Introduction
BCLAF1 was originally identified as a binding
protein that interacted with adenoviral Bcl-2 homolog E1B19K, and
served as an inducer of apoptosis and suppressor of transcription
(1). Subsequent studies have shown
that BCLAF1 also played a key role in a wide range of biological
processes, including pre-mRNA splicing and processing (2–6), DNA
damage response (DDR) (3,4,7–11),
lung development (12), viral
infection (13–16), muscle cell proliferation and
differentiation (17–20), autophagy (21,22),
ischemia-reperfusion (I/R) injury (23), and T-cell activation (12,24,25).
BCLAF1 primarily localizes to the dot-like
structures throughout the nucleus and at a lower number in the
cytoplasm (1). The salient
features of the BCLAF1 structure include an arginine-serine (RS)
rich domain, a basic-leucine zipper (bZIP) domain located within
the RS domain, and a MYB DNA-binding domain (Fig. 1A) (1,12).
Studies have shown that proteins containing the RS domain are
usually involved in post-transcriptional events, such as pre-mRNA
splicing and processing (26–28).
In addition, the MYB DNA-binding and bZIP domains have also been
found to be required for the transcriptional regulatory function of
BCLAF1 (29,30). Therefore, these structural features
are necessary for the regulatory function of BCLAF1 at the
transcriptional and post-transcriptional levels. For example,
BCLAF1 was found to be associated with apoptosis, DDR and
tumorigenesis by activating the transcription of its downstream
target genes [such as TP53, Bax, HIF-1α and long
non-coding (lnc) RNA NEAT1] or inhibiting the transcription of its
downstream genes (such as MDM2) (7,9,30–32).
In addition, BCLAF1 also exerted important functions in the cell
cycle, DDR, tumorigenesis and T-cell differentiation by regulating
pre-mRNA splicing and mRNA processing (4,5,30,33,34).
By contrast, BCLAF1 serves as a downstream target, regulated by
numerous molecules at the transcriptional, translational and
post-translational levels, thus influencing several biological
processes. For example, the type III histone deacetylase, Sirt1,
histone methyltransferase SET and MYND domain-containing protein 3
(SMYD3) and NF-κB have all been associated with tumorigenesis by
regulating the transcription of BCLAF1 (8,22,24,35).
Furthermore, micro (mi)RNA-194-5p exerted its anticancer activity
by repressing the translation of BCLAF1 (36). Furthermore, DNA-protein kinase C
(DNA-PKC) was associated with DDR and apoptosis by directly
phosphorylating serine151 and tyrosine150 in
the RS domain of BCLAF1 (Fig.
1B-a) (7). Notably, increasing
evidence has demonstrated that BCLAF1 was associated with
tumorigenesis as either a tumor promotor (11,22,30,31,34,37–42)
or a tumor suppressor (7,21,32,35,43)
in a context-dependent manner. In addition to for tumorigenesis and
viral replication, BCLAF1 has also been associated with cardiac I/R
injury (23). A previous study
showed that BCLAF1 could promote the apoptosis of cardiomyocytes
via activating apoptosis regulatory proteins such as TP53 and BAX,
and then aggravated the cardiac I/R injury (Fig. 1B-k) (23).
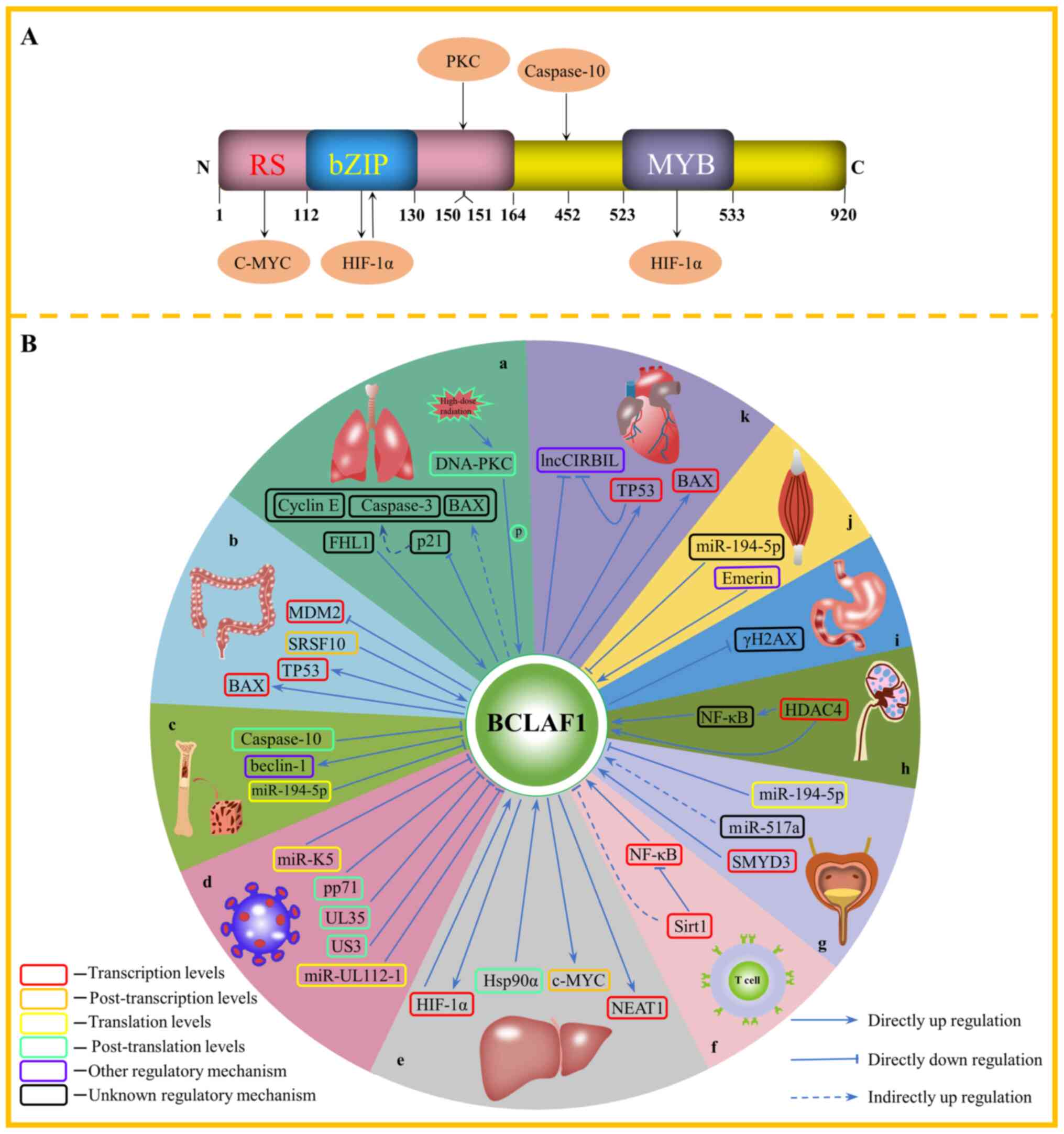 | Figure 1.(A) Structure of the BCLAF1 protein
and the molecules that interact with BCLAF1 and their binding
sites. The structure of BCLAF1 contains multiple domains, including
an N-terminal RS domain, which contains the bZIP domain, and a
C-terminal MYB DNA-binding domain. The molecules that interact with
BCLAF1 include c-Myc, HIF-1α, PKC and caspase-10. BCLAF1 can
protect mature c-Myc mRNA from degradation by the RS domain
in HCC. In addition, the bZIP domain of BCLAF1 can bind to HIF-1α
and promote transcription of each other in HCC. Furthermore, BCLAF1
reduces HIF-1α ubiquitination and subsequent degradation via its
MYB domain, binding to the helix-loop-helix domain of HIF-1α. In
addition, DNA-PKC is involved in the DNA damage response and
apoptosis by directly phosphorylating serine151 and
tyrosine150 in the RS domain. Furthermore, caspase-10
protects MM cells from autophagic death induced by BCLAF1 via
cleavage of the aspartic acid at position 452. (B) The role of
BCLAF1 in different diseases and pathological processes. (a) Under
high-dose radiation, DNA-PKC is activated and phosphorylates
BCLAF1, which then initiates DNA damage repair. BCLAF1 antagonizes
p21-dependent downregulation of cyclin E expression and inhibition
of p21-dependent pro-apoptotic factors, caspase-3 and BAX by
repressing the expression level of p21; however, downregulation of
BCLAF1 levels may contribute to the tumorigenesis of LC.
Phosphorylated FHL1 can interact with BCLAF1 and promote its
expression, then promote the proliferation of the LC cells. (b)
BCLAF1 mediates the apoptosis of colon cancer cells by activating
the transcription of TP53 and BAX, and inhibiting the transcription
of MDM2. Splicing factor, SRSF10 is involved in the
post-transcriptional splicing of BCLAF1 and forms the L isoform,
thereby promoting the progression of colorectal cancer. (c)
miR-194-5p binds to the 3′-UTR of BCLAF1 and inhibits the
translation of BCLAF1 in acute myelocytic leukemia. Caspase-10
protects MM cells from autophagic death induced by BCLAF1 by
phosphorylating aspartic acid at position 452, which leads to
BCLAF1 replacing beclin-1, thereby promoting autophagy. (d) miR-K5
and miR-UL112-1 inhibit the translation of BCLAF1 by binding to its
3′-UTR. After HCMV and PRV infect host cells, they promote the
degradation of BCLAF1 protein by releasing viral proteins, pp71,
UL35 and US3. (e) BCLAF1 can activate the transcription of HIF-1α
and NEAT1 and promote the occurrence and development of HCC. At the
same time, HIF-1α can also activate the transcription of BCLAF1 in
HCC. Hsp90α interacts with BCLAF1 and inhibits its degradation by
the proteasome, and BCLAF1 contributes to the occurrence and
development of HCC by protecting mature oncogene, c-Myc mRNA from
degradation. (f) Sirt1 binds to NF-κB, translocates to the BCLAF1
promoter and deacetylates histone H3K56 to inhibit the
NF-κB-dependent transcription of BCLAF1, ultimately inhibiting the
activation of T cells. (g) miR-517a may inhibit cell proliferation
and promote cell apoptosis by indirectly upregulating the
expression level of BCLAF1 in BC. miR-194-5p binds to the 3′-UTR of
BCLAF1 and inhibits the translation of BCLAF1, thereby repressing
the malignant phenotype of BC. SMYD3 activates the transcription of
BCLAF1 by increasing the methylation of H3K4, and SMYD3 promotes
the progression of BC by targeting BCLAF1 to activate autophagy.
(h) HDAC4 inhibits the transcription of BCLAF1 by deacetylating the
BCLAF1 promoter. HDAC4 may indirectly inhibit the expression of
BCLAF1 by upregulating NF-κB. (i) Gastric cancer cells with BCLAF1
knocked down display decreased cell proliferation and increased
basal γH2AX, and are more vulnerable to I/R-induced DNA damage and
apoptosis. (j) An Emerin mutation disrupts the interaction between
Emerin and BCLAF1, which in turn leads to abnormal muscle cell
proliferation and differentiation. miR-194-5p promoted the myogenic
differentiation of mouse muscle cells by downregulating BCLAF1. (k)
Translocation of BCLAF1 to the nucleus activates the transcription
of TP53 and BAX, which promotes the apoptosis of cardiomyocytes
induced by I/R. lncCIRBIL binds to the BCLAF1 protein in the
cytoplasm of cardiomyocytes to prevent its translocation to the
nucleus, thus repressing the cardiac I/R injury. BCLAF1, B-cell
lymphoma-2-associated transcription factor 1; RS rich domain,
arginine-serine rich domain; bZIP domain, basic-leucine zipper
domain; HIF-1α, hypoxia inducible factor-1α; PKC, protein kinase C;
HCC, hepatocellular carcinoma; MM, multiple myeloma; FHL1,
four-and-a-half LIM protein 1; miR, microRNA; UTR, untranslated
region; HCMV, human cytomegalovirus; PRV, pseudorabies virus; NEAT,
nuclear enrichment-rich transcription factor 1; BC, bladder cancer;
SMYD3, histone methyltransferase SET and MYND domain-containing
protein 3; HDAC, histone deacetylase; γH2AX, H2AX phosphorylated on
serine 139; I/R injury, ischemia-reperfusion injury; lnc, long
non-coding. |
In consideration of recent advances in the
understanding of multiple biological functions of BCLAF1,
particularly in tumorigenesis, BCLAF1 exerts an important role in
different human cancers; therefore, the aim of the present review
was to introduce the subcellular localization, structural features,
expression and mutations, regulation, biological functions and
pathological functions of BCLAF1, then describe the roles of BCLAF1
in tumorigenesis. Lastly, BCLAF1 may represent a potential cancer
therapeutic target in the future.
Subcellular localization of BCLAF1
BCLAF1 is predominantly located in the nuclear
dot-like structures and secondarily located in the cytoplasm
(1). Previous studies have
discovered that the diverse biological functions of BCLAF1 have
been associated with its different cellular locations. For example,
the anti-apoptotic members of the Bcl-2 family inhibited the
pro-apoptotic effect of BCLAF1 by isolating BCLAF1 in the cytoplasm
(1). In another study, in the HeLa
cell line, which were induced to undergo apoptosis, BCLAF1
relocated from the nuclear dot-like structures to a position near
the nuclear envelope (18).
Similarly, Lee et al (7)
also observed the same translocation phenomenon of BCLAF1 when the
293T cell line was exposed to high-dose radiation. Furthermore, a
recent report showed that the nuclear translocation of BCLAF1
significantly increased following the induction of cardiac I/R
injury (23).
Structural features of BCLAF1
The BCLAF1 gene is located on human
chromosome 6q23.3, encoding 17 transcription variants of different
isoforms (44). The L isoform of
BCLAF1 (a large protein containing 920 amino acids) will be
described. BCLAF1 contains multiple domains, including a N-terminal
RS domain, which contains a bZIP domain and a C-terminal MYB
DNA-binding domain (Fig. 1A)
(1,12). The RS domain is involved in the
biogenesis and splicing of pre-mRNA (26–28),
and the formation of the ribonucleoprotein (RNP) complex, which is
an important part of the pre-mRNA spliceosome (6,45,46).
