Introduction
The anti-leukemia role of natural killer (NK) cells
has been indicated by study of the prevention of relapse by
alloreactive NK cells in hematopoietic stem cell transplantation
(SCT) (1). A recent study
demonstrated the involvement of impaired NK cell function in the
immune evasion of leukemic cells (2). NK cells are one of the key players in
the innate immune response characterized by tumor cell destruction
(3). The balance between
activation and inhibition mediated by different receptors controls
NK cell activation (4). This
balance is supposed to be regulated by the Nectins and Nectin-like
molecules (Necls) family, including the activating receptor DNAX
accessory molecule-1 (DNAM-1; also known as CD226) and the
inhibitory axis comprising T cell immunoglobulin and immunoreceptor
tyrosine-based inhibitory motif domain (TIGIT) (5). DNAM-1 and TIGIT share the same
ligands, CD155 (Necl-5) and/or CD112 (Nectin-2), and these
molecules are immune checkpoints, such as programmed cell death
protein-1 (PD-1) and programmed death-ligand 1 (PD-L1) (3,6,7).
FMS-like tyrosine kinase 3 (FLT3), a receptor
tyrosine kinase, serves a major role in the regulation of
hematopoiesis (8). Mutations in
FLT3, including the internal tandem duplication (ITD), which
is the most common type of FLT3 mutation, occurs in ~30% of
all acute myeloid leukemia (AML) cases (9,10).
FLT3-ITD leads to a high leukemic burden and confers a poor
prognosis in patients with AML (10). First-generation FLT3
inhibitors developed for clinical use in patients with mutated
FLT3 are broad-spectrum, multikinase inhibitors and lack
specificity for the mutated FLT3-ITD, which may explain
their transient anti-leukemic activity, particularly when used as
monotherapy in patients with relapsed disease (11). Next-generation FLT3
inhibitors, including quizartinib and gilteritinib, have greater
specificity for FLT3 and higher potency compared with the
first-generation FLT3 inhibitors (12). These FLT3 inhibitors have
shown promising anti-leukemia effects in single agent in clinical
trials, and their clinical use has been approved by the US Food and
Drug Administration (13,14).
Regarding the relevance of FLT3 mutations and
TIGIT, the frequencies of CD8+ T cells expressing TIGIT
and PD-1 without DNAM-1 were higher in patients with
FLT3-ITD mutations than in those without these mutations
(15). However, to the best of our
knowledge, the relevance of CD155 and CD112, cognate ligands of
TIGIT, in AML prognosis and their effect on NK cell function remain
unclear.
The present study analyzed the mRNA expression of
CD155 and CD112 via reverse transcription-quantitative (RT-q)PCR in
AML cells with FLT3 mutations treated with FLT3
inhibitors. Surface expression of CD155 and CD 112 in AML cells
with or without FLT3 mutations treated with FLT3
inhibitors was also analyzed by flow cytometry. Furthermore, the
present study investigated whether the anti-leukemic effect of NK
and γδ T cells was affected by treatment of AML cells with
FLT3 inhibitors.
Materials and methods
Cell lines and culture conditions
Human AML cell lines (MOLM-13, MV-4-11, THP-1, NB-4
and KG-1) and a chronic myeloid leukemia (CML) cell line (K562)
were cultured in RPMI-1640 medium (FUJIFILM Wako Pure Chemical
Corporation) 10% fetal bovine serum (FBS; Sigma-Aldrich; Merck
KGaA) and 1% penicillin-streptomycin at 37°C in an atmosphere with
5% CO2. KHYG-1 cells were cultured in RPMI-1640 medium
supplemented with 10% FBS and 1% penicillin/streptomycin in the
presence of 2–20 ng/ml recombinant human IL-2 (rhIL-2, PeproTech)
at 37°C with 5% CO2. MV-4-11, THP-1, KG-1, K562 and NB-4
cells were obtained from American Type Culture Collection. KHYG-1
cells were obtained from Japanese Collection of Research
Bioresources Cell Bank, whereas MOLM-13 cells were obtained from
Leibniz Institute DSMZ (German Collection of Microorganisms and
Cell Cultures).
Primary cells
NK cells were purified from peripheral blood
mononuclear cells (PBMCs) obtained from healthy donors (age, 20–65
years, three males and one female) from October 2019 to September
2021 at Research Hospital, The Institute of Medical Science, The
University of Tokyo, Tokyo, Japan, using a human NK Cell Isolation
Kit (Miltenyi Biotec GmbH). Cell counting was performed using a
hemocytometer (Erma Inc.). The γδ T cell isolation protocol as
described by Cui et al (16) was followed. Briefly, γδ T cells
were isolated from PBMCs under stimulation with zoledronic acid
(Selleck Chemicals) at 1 µM, in combination with 50 IU/ml rhIL-2 at
37°C with 5% CO2 for 7 days. The culture media were
changed every 3 days. After 1 week of culture, the cells were
harvested and CD3+Vd2T cell receptor (TCR)+
cells were determined by flow cytometry.
Lentiviral production and
transduction
Lenti-X293T cells (Clontech; Takara Bio USA) were
cultured in DMEM (FUJIFILM Wako Pure Chemical Corporation) with 10%
FBS and 1% penicillin-streptomycin, at 37°C with 5% CO2.
Lentiviral plasmid (CSII-EF-MCS) was purchased from National
BioResource Project and 3rd generation system was used. Lentiviral
plasmid (CSII-EF-fLuc-2A-EGFP) was produced as described previously
(17). Lentiviral vector
(CSII-EF-fLuc-2A-EGFP) particles (5 µg) were produced by
cotransfection of Lenti-X293T cells with a transfer plasmid, and
packaging plasmids pMDLg/p.RRE (3 µg), pRSV-rev (1 µg) and pMD.G (1
µg) at 37°C with 5% CO2 for 2 days. The lentiviral
particles were obtained by centrifugation at 400 × g and 4°C for 5
min and collection of supernatant. Then the lentiviral vector
particles were titrated in HeLa cells as described previously
(18). A total of 1×106
Target cells (MOLM-13, MV-4-11 and THP-1) were transduced with the
CSII-EF-fLuc-2A-EGFP lentiviral vector at a multiplicity of
infection of 5. Two days after transduction, target cells
(EGFP+) were harvested by fluorescence-activated cell
sorting using a cell sorter SH800s (Sony Corporation) and expanded
for an additional 5 days.
Reagents
Quizartinib (AC220) was obtained from Selleck
Chemicals and Daiichi Sankyo Co., Ltd. Gilteritinib (ASP2215) was
obtained from Selleck Chemicals. Trametinib (cat. no. GSK-1120212)
was obtained from MedChemExpress. Each chemical was dissolved in
DMSO and added to the culture medium at 1–100 nM for in
vitro experiments. Daratumumab for in vitro experiments
was purchased from Janssen Pharmaceutical K.K.
Flow cytometry (AML and CML)
AML cell lines (MOLM-13, MV-4-11, THP-1, NB-4 and
KG-1) and a CML cell line (K562) were exposed to FLT3
inhibitors (quizartinib and gilteritinib) or MEK inhibitor
(trametinib) at 37°C for 48 h. The cells were subsequently
harvested, and changes in CD155 and CD112 expression were analyzed.
AML cell lines (MOLM-13, MV-4-11, THP-1, NB-4 and KG-1) and a CML
cell line (K562) were stained with phycoerythrin (PE) anti-human
CD155 (cat. no. 337508; BioLegend, Inc.), PE anti-human CD112 (cat.
no. 337410; BioLegend, Inc.) and PE anti-human CD38 (cat. no.
356604; BioLegend, Inc.) antibodies. Changes in marker expression
were assessed by comparing the mean fluorescence intensity of CD155
and CD112 between inhibitor-exposed and DMSO-exposed cell lines.
Three independent experiments were performed. PE and FITC were
detected by blue laser (488 nm). Flow cytometric analysis was
performed using a BD FACSCelesta Flow Cytometer (BD Biosciences).
Data were analyzed using FlowJo software (ver. 10.8.0, FlowJo
LLC.).
