptosis by downregulating the NF-κB signaling pathway. Oncol
Lett. 13:4427–4432. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Azad N, Vallyathan V, Wang L,
Tantishaiyakul V, Stehlik C, Leonard SS and Rojanasakul Y:
S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal
degradation. A novel antiapoptotic mechanism that suppresses
apoptosis. J Biol Chem. 281:34124–34134. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Esparza-López J, Escobar-Arriaga E,
Soto-Germes S and Ibarra-Sánchez MJ: Breast cancer intra-tumor
heterogeneity: One tumor, different entities. Rev Invest Clin.
69:66–76. 2017.PubMed/NCBI
|
|
58
|
Esparza-López J, Martinez-Aguilar JF and
Ibarra-Sánchez MJ: Deriving primary cancer cell cultures for
personalized therapy. Rev Invest Clin. 71:369–380. 2019.PubMed/NCBI
|
|
59
|
Rosenbluth JM, Schackmann RCJ, Gray GK,
Selfors LM, Li CM, Boedicker M, Kuiken HJ, Richardson A, Brock J,
Garber J, et al: Organoid cultures from normal and cancer-prone
human breast tissues preserve complex epithelial lineages. Nat
Commun. 11:17112020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kim J, Koo BK and Knoblich JA: Human
organoids: Model systems for human biology and medicine. Nat Rev
Mol Cell Biol. 21:571–584. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Grisanzio C, Seeley A, Chang M, Collins M,
Napoli AD, Cheng SC, Percy A, Beroukhim R and Signoretti S:
Orthotopic xenografts of RCC retain histological, immunophenotypic
and genetic features of tumours in patients. J Pathol. 225:212–221.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jager W, Moskalev I, Janssen C, Hayashi T,
Awrey S, Gust KM, So AI, Zhang K, Fazli L, Li E, et al:
Ultrasound-guided intramural inoculation of orthotopic bladder
cancer xenografts: A novel high-precision approach. PLoS One.
8:e595362013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Murayama T and Gotoh N: Patient-derived
xenograft models of breast cancer and their application. Cells.
8:6212019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Schmidt KF, Ziu M, Schmidt NO, Vaghasia P,
Cargioli TG, Doshi S, Albert MS, Black PM, Carroll RS and Sun Y:
Volume reconstruction techniques improve the correlation between
histological and in vivo tumor volume measurements in mouse models
of human gliomas. J Neurooncol. 68:207–215. 2004. View Article : Google Scholar : PubMed/NCBI
|