Thus, BCLAF1 also exerts its post-transcriptional splicing function
by participating in the formation of the spliceosome (2–6). It
has been found that the RS domain can mediate protein-protein
interactions, such as RNP, snRNP U1-70K and the splicing factor,
U2AF (47,48). Notably, three of the five protein
binding sites, that interact with BCLAF1, are located in the RS
domain (7,30,39),
suggesting that interactions between BCLAF1 and other proteins may
be mediated by the RS domain; however, this requires further
research. It is well-known that the bZIP and MYB DNA-binding
domains are characteristic components of eukaryotic transcription
factor families (49–51). These structural features of BCLAF1
allow it to act as a transcription factor and mRNA splicing factor,
at the transcriptional and post-transcriptional levels, to
participate in the regulation of expression of downstream target
genes (4,5,7,9,26–28,30–34).
Expression and mutations in BCLAF1 in
cancer
To the best of our knowledge, the expression level
and mutations in BCLAF1 in human normal tissues and tumor tissues
have not been systematically studied or summarized in the
literature, particularly the mutations in BCLAF1. Therefore, the
expression level of BCLAF1 in human normal and cancer tissues, and
the mutation status of BCLAF1 in patients with malignant tumors was
found using the Human Protein Atlas database (https://www.proteinatlas.org/ENSG00000029363-BCLAF1)
(Fig. 2) and the International
Cancer Genome Consortium database (ICGC; http://dcc.icgc.org/genes/ENSG00000029363) (Table I), respectively. In addition,
survival analysis was performed using the GEPIA2 database
(http://gepia2.cancer-pku.cn/#survival) with
Kaplan-Meier analysis (Fig.
3).
 | Table I.Mutations in B-cell
lymphoma-2-associated transcription factor 1 in different types of
cancer. |
Table I.
Mutations in B-cell
lymphoma-2-associated transcription factor 1 in different types of
cancer.
| Cancer type | Mutation frequency
in cancer, % | Mutations in the RS
domain | Mutations in the
bZIP domain | Mutations in the
MYB domain | Mutations in other
regions |
|---|
| Endometrial | 2.45 | E42a, E121a, R30a, A79Vb, A31Vb, E41a, M1I, R22a, R107a, R105a | E121a | E523a | E776a, E774a, E603a, E504a, E677a, E675a, E676a, E901a, E854a, E903a, E730a, E169a, E852a, E359a, E357a, E411a, E584a, E582a, E350a, E521a, R762a, R764a, R591a, A813Vb, A640Vb, A811Vb, E775a, E602a, E773a, E372a, E374a |
| Blood | 1.40 | R159a, R157a | - | - | W213a, W211a, R296a, R298a |
| Skin | 1.40 | R88a, R90a, W95a, W97a, R76a, R78a, M1I, W153a, W105a, R30a, R107a, R105a | - | - | Q463a, Q636a, Q634a, W887a, W836a, W838a, W714a, W885a, R762a, R764a, R591a, Q326a, Q328a |
| Ovarian | 1.08 | - | - | - | W375a, W373a. |
| Bladder | 0.97 | S11a, S13a | - | - | Q617a, Q446a, Q619a, Q266a, Q264a. |
| Colorectal | 0.92 | E51a, E3a | - | E523a | G363a, G365a, E785a, E612a, E783a, E454a, E456a, E480a, E478a, E501a, E503a, E350a, E521a |
| Brain tumor | 0.70 | R12a, R60a, R69a, R67a | - | - | R794a, R621a, R792a, Y447a, Y620a, Y618a, R296a, R298a |
| Hepatocellular
carcinoma | 0.63 | R62a, R14a, Y34a | - | - | R794a, R796a, R623a, S750a, S579a, S752a, Y447a, Y620a, Y618a, D400, D398 |
| Pancreatic | 0.56 | R69a, R67a | - | - | - |
| Gastric | 0.53 | R159a, R157a, E51a, E3a | - | - | R281a, R283a, E785a, E612a, E783a |
| Thyroid | 0.41 | R22a | - | - | - |
| Lung | 0.41 | - | - | - | G278a, G276a. |
| Head and neck | 0.39 | Y71a, Y73a | - | - | K419a, K421a. |
| Prostate | 0.20 | R69a, R67a | - | - | - |
| Breast | 0.20 | R159a, R157a, KE138 | - | - | S750a, S579a, S752a, KE918, KE747, KE186, KE869,
KE871, KE920 |
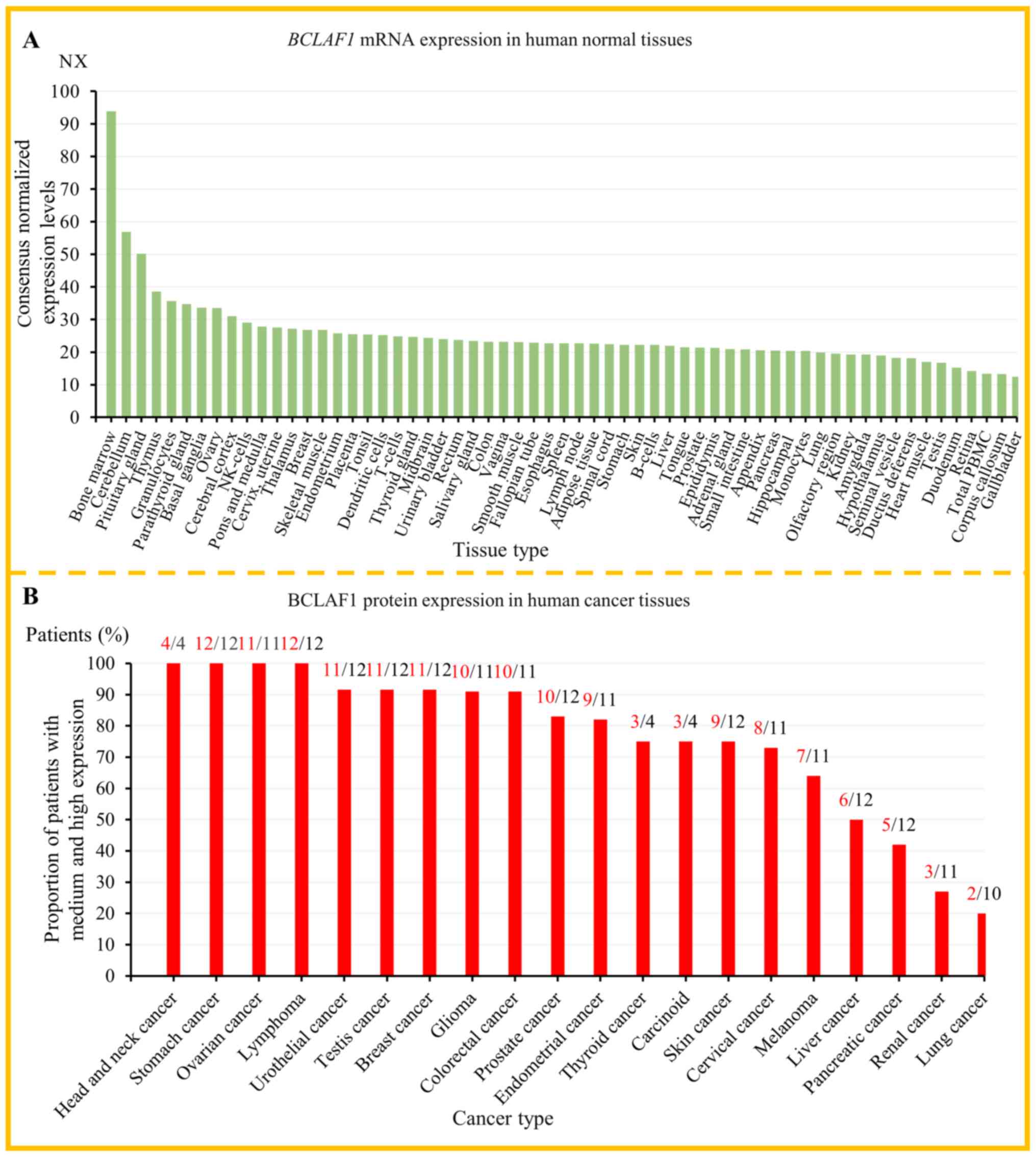
According to the data from the Human Protein Atlas
database, BCLAF1 was expressed at different levels in all normal
tissues (Fig. 2A) and most cancer
tissues (Fig. 2B), and its
expression has low tissue specificity and tumor specificity. In
addition, survival analysis, into the association between the
expression level of BCLAF1 in multiple types of cancer and the
survival rate of patients, indicated that BCLAF1 could not be used
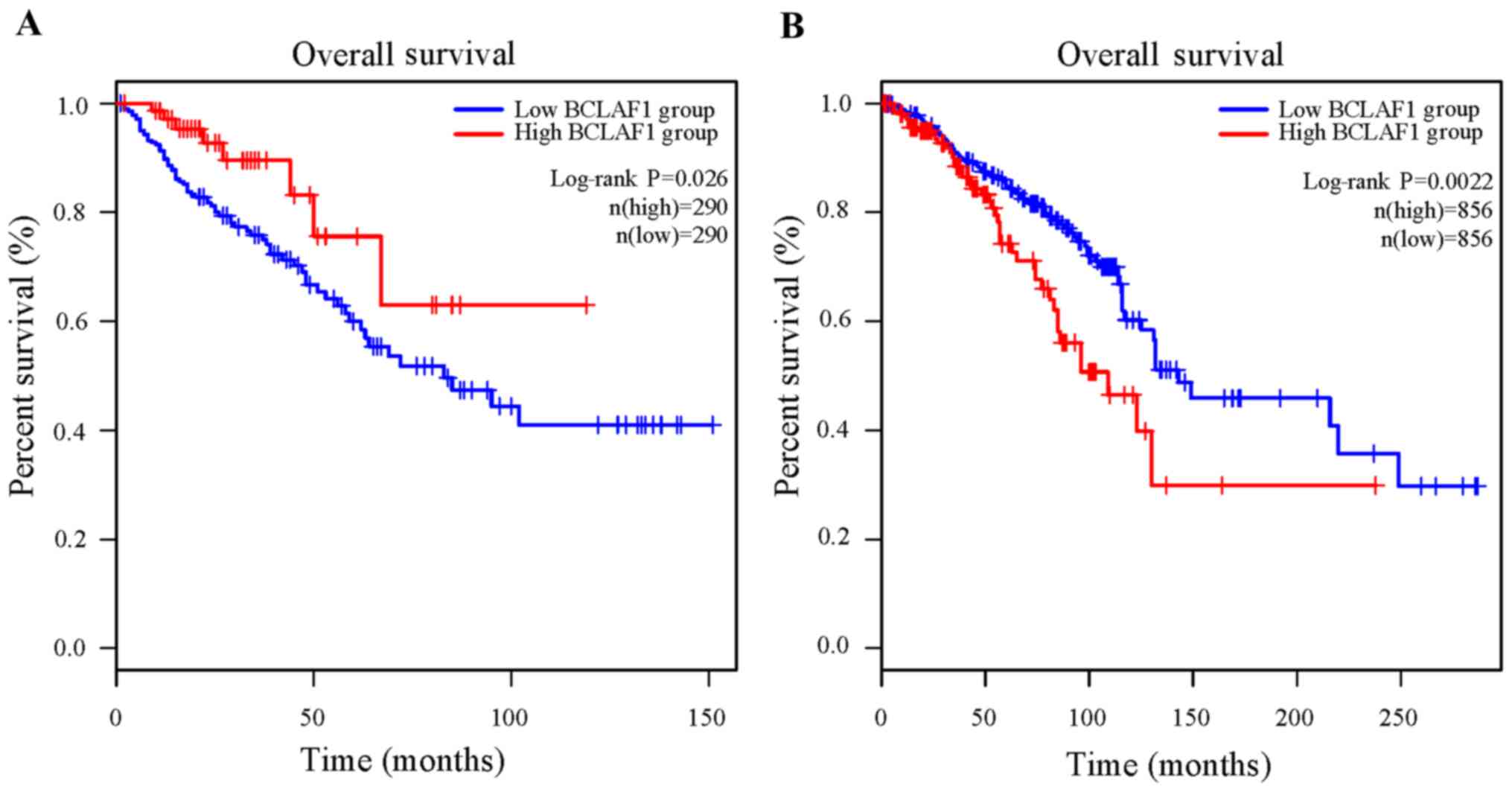
as a clear prognostic marker in most cancers, except for colorectal
cancer and breast cancer (Fig. 3).
In colorectal cancer, the high mRNA expression level of
BCLAF1 was associated with favorable overall survival rate
in 290 patients and low mRNA expression level of BCLAF1 was
associated with poor overall survival in 290 patients (P=0.026)
(Fig. 3A). In breast cancer, the
high mRNA expression level of BCLAF1 was associated with
poor overall survival in 856 patients and the low mRNA expression
level of BCLAF1 was associated with favorable overall
survival rate in 856 patients (P=0.0022) (Fig. 3B).
Furthermore, the ICGC database showed that different
mutations were found in BCLAF1 in various types of cancer. The
mutations were primarily located in the RS domain and other regions
(sites other than the known domains of BCLAF1), while there were
fewer mutations in the bZIP and the MYB domains (Table I). In addition, the majority of the
mutations in BCLAF1 were stop-gain mutations, with fewer frameshift
and start-loss mutations (a subtype of non-sense mutations)
(Table I). The mutations in BCLAF1
have been detected in numerous cancer samples; however, the
clinical significance is unknown; therefore, additional research is
required.
Taken together, from the analysis of data from TCGA
and ICGC, it can be concluded that the existing pathological
evidence (human cancer specimens) is still insufficient to clarify
the specific function of BCLAF1 in tumorigenesis. It is necessary
to further expand the sample size to obtain more evidence, such as
physiological evidence (animal models) and biochemical evidence
(upstream and downstream molecules of BCLAF1).
Regulation of BCLAF1 expression and
function
The expression and function of BCLAF1 is affected by
numerous processes at multiple levels of regulation, including the
transcriptional, post-transcriptional, translational and
post-translational levels. A previous study showed that a NF-κB
binding site was found in the promoter region of BCLAF1 and NF-κB
activates its transcription by binding to the promoter of BCLAF1 in
T-cell activation (24).
Similarly, another study also confirmed this process in senescent
cells induced by the chemotherapeutic agent, doxorubicin (8). However, Li et al (35) found the opposite results. They
demonstrated that the mRNA expression level of BCLAF1 was
increased in the diffuse large B-cell lymphoma (DLBCL) cell lines
treated with the NF-κB inhibitor, Bay11-7082, indicating that NF-κB
may downregulate the expression level of BCLAF1; however, the
specific mechanism involved requires further investigation.
Furthermore, hypoxia inducible factor-1α (HIF-1α)
has been shown to bind to the promoter region of BCLAF1 and
directly activate its transcription in hepatocellular carcinoma
(HCC) (Fig. 1A and B-e) (30). In addition to regulating the
expression level of BCLAF1, through the combination of
transcription factors and promoters, epigenetic modifications have
also been found to be involved in the regulation of BCLAF1
(22,24,35).