Flow cytometry (γδ T cells)
γδ T cells were stained with FITC anti-human CD3
(cat. no. 300405; BioLegend, Inc.), allophycocyanin (APC)
anti-human TCR Vd2 (cat. no. 331417; BioLegend, Inc.) and PE
anti-human TIGIT (cat. no. 372704; BioLegend, Inc.) antibodies. PE
and FITC were detected by blue laser (488 nm). APC was detected by
red laser (638 nm). Flow cytometric analysis was performed using a
BD FACSCelesta Flow Cytometer (BD Biosciences). Data were analyzed
using FlowJo software (ver. 10.8.0; FlowJo LLC.).
Flow cytometry (KHYG-1 cells)
KHYG-1 cells were stained with PE anti-human TIGIT
antibodies (cat. no. 372704; BioLegend, Inc.). PE was detected by
blue laser (488 nm). Flow cytometric analysis was performed using a
BD FACSCelesta Flow Cytometer (BD Biosciences). Data were analyzed
using FlowJo software (ver. 10.8.0, FlowJo LLC.).
Reverse transcription-quantitative PCR
(RT-qPCR)
All AML cell lines were exposed to FLT3
inhibitors (quizartinib and gilteritinib) at 10 nM and DMSO at 37°C
for 0, 6, 12 or 24 h. After treatment, total RNA was extracted from
AML cells using RNeasy Mini Kit (cat. no. 74104; Qiagen GmbH). cDNA
was synthesized from total RNA using the SuperScript III
First-Strand Synthesis System (cat. no. 18080051; Thermo Fisher
Scientific, Inc.). Using the SuperScript III First-Strand Synthesis
System, RT was performed at 25°C for 10 min, 50°C for 50 min, and
85°C for 5 min. qPCR thermocycling conditions were as follows:
Initial denaturation at 95°C for 10 min, followed by 39 cycles of
95°C for 15 sec and 60°C for1 min. Using CFX Manager Software (ver.
2.1; Bio-Rad Laboratories, Inc.), the target gene was quantified by
the 2−ΔΔCq method (19). All RT-qPCR assays were performed in
triplicate. 18S ribosomal RNA was used as an internal control,
confirming that its expression was consistent in tumor cell lines
and was not affected by FLT3 inhibitors. CD155 and CD112
expression in AML cell lines was normalized to that of 18S
ribosomal RNA. The expression levels of CD155 and CD112 after each
0, 6, 12, and 24 h exposure to DMSO or FLT3 inhibitor were
divided by the expression levels of CD155 or CD112 after 0 h of
DMSO administration and expressed as a ratio. RT-qPCR experiments
were performed using TaqMan Universal Master Mix II, no UNG (Thermo
Fisher Scientific, Inc.) and the CFX Connect Real-Time PCR
Detection System (Bio-Rad Laboratories, Inc.). The following
primers were used: TaqMan Gene Expression Assays (Thermo Fisher
Scientific, Inc.) for CD155 (Hs.00197846), CD112 (Hs.01071562) and
18S (Hs.99999901). Details of the primer sequences were not
provided by the company.
Antibody-dependent cellular
cytotoxicity (ADCC) and NK cell direct cytotoxicity assays
Luciferase-expressing AML cell lines were exposed to
FLT3 inhibitor (10 nM), MEK inhibitor (100 nM) or DMSO at
37°C for 24 h. For the ADCC assay, AML cells were treated with 0.1
or 10 µg/ml daratumumab or control (IgG) and cocultured with NK
cells at a ratio of 10:1 at 37°C for 4 h. For the direct
cytotoxicity assay, AML cells were coincubated with NK cells at an
effector/tumor ratio of 0–30:1 at 37°C for 72 h. Cell death was
calculated from the decrease in luciferase activity, which was
detected using Steady Glo (Promega Corporation). Luciferase
luminescence in the samples was evaluated using a Nivo
spectrophotometer (PerkinElmer, Inc.). Both assays were repeated at
least thrice.
Cancer genome atlas program
analysis
Data from The Cancer Genome Atlas (TCGA) program
[https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga,
(20)] were analyzed using
cBioPortal (ver. 3.7.16) for Cancer Genomics (https://www.cbioportal.org/). The mRNA expression of
TCGA data was set to a high value with a threshold Z-score of 1.8.
The overall survival (OS) curve by Kaplan-Meier method was analyzed
using the log-rank test by cBioPortal.
Statistical analysis
All data are presented as the mean ± SD. Differences
between the two groups of samples were analyzed by a one-tailed
unpaired t-test using GraphPad Prism version 9.1.2 (GraphPad
Software, Inc.). All experiments were repeated ≥3 times. P<0.05
was considered to indicate a statistically significant difference.
The statistical significance of differences in the responses due to
dose variation was evaluated by P-values for the interaction term
of dose and treatment in a multiple linear regression model (SPSS
25; IBM Corp.).
Results
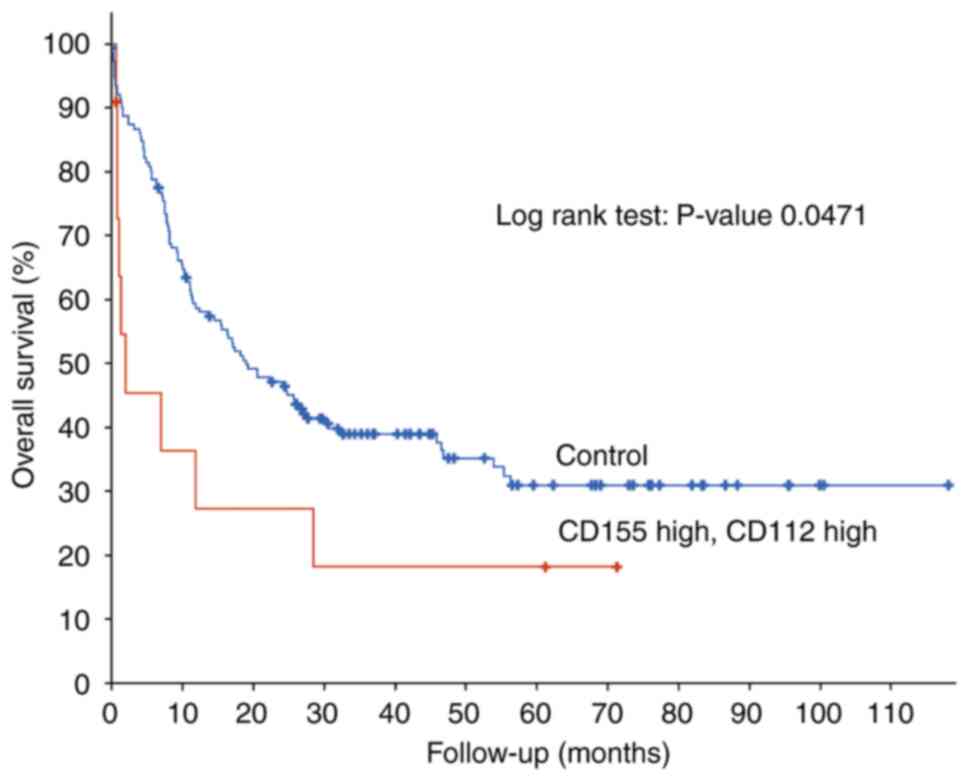
High mRNA expression of CD155/CD112 is
a prognostic marker of poor OS in patients with AML
Analysis of gene expression profiles from patients
with newly diagnosed AML deposited in TCGA revealed that high
CD155/CD112 mRNA expression was significantly associated with poor
OS [n=162 (CD155/CD112 high group, n=11; control group, n=151);
P=0.0471; Fig. 1]. Therefore,
expression of CD155 and CD112, novel immune checkpoints, may be
novel biomarkers for poor OS in patients with AML.