In one study it was found that Sirt1 repressed BCLAF1 transcription
by deacetylating the histone 3 lysine 56 residues (H3K56) on
conserved non-coding sequences (CNS) 3 and CNS4 in the promoter
region and the loss of Sirt1, using RNA interference resulted in
increased transcription of BCLAF1, which was responsible for T-cell
activation (Fig. 1B-f) (24). Specifically, Sirt1 was combined
with Rel-A (a subunit of NF-κB) and recruited to the BCLAF1
promoter, which initiated the deacetylation modification of BCLAF1
(Fig. 1B-f) (24). Furthermore, in another study, a
HDAC inhibitor (HDACi), LMK-235 upregulated BCLAF1 mRNA
expression and promoted apoptosis in DLBCL (35) Mechanistically, LMK-235 upregulated
BCLAF1 expression and BCLAF1-mediated apoptosis by inhibiting the
NF-κB pathway (35). In addition,
Shen et al (22) confirmed
that the histone methyltransferase, SMYD3 physically interacted
with the promoter of BCLAF1 and upregulated its mRNA expression by
the increase in the dimethylation and trimethylation of histone H3
lysine 4 (H3K4) at the BCLAF1 locus (Fig. 1B-g). In addition, Zhou et al
(34) demonstrated that the
splicing factor, SRSF10 was a direct upstream regulatory molecule
of BCLAF1 and participated in the production of the BCLAF-L isoform
via the post-transcriptional splicing of BCLAF1 in colorectal
cancer (Fig. 1B-b).
miRNAs are small non-coding RNAs, that negatively
regulate gene expression (52).
BCLAF1 has also been found to be regulated as a direct downstream
target of miR-194-5p, miR-K5 and miR-UL112-1 (13,15,20,36,38).
Dell'Aversana et al (38)
discovered that miR-194-5p binds the 3′-untranslated region
(3′-UTR) of BCLAF1 to suppress its gene expression, proving that
the dysregulation of this regulatory pathway results in the
occurrence of acute myelocytic leukemia (AML) (Fig. 1B-c). Similarly, two other studies
reported that miR-194-5p could inhibit the translation of BCLAF1 in
bladder cancer and muscle cells (Fig.
1B-g and -j) (20,36). Furthermore, similar inhibitory
effects of miRNAs on BCLAF1 have also been observed in viral
infections (13,15). For example, miR-K5, encoded by
Kaposi's sarcoma-associated herpes virus (KSHV) (15) and miR-UL112-1, encoded by human
cytomegalovirus (HCMV) (13) both
inhibited the translation of BCLAF1 by binding to its 3′-UTR
(Fig. 1B-d). In addition, Liu
et al (9) showed that after
induction of DNA damage, PKCδ and BCLAF1 formed a complex to occupy
TP53 core promoter element (CPE-TP53) and promoted TP53-mediated
apoptosis in response to DNA damage. A subsequent study further
validated this conclusion. Lee et al (7) indicated that DNA-PKC was associated
with DDR and apoptosis by directly phosphorylating
serine151 and tyrosine150 in the RS domain in
BCLAF1 (Fig. 1A and B-a).
Similarly, a recent study proved that the phosphorylation of BCLAF1
at serine290 participated in DDR mediated by H2AX
phosphorylated on serine139 (γH2AX) (11). Lastly, heat shock protein 90α
(Hsp90α) was found to bind to BCLAF1 and stabilize its protein
structure, thus preventing BCLAF1 from degradation via the
ubiquitin-proteasome system (UPS) in HCC (Fig. 1B-e) (39).
It is worth noting that 20 high-frequency
transcription factors, that might bind to the BCLAF1 promoter
region were predicted using the PROMO database (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3)
(Table II). It predicted
high-frequency transcription factors may participate in the
regulation of BCLAF1 expression by activating or inhibiting the
transcription of BCLAF1.
| Table II.Predicted transcription factors of
B-cell lymphoma-2-associated transcription factor 1. |
Table II.
Predicted transcription factors of
B-cell lymphoma-2-associated transcription factor 1.
| Name of
transcription factor | Predicted number of
binding sites |
|---|
| C/EBPβ | 37 |
| GR-β | 26 |
| YY1 | 14 |
| TFIID | 9 |
| GR-α | 7 |
| TFII–I | 4 |
| FOXP3 | 3 |
| HNF-3α | 2 |
| GR | 2 |
| NF-AT2 | 1 |
| AP-2αA | 1 |
| HNF-1A | 1 |
| NF-AT1 | 1 |
| HOXD9 | 1 |
| HOXD10 | 1 |
| C/EBPα | 1 |
| c-Jun | 1 |
| STAT4 | 1 |
| PR B | 1 |
| PR A | 1 |
In summary, multiple studies have revealed that the
expression and function of BCLAF1 is regulated at different levels
and in different biological processes (8,13,15,20,22,24,30,34–36,38).
Compared to the transcriptional and translational levels, studies
on the post-translational modification of BCLAF1 are limited, such
as ubiquitination, which has not yet been reported. In addition,
the high-frequency transcription factors predicted by the PROMO
database, that can regulate the transcription of BCLAF1, require
further verification, such as from dual-luciferase or chromatin
immunoprecipitation assays.
Downstream targets regulated by BCLAF1
BCLAF1 has numerous downstream targets and
participates in a series of biological processes. Previous research
has shown that BCLAF1 positively regulated TP53 expression by
interacting with the CPE-TP53 during DNA damage (9). Analogously, another study showed that
high doses of ionizing radiation (IR) triggered rapid
phosphorylation of BCLAF1 and enhanced the binding of BCLAF1 to
CPE-TP53 (Fig. 1B-a) (7). Furthermore, Rénert et al
(32) confirmed that BCLAF1 played
an important role in ceramide-mediated apoptosis of human HCT-116
colon carcinoma cells. BCLAF1 not only promoted the transcription
of apoptosis-related proteins, such as TP53 and BAX, but also
inhibited the transcription of the TP53 inhibitor, MDM2 (Fig. 1B-b) (32). All these results indicated that
BCLAF1 participated in apoptosis and DNA damage in a TP53-dependent
manner.
Hypoxia-inducible factors (HIFs) serve an
indispensable role in the response of cancer cells to hypoxic
stress, maintaining cell survival and growth by activating gene
transcription containing hypoxia-responsive elements (53,54).
In hypoxic conditions, BCLAF1 binds the promoter region of HIF-1α
via the bZIP domain, directly activating its transcription and
promoting HIF-1α-mediated angiogenesis in HCC (Fig. 1A and B-e) (30). In addition, BCLAF1 could reduce
HIF-1α ubiquitination and subsequent degradation by the binding of
the MYB domain to the helix-loop-helix domain of HIF-1α (Fig. 1A) (55). In addition, studies have shown that
lncRNA NEAT1 promotes the resistance of cancer cells to astib,
bortezomib, paclitaxel, doxorubicin and cisplatin in ovarian
cancer, leukemia and gastric cancer cell lines (56–58).
Mou et al (31) revealed
that BCLAF1 interacted with the promoter region of lncRNA NEAT1 and
activated its transcription, thereby promoting the proliferation,
invasion and resistance of HCC cells to 5-fluorouracil (5-Fu)
(Fig. 1B-e). Furthermore, BCLAF1
has also been reported to be involved in post-transcriptional
regulation. Zhou et al (39) demonstrated that BCLAF1 was a key
post-slicing regulator of c-Myc mRNA stability and it could
protect mature c-Myc mRNA from degradation by the RS domain
in HCC; however, the specific regulatory mechanism involved is
unclear (Fig. 1A and B-e).
BCLAF1, HIF-1α and NF-κB in cancer
BCLAF1 and NF-κB in cancer. NF-κB is an important
transcription factor that regulates a variety of pathophysiological
processes involved in cell survival and death (59–62),
particularly by inhibiting apoptosis in the process of
carcinogenesis and cancer progression, thereby promoting tumor
growth (63,64). However, BCLAF1 acts an inducer of
apoptosis (1,7,9,23,32,35,38)
and has an opposing effect to NF-κB in the regulation of apoptosis.
A study showed that following stimulation by T cell receptor and
CD28 signals, Rel-A could be significantly induced to bind to the
BCLAF1 promoter and promote its transcription, leading to the
activation and maturation of immature CD4+T cells
(24). In another study, BCLAF1
was found to be directly downstream of NF-κB, which was bound to
the BCLAF1 promoter and activated transcription of BCLAF1 in DNA
damage-induced senescence (8). Li
et al (35) demonstrated
that NF-κB inhibited BCLAF1 expression and BCLAF1-mediated
apoptosis in DLBCL cells; however, the specific mechanism is not
clear (Fig. 1B-h).
Overall, these studies collectively indicated that
NF-κB is directly upstream of BCLAF1, but the regulatory effect of
NF-κB on BCLAF1 was observed with different results. NF-κB could
act as a positive regulator to activate the transcription of BCLAF1
and it has also been found that NF-κB could repress the mRNA and
protein expression level of BCLAF1. Given the insufficient research
on the interaction between NF-κB and BCLAF1 in cancer, further
investigation is required.
BCLAF1 and HIF-1α in cancer. HIF-1α is a
well-defined hypoxia response factor, which can activate a variety
of pathways regulating angiogenesis, cell metabolism, proliferation
and drug resistance (54,65). Wen et al (30) reported that the mRNA and protein
expression levels of HIF-1α and BCLAF1 in tumor tissues were
significantly higher compared with that in the adjacent normal
tissues and were positively correlated. Furthermore, in the nude
mouse xenograft tumor model, it was also confirmed that BCLAF1
significantly promoted HCC angiogenesis and tumor growth (30). Mechanistic studies have shown that
BCLAF1 promotes angiogenesis by activating the transcription of
HIF-1α, which in turn promotes the proliferation, angiogenesis, and
metastasis of HCC cells (Fig.
1B-e) (30). Notably, HIF-1α
could in turn activate the transcription of BCLAF1 and form a
closed loop with BCLAF1 (30). In
another study, BCLAF1 also maintained HIF-1α activity by binding to
HIF-1α and protecting HIF-1α from degradation under long-term
hypoxia, and BCLAF1 may enhance the stability of HIF-1α to promote
tumor progression (55). In
addition, Zhang et al (42)
demonstrated that the ginsenoside, compound K (CK; a ginsenoside
diol type saponins, which has a variety of pharmacological
activities, including anti-inflammatory, hepatoprotective and
antitumor effects) (66),
inhibited the HIF-1α-mediated glycolysis pathway by downregulating
the protein expression level of BCLΑF1, thereby inhibiting the
proliferation of liver cancer cells.
In summary, several studies have confirmed that
BCLAF1 upregulates the expression level of HIF-1α; therefore,
participates in different biological processes, such as cell
proliferation, angiogenesis and cell metabolism (30,42,55).
In particular, in HCC, which is characterized by a hypoxic
microenvironment, HIF-1α-mediated angiogenesis and anaerobic
glycolysis notably promoted the occurrence and development of HCC.
Therefore, targeting the BCLAF1-HIF-1α pathway may represent a
preclinical treatment strategy for HCC.
Physiological function of BCLAF1
BCLAF1 is involved in mediating numerous
physiological processes, such as DDR (3,4,7,8,9),
pre-mRNA splicing and processing (2–6),
apoptosis (1,7,9,12,23,32,35,37,38),
cell cycle (1,7), lung development (10), T-cell activation (12,24,25),
and muscle cell proliferation and differentiation (17–20).
The current knowledge arises from physiological evidence obtained
from mouse and cell models, pathological evidence from human tumor
cell lines and disease cell models, and biochemical evidence from
in vitro studies of substrates that can interact with BCLAF1
and participate in several physiological processes (Table III). Loss of functional Emerin, a
nuclear membrane protein, causes X-linked recessive Emery-Dreifuss
muscular dystrophy (EDMD) (17).
Haraguchi et al (18) found
that Emerin could bind to BCLAF1 and co-locate to the nuclear
membrane, and participate in the regulation of apoptosis, and the
proliferation and differentiation of muscle cells (Fig. 1B-j). In addition, the loss of
binding of Emerin to BCLAF1 may be associated with muscle atrophy
in EDMD (Table III and Fig. 1B-j) (18). Furthermore, Wang et al
(20) demonstrated that circular
RNA Zfp609 (circZfp609) regulated muscle cell differentiation by
sponging miR-194-5p. Specifically, circZfp609 could sponge
miR-194-5p to sequester its inhibition on BCLAF1 and repress
myogenic differentiation; however, the specific mechanism is
unclear. Notably, numerous studies have shown that BCLAF1 is a key
apoptosis regulator; however, its specific role is still
controversial. At present, most reports indicate that BCLAF1
promotes apoptosis via various mechanisms, such as antagonizing the
anti-apoptosis effect of Bcl-2 (1)
and upregulating the expression of apoptosis-related proteins, such
as TP53, BAX and caspase-3 (Fig.
1B-a) (7,9,23,32).
However, in one study, after a variety of chemical apoptosis
inducers (such as anisomycin, etoposide and staurosporine) and
γ-irradiation were used, BCLAF1-deficient T cells and B cells did
not show notable apoptotic disorders, suggesting that BCLAF1 was
not necessary for apoptosis (12).
In another study, BCLAF1-mediated autophagy of HCC cells could
enhance cell proliferation and prevent cell apoptosis under stress
conditions (37). Similar to
tumorigenesis, the regulation of BCLAF1 on apoptosis may be
dependent on cell type and cell context; however, further
investigation is required.
| Table III.Physiological functions of
BCLAF1. |
Table III.
Physiological functions of
BCLAF1.
| Physiological
function | Physiological
evidence | Pathological
evidence | Biochemical
evidence | Ref. |
|---|
| DNA damage
response | In the BCLAF1
knockdown 293T cell model, the sensitivity of the cells to DNA
damage and DNA repair defects was increased. | In anti-IR cancer
cells, BCLAF1 was intrinsically inhibited, resulting in the
weakening of the Ku70/DNA-PKC complex; however, the binding of
Ku70/Bcl-2/p18-Cyclin E was enhanced, which subsequently reduced
the DSB repair activity. Overexpression of BCLAF1 induced severe
sensitivity of cells to IR. | After ChIP analysis
of the TP53 promoter, it was found that PKCδ forms a complex with
BCLAF1 to form CPE-TP53 and activate TP53 transcription to promote
DNA damage repair. | (3,4,7–9) |
| Pre-mRNA splicing
and processing | BCLAF1 and TRAP150
control the abundance of transcripts encoding mitotic regulators,
thereby affecting the process of mitosis in human cells. | Knockdown of BCLAF1
in U2OS-DR-GFP cells can lead to susceptibility of DNA damaging
agents, DNA repair defects and genome instability. | The spliced human
mRNP was affinity-purified from HeLa nuclear extracts and BCLAF1
was identified by mass spectrometry to participate in the formation
of mRNP. BCLAF1, SNIP1 and RNA processing factors together form the
SNARP complex and play a key role in the stability of cyclin D1
mRNA | (2–6) |
| Apoptosis | After BCLAF1 was
transiently transfected into HeLa cells, it induced its apoptosis.