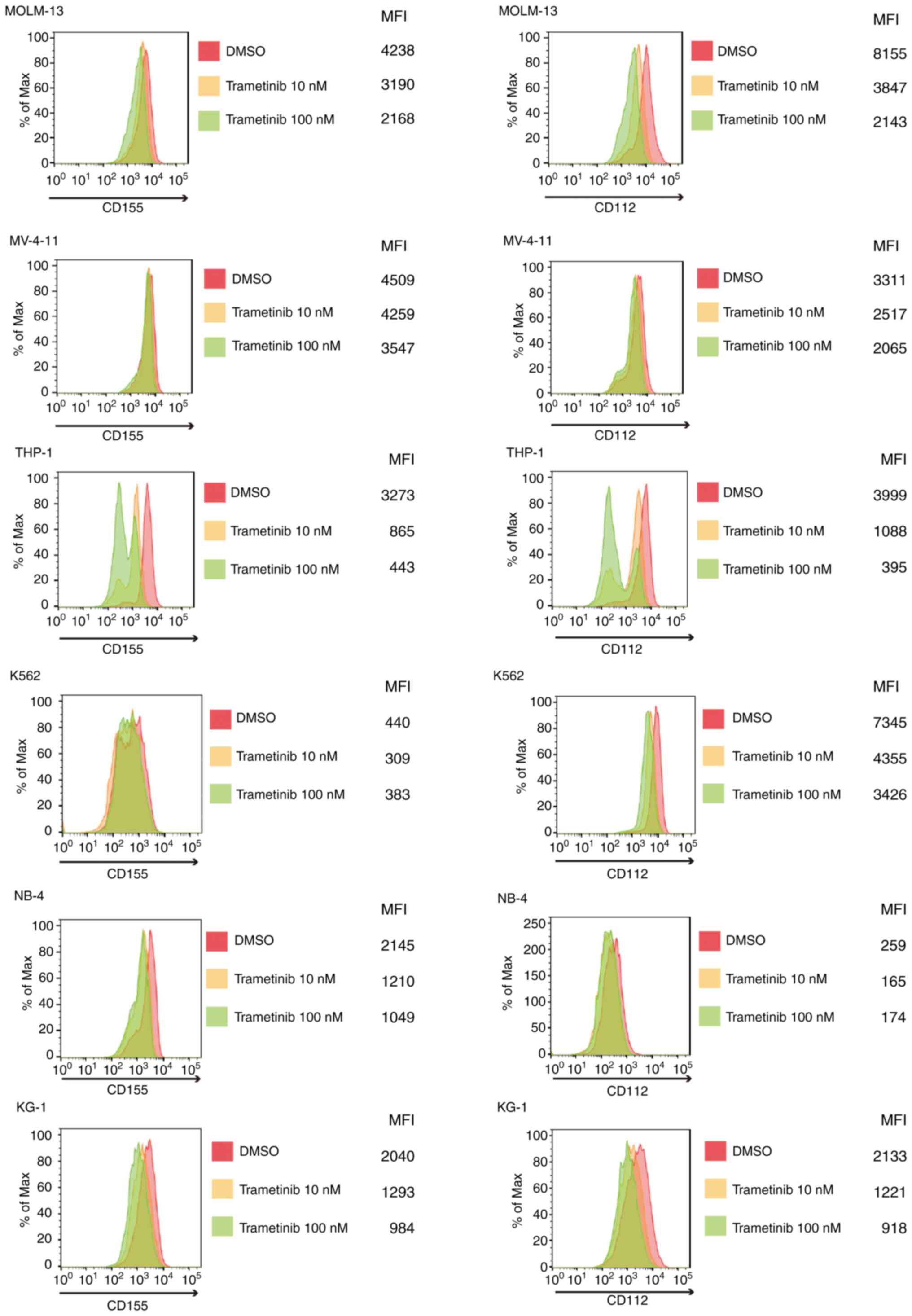
CD155/CD112 expression is
downregulated by trametinib in AML cells
The present study subsequently investigated how
CD155/CD112 expression in AML cells can be manipulated. CD155
expression is induced by fibroblast growth factor via the
Raf-MEK-ERK signaling pathway in NIH3T3 cells (21). It was hypothesized that CD155
expression could be suppressed by Raf-MEK-ERK signaling pathway
inhibition in AML cells. The change in CD155 surface expression was
investigated following treatment with trametinib, an MEK inhibitor,
in AML cells with and without FLT3 mutation (22) and was shown to be suppressed by
trametinib in all of the examined cell lines. In addition, CD112
surface expression was downregulated by trametinib in AML cells
with or without FLT3 mutations (Fig. 2). However, no drug
concentration-dependent changes were observed for CD155 expression
in K562 cells or CD112 expression in NB4 cells. These results may
be due to low expression of CD155 or CD112 at baseline.
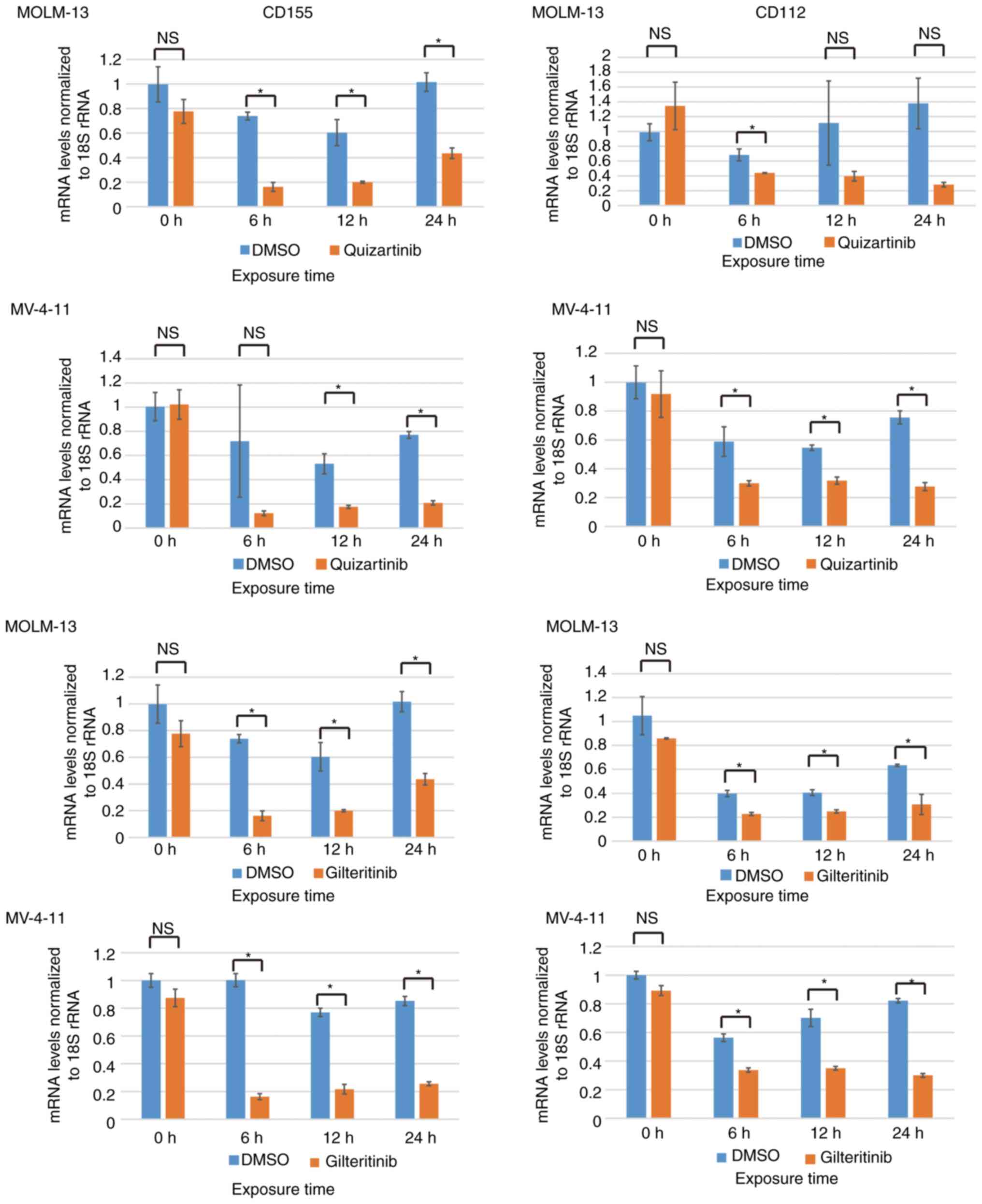
CD155 and CD112 mRNA expression is
downregulated by FLT3 inhibitors in AML cells with FLT3
mutations
The Raf/MEK/ERK signaling pathway can be activated
by mutations/amplifications of FLT3 kinase, and it is
considered to be located downstream of the FLT3 signaling
pathway (9). CD155 and CD112
downregulation by trametinib suggested the possibility of their
downregulation by FLT3 inhibitors in AML cells containing
FLT3-ITD mutations. The present study investigated the
changes in CD155/CD112 mRNA expression after treatment of MOLM-13
and MV-4-11 cells with FLT3 inhibitors. Quizartinib is a
type II FLT3 inhibitor, which targets only mutated
FLT3 with an inactive conformation, whereas gilteritinib is
a type I FLT3 inhibitor targeting mutated FLT3 with
both active and inactive conformations (10). Quizartinib decreased CD155 mRNA
expression in MOLM-13 (6, 12 and 24 h) and MV-4-11 cells (12 and 24
h). Quizartinib decreased CD112 mRNA expression in MOLM-13 (6 h)
and MV-4-11 cells (6, 12 and 24 h). Gilteritinib decreased CD155
mRNA expression in MOLM-13 (6, 12 and 24 h) and MV-4-11 cells (6,
12 and 24 h). Gilteritinib decreased CD112 mRNA expression in
MOLM-13 (6, 12 and 24 h) and MV-4-11 cells (6, 12 and 24 h;
Fig. 3).