Knockdown of BCLAF1 significantly attenuates DNA damage-induced
apoptosis in U2OS cells and SaOS-2 cells. | After IR induced
apoptosis, it was found that BCLF1 promotes the pro-apoptotic
activity of caspase-3 by regulating cyclin E and the
p53/p21-dependent mitochondrial pathway. | Using
immunofluorescence technology, it was found that high-dose IR
triggers the translocation of BCLAF1 to γH2AX foci and enhances the
binding of BCLAF1 to CPE-TP53, thereby promoting TP53 transcription
and TP53-dependent pro-apoptotic effects. | (1,7,9) |
| Cell cycle | Knockdown of BCLAF1
in HeLa cells increased chromosomal misalignment and caused mitotic
defects. In BCLAF1 knockout cells, the expression level of key
mitotic regulation transcripts and the mitotic index increased
significantly. | Compared with
BCLAF1 overexpression, BCLAF1 knockdown increased the level of p21
protein, downregulated the level of the cyclin D1 protein, and
caused significant G1 phase arrest in cells that were
induced to DNA damage and apoptosis. | None | (1,7) |
| Lung
development | BCLAF1-knockout
mouse die mainly in the neonatal period and exhibit end-stage lung
development defects that affect survival. | None | None | (12) |
| T-cell
activation | The total number of
cells (splenocytes, T-cells and B-cells) in all recombinant mice
models was equal. BCLAF1-knockout mice will not only increase the
steady-state number of peripheral B cells, but also damage the
steady-state number of peripheral T-cells, leading to T-cells and
B-cells homeostasis defects. Knockdown of BCLAF1 expression
inhibits Sirt1-null T-cell activation. | None | BCLAF1 was screened
by proteomics methods and found to bind to Jurkat T-cell nucleolus.
It is also a transcriptional inhibitor that is essential for T-cell
function and differentiation. | (12,24,25) |
| Muscle cell
proliferation and differentiation | In the C2C12 cell
model (mouse myoblast cell line) with double knockdown of BCLAF1
and Cry2, the stability of cyclin D1 mRNA and Tmem176b mRNA
decreased and inhibited circadian myoblast proliferation and
myogenic fusion. miR-194-5p mimic transfection reduced protein
expression of BCLAF1 and increased the expression of myogenic
markers (Myf5 and MyoG) in the C2C12 cell model, thereby promoting
myogenic differentiation. | The constructed
emerin-S54F pathogenic mutant loses its ability to bind to BCLAF1
and is associated with muscle atrophy in Emery-Dreifuss muscular
dystrophy. | None | (17–20) |
Roles of BCLAF1 in human cancer
BCLAF1 is a nuclear protein, which was first found
to interact with Bcl-2 and promote apoptosis (1). Furthermore, BCLAF1 has been found to
induce apoptosis by activating the transcription of TP53 (9). Subsequently, BCLAF1 was identified as
a potential tumor suppressor by promoting the apoptosis of colon
adenocarcinoma cells and bladder cancer (BC) cells (32,43).
Collectively, another three studies also confirmed that BCLAF1
acted as a tumor suppressor in lung cancer (LC), multiple myeloma
(MM) and DLBCL (7,21,35).
However, recently, BCLAF1 has become a hot topic due to its
carcinogenic characteristics in certain types of human cancer,
including colorectal cancer (CRC) (34), BC (22,36),
AML (38), LC (40), HCC (30,31,37,39,42)
and gastric cancer (11). These
differences indicated that the specific role of BCLAF1 in tumor
progression may depend on the cellular context and type of cancer
(Table IV).
| Table IV.Pathological function of BCLAF1 in
vitro and in vivo models of disease. |
Table IV.
Pathological function of BCLAF1 in
vitro and in vivo models of disease.
| A, Animal models of
cancer |
|---|
|
|---|
| Authors, year | Disease type | Pathological
evidence | Role | Effect | Ref. |
|---|
| Wang et al,
2018 | LC | LC model | Oncogene | BCLAF1 knockout
mice had a smaller tumor volume and lighter weight by inhibiting
cell proliferation, G1/S phase transition and clone
formation. | (40) |
| Chen et al,
2020 | BC | BC model | Oncogene | High expression of
BCLAF1 was associated with higher tumor weight and volume in
xenograft model | (36) |
| Zhou et al,
2014 | CRC | CRC model | Oncogene | Knockdown of BCLAF1
in the mouse subcutaneous tumor model reduced the tumor growth rate
and tumor size. | (34) |
| Wen et al,
2019 | HCC | HCC model | Oncogene | Knockdown of BCLAF1
significantly inhibits HCC angiogenesis and tumor growth by
inhibiting HIF-1α transcription. | (30) |
| Zhou et al,
2019 | HCC |
| Oncogene | Knockdown of BCLAF1
significantly reduced tumor size and tumor growth rate by
inhibiting the stability of oncogene c-Myc mRNA. | (39) |
| Yu et al,
2019 | HCC |
| Oncogene | Animal experiments
show that BCLAF1 was associated with the tumorigenicity of HCC by
inducing autophagy. | (37) |
| Zhang et al,
2020 | HCC |
| Oncogene | In the mouse model
of HCC, BCLAF1 knockdown promotes the ubiquitination and
degradation of HIF-1α activated by CK, thereby inhibiting
glycolysis in hypoxic HCC cells, and ultimately inhibiting the
proliferation of HCC cells. | (42) |
|
| B, Human tumor
tissue sample and cancer cell lines |
|
| Authors,
year | Disease
type | Pathological
evidence | Role | Effect | Ref. |
|
| Yoshitomi et
al, 2011 | BC | BC cell lines | Tumor
suppressor | SMYD can
significantly inhibit the proliferation of BC cells and induce cell
apoptosis. In the BC cells transfected with miR-517a, the
BCLAF1 gene was found to be significantly upregulated. | (43) |
| Chen et al,
2020 | BC |
| Oncogene | Knockdown of BCLAF1
inhibited proliferation and promoted apoptosis of the BC cells | (36) |
| Shen et al,
2016 | BC | BC cell lines and
tissues sample | Oncogene | BCLAF1 is increased
in the BC cell and tissue samples, and promotes the progression of
BC by activating autophagy. | (22) |
| Lee et al,
2012 | LC | LC cell lines | Tumor
suppressor | In IR-resistant LC
cells, BCLAF1 is inhibited, leading to the formation of the
anti-apoptotic Ku70-Bax complex and the destruction of the
Ku70/DNA-PKC complex, thereby promoting the apoptosis resistance
and survival of LC cells. |
(7) |
| Jiang et al,
2020 | LC |
| Oncogene | BCLAF1 is increased
in LC cells. It can promote LC cell resistance to cisplatin by
promoting DNA damage repair and promoting G1 cell cycle
arrest. | (41) |
| Wang et al,
2018 | LC | LC cell lines and
tissues sample | Oncogene | BCLAF1 is increased
in LC cells and tissue samples. Phosphorylated FHL1 translocates
into the nucleus and interacts with BCLAF1, promoting the
proliferation of LC cells. | (40) |
| Lamy et al,
2013 | MM | MM cell lines and
tissues sample | Tumor
suppressor | Overexpression of
BCLAF1 induces autophagic death of MM cells. | (21) |
| Li et al,
2018 | DLBCL | DLBCL cell
lines | Tumor
suppressor | BCLAF1 promotes the
apoptosis of DLBCL mediated by HDAC inhibitor LMK235 via the NF-κB
signaling pathway. | (35) |
| Zhou et al,
2014 | CRC | CRC cell lines and
tissues sample | Oncogene | BCLAF1 is highly
expressed in CRC cells and tissues. BCLAF1 gene knockout
significantly reduces cell proliferation and colony formation. | (34) |
| Rénert et
al, 2009 | CRC | CRC cell lines | Tumor
suppressor | Knockdown of BCLAF1
in CRC cells have an increased survival rate and a stronger
resistance to death caused by C16-ceramide. After
C16-ceramide-induced apoptosis, knockdown of BCLAF1 reduced
caspase-3/7 activity in CRC cells. | (32) |
| Dell'Aversana et
al, 2017 | AML | AML cell lines and
tissues sample | Oncogene | BCLAF1 is highly
expressed in AML cells and is overexpressed in AML cells to promote
cell proliferation, inhibit the differentiation and maturation of
dendritic cells, and inhibit the sensitivity of anti-cancer
treatment. | (38) |
| Wen et al,
2019 | HCC | Human HCC cell
lines and tissues sample | Oncogene | BCLAF1 is highly
expressed in HCC cells and tissues, and promotes HCC angiogenesis
and cell proliferation by activating HIF-1α transcription. | (30) |
| Zhou et al,
2019 | HCC |
| Oncogene | BCLAF1 is highly
expressed in HCC cells and tissues, and promotes the proliferation
of HCC by enhancing the stability of the oncogene c-Myc
mRNA. | (39) |
| Yu et al,
2019 | HCC |
| Oncogene | BCLAF1 is highly
expressed in HCC cells and tissues, and promotes the progression of
HCC and the acquired resistance of sorafenib via autophagy
induction. | (37) |
| Mou et al,
2020 | HCC |
| Oncogene | BCLAF1 is highly
expressed in HCC cells and tissues, and targets NEAT1 to promote
HCC cell proliferation, invasion, and resistance to 5-Fu. | (31) |
| Zhang et al,
2020 | HCC | HCC cell lines | Oncogene | BCLAF1 knockdown
promotes the ubiquitination and degradation of HIF-1α activated by
CK, thereby inhibiting glycolysis in hypoxic HCC cells, and
ultimately inhibiting the proliferation of HCC cells. | (42) |
| Liu et al,
2021 | GC | GC cell lines and
tissues sample | Oncogene | The
serine290 phosphorylation of BCLAF1 was significantly
increased in GC tissues compared with that in paracancerous
tissues. Knockdown of BCLAF1 significantly delayed gastric cancer
cell proliferation and GC cells with BCLAF1 serine290
phosphorylation showed the highest viability at all time
points. | (11) |
|
| C, Animal
models |
|
| Authors,
year | Disease
type | Pathological
evidence | Role | Effect | Ref. |
|
| Shen et al,
2016 | Cardiac I/R
injury | Mouse model of
cardiac I/R injury | Increases cardiac
I/R injury | Overexpression of
BCLAF1 increases cardiac I/R damage and knockdown of BCLAF1 can
reduce cardiac I/R damage. | (22) |
| Qin et al,
2019 | PRV infection | PRV infection mouse
model | Inhibits the
replication of PRV | BCLAF1 knockout
mice are more sensitive to PRV infection and have greater
inflammatory damage in the lungs. | (14) |
|
| D, Cell
models |
|
| Authors,
year | Disease
type | Pathological
evidence | Role | Effect | Ref. |
|
| Ziegelbauer et
al, 2009 | KSHV infection | KSHV-infected cell
model | Inhibits the
replication of KSHV | BCLAF1 inhibits the
replication of KSHV. | (15) |
| Lee et al,
2012 | HCMV infection | HCMV-infected cell
model | Inhibits the
replication of HCMV | Decreased BCLAF1
enhances HCMV gene expression, while increased BCLAF1 inhibits HCMV
replication. | (13) |
| Qin et al,
2019 | PRV, HSV
infection | PRV and
HSV-1-infected cell model | Inhibits the
replication of PRV and HSV-1 | Knockdown of BCLAF1
can significantly promote the replication of US3-deficient
α-herpesvirus PRV and HSV-1. | (14) |
| Nilsson et
al, 2018 | HPV infection | HPV16- infected
cell model | Promotes the
replication of HPV16. | BCLAF1 is recruited
onto HPV16 DNA by co-recruitment with other DDR factors and RNA
processing factors, then induces replication of HPV DNA and HPV
late gene expression. | (16) |
BC
Human BC is one of the most common malignant tumors
and was the second leading cause of mortality in patients with
cancer of the urogenital tract worldwide in 2018 (67,68).
Yoshitomi et al (43) found
that miR-517a, as a tumor suppressor, was downregulated in BC
cells, and overexpression of miR-517a could significantly inhibit
cell proliferation and induce apoptosis. Furthermore, following
transfection of miR-517a mimics in BC cells, oligonucleotide array
analysis identified significant upregulation of BCLAF1; however,
the mechanism involved is not clear (43). It is well-known that miRNAs can
inhibit transcription by binding to the 3′-UTR of a target molecule
(52). For example, miR-K5
(15) and miR-UL112-1 (13) both inhibited the translation of
BCLAF1 by binding to its 3′-UTR (Fig.
1B-d). Therefore, these results suggested that miR-517a may
inhibit cell proliferation and promote cell apoptosis by indirectly
upregulating the expression of BCLAF1 in the BC, and that BCLAF1
acted as a potential tumor suppressor in BC.
However, certain subsequent studies have reported
different conclusions. In one study, compared with that in
para-carcinoma tissues, the relative expression level of BCLAF1 was
significantly increased (~1.82 times) in BC samples (P=0.002)
(36). Furthermore, the knockdown
of BCLAF1 inhibited the proliferation of BC cells and
promoted the apoptosis of BC cells (36). In addition, in a mouse xenograft
tumor model, BCLAF1facilitated the tumorigenicity of BC (36). Mechanistically, miR-194-5p bound to
the 3′-UTR of BCLAF1 and inhibited the translation of BCLAF1. In
addition, lncRNA PVT1 acted as a miRNA sponge to actively promote
the expression level of BCLAF1 by sponging miR-194-5p and
subsequently increased the malignant phenotype of BC (Fig. 1B-g) (36). Epigenetic studies have shown that
the dysfunction of histone methylation and its modifications may
serve an important role in tumorigenesis and development (69,70).
SMYD3 is a histone methyltransferase, which has been reported to be
involved in tumorigenesis, cancer cell proliferation, invasion and
migration (71,72). Shen et al (22) discovered that compared with that in
adjacent normal controls, SMYD3 and BCLAF1 mRNA and protein
expression levels were significantly upregulated in 77% (17/22) of
BC tissues. Mechanistic research showed that the overexpression of
SMYD3 activated the transcription of BCLAF1 by increasing the
methylation of histone H3K4 and SMYD3 promoted the progression of
BC by targeting BCLAF1 to activate autophagy (Fig. 1B-g) (22). Notably, there have been reports
that BCLAF1 was a strong autophagy inducer by replacing bcline-1 in
Bcl-2 (21,73). As the heterogeneity of tumors is
considered to be a sign of BC (74), it could be hypothesized that the
differential expression and different roles of BCLAF1 in BC may be
associated with the significant heterogeneity of BC.
LC
LC was the leading cause of cancer worldwide and
cancer-associated mortality in 2020, with an ~1.8 million (18%)
deaths (75). In one study,
DNA-PKC was specifically activated and phosphorylated BCLAF1 at
tyrosine150 and serine151 in IR-reactive
cells (293T, MRC-5 and WI-38 cell lines) under a high dose of IR,
which resulted in BCLAF1 relocating to the nuclear envelope, then
binding with γH2AX foci, which in turn stabilized the Ku70/DNA-PKC
complex to facilitate the non-homologous end-joining (NHEJ) pathway
of double-strand break (DSB) repair (Fig. 1B-a) (7). Ku70 is a DNA repair factor in the
cell nucleus, forming a heterodimer with Ku80 via the central
domain to bind double-stranded (ds) DNA, which is essential for the
DSB repair of the NHEJ pathway of dsDNA (76–78).
At the same time, BCLAF1 prevents p21-dependent G1 cell
cycle arrest, downregulation of cyclin E protein expression level,
and inhibition of the pro-apoptotic factors, caspase-3 and BAX, by
repressing the p21 protein expression level, thereby promoting cell
apoptosis and cell cycle progression (Fig. 1B-a) (7). However, BCLAF1 was inhibited in LC
cells with natural radioresistance. On the one hand, the formation
of the Ku70/DNA-PKC complex decreased and DNA repair was impaired;
on the other hand, p21-dependent cell cycle arrest was increased,
and the caspase-dependent apoptosis pathway, mediated by Ku70, was
inhibited, which eventually resulted in carcinogenesis (Fig. 1B-a) (7). Overall, BCLAF1 served as a tumor
suppressor in LC by promoting DNA repair, apoptosis and inhibiting
cell cycle arrest.
However, subsequent studies have discovered that
BCLAF1 may serve a different role in LC. Four-and-a-half LIM
protein 1 (FHL1) was identified in early studies as a tumor
suppressor factor and its expression level was significantly
decreased in a variety of cancers, including LC (79), HCC (80) and breast cancer (81). Conversely, a previous study found
that the role of FHL1 in cancer progression could be to promote
tumorigenesis (82). In addition,
Wang et al (40) confirmed
this conclusion, as they found that phosphorylation of FHL1
promoted LC cell proliferation. Specifically, phosphorylated FHL1
translocated into the nucleus and interacted with BCLAF1, then
promoted the proliferation of the H1299 non-small cell lung cancer
(NSCLC) cell line (Fig. 1B-a)
(40). Furthermore, in a nude
mouse xenograft tumor model, BCLAF1 knockout cells exhibited a
smaller tumor volume and weight (40). These results indicated that BCLAF1
could be a potential tumorigenic factor in LC. Furthermore, another
group revealed that the protein and mRNA expression levels of
BCLAF1 were higher in the cisplatin-resistant A549 NSCLC
cell line (41). Cisplatin is a
widely used small molecule chemotherapeutic drug (83), it can bind and cross-link with DNA,
thereby destroying DNA function, repressing mitosis and
subsequently inducing cell apoptosis (84,85).
Further research found that BCLAF1 may induce cisplatin resistance
in two ways; i) BCLAF1 could increase ubiquitin-specific peptidase
22 (USP22) mRNA expression to promote DNA repair, thereby inducing
cisplatin resistance (41). It has
been previously reported that USP22 overexpression could promote
cisplatin resistance in the A549 cell line, then regulated
Ku70/BAX-dependent apoptosis and γH2AX-mediated DNA damage repair
(86); and ii) BCLAF1 induced
G1 cell cycle arrest by increasing the p21 protein
expression level and decreasing the cyclin D1 protein expression
level, which resulted in cisplatin resistance (41). These studies indicated that BCLAF1
could promote LC cell proliferation and resistance to cisplatin by
targeting FHL1, promoting DNA damage repair and G1 cell
cycle arrest. However, the exact role and mechanism of action of
BCLAF1 in LC requires further investigation.
CRC
CRC was the second most common cause of
cancer-associated mortality worldwide in 2018, with a high
incidence rate in Westernized countries, while the incidence rate
is currently on the rise in Asian countries (87). There are a few studies on the role
of BCLAF1 in CRC. In 2009, Rénert et al (32) first reported the pro-apoptotic
effect of BCLAF1 in colon cancer cell lines. It was found that
C16-ceramide, the core molecule involved in sphingomyelin
metabolism relied on BCLAF1 to promote the apoptosis of colon
cancer cells (32). Furthermore a
mechanistic study reported that BCLAF1 promoted the apoptosis of
colon cancer cells by regulating apoptosis-related proteins
(activating the transcription of TP53 and BAX, and inhibiting the
transcription of MDM2) (Fig. 1B-b)
(32). Notably, in the present
review the survival prediction analysis of BCLAF1 in patients with
CRC using the GEPIA2 database, showed that high mRNA expression
level of BCLAF1 was associated with a favorable overall
survival rate in 290 patients, while low mRNA expression level was
associated with poor overall survival in 290 patients (P=0.026)
(Fig. 3A). Collectively, these
results indicate that BCLAF1 could be a potential tumor suppressor
in CRC.
However, another group strongly challenged this
conclusion. Zhou et al (34) discovered that the expression of the
L isoform of BCLAF1 was increased in CRC cell lines, suggesting
that the L isoform may serve a key role in colorectal tumorigenesis
(34). Furthermore, the
overexpression of the L isoform promoted the proliferation and
colony formation of CRC cells (34). In addition, the splicing factor,
SRSF10 was found to be a direct upstream regulatory molecule of
BCLAF1 and participated in the production of the L isoform via the
inclusion of exon 5a, which promoted the tumorigenesis of CRC
(Fig. 1B-b) (34). In addition, the knockdown of the L
isoform in a mouse subcutaneous tumor model reduced the tumor
growth rate and tumor size, which also confirmed the role of the L
isoform in promoting tumorigenesis and development of CRC (34). Overall, these results indicated
that the role of BCLAF1 in CRC could be contradictory, possibly as
either a tumor suppressor or tumor-promoting factor. Therefore, an
in-depth investigation is required to determine the role of BCLAF1
in CRC.
HCC
HCC was the fourth leading cause of cancer-related
mortality worldwide and the leading cause of death among patients
with cirrhosis in 2019 (88).
However, studies focusing on the role of BCLAF1 in the
tumorigenesis of HCC have not been reported until recently. As
aforementioned, multiple studies have shown that BCLAF1 was
associated with the regulation of angiogenesis, cell proliferation
and drug resistance in HCC under hypoxic conditions by activating
the transcription of HIF-1α (Fig.
1B-e) (30,42,55).
Furthermore, Zhou et al (39) found that, compared with that in
adjacent tissues and normal hepatocytes, the BCLAF1 protein
expression level in HCC tissues and cell lines was significantly
increased and in a nude mouse xenograft tumor model, knockdown of
BCLAF1 significantly reduced tumor size and tumor growth rate.
Mechanistically, Hsp90α interacted with BCLAF1 and prevented its
subsequent degradation via the proteasome pathway (Fig. 1B-e) (39). Furthermore, BCLAF1 protected the
mature mRNA of the oncogene c-Myc from degradation via its
RS domain, thereby promoting the occurrence and progression of HCC
(Fig. 1A and B-e) (39). In addition, another group also
confirmed that BCLAF1 was highly expressed in HCC specimens
compared with that in adjacent normal tissues, and higher BCLAF1
protein expression levels were associated with higher TNM stage,
poorer differentiation and a less favorable prognosis in patients
with HCC (6). In addition, BCLAF1
induced autophagy in response to starvation of the HCC cells, and
BCLAF1 might increase cell proliferation and prevent cell apoptosis
under stress conditions by inducing autophagy (6). Animal experiments also showed that
BCLAF1 facilitated the tumorigenicity of HCC cells in vivo
(37). In addition, the high
BCLAF1 protein expression levels may lead to sorafenib resistance
in patients with HCC (37).
Consistent with the two aforementioned studies, Mou et al
(31) also verified that BCLAF1
was highly expressed in HCC cells and tissues. Furthermore,
additional research also found that BCLAF1 could bind to the lncRNA
NEAT1 promoter and activate its transcription, thereby promoting
cell proliferation, invasion and resistance to 5-Fu (Fig. 1B-e) (31). A recent study found that BCLAF1
might play a regulatory role in HCC by mediating the glycolytic
pathway: Zhang et al (42)
discovered that knockdown of BCLAF1 could enhance the ability of
ginsenoside CK to activate HIF-1α ubiquitination and inhibit the
glycolysis pathway, mediated by HIF-1α, leading to inhibition of
HCC cell proliferation. In addition, in a mouse model of HCC,
induced by diethylnitrosamine, after the administration of CK, both
the volume and the glucose uptake ability of the tumor was reduced
(42). Multiple studies have also
confirmed that enhanced glycolysis was associated with the
tumorigenesis and progression of tumors (89,90).
Collectively, these studies showed that BCLAF1 was a tumor-promotor
in HCC, promoting the proliferation, migration and invasion,
angiogenesis and drug resistance of HCC via different
mechanisms.
MM
MM is a clonal malignant tumor caused by the
uncontrolled proliferation of immunoglobulin-secreting plasma cells
in the bone marrow and it accounts for 10% of all hematological
malignancies (91). Lamy et
al (21) used short hairpin
RNAs in a retroviral library to identify essential pathways in MM
and to determine potential therapeutic targets in MM. It was found
that caspase-10 was essential for MM viability and did not induce
apoptosis, but instead blocked autophagy-dependent cell death
pathways in MM (21). Caspase-10
has been reported to be involved in the formation of death-inducing
signaling complex (DISC) and induced the proteolysis of their
downstream targets, which resulted in the initiation of extrinsic
apoptosis (92). However, it has
been reported that caspase-10 can change the apoptotic cell death
response following the formation of DISC to activate NF-κB and cell
survival (93). Furthermore, the
gene expression analysis following caspase-10 knockdown revealed
that BCLAF1 was one of the upregulated genes. In particular,
the level of BCLAF1 protein induction was greater than that for the
mRNA expression level (21). This
suggests that BCLAF1 may be a substrate of caspase-10. Further
in vitro experiments indicated that caspase-10 protected MM
cells from autophagic death, induced by BCLAF1 from the cleavage of
aspartic acid at position 452 in BCLAF1 (Fig. 1A and B-c) (21). As aforementioned, BCLAF1 is a
strong autophagy inducer by replacing beclin-1 in Bcl-2 (Fig. 1B-c) (21,72).
These results verified that the interaction between BCLAF1 and
Bcl-2 increased following caspase-10 inhibition, leading to the
dissociation of beclin-1 from Bcl-2, thereby initiating autophagy.
Overall, BCLAF1 could act as a tumor suppressor by inducing the
autophagic death of MM cells.
DLBCL
DLBCL is a biologically and clinically
heterogeneous B-cell tumor. Its morphology is characterized by
large lymphocytes and B-cell markers proliferating rapidly and in a
diffuse pattern (94). HDACi are a
new type of drug for the treatment of hematological malignancies,
which can increase histone acetylation, induce apoptosis and
differentiation, and inhibit the proliferation of tumor cells
(95–97). LMK-235 is a specific inhibitor of
HDAC4 and HDAC5 (98). A study has
shown that after treating DLBCL cells with LMK-235, both the mRNA
and protein expression level of BCLAF1 and the rate of apoptosis
increased (35). Similar results
were observed after the HDAC4 gene was knocked down
(35). In addition, LMK-235
increased the BCLAF1 protein expression level by inhibiting the
NF-κB pathway (35). Furthermore,
both LMK-235 and small interfering (si)RNA-HDAC4 inhibited the
activation of NF-κB and increased the BCLAF1 protein expression
level, and the NF-κB inhibitor, Bay11-7082 increased the BCLAF1
protein expression level (Fig.
1B-h) (35).
In summary, BCLAF1 may represent a potential tumor
suppressor in DLBCL, and LMK-235 increases the BCLAF1 protein
expression level by inhibiting the activation of NF-κB, then
promoting the apoptosis of DLBCL cells. Notably, previous studies
found that NF-κB inhibited the BCLAF1 mRNA expression level
in different cellular processes. Shao et al (8) showed that BCLAF1 was a direct
downstream target of NF-κB, which bound to the BCLAF1 promoter and
activated its transcription during therapeutic drug
doxorubicin-induced senescence. Similarly, another study showed
that in the regulation of BCLAF1 histone acetylation, Rel-A bound
to the BCLAF1 promoter and activated its transcription (24). A recent study suggested that BCLAF1
might play a pro-apoptotic effect in DLBCL through negative
regulation by NF-κB (Fig. 1B-h)
(35); however, the specific
regulatory mechanism is still unclear. As it has been previously
reported that NF-κB could activate the transcription of BCLAF1,
further studies are required to verify whether BCLAF1 could also be
involved in the development of DLBCL via the transcriptional
regulation of NF-κB.
AML
AML is a type of cancer derived from the myeloid
lineage of blood cells. It is characterized by the excessive
production of leukemic mother cells, but the survival rate is still
low due to the high recurrence rate (99). BCLAF1 mRNA and protein
expression level was found to be highly expressed in AML cells
compared with that in normal CD34+ cells, while the
opposite effect was observed with respect to the level of
miR-194-5p (38). In addition,
lower BCLAF1 mRNA and protein expression levels (and higher
miR-194-5p expression levels) were associated with a favorable
prognosis based on the 5-year survival rate (38). It was further found that miR-194-5p
bound to the 3′-UTR of BCLAF1 and inhibited the translation of
BCLAF1 in AML (Fig. 1B-c)
(38). Furthermore, overexpression
of miR-194-5p and the accompanying decrease in the expression of
BCLAF1 resulted in G1 cell cycle arrest and caspase
9-dependent activation of apoptosis in AML cells (38). In addition, BCLAF1 knockdown cells
were more sensitive to the AML treatment drug, suberoylanilide
hydroxamic acid (a HDACi), and the proliferation and number of AML
colonies formed were notably reduced (38). All these results showed that BCLAF1
might be a potential tumor-promoting factor in AML.