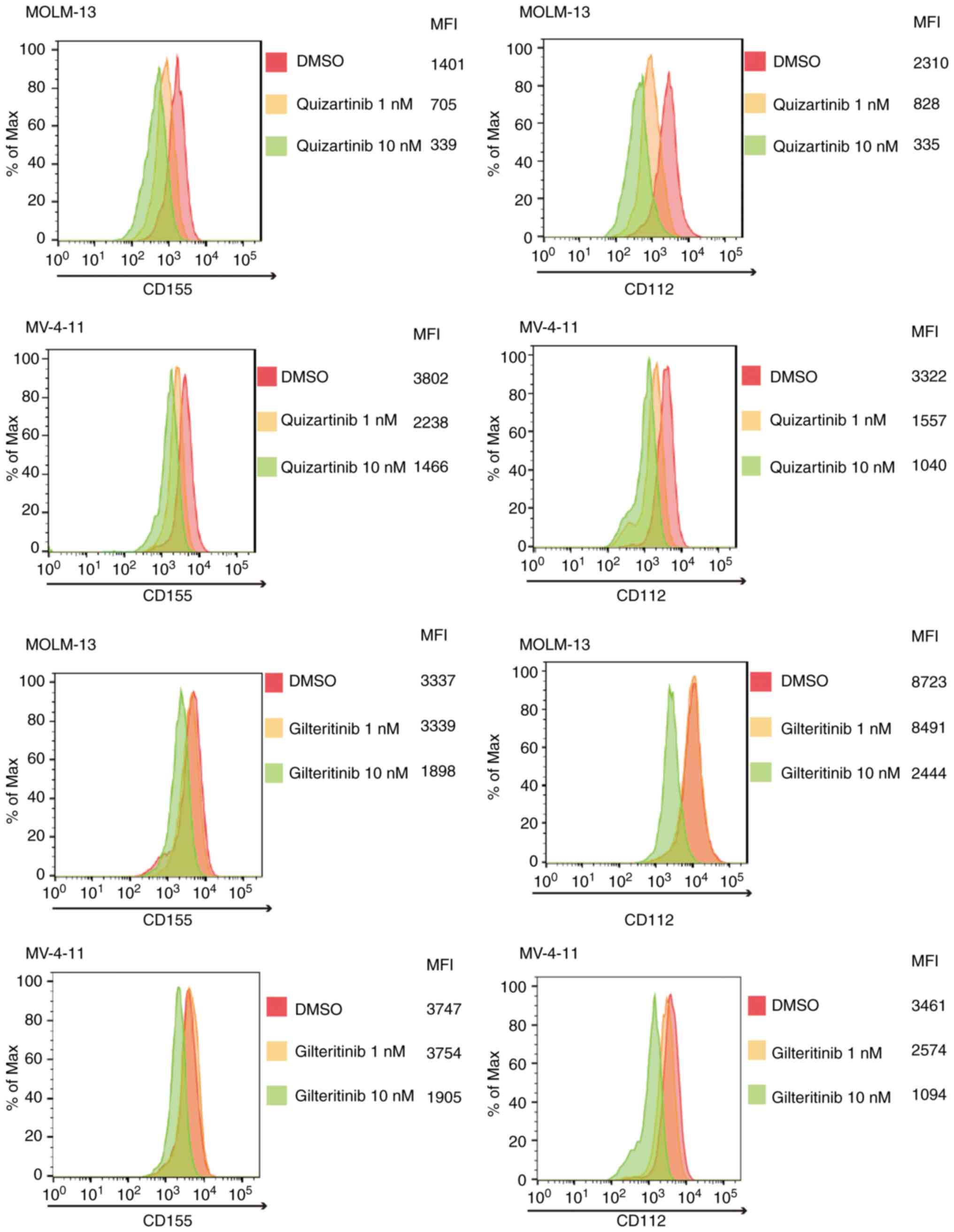
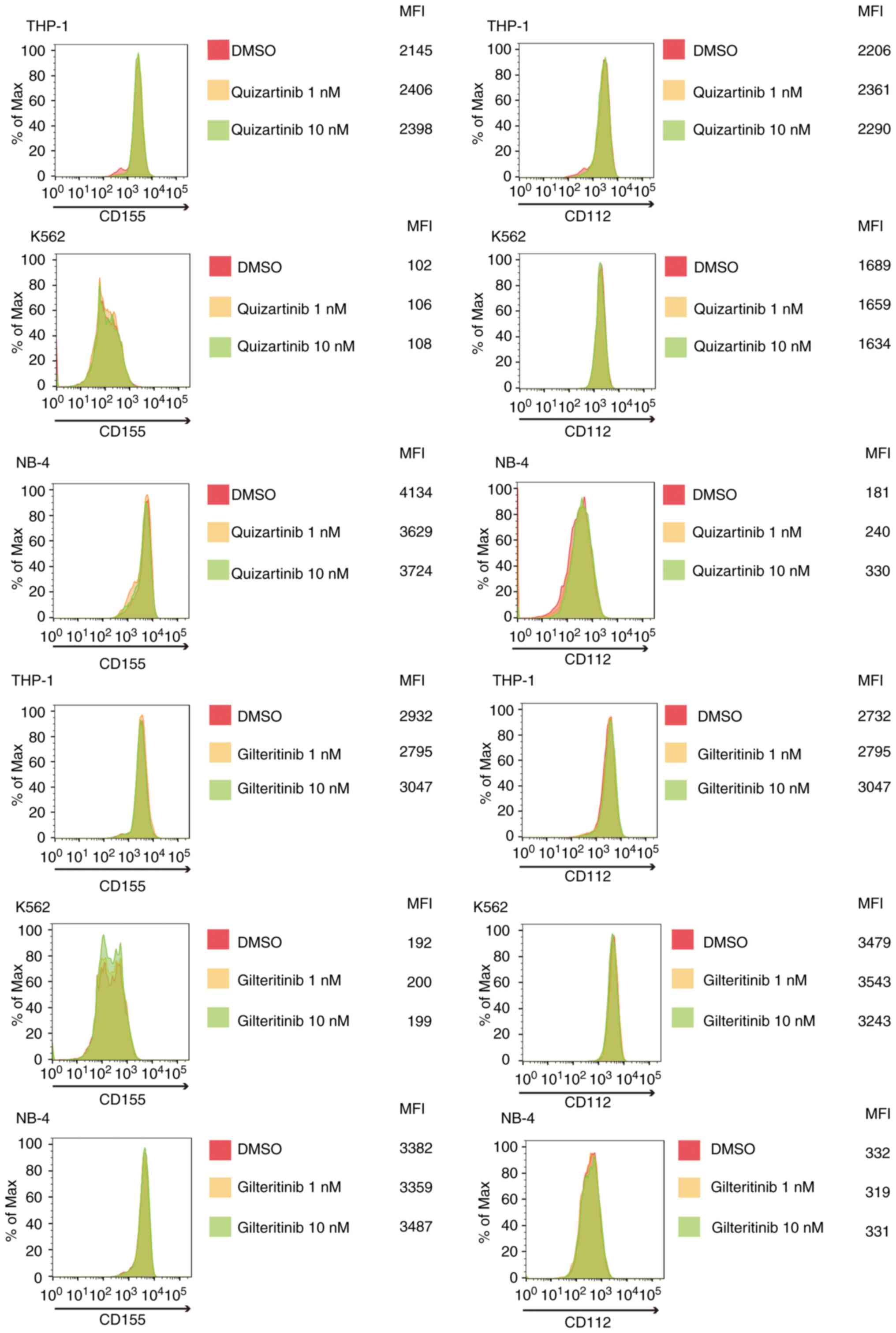
FLT3 mutation-specific downregulation
of CD155 and CD112 surface expression in AML cells by FLT3
inhibitors
As shown in Fig. 4,
quizartinib and gilteritinib downregulated CD155 and CD112 surface
expression in both MOLM-13 and MV-4-11 cells. By contrast, the
treatment of AML cells without FLT3 mutations, including
THP-1, K562 and NB-4 cells, with quizartinib or gilteritinib did
not affect the surface expression of CD155 and CD112 (Fig. 5). These results suggested that
downregulation of CD155 and CD112 surface expression in AML cells
by FLT3 inhibitors is dependent on the presence of
FLT3 mutations.