BCLAF1 and RT
RT is an important treatment modality for localized
tumors (100). RT produces DNA
DSBs using high-energy IR, which induces cell cycle arrest,
senescence and various tumor cell death modes, including apoptosis,
autophagy, necrosis and mitotic catastrophes (101). After IR-induced cellular DNA
damage, H2AX responds to DSBs by phosphorylating
serine139 (γh2AX) and subsequently recruits various
proteins, that are associated with apoptosis and repair of DNA
damage, such as BRCA1 and RAD51 (102). A number of studies have
associated BCLAF1 with DDR. Liu et al (9) first reported that BCLAF1 promoted the
transcription of TP53 by forming a complex with PKCδ in response to
DNA damage. Lee et al (7)
found that the complex formed by BCLAF1 and γH2AX was co-localized
to the damaged DNA and could stabilize the Ku70/DNA-PKC complex to
facilitate the NHEJ pathway of DSB repair (Fig. 1B-a). Similarly, in the latest
research, IR induced phosphorylation of serine290 in
BCLAF1 and the co-localization of BCLAF1 with γH2AX, which promoted
DDR and radiotherapy resistance of gastric cancer cells (Fig. 1B-i) (11). In addition, Savage et al
(3) discovered that BCLAF1 was
identified in the protein interacting with BRCA1, which mediated
the formation of the BRCA1-mRNA splice complex after DNA damage,
and knockdown of BCLAF1 increased cell sensitivity to IR. Similar
results were found in a comparable study (4). Notably, a recent report showed that
after IR induction, BCLAF1 increased the mRNA expression level of
programmed cell death ligand 1 (PD-L1) via the DNA DSB repair
pathway, thereby increasing the resistance of the cells to IR
(10). It is well-known that PD-L1
is a negative regulator of the immune response and blocking
antibodies targeting PD-L1 can reactivate immune surveillance
(103,104). In addition, mounting evidence has
revealed that the synergistic combination of radiotherapy and
immunotherapy could provide significant therapeutic effects in
NSCLC (105,106). Therefore, it would be important
to further investigate the role of BCLAF1 in the combined treatment
of tumors with radiotherapy and chemotherapy.
Taken together, numerous studies have shown that
BCLAF1 is a DDR-associated protein and serves an important role in
DDR (3,4,7,9,10,11).
In addition, for normal cells, DDR is a key pathway for maintaining
genomic stability and preventing oncogenic transformation (100). Whereas, for tumor cells, high
levels of DDR may increase IR-induced DSB, promote tumor cell
resistance to RT and eventually lead to treatment failure (3,4,10,11).
Therefore, targeting BCLAF1 may be an effective therapeutic
strategy against radioresistance in the future.
Roles of BCLAF1 in virus replication
In addition to participation in the occurrence and
development of several types of malignant tumor, studies have also
found that BCLAF1 serves an important role in virus replication.
Ziegelbauer et al (15)
discovered, for the first time, that BCLAF1 had a negative
regulatory role in the replication of KSHV. Further analysis showed
that BCLAF1 was the target of KSHV-encoded miR-K5, by binding to
the 3′-UTR of BCLAF1, thus decreasing the mRNA expression level of
BCLAF1 (15). KSHV, as a
hemolytic virus, is the etiological agent of some malignant tumors
associated with acquired immunodeficiency syndrome, such as
Kaposi's sarcoma, variants of multicentric Castleman disease and
primary effusion lymphoma (107–111). Furthermore, another study
demonstrated that decreased BCLAF1 protein expression level
enhanced HCMV gene expression, while increased BCLAF1
protein expression level inhibited virus replication (13). Further analysis found that the
proteasome degradation of BCLAF1 was induced by releasing viral
particle proteins in the early stage after HCMV infection, such as
pp71 and UL35 proteins (Fig. 1B-d)
(13). In the later stage of HCMV
infection, the miR-UL112-1 encoded by HCMV, bound to the 3′-UTR of
BCLAF1 and inhibited its translation (13). HCMV is a widespread β-herpes virus
that can cause diseases in individuals with immature or impaired
immune systems (112), such as
atherosclerosis, vascular disease and immune aging as well as
increase the risk in developing different types of tumor (113,114). Furthermore, a recent study
indicated that knockdown of BCLAF1 could significantly promote the
replication of US3-deficient α-herpesvirus pseudorabies virus (PRV)
and herpes simplex virus type 1 (HSV-1) (14). Mechanistic analysis found that
BCLAF1 could repress viral replication by enhancing type I
interferon signaling (14).
Conversely, PRV induced the degradation of BCLAF1 in host cells
during infection via its expressed viral protein, US3 (Fig. 1B-d) (14). Type I interferon response serves an
important role in combating viral infections and it is often
dysregulated in viral infections (115). In a similar manner to KSHV and
HCMV, both PRV and HSV-1 belong to the α-herpesvirus subfamily.
In summary, the aforementioned studies have
uncovered the role of BCLAF1 in herpes virus defense, and more
importantly, that BCLAF1 is also the target of multiple viral
components. However, BCLAF1 may play a different role in human
papillomaviruses (HPV)16. HPV16 is a small DNA virus that infects
keratinocytes of the squamous and mucosal epithelium (116,117). In one study, in cells with
induced DNA damage, BCLAF1 was recruited to the HPV16 DNA by
co-recruitment with other DDR factors (such as BRCA1 and BARD1) and
RNA processing factors (such as U2AF65 and SF3b), then induced
replication of HPV DNA and HPV late gene expression (16), indicating the role of BCLAF1 in
promoting HPV replication. Taken together, these studies showed
that BCLAF1 may serve a contradictory role in viral infections. On
the one hand, BCLAF1 represses the replication and infection of
some viruses, such as herpes viruses, including KSHV, HCMV, PRV and
HSV-1; on the other hand, BCLAF1 promotes the DNA replication and
gene expression of HPV16 (Table IV).
Roles of BCLAF1 in cardiac I/R injury
I/R injury, characterized by oxidative stress,
inflammation and organ dysfunction (118,119), is the initial temporary blockage
of blood supply to tissues and organs, followed by the recovery of
perfused blood supply and accompanying reoxygenation, known as
reperfusion injury of tissues and organs. It has recently been
reported that overexpression of BCLAF1 increased cardiac I/R injury
and increased the infarct size in the heart of a I/R mouse model,
and knockdown of BCLAF1 reduced cardiac I/R injury (23). Mechanistically, the translocation
of BCLAF1 to the nucleus increased the expression and activity of
apoptosis-related proteins, such as TP53, BAX and caspase-3 induced
by I/R injury, then promoted the apoptosis of cardiomyocytes
(Fig. 1B-k) (23). In addition, further research has
demonstrated that lncCIRBIL was bound to the BCLAF1 protein in the
cytoplasm of cardiomyocytes to prevent its translocation to the
nucleus, thus repressing cardiac I/R injury (Fig. 1B-k) (23). In summary, BCLAF1 could act as a
cardiomyocyte protective factor in cardiac I/R injury and mediate
the regulation of lncCIRBIL in cardiac I/R injury (Table IV).
Conclusions
In conclusion, BCLAF1 has complex physiological and
pathological functions, and exerts these biological functions from
the regulation of its upstream molecules and the downstream target
molecules. On the one hand, BCLAF1, as a downstream molecule, is
mostly regulated at the transcriptional and post-transcriptional
level. For example, NF-κB can regulate BCLAF1 transcription by
binding to the promoter of BCLAF1 (Fig. 1B-f and h) (8,24,35)
and Sirt1 can inhibit BCLAF1 transcription by deacetylating the
H3K56 at the BCLAF1 promoter (Fig.
1B-f) (24). In addition,
several studies have found that BCLAF1 was phosphorylated at
different sites associated with the repair of DNA damage (7,31).
Notably, mass spectrometry found >30 phosphorylation sites
within BCLAF1 (120); however,
the significance and function of these sites have not been
reported. Therefore, post-translational modification (such as
phosphorylation) of BCLAF1 may be one of the key forms for
regulating its function, and further research is required.
On the other hand, BCLAF1 regulates its downstream
targets at the transcriptional and post-transcriptional level to
participate in numerous physiological and pathological processes.
For example, BCLAF1 participates in DDR and apoptosis by activating
the transcription of TP53 and BAX (Fig. 1B-a) (7,9).
Furthermore, BCLAF1 has been associated with colon cancer and found
to activate the transcription of TP53 and BAX, and inhibit MDM2
(Fig. 1B-b) (32). In addition, BCLAF1 was associated
with the development of HCC by activating the transcription of
HIF-1α and lncRNA NEAT1 (Fig.
1B-e) (30,31). Lastly, BCLAF1 was found to be an
important splicing factor to maintain the stability of c-Myc
mRNA expression in HCC (Fig. 1A and
B-e) (39).
Given the subcellular localization of BCLAF1, most
of the previous research found that BCLAF1 primarily localizes in
the nucleus and in a small amount in the cytoplasm (1,7,18).
Under different conditions, the localization of BCLAF1 can change,
which is accompanied by functional changes. For example, the
anti-apoptotic members of the Bcl-2 family inhibit the
pro-apoptotic effect of BCLAF1 by isolating BCLAF1 in the cytoplasm
(1). Similarly, in
apoptosis-induced cells, BCLAF1 is translocated to dot-like
structures in the nucleus to exert its pro-apoptotic function
(18). However, further research
is required to reveal the association between the subcellular
localization of BCLAF1 and its complex functions.
An increasing amount of evidence has shown that
BCLAF1 can exert carcinogenic and antitumor effects in specific
types of cancer and in a cell background-specific manner. For
example, BCLAF1 has been found to plays a potential tumor
suppressor effect in MM (21) and
DLBCL (35), but has a
tumor-promoting effect in AML (38), HCC (30,31,37,39,42)
and gastric cancer (11). Even in
some tumor types (such as BC, LC and CRC), it has been observed
that BCLAF1 is both a tumor suppressor (7,32,43)
and a tumor promoter (22,34,40,41).
The reasons for this may be due to the tumor microenvironment or
tumor heterogeneity, or differences in biological samples. Given
multiple tumors are characterized by significant heterogeneity and
complex microenvironment (73,74,87),
these studies may use different biological samples (human cancer
tissues, tumor cell lines and animal models). In addition, the
human BCLAF1 gene encodes several transcriptional variants;
therefore, the complex structure of the BCLAF1 protein itself may
also mediate its complex and even contradictory effects in
tumorigenesis. Thus, the specific mechanism requires further
investigation to clarify its different roles. For example, the
establishment of a systemic and tissue-specific BCLAF1 gene
knockout mouse model may provide a comprehensive understanding of
the physiological and pathological functions of BCLAF1,
particularly in tumorigenesis (121). In addition, attention should be
paid to the role of BCLAF1 in pathological processes other than
tumorigeneses, such as viral infection and cardiac I/R injury. Some
progress has been made in the investigation into the role and
mechanisms of BCLAF1 in virus infection; however, contradictory
results have been observed. For example, BCLAF1 acted as a viral
inhibitor in viral infections, such as KSHV, HCMV and PRV, but
acted as a viral replication-promoting factor in HPV16 (13–16).
Notably, BCLAF1 has recently been found to promote cardiac I/R
injury by mediating cardiomyocyte apoptosis (23), which was the first time to
associate the regulation of BCLAF1 in apoptosis with cardiac I/R
injury.
Given the complex and even contradictory roles of
BCLAF1 in tumorigenesis, BCLAF1 has been classified and discussed
based on its roles in tumorigenesis to identify targets for
specific therapy. In tumor types in which BCLAF1 acts as a tumor
promoter (such as HCC and AML), the pathogenic pathway of BCLAF1
may be blocked by targeting some key signaling molecules. For
example, targeted inhibition of HSP90, HIF-1α and lncRNA NEAT1 in
HCC may be considered, while in AML, upregulation of miR-194-5p
could be considered. By contrast, in other tumor types in which
BCLAF1 acts as a tumor suppressor (such as DLBCL and MM), the
anti-cancer pathway of BCLAF1 may be activated by targeting some
key signaling molecules. For example, in DLBCL, inhibition of NF-κB
may be effective, while in MM, caspase-10 could be inhibited. Of
course, more in-depth research is required to determine the exact
roles of BCLAF1 in tumorigenesis, particularly in different types
of cancer with paradoxical effects of BCLAF1 (including BC, LC and
CRC), thus making it possible to perform targeted therapy. Notably,
although survival analysis of data from the GEPIA2 database showed
established prognostic value of BCLAF1 in BC (high expression of
BCLAF1 was associated with a less favorable prognosis) (Fig. 3B), there are no reports of the
association between BCLAF1 and BC; therefore, more studies into the
mechanism of BCLAF1 in BC is required.
Taken together, in vitro, in vivo and
studies using clinical samples have aided in the understanding of
the role of BCLAF1 in the occurrence, progression and resistance of
different types of cancer. As BCLAF1 plays an important role in key
cancer-related signaling pathways, regulating the activity or
abundance of this key molecule may provide a new therapeutic
strategy for patients with cancer in the future.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural Science
Foundation of Zhejiang Province (grant no. LY20C070001), the
Natural Science Foundation of Ningbo (grant nos. 202003N4197 and
2021J294), the National Natural Science Foundation of China (grant.
no. 31801165) and the K.C. Wong Magna Fund in Ningbo
University.
Availability of data and materials
The datasets used during the current study are
available in the Human Protein Atlas (https://www.proteinatlas.org/ENSG00000029363-BCLAF1),
GEPIA2 (http://gepia2.cancer-pku.cn/#survival), ICGC
(https://dcc.icgc.org/genes/ENSG00000029363) and the
PROMO databases (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3).