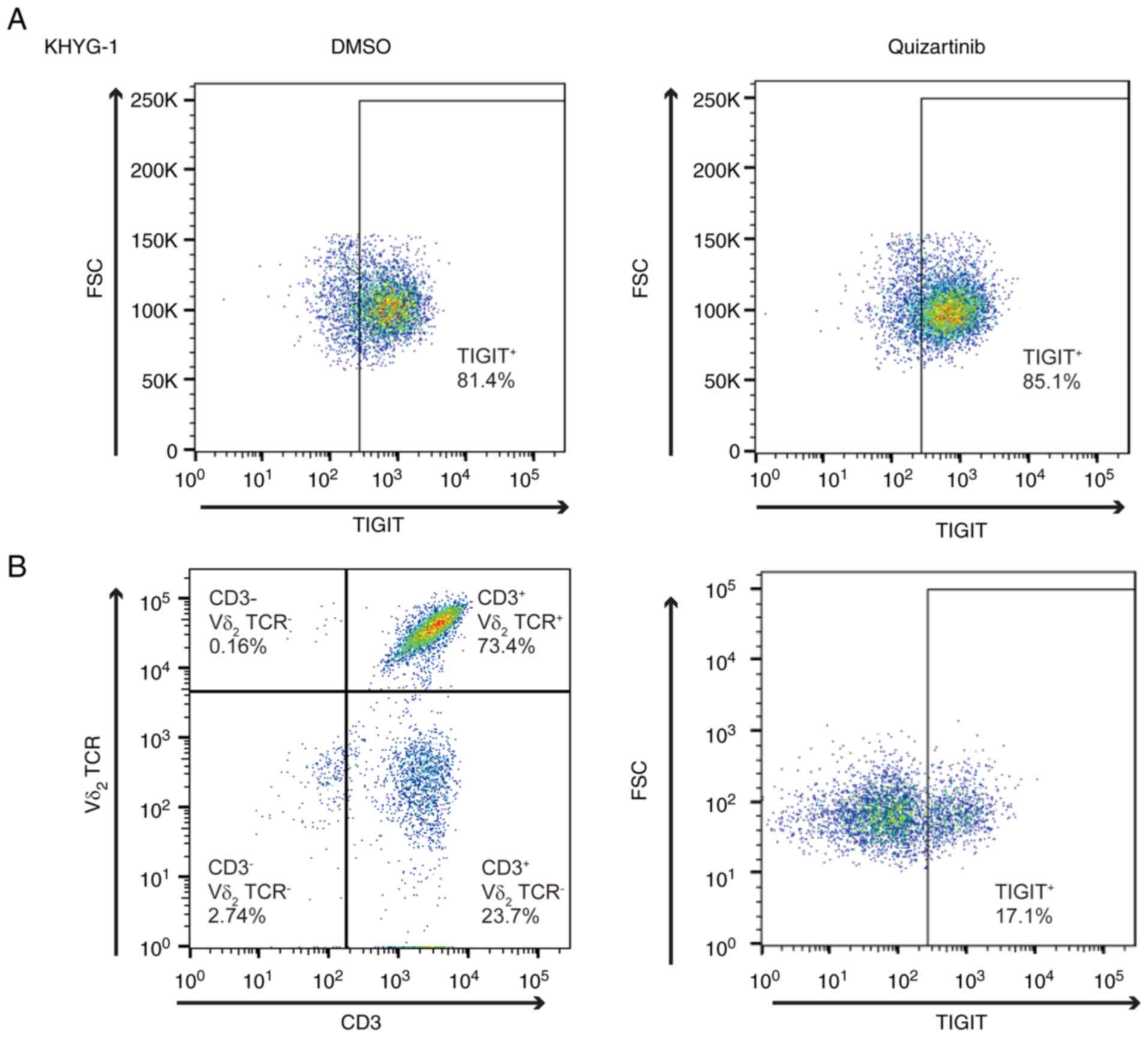
TIGIT expression in NK and γδ T
cells
The present study subsequently examined how CD155
and CD112 downregulation in AML cells under FLT3 inhibition
affects the cytotoxic effects of effector cells, including NK and T
cells. Poor OS in patients with AML with high CD155 and CD112
expression in leukemic cells indicates that the interaction between
TIGIT and CD155/CD112 blocks the cytotoxicity of effector cells. To
test our hypothesis, TIGIT expression in NK cells was analyzed.
KHYG-1 is a cell line derived from human primary NK cells (23), and flow cytometric analysis
revealed that 81.4% of the cells were positive for TIGIT (Fig. 6A). Furthermore, TIGIT expression in
KHYG-1 cells was unaffected by FLT3 inhibition by flow
cytometry (85.1%). γδ T cells are CD3+ Vd2+
and highly cytotoxic, and the activation of γδ T cells does not
necessarily depend on major histocompatibility complex antigen
presentation, which makes γδ T cells a promising target for
allogeneic cell transfer therapy (16,24).
Flow cytometric analysis of γδ T cells revealed that TIGIT
expression was restricted in 17.1% of the analyzed cells (Fig. 6B).