Authors' contributions
XJ and HL conceived the study. ZY and JZ drafted
the manuscript. HW made substantial contributions to the
interpretation, drafting the manuscript and revising it critically
for important intellectual content. Data sharing is not applicable.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
BCLAF1
|
B-cell lymphoma-2-associated
transcription factor 1
|
|
RS rich domain
|
arginine-serine rich domain
|
|
bZIP domain
|
basic-leucine zipper domain
|
|
DDR
|
DNA damage response
|
|
SMYD3
|
histone methyltransferase SET and
MYND domain-containing protein 3
|
|
miR
|
microRNA
|
|
DNA-PKC
|
DNA-protein kinase C
|
|
RNP
|
ribonucleoprotein
|
|
DLBCL
|
diffuse large B-cell lymphoma
|
|
HIF-1α
|
hypoxia inducible factor-1α
|
|
HCC
|
hepatocellular carcinoma
|
|
H3K56
|
histone 3 lysine 56 residues
|
|
HDACi
|
histone deacetylase inhibitor
|
|
CNS
|
conserved non-coding sequences
|
|
EDMD
|
Emery–Dreifuss muscular dystrophy
|
|
AML
|
acute myelocytic leukemia
|
|
KSHV
|
Kaposi's sarcoma-associated
herpesvirus
|
|
HCMV
|
human cytomegalovirus
|
|
Hsp90α
|
heat shock protein 90α
|
|
CPE-TP53
|
TP53 core promoter element
|
|
IR
|
ionizing radiation
|
|
DSB
|
double-strand break
|
|
γH2AX
|
H2AX phosphorylated on serine 139
|
|
lncRNA NEAT1
|
long non-coding RNA nuclear
enrichment-rich transcription factor 1
|
|
5-Fu
|
5-fluorouracil
|
|
BC
|
bladder cancer
|
|
H3K4
|
histone H3 lysine 4
|
|
Bcl-2
|
B-cell lymphoma-2
|
|
LC
|
lung cancer
|
|
NHEJ
|
non-homologous end-joining
|
|
dsDNA
|
double-stranded DNA
|
|
FHL1
|
four-and-a-half LIM protein 1
|
|
NSCLC
|
non-small cell lung cancer
|
|
USP22
|
ubiquitin-specific peptidase 22
|
|
MM
|
multiple myeloma
|
|
DISC
|
death-inducing signaling complex
|
|
CRC
|
colorectal cancer
|
|
L isoform
|
large protein of 920 amino acids
|
|
CK
|
compound K
|
|
PRV
|
pseudorabies virus
|
|
HSV-1
|
herpes simplex virus type 1
|
|
HPV16
|
human papillomaviruses 16
|
|
I/R injury
|
ischemia-reperfusion injury
|
|
PD-L1
|
programmed cell death ligand 1
|
|
3′UTR
|
3′-untranslated region
|
References
|
1
|
Kasof GM, Goyal L and White E: Btf, a
novel death-promoting transcriptional repressor that interacts with
Bcl-2-related proteins. Mol Cell Biol. 19:4390–4404. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bracken CP, Wall SJ, Barré B, Panov KI,
Ajuh PM and Perkins ND: Regulation of cyclin D1 RNA stability by
SNIP1. Cancer Res. 68:7621–7628. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Savage KI, Gorski JJ, Barros EM, Irwin GW,
Manti L, Powell AJ, Pellagatti A, Lukashchuk N, McCance DJ,
McCluggage WG, et al: Identification of a BRCA1-mRNA splicing
complex required for efficient DNA repair and maintenance of
genomic stability. Mol Cell. 54:445–459. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vohhodina J, Barros EM, Savage AL,
Liberante FG, Manti L, Bankhead P, Cosgrove N, Madden AF, Harkin DP
and Savage KI: The RNA processing factors THRAP3 and BCLAF1 promote
the DNA damage response through selective mRNA splicing and nuclear
export. Nucleic Acids Res. 45:12816–12833. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Varia S, Cheedu D, Markey M, Torres-Shafer
K, Battini VP, Bubulya A and Bubulya PA: Alignment of Mitotic
Chromosomes in Human Cells Involves SR-Like Splicing Factors Btf
and TRAP150. Int J Mol Sci. 18:19562017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Merz C, Urlaub H, Will CL and Lührmann R:
Protein composition of human mRNPs spliced in vitro and
differential requirements for mRNP protein recruitment. RNA.
13:116–128. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee YY, Yu YB, Gunawardena HP, Xie L and
Chen X: BCLAF1 is a radiation-induced H2AX-interacting partner
involved in γH2AX-mediated regulation of apoptosis and DNA repair.
Cell Death Dis. 3:e3592012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shao AW, Sun H, Geng Y, Peng Q, Wang P,
Chen J, Xiong T, Cao R and Tang J: Bclaf1 is an important NF-κB
signaling transducer and C/EBPβ regulator in DNA damage-induced
senescence. Cell Death Differ. 23:865–875. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu H, Lu ZG, Miki Y and Yoshida K:
Protein kinase C delta induces transcription of the TP53 tumor
suppressor gene by controlling death-promoting factor Btf in the
apoptotic response to DNA damage. Mol Cell Biol. 27:8480–8491.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma Z, Wang H, Meng F, Han Y, Chen Y, Xiao
M, Jiang H, Yu Z and Xu B: Role of BCLAF-1 in PD-L1 stabilization
in response to ionizing irradiation. Cancer Sci. 112:4064–4074.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu J, Li J, Sun Z, Duan Y, Wang F, Wei G
and Yang JH: Bcl-2-associated transcription factor 1
Ser290 phosphorylation mediates DNA damage response and
regulates radiosensitivity in gastric cancer. J Transl Med.
19:3392021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McPherson JP, Sarras H, Lemmers B, Tamblyn
L, Migon E, Matysiak-Zablocki E, Hakem A, Azami SA, Cardoso R, Fish
J, et al: Essential role for Bclaf1 in lung development and immune
system function. Cell Death Differ. 16:331–339. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee SH, Kalejta RF, Kerry J, Semmes OJ,
O'Connor CM, Khan Z, Garcia BA, Shenk T and Murphy E: BclAF1
restriction factor is neutralized by proteasomal degradation and
microRNA repression during human cytomegalovirus infection. Proc
Natl Acad Sci USA. 109:9575–9580. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qin C, Zhang R, Lang Y, Shao A, Xu A, Feng
W, Han J, Wang M, He W, Yu C, et al: Bclaf1 critically regulates
the type I interferon response and is degraded by alphaherpesvirus
US3. PLoS Pathog. 15:e10075592019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ziegelbauer JM, Sullivan CS and Ganem D:
Tandem array-based expression screens identify host mRNA targets of
virus-encoded microRNAs. Nat Genet. 41:130–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nilsson K, Wu C and Schwartz S: Role of
the DNA Damage Response in Human Papillomavirus RNA Splicing and
Polyadenylation. Int J Mol Sci. 19:17352018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Meinke P, Nguyen TD and Wehnert MS: The
LINC complex and human disease. Biochem Soc Trans. 39:1693–1697.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Haraguchi T, Holaska JM, Yamane M, Koujin
T, Hashiguchi N, Mori C, Wilson KL and Hiraoka Y: Emerin binding to
Btf, a death-promoting transcriptional repressor, is disrupted by a
missense mutation that causes Emery-Dreifuss muscular dystrophy.
Eur J Biochem. 271:1035–1045. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lowe M, Lage J, Paatela E, Munson D,
Hostager R, Yuan C, Katoku-Kikyo N, Ruiz-Estevez M, Asakura Y,
Staats J, et al: Cry2 Is Critical for Circadian Regulation of
Myogenic Differentiation by Bclaf1-Mediated mRNA Stabilization of
Cyclin D1 and Tmem176b. Cell Rep. 22:2118–2132. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Y, Li M, Wang Y, Liu J, Zhang M, Fang
X, Chen H and Zhang C: A Zfp609 circular RNA regulates myoblast
differentiation by sponging miR-194-5p. Int J Biol Macromol.
121:1308–1313. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lamy L, Ngo VN, Emre NC, Shaffer AL III,
Yang Y, Tian E, Nair V, Kruhlak MJ, Zingone A, Landgren O, et al:
Control of autophagic cell death by caspase-10 in multiple myeloma.
Cancer Cell. 23:435–449. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shen B, Tan M, Mu X, Qin Y, Zhang F, Liu Y
and Fan Y: Upregulated SMYD3 promotes bladder cancer progression by
targeting BCLAF1 and activating autophagy. Tumour Biol.
37:7371–7381. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Y, Zhang X, Cai B, Li Y, Jiang Y, Fu
X, Zhao Y, Gao H, Yang Y, Yang J, et al: The long noncoding RNA
lncCIRBIL disrupts the nuclear translocation of Bclaf1 alleviating
cardiac ischemia-reperfusion injury. Nat Commun. 12:5222021.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kong S, Kim SJ, Sandal B, Lee SM, Gao B,
Zhang DD and Fang D: The type III histone deacetylase Sirt1 protein
suppresses p300-mediated histone H3 lysine 56 acetylation at Bclaf1
promoter to inhibit T cell activation. J Biol Chem.
286:16967–16975. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jarboui MA, Wynne K, Elia G, Hall WW and
Gautier VW: Proteomic profiling of the human T-cell nucleolus. Mol
Immunol. 49:441–452. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Philipps D, Celotto AM, Wang QQ, Tarng RS
and Graveley BR: Arginine/serine repeats are sufficient to
constitute a splicing activation domain. Nucleic Acids Res.
31:6502–6508. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schaal TD and Maniatis T: Multiple
distinct splicing enhancers in the protein-coding sequences of a
constitutively spliced pre-mRNA. Mol Cell Biol. 19:261–273. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Smith CW and Valcárcel J: Alternative
pre-mRNA splicing: The logic of combinatorial control. Trends
Biochem Sci. 25:381–388. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sarras H, Alizadeh Azami S and McPherson
JP: In search of a function for BCLAF1. ScientificWorldJournal.
10:1450–1461. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wen Y, Zhou X, Lu M, He M, Tian Y, Liu L,
Wang M, Tan W, Deng Y, Yang X, et al: Bclaf1 promotes angiogenesis
by regulating HIF-1α transcription in hepatocellular carcinoma.
Oncogene. 38:1845–1859. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mou SJ, Yang PF, Liu YP, Xu N, Jiang WW
and Yue WJ: BCLAF1 promotes cell proliferation, invasion and
drug-resistance though targeting lncRNA NEAT1 in hepatocellular
carcinoma. Life Sci. 242:1171772020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rénert AF, Leprince P, Dieu M, Renaut J,
Raes M, Bours V, Chapelle JP, Piette J, Merville MP and Fillet M:
The proapoptotic C16-ceramide-dependent pathway requires the
death-promoting factor Btf in colon adenocarcinoma cells. J
Proteome Res. 8:4810–4822. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Orieux G, Picault L, Slembrouck A, Roger
JE, Guillonneau X, Sahel JA, Saule S, McPherson JP and Goureau O:
Involvement of Bcl-2-associated transcription factor 1 in the
differentiation of early-born retinal cells. J Neurosci.
34:1530–1541. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhou X, Li X, Cheng Y, Wu W, Xie Z, Xi Q,
Han J, Wu G, Fang J and Feng Y: BCLAF1 and its splicing regulator
SRSF10 regulate the tumorigenic potential of colon cancer cells.
Nat Commun. 5:45812014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li X, He Z, Cheng B, Fang Q, Ma D, Lu T,
Wei D, Kuang X, Tang S, Xiong J, et al: Effect of BCLAF1 on HDAC
inhibitor LMK-235-mediated apoptosis of diffuse large B cell
lymphoma cells and its mechanism. Cancer Biol Ther. 19:825–834.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen M, Zhang R, Lu L, Du J, Chen C, Ding
K, Wei X, Zhang G, Huang Y and Hou J: lncRNA PVT1 accelerates
malignant phenotypes of bladder cancer cells by modulating
miR-194-5p/BCLAF1 axis as a ceRNA. Aging (Albany NY).
12:22291–22312. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yu S, Wang X, Dou N, Zhou J, Gao Y and Li
Y: B-cell lymphoma-2-associated transcription factor 1 is
overexpressed and contributes to sorafenib resistance in
hepatocellular carcinoma. Hepatol Res. 49:1329–1340. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dell'Aversana C, Giorgio C, D'Amato L,
Lania G, Matarese F, Saeed S, Di Costanzo A, Belsito Petrizzi V,
Ingenito C, Martens JHA, et al: miR-194-5p/BCLAF1 deregulation in
AML tumorigenesis. Leukemia. 31:2315–2325. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhou X, Wen Y, Tian Y, He M, Ke X, Huang
Z, He Y, Liu L, Scharf A, Lu M, et al: Heat Shock Protein
90α-Dependent B-Cell-2-Associated Transcription Factor 1 Promotes
Hepatocellular Carcinoma Proliferation by Regulating MYC
Proto-Oncogene c-MYC mRNA Stability. Hepatology. 69:1564–1581.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang X, Wei X, Yuan Y, Sun Q, Zhan J,
Zhang J, Tang Y, Li F, Ding L, Ye Q, et al: Src-mediated
phosphorylation converts FHL1 from tumor suppressor to tumor
promoter. J Cell Biol. 217:1335–1351. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jiang T, Liu B, Wu D and Zhang F: BCLAF1
induces cisplatin resistance in lung cancer cells. Oncol Lett.
20:2272020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang S, Zhang M, Chen J, Zhao J, Su J and
Zhang X: Ginsenoside Compound K Regulates HIF-1α-Mediated
Glycolysis Through Bclaf1 to Inhibit the Proliferation of Human
Liver Cancer Cells. Front Pharmacol. 11:5833342020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yoshitomi T, Kawakami K, Enokida H,
Chiyomaru T, Kagara I, Tatarano S, Yoshino H, Arimura H, Nishiyama
K, Seki N, et al: Restoration of miR-517a expression induces cell
apoptosis in bladder cancer cell lines. Oncol Rep. 25:1661–1668.
2011.PubMed/NCBI
|
|
44
|
The National Center for Biotechnology
Information [OL], BCLAF1 BCL2 associated transcription factor 1
[Homo sapiens (human)]. https://www.ncbi.nlm.nih.gov/gene/9774November
7–2021.
|
|
45
|
Black DL: Mechanisms of alternative
pre-messenger RNA splicing. Annu Rev Biochem. 72:291–336. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wahl MC, Will CL and Lührmann R: The
spliceosome: Design principles of a dynamic RNP machine. Cell.
136:701–718. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wu JY and Maniatis T: Specific
interactions between proteins implicated in splice site selection
and regulated alternative splicing. Cell. 75:1061–1070. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kohtz JD, Jamison SF, Will CL, Zuo P,
Lührmann R, Garcia-Blanco MA and Manley JL: Protein-protein
interactions and 5′-splice-site recognition in mammalian mRNA
precursors. Nature. 368:119–124. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ramsay RG and Gonda TJ: MYB function in
normal and cancer cells. Nat Rev Cancer. 8:523–534. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Shaulian E and Karin M: AP-1 as a
regulator of cell life and death. Nat Cell Biol. 4:E131–E136. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Nerlov C: The C/EBP family of
transcription factors: A paradigm for interaction between gene
expression and proliferation control. Trends Cell Biol. 17:318–324.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
LaGory EL and Giaccia AJ: The
ever-expanding role of HIF in tumour and stromal biology. Nat Cell
Biol. 18:356–365. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Keith B, Johnson RS and Simon MC: HIF1α
and HIF2α: Sibling rivalry in hypoxic tumour growth and
progression. Nat Rev Cancer. 12:9–22. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Shao A, Lang Y, Wang M, Qin C, Kuang Y,
Mei Y, Lin D, Zhang S and Tang J: Bclaf1 is a direct target of
HIF-1 and critically regulates the stability of HIF-1α under
hypoxia. Oncogene. 39:2807–2818. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Liu F, Tai Y and Ma J: lncRNA
NEAT1/let-7a-5p axis regulates the cisplatin resistance in
nasopharyngeal carcinoma by targeting Rsf-1 and modulating the
Ras-MAPK pathway. Cancer Biol Ther. 19:534–542. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhang J, Zhao B, Chen X, Wang Z, Xu H and
Huang B: Silence of Long Noncoding RNA NEAT1 Inhibits Malignant
Biological Behaviors and Chemotherapy Resistance in Gastric Cancer.
Pathol Oncol Res. 24:109–113. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
An J, Lv W and Zhang Y: lncRNA NEAT1
contributes to paclitaxel resistance of ovarian cancer cells by
regulating ZEB1 expression via miR-194. OncoTargets Ther.
10:5377–5390. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Maloney A and Workman P: HSP90 as a new
therapeutic target for cancer therapy: The story unfolds. Expert
Opin Biol Ther. 2:3–24. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sirvent N, Imbert V, Frelin C, Griessinger
E and Peyron JF: Fighting cancer via NF-kappa B inhibition. Arch
Pediatr. 10:632–634. 2003.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Haefner B: NF-κB: Arresting a major
culprit in cancer. Drug Discov Today. 7:653–663. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Barkett M and Gilmore TD: Control of
apoptosis by Rel/NF-kappaB transcription factors. Oncogene.
18:6910–6924. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Karin M, Cao Y, Greten FR and Li Z-W:
NF-kappaB in cancer: From innocent bystander to major culprit. Nat
Rev Cancer. 2:301–310. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
64
|
Karin M and Lin A: NF-kappaB at the
crossroads of life and death. Nat Immunol. 3:221–227. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wilson GK, Tennant DA and McKeating JA:
Hypoxia inducible factors in liver disease and hepatocellular
carcinoma: Current understanding and future directions. J Hepatol.
61:1397–1406. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kim DY, Yuan HD, Chung IK and Chung SH:
Compound K, intestinal metabolite of ginsenoside, attenuates
hepatic lipid accumulation via AMPK activation in human hepatoma
cells. J Agric Food Chem. 57:1532–1537. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Luo M, Li Z, Wang W, Zeng Y, Liu Z and Qiu
J: Long non-coding RNA H19 increases bladder cancer metastasis by
associating with EZH2 and inhibiting E-cadherin expression. Cancer
Lett. 333:213–221. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Varier RA and Timmers HT: Histone lysine
methylation and demethylation pathways in cancer. Biochim Biophys
Acta. 1815:75–89. 2011.PubMed/NCBI
|
|
70
|
Bian Y, Li W, Kremer DM, Sajjakulnukit P,
Li S, Crespo J, Nwosu ZC, Zhang L, Czerwonka A, Pawłowska A, et al:
Cancer SLC43A2 alters T cell methionine metabolism and histone
methylation. Nature. 585:277–282. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Luo XG, Zhang CL, Zhao WW, Liu ZP, Liu L,
Mu A, Guo S, Wang N, Zhou H and Zhang TC: Histone methyltransferase
SMYD3 promotes MRTF-A-mediated transactivation of MYL9 and
migration of MCF-7 breast cancer cells. Cancer Lett. 344:129–137.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Fenizia C, Bottino C, Corbetta S,
Fittipaldi R, Floris P, Gaudenzi G, Carra S, Cotelli F, Vitale G
and Caretti G: SMYD3 promotes the epithelial-mesenchymal transition
in breast cancer. Nucleic Acids Res. 47:1278–1293. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
da Costa JB, Gibb EA, Nykopp TK, Mannas M,
Wyatt AW and Black PC: Molecular tumor heterogeneity in muscle
invasive bladder cancer: Biomarkers, subtypes, and implications for
therapy. Urol Oncol. 2018:S1078-1439(18)30463-0. 2018.
|
|
75
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global Cancer Statistics 2020:
GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36
Cancers in 185 Countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Burma S, Chen BP and Chen DJ: Role of
non-homologous end joining (NHEJ) in maintaining genomic integrity.
DNA Repair (Amst). 5:1042–1048. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Wang Q, Gao F, Wang T, Flagg T and Deng X:
A nonhomologous end-joining pathway is required for protein
phosphatase 2A promotion of DNA double-strand break repair.
Neoplasia. 11:1012–1021. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Li Z, Owonikoko TK, Sun SY, Ramalingam SS,
Doetsch PW, Xiao ZQ, Khuri FR, Curran WJ and Deng X: c-Myc
suppression of DNA double-strand break repair. Neoplasia.
14:1190–1202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Niu C, Liang C, Guo J, Cheng L, Zhang H,
Qin X, Zhang Q, Ding L, Yuan B, Xu X, et al: Downregulation and
growth inhibitory role of FHL1 in lung cancer. Int J Cancer.
130:2549–2556. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ding L, Wang Z, Yan J, Yang X, Liu A, Qiu
W, Zhu J, Han J, Zhang H, Lin J, et al: Human four-and-a-half LIM
family members suppress tumor cell growth through a TGF-beta-like
signaling pathway. J Clin Invest. 119:349–361. 2009.PubMed/NCBI
|
|
81
|
Ding L, Niu C, Zheng Y, Xiong Z, Liu Y,
Lin J, Sun H, Huang K, Yang W, Li X, et al: FHL1 interacts with
oestrogen receptors and regulates breast cancer cell growth. J Cell
Mol Med. 15:72–85. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Xu X, Fan Z, Liang C, Li L, Wang L, Liang
Y, Wu J, Chang S, Yan Z, Lv Z, et al: A signature motif in LIM
proteins mediates binding to checkpoint proteins and increases
tumour radiosensitivity. Nat Commun. 8:140592017. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Fennell DA, Summers Y, Cadranel J, Benepal
T, Christoph DC, Lal R, Das M, Maxwell F, Visseren-Grul C and Ferry
D: Cisplatin in the modern era: The backbone of first-line
chemotherapy for non-small cell lung cancer. Cancer Treat Rev.
44:42–50. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Perše M and Večerić-Haler Ž:
Cisplatin-Induced Rodent Model of Kidney Injury: Characteristics
and Challenges. BioMed Res Int. 2018:14628022018. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wang A, Ning Z, Lu C, Gao W, Liang J, Yan
Q, Tan G and Liu J: USP22 Induces Cisplatin Resistance in Lung
Adenocarcinoma by Regulating γH2AX-Mediated DNA Damage Repair and
Ku70/Bax-Mediated Apoptosis. Front Pharmacol. 8:2742017. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Park CH, Eun CS and Han DS: Intestinal
microbiota, chronic inflammation, and colorectal cancer. Intest
Res. 16:338–345. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Kulik L and El-Serag HB: Epidemiology and
management of hepatocellular carcinoma. Gastroenterology.
156:477–491.e1. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Burns JS and Manda G: Metabolic Pathways
of the Warburg Effect in Health and Disease: Perspectives of
Choice, Chain or Chance. Int J Mol Sci. 18:27552017. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Yu L, Chen X, Sun X, Wang L and Chen S:
The Glycolytic Switch in Tumors: How Many Players Are Involved? J
Cancer. 8:3430–3440. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Turesson I, Bjorkholm M, Blimark CH,
Kristinsson S, Velez R and Landgren O: Rapidly changing myeloma
epidemiology in the general population: Increased incidence, older
patients, and longer survival. Eur J Haematol. 101:237–244. 2018.
View Article : Google Scholar
|
|
92
|
Wang J, Chun HJ, Wong W, Spencer DM and
Lenardo MJ: Caspase-10 is an initiator caspase in death receptor
signaling. Proc Natl Acad Sci USA. 98:13884–13888. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Horn S, Hughes MA, Schilling R, Sticht C,
Tenev T, Ploesser M, Meier P, Sprick MR, MacFarlane M and Leverkus
M: Caspase-10 Negatively Regulates Caspase-8-Mediated Cell Death,
Switching the Response to CD95L in Favor of NF-κB Activation and
Cell Survival. Cell Rep. 19:785–797. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Caimi PF, Hill BT, Hsi ED and Smith MR:
Clinical approach to diffuse large B cell lymphoma. Blood Rev.
30:477–491. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Mensah AA, Kwee I, Gaudio E, Rinaldi A,
Ponzoni M, Cascione L, Fossati G, Stathis A, Zucca E, Caprini G, et
al: Novel HDAC inhibitors exhibit pre-clinical efficacy in lymphoma
models and point to the importance of CDKN1A expression levels in
mediating their anti-tumor response. Oncotarget. 6:5059–5071. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Ganai SA: Histone deacetylase inhibitor
givinostat: The small-molecule with promising activity against
therapeutically challenging haematological malignancies. J
Chemother. 28:247–254. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Adams CM, Hiebert SW and Eischen CM: Myc
Induces miRNA-Mediated Apoptosis in Response to HDAC Inhibition in
Hematologic Malignancies. Cancer Res. 76:736–748. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Marek L, Hamacher A, Hansen FK, Kuna K,
Gohlke H, Kassack MU and Kurz T: Histone deacetylase (HDAC)
inhibitors with a novel connecting unit linker region reveal a
selectivity profile for HDAC4 and HDAC5 with improved activity
against chemoresistant cancer cells. J Med Chem. 56:427–436. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Culp-Hill R, D'Alessandro A and Pietras
EM: Extinguishing the Embers: Targeting AML Metabolism. Trends Mol
Med. 27:332–344. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Safi S, Beckhove P, Warth A, Benner A,
Roeder F, Rieken S, Debus J, Dienemann H, Hoffmann H and Huber PE:
A randomized phase II study of radiation induced immune boost in
operable non-small cell lung cancer (RadImmune trial). BMC Cancer.
15:9882015. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Baidoo KE, Yong K and Brechbiel MW:
Molecular pathways: Targeted α-particle radiation therapy. Clin
Cancer Res. 19:530–537. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Matsuoka S, Ballif BA, Smogorzewska A,
McDonald ER III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini
N, Lerenthal Y, et al: ATM and ATR substrate analysis reveals
extensive protein networks responsive to DNA damage. Science.
316:1160–1166. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Keir ME, Liang SC, Guleria I, Latchman YE,
Qipo A, Albacker LA, Koulmanda M, Freeman GJ, Sayegh MH and Sharpe
AH: Tissue expression of PD-L1 mediates peripheral T cell
tolerance. J Exp Med. 203:883–895. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Schreiner B, Bailey SL, Shin T, Chen L and
Miller SD: PD-1 ligands expressed on myeloid-derived APC in the CNS
regulate T-cell responses in EAE. Eur J Immunol. 38:2706–2717.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Antonia SJ, Villegas A, Daniel D, Vicente
D, Murakami S, Hui R, Kurata T, Chiappori A, Lee KH, de Wit M, et
al PACIFIC Investigators, : Overall survival with durvalumab after
chemoradiotherapy in stage III NSCLC. N Engl J Med. 379:2342–2350.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Shaverdian N, Lisberg AE, Bornazyan K,
Veruttipong D, Goldman JW, Formenti SC, Garon EB and Lee P:
Previous radiotherapy and the clinical activity and toxicity of
pembrolizumab in the treatment of non-small-cell lung cancer: A
secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol.
18:895–903. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Chang Y, Cesarman E, Pessin MS, Lee F,
Culpepper J, Knowles DM and Moore PS: Identification of
herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma.
Science. 266:1865–1869. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
1Gao SJ, Kingsley L, Hoover DR, Spira TJ,
Rinaldo CR, Saah A, Phair J, Detels R, Parry P, Chang Y, et al:
Seroconversion to antibodies against Kaposi's sarcoma-associated
herpesvirus-related latent nuclear antigens before the development
of Kaposi's sarcoma. N Engl J Med. 335:233–241. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Cesarman E, Chang Y, Moore PS, Said JW and
Knowles DM: Kaposi's sarcoma-associated herpesvirus-like DNA
sequences in AIDS-related body-cavity-based lymphomas. N Engl J
Med. 332:1186–1191. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Soulier J, Grollet L, Oksenhendler E,
Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael
M, Degos L, et al: Kaposi's sarcoma-associated herpesvirus-like DNA
sequences in multicentric Castleman's disease. Blood. 86:1276–1280.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Boshoff C and Weiss R: AIDS-related
malignancies. Nat Rev Cancer. 2:373–382. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Griffante G, Gugliesi F, Pasquero S,
Dell'Oste V, Biolatti M, Salinger AJ, Mondal S, Thompson PR,
Weerapana E, Lebbink RJ, et al: Human cytomegalovirus-induced host
protein citrullination is crucial for viral replication. Nat
Commun. 12:39102021. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Halenius A and Hengel H: Human
cytomegalovirus and autoimmune disease. BioMed Res Int.
2014:4729782014. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Herbein G: The Human Cytomegalovirus, from
Oncomodulation to Oncogenesis. Viruses. 10:4082018. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Sadler AJ and Williams BR:
Interferon-inducible antiviral effectors. Nat Rev Immunol.
8:559–568. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
zur Hausen H: Papillomaviruses and cancer:
From basic studies to clinical application. Nat Rev Cancer.
2:342–350. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
117
|
You J, Srinivasan V, Denis GV, Harrington
WJ Jr, Ballestas ME, Kaye KM and Howley PM: Kaposi's
sarcoma-associated herpesvirus latency-associated nuclear antigen
interacts with bromodomain protein Brd4 on host mitotic
chromosomes. J Virol. 80:8909–8919. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion–from mechanism to translation. Nat Med. 17:1391–1401.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Kalogeris T, Baines CP, Krenz M and
Korthuis RJ: Ischemia/Reperfusion. Compr Physiol. 7:113–170. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Diella F, Cameron S, Gemünd C, Linding R,
Via A, Kuster B, Sicheritz-Pontén T, Blom N and Gibson TJ: Phospho.
ELM: A database of experimentally verified phosphorylation sites in
eukaryotic proteins. BMC Bioinformatics. 5:792004. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Wang Z, Song Y, Ye M, Dai X, Zhu X and Wei
W: The diverse roles of SPOP in prostate cancer and kidney cancer.
Nat Rev Urol. 17:339–350. 2020. View Article : Google Scholar : PubMed/NCBI
|