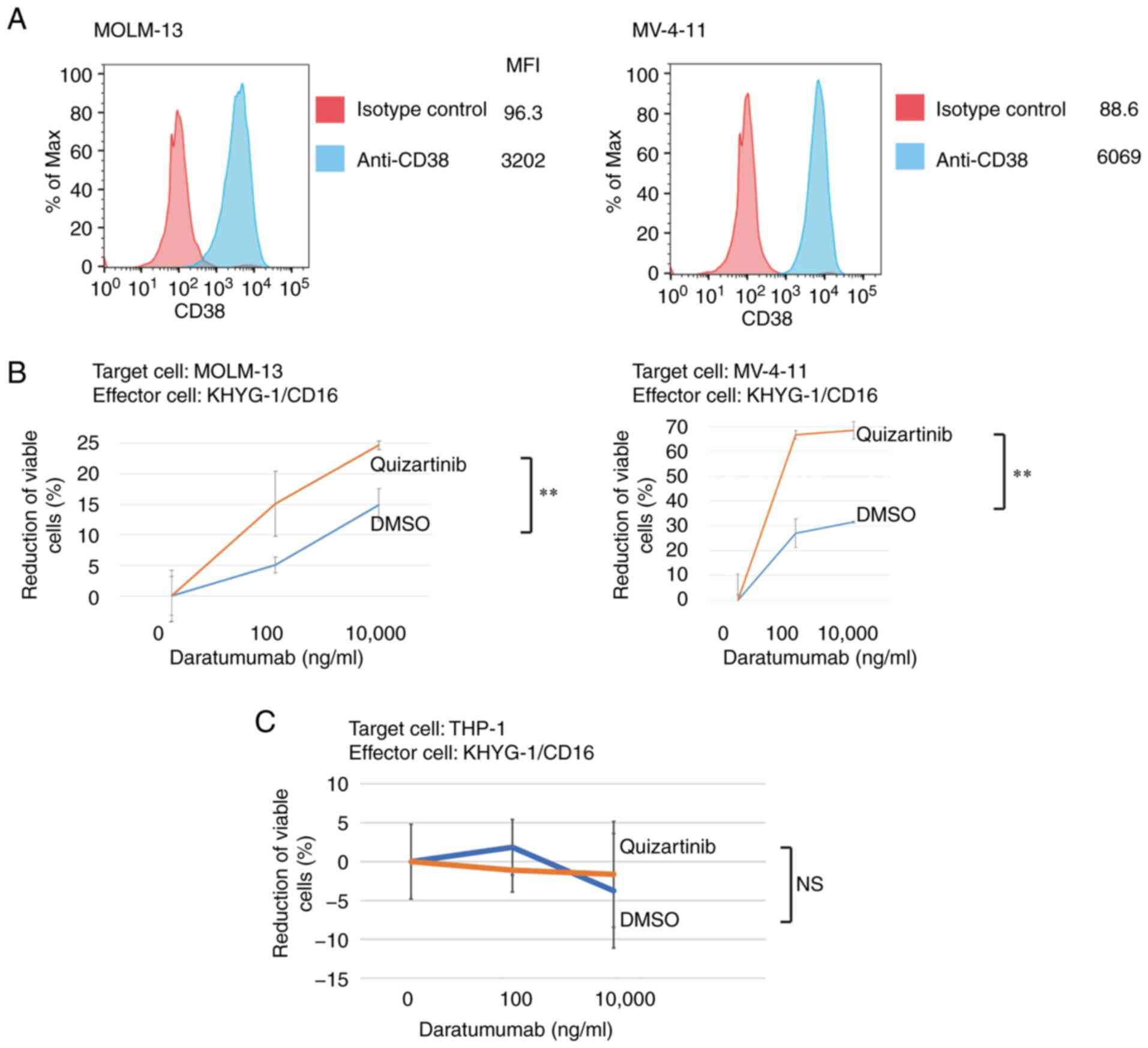
Enhanced ADCC activity of daratumumab
following FLT3 inhibitor treatment in AML cells with FLT3-ITD
mutations
As shown in Fig.
6A, 81.4% of the analyzed KHYG-1 cells were positive for TIGIT.
These results suggested the possibility that cytotoxicity of KYHG-1
cells against AML cells with FLT3-ITD mutations treated with
FLT3 inhibitors could be affected by the reduction of
CD155/CD112. Daratumumab is a therapeutic antibody for multiple
myeloma with ADCC activity that targets CD38 expressed in myeloma
cells (25). The present study
demonstrated CD38 expression in MV-4-11 and MOLM-13 cells (Fig. 7A) and analyzed the ADCC activity of
daratumumab in these cells with or without treatment with
quizartinib. In both MV-4-11 and MOLM-13 cells, the ADCC activity
of daratumumab was enhanced by FLT3 inhibition (Fig. 7B). Conversely, the ADCC activity of
daratumumab was unaffected in THP-1 cells without FLT3
mutations under FLT3 inhibition (Fig. 7C). These results suggested that the
suppression of anti-leukemic activity of NK cells is mediated by
the interaction between CD155/CD112 and TIGIT, which is
de-repressed by FLT3 inhibition in AML cells with
FLT3 mutations.
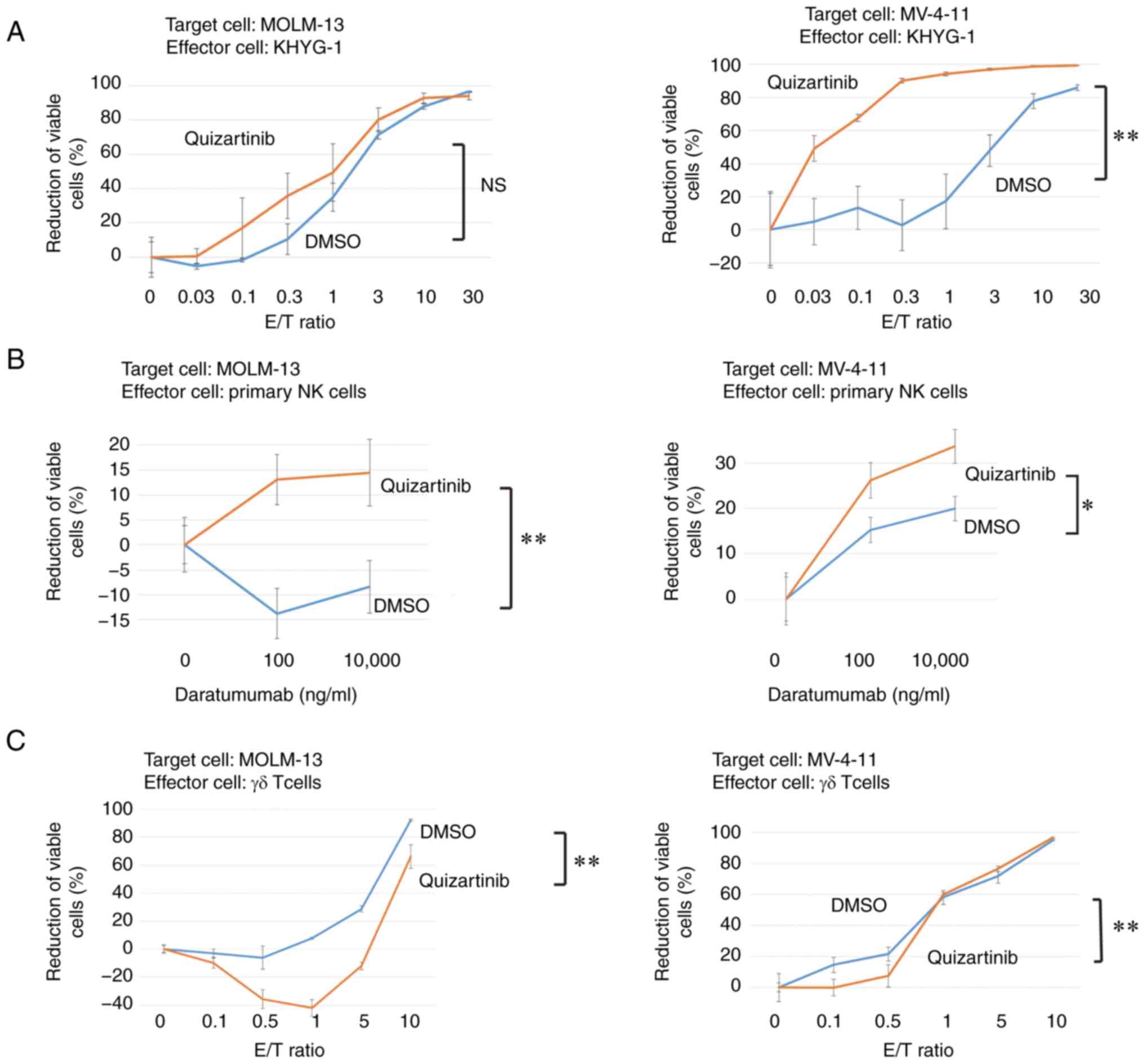
Enhancement of direct activity of NK
cells and ADCC activity of primary NK cells against AML cells with
FLT3-ITD mutations under FLT3 inhibition
The direct cytotoxicity of KHYG-1 cells against
MOLM-13 and MV-4-11 cells under FLT3 inhibition was
investigated. Direct activity was confirmed in both cell lines. In
MOLM-13 cells, direct activity was enhanced, but this was not
significant. In MV-4-11 cells, direct activity was significantly
enhanced under FLT3 inhibition (Fig. 8A). Primary NK cells were purified
from peripheral blood and the ADCC activity of primary NK cells
against AML cells with FLT3 mutations under FLT3
inhibition was estimated. The ADCC of primary NK cells against
MOLM-13 and MV-4-11 cells was also enhanced by FLT3
inhibition (Fig. 8B).
γδ T cell cytotoxicity against AML
cells with FLT3 mutations is not enhanced by FLT3 inhibition
As shown in Fig.
6B, TIGIT-positive rate (17.1%) of γδ T cells was lower than
that of KHYG-1 cells (81.4%). The cytotoxicity of γδ T cells
against AML cells with FLT3-ITD mutations was assessed. The
cytotoxicity was significantly diminished by FLT3 inhibition
(Fig. 8C).
Discussion
The present study suggested that CD155 and CD112
expression in AML cells with FLT3-ITD mutations was
decreased, and cytotoxicity against these cells by NK cells was
enhanced following FLT3 inhibition. The possibility of CD155
and CD112 as biomarkers of poor OS in patients with AML was also
suggested.
CD155 and CD112 are immune checkpoints, and the
involvement of increased expression of immune checkpoints in poor
prognosis of AML has been reported (26). B7-H2 positivity in leukemic cells
has a strong prognostic value for shorter survival (27). Higher expression of the checkpoint
molecules cytotoxic T-lymphocyte associated protein 4, PD-1 and
lymphocyte activating 3 in leukemic cells compared with patients
with normal prognosis is also a marker of poor prognosis (28). As for the association between
prognosis and specific mutations found in AML, the expression of
PD-1 ligand PD-L1 is increased in hematopoietic stem cells of
patients with TP53 mutations (29). A recent study analyzed the
expression of immune checkpoints in NK cells from patients with AML
and found that PD-1, TIGIT and T cell immunoglobulin and
mucin-domain containing-3 expression are increased in these cells
compared with those from healthy donors (30). Furthermore, a high frequency of
TIGIT+NK cells is associated with a poor prognosis in
patients with AML (30). To the
best of our knowledge, the present study was the first to report
the expression of TIGIT ligands as a marker of poor prognosis in
AML as well as a target of FLT3 inhibition.
Although therapies using FLT3 inhibitors have
improved the historically poor outcomes of FLT3-mutated AML,
the clinical response to an FLT3 inhibitor is temporary in
most cases of relapse/refractory disease (31). Resistance to FLT3 inhibitors
is derived from a number of mechanisms, including inherent
mutations insensitive to FLT3 inhibitors, acquisition of
mutations in the ATP-binding pocket and activation of alternative
survival pathways (32).
Enhancement of allogeneic immunity has received attention as a
strategy to overcome resistance. Increased activity of sorafenib,
an FLT3 inhibitor, which synergizes with allogeneic SCT in
FLT3-ITD-positive AML suggests the possibility of alloimmune
effects to overcome the resistance to FLT3 inhibitors
(33). Furthermore, it has been
reported that promotion of graft-vs.-leukemia activity by sorafenib
through IL-15 production in FLT3-ITD mutant leukemia has the
potential to cure FLT3-ITD AML (34).
In addition to allogeneic immunity, the inhibition
of immune evasion mediated by immune checkpoints is expected to
have a cytotoxic effect (35). The
response of leukemic cells to T cell activation leads to the
downregulation of T cell costimulatory ligand B7-H2 along with the
upregulation of coinhibitory ligand PD-L1 to shut down T cell
activation (36). It was
hypothesized that this immune phenotypic switch would cause immune
evasion by AML cells.
The association of DNAM-1/TIGIT, another immune
checkpoint, with immune evasion has been mainly reported in T cells
(37). In addition to the presence
of high frequencies of CD8+ T cells expressing TIGIT in
patients with FLT3-ITD mutations, a decrease in
CD226+ γδ T cells and an increase in TIGIT+
γδ T cells in patients with de novo AML have been reported
(38). It has also been reported
that TIGIT−DNAM-1+ γδ T cells are restored in
patients with AML who achieve complete remission after
chemotherapy. Furthermore, the high expression of
TIGIT+DNAM-1− in γδ T cells may be a
biomarker of poor OS (38).
As for the anti-leukemic effect of NK cells, relapse
prevention by killer cell immunoglobulin-like receptor
(KIR)-mismatched NK cells in allogeneic SCT (KIR mismatched) has
been reported (1). However, to the
best of our knowledge, the role of immune checkpoints in NK cells
has not been previously described in patients with AML. The results
of the present study indicated the possible usefulness of blocking
the interaction between CD155/CD112 and TIGIT for NK cell therapy
and the inhibitory role of TIGIT in the anti-leukemic effect of NK
cells.
The lower effect of CD155/CD112 downregulation on
the anti-leukemic effect of γδ T cells compared with NK cells was
considered to be due to low TIGIT expression in γδ T cells. As
previously described, TIGIT+ γδ T cells are increased in
patients with de novo AML, and there is a possibility that
CD155 and CD112 downregulation by FLT3 inhibitors in
FLT3-mutated AML cells may stimulate the cytotoxicity of γδ
T cells in these patients (38).
In conclusion, the present study revealed that CD155
and CD112 downregulation in AML cells by FLT3 inhibitors is
dependent on FLT3 mutations. Although the treatment of AML
cells with trametinib also reduced CD155 and CD112 expression, the
downregulation was independent of FLT3 mutations. In
addition, the cytotoxic effect of FLT3 inhibitors was
restricted to AML cells with FLT3 mutations. Therefore,
blocking immune evasion through CD155 and CD112 downregulation by
FLT3 inhibitors is expected to lead to therapeutic effects
with fewer side effects compared with the use of trametinib in
patients with FLT3 mutations. The present finding that
FLT3 inhibitors displayed cytotoxic effects as well as
enhanced NK cell activity suggested the possible usefulness of
FLT3 inhibitors in combination with adaptive NK cell therapy
as a novel strategy for immunotherapy of AML.
Acknowledgements
Quizartinib was provided by Daiichi Sankyo Co.,
Ltd., Tokyo, Japan.
Funding
The present study was funded by Japan Society for the Promotion
of Science Kagaku Kenkyu hi (grant no. 20K08726).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YK, MN and YI performed the research, analyzed and
interpreted the data, and wrote the manuscript. YI designed the
research and edited the manuscript. MH, MF, HT and AT analyzed and
interpreted the data and revised the manuscript. YI supervised the
projects. YK and YI confirm the authenticity of all the raw data.
All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board at the Institute of Medical Science of the University
of Tokyo (approval no. 30–72-A0222; Tokyo, Japan). Primary cells
derived from the patients who signed informed consent form were
used.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ruggeri L, Capanni M, Urbani E, Perruccio
K, Shlomchik WD, Tosti A, Posati S, Rogaia D, Frassoni F, Aversa F,
et al: Effectiveness of donor natural killer cell alloreactivity in
mismatched hematopoietic transplants. Science. 295:2097–2100. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Paczulla AM, Rothfelder K, Raffel S,
Konantz M, Steinbacher J, Wang H, Tandler C, Mbarga M, Schaefer T,
Falcone M, et al: Absence of NKG2D ligands defines leukaemia stem
cells and mediates their immune evasion. Nature. 572:254–259. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sanchez-Correa B, Valhondo I, Hassouneh F,
Lopez-Sejas N, Pera A, Bergua JM, Arcos MJ, Bañas H, Casas-Avilés
I, Durán E, et al: DNAM-1 and the TIGIT/PVRIG/TACTILE Axis: Novel
immune checkpoints for natural killer cell-based cancer
immunotherapy. Cancers (Basel). 11:8772019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Long EO, Kim HS, Liu D, Peterson ME and
Rajagopalan S: Controlling natural killer cell responses:
Integration of signals for activation and inhibition. Annu Rev
Immunol. 31:227–258. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pende D, Spaggiari GM, Marcenaro S,
Martini S, Rivera P, Capobianco A, Falco M, Lanino E, Pierri I,
Zambello R, et al: Analysis of the receptor-ligand interactions in
the natural killer-mediated lysis of freshly isolated myeloid or
lymphoblastic leukemias: Evidence for the involvement of the
poliovirus receptor (CD155) and nectin-2 (CD112). Blood.
105:2066–2073. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tahara-Hanaoka S, Shibuya K, Onoda Y,
Zhang H, Yamazaki S, Miyamoto A, Honda S, Lanier LL and Shibuya A:
Functional characterization of DNAM-1 (CD226) interaction with its
ligands PVR (CD155) and nectin-2 (PRR-2/CD112). Int Immunol.
16:533–538. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stanietsky N, Simic H, Arapovic J, Toporik
A, Levy O, Novik A, Levine Z, Beiman M, Dassa L, Achdout H, et al:
The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell
cytotoxicity. Proc Natl Acad Sci USA. 106:17858–17863. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gilliland DG and Griffin JD: The roles of
FLT3 in hematopoiesis and leukemia. Blood. 100:1532–1542. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Steelman LS, Abrams SL, Whelan J, Bertrand
FE, Ludwig DE, Bäsecke J, Libra M, Stivala F, Milella M, Tafuri A,
et al: Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and
Jak/STAT pathways to leukemia. Leukemia. 22:686–707. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Daver N, Schlenk RF, Russell NH and Levis
MJ: Targeting FLT3 mutations in AML: Review of current knowledge
and evidence. Leukemia. 33:299–312. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fischer T, Stone RM, Deangelo DJ, Galinsky
I, Estey E, Lanza C, Fox E, Ehninger G, Feldman EJ, Schiller GJ, et
al: Phase IIB trial of oral Midostaurin (PKC412), the FMS-like
tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase
inhibitor, in patients with acute myeloid leukemia and high-risk
myelodysplastic syndrome with either wild-type or mutated FLT3. J
Clin Oncol. 28:4339–4345. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wander SA, Levis MJ and Fathi AT: The
evolving role of FLT3 inhibitors in acute myeloid leukemia:
Quizartinib and beyond. Ther Adv Hematol. 5:65–77. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Perl AE, Martinelli G, Cortes JE, Neubauer
A, Berman E, Paolini S, Montesinos P, Baer MR, Larson RA, Ustun C,
et al: Gilteritinib or chemotherapy for relapsed or refractory
FLT3-mutated AML. N Engl J Med. 381:1728–1740. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cortes JE, Khaled S, Martinelli G, Perl
AE, Ganguly S, Russell N, Krämer A, Dombret H, Hogge D, Jonas BA,
et al: Quizartinib versus salvage chemotherapy in relapsed or
refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): A
multicentre, randomised, controlled, open-label, phase 3 trial.
Lancet Oncol. 20:984–997. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang M, Bu J, Zhou M, Sido J, Lin Y, Liu
G, Lin Q, Xu X, Leavenworth JW and Shen E: CD8+T cells
expressing both PD-1 and TIGIT but not CD226 are dysfunctional in
acute myeloid leukemia (AML) patients. Clin Immunol. 190:64–73.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cui Q, Shibata H, Oda A, Amou H, Nakano A,
Yata K, Hiasa M, Watanabe K, Nakamura S, Miki H, et al: Targeting
myeloma-osteoclast interaction with Vγ9Vδ2 T cells. Int J Hematol.
94:63–70. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Futami M, Suzuki K, Kato S, Ohmae S,
Tahara Y, Nojima M, Imai Y, Mimura T, Watanabe Y and Tojo A: The
novel multi-cytokine inhibitor TO-207 specifically inhibits
pro-inflammatory cytokine secretion in monocytes without affecting
the killing ability of CAR T cells. PLoS One. 15:e02318962020.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bai Y, Soda Y, Izawa K, Tanabe T, Kang X,
Tojo A, Hoshino H, Miyoshi H, Asano S and Tani K: Effective
transduction and stable transgene expression in human blood cells
by a third-generation lentiviral vector. Gene Ther. 10:1446–1457.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cancer Genome Atlas Research Network, .
Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A,
Hoadley K, Triche TJ Jr, Laird PW, et al: Genomic and epigenomic
landscapes of adult de novo acute myeloid leukemia. N Engl J Med.
368:2059–2074. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hirota T, Irie K, Okamoto R, Ikeda W and
Takai Y: Transcriptional activation of the mouse
Necl-5/Tage4/PVR/CD155 gene by fibroblast growth factor or
oncogenic Ras through the Raf-MEK-ERK-AP-1 pathway. Oncogene.
24:2229–2235. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Quentmeier H, Reinhardt J, Zaborski M and
Drexler HG: FLT3 mutations in acute myeloid leukemia cell lines.
Leukemia. 17:120–124. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yagita M, Huang CL, Umehara H, Matsuo Y,
Tabata R, Miyake M, Konaka Y and Takatsuki K: A novel natural
killer cell line (KHYG-1) from a patient with aggressive natural
killer cell leukemia carrying a p53 point mutation. Leukemia.
14:922–930. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Deseke M and Prinz I: Ligand recognition
by the γδ TCR and discrimination between homeostasis and stress
conditions. Cell Mol Immunol. 17:914–924. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hirano M, Imai Y, Kaito Y, Murayama T,
Sato K, Ishida T, Yamamoto J, Ito T, Futami M, Ri M, et al:
Small-molecule HDAC and Akt inhibitors suppress tumor growth and
enhance immunotherapy in multiple myeloma. J Exp Clin Cancer Res.
40:1102021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stamm H, Klingler F, Grossjohann EM,
Muschhammer J, Vettorazzi E, Heuser M, Mock U, Thol F, Vohwinkel G,
Latuske E, et al: Immune checkpoints PVR and PVRL2 are prognostic
markers in AML and their blockade represents a new therapeutic
option. Oncogene. 37:5269–5280. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tamura H, Dan K, Tamada K, Nakamura K,
Shioi Y, Hyodo H, Wang SD, Dong H, Chen L and Ogata K: Expression
of functional B7-H2 and B7.2 costimulatory molecules and their
prognostic implications in de novo acute myeloid leukemia. Clin
Cancer Res. 11:5708–5717. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dao FT, Wang J, Yang L and Qin YZ:
Development of a poor-prognostic-mutations derived immune
prognostic model for acute myeloid leukemia. Sci Rep. 11:48562021.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sallman DA, McLemore AF, Aldrich AL,
Komrokji RS, McGraw KL, Dhawan A, Geyer S, Hou HA, Eksioglu EA,
Sullivan A, et al: TP53 mutations in myelodysplastic syndromes and
secondary AML confer an immunosuppressive phenotype. Blood.
136:2812–2823. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu G, Zhang Q, Yang J, Li X, Xian L, Li
W, Lin T, Cheng J, Lin Q, Xu X, et al: Increased TIGIT expressing
NK cells with dysfunctional phenotype in AML patients correlated
with poor prognosis. Cancer Immunol Immunother. Jun 15–2021.(Epub
ahead of print).
|
|
31
|
Antar AI, Otrock ZK, Jabbour E, Mohty M
and Bazarbachi A: FLT3 inhibitors in acute myeloid leukemia: Ten
frequently asked questions. Leukemia. 34:682–696. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chu SH and Small D: Mechanisms of
resistance to FLT3 inhibitors. Drug Resist Updat. 12:8–16. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Metzelder SK, Schroeder T, Finck A, Scholl
S, Fey M, Götze K, Linn YC, Kröger M, Reiter A, Salih HR, et al:
High activity of sorafenib in FLT3-ITD-positive acute myeloid
leukemia synergizes with allo-immune effects to induce sustained
responses. Leukemia. 26:2353–2359. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mathew NR, Baumgartner F, Braun L,
O'Sullivan D, Thomas S, Waterhouse M, Müller TA, Hanke K, Taromi S,
Apostolova P, et al: Sorafenib promotes graft-versus-leukemia
activity in mice and humans through IL-15 production in
FLT3-ITD-mutant leukemia cells. Nat Med. 24:282–291. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Darvin P, Toor SM, Sasidharan Nair V and
Elkord E: Immune checkpoint inhibitors: Recent progress and
potential biomarkers. Exp Mol Med. 50:1–11. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yao S and Chen L: Adaptive resistance: A
tumor strategy to evade immune attack. Eur J Immunol. 43:576–579.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Qin S, Xu L, Yi M, Yu S, Wu K and Luo S:
Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol
Cancer. 18:1552019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jin Z, Lan T, Zhao Y, Du J, Chen J, Lai J,
Xu L, Chen S, Zhong X, Wu X and Li Y: Higher
TIGIT+CD226-γδ T cells in patients with acute myeloid
leukemia. Immunol Invest. Aug 20–2020.(Epub ahead of print).
View Article : Google Scholar